CompositionPrincipes actifs
Acide obéticholique
Excipients
Cellulose microcristalline (E 460), glycolate d’amidon sodique (type A), stéarate de magnésium, alcool polyvinylique partiellement hydrolysé (E 1203), macrogol 3350 (E 1521), talc (E 553b), colorants dioxyde de titane (E 171) et oxyde de fer jaune (E 172).
Indications/Possibilités d’emploiOCALIVA, un agoniste du récepteur farnésoïde X (FXR), est indiqué dans le traitement de la cholangite biliaire primitive (CBP)
§sans cirrhose ou
§avec une cirrhose compensée sans signes d’hypertension portale
en association avec l’acide ursodésoxycholique (AUDC) chez les adultes présentant une réponse insuffisante à l’AUDC ou en monothérapie chez les adultes qui ne tolèrent pas l’AUDC.
Cette indication a été autorisée en raison de la baisse du taux de phosphatases alcalines (PAL).
Une amélioration du taux de survie ou des symptômes liés à la maladie n’a pas été établie. Le maintien de l’autorisation pour cette indication pourra dépendre du bénéfice clinique testé et décrit dans des études de validation.
Posologie/Mode d’emploiPosologie initiale et titration de la dose
Avant le début du traitement par OCALIVA, il convient de déterminer si le patient présente une cirrhose décompensée (y compris classe Child-Pugh B ou C) ou a présenté antérieurement un événement de type décompensation, s’il présente une cirrhose compensée avec des signes cliniques d’hypertension portale (par ex. ascite, varices œsophagiennes, thrombopénie chronique < 150 × 109/l), car OCALIVA est contre-indiqué chez ces patients (voir « Contre-indications » et « Mises en garde et précautions »).
Posologie recommandée
La posologie recommandée d’OCALIVA pour les patients atteints de CBP sans cirrhose ou avec une cirrhose compensée sans signes d’hypertension portale, qui n’ont pas obtenu une réponse biochimique adéquate avec un traitement par AUDC à une posologie adaptée pendant au moins 1 an ou qui n’ont pas toléré l’AUDC (voir « Efficacité clinique ») est précisée ci-après.
§La dose initiale recommandée d’acide obéticholique est de 5 mg une fois par jour pendant les 3 premiers mois.
§Après les 3 premiers mois, augmenter la dose à 10 mg maximum une fois par jour chez les patients qui n’ont pas obtenu une réduction adéquate des phosphatases alcalines (PAL) et/ou de bilirubine totale et qui tolèrent l’acide obéticholique.
Surveillance en vue d’évaluer la sécurité, nécessité de suspendre le traitement par OCALIVA
Les patients sous traitement par OCALIVA doivent subir une surveillance de routine en termes de réponse biochimique, tolérance et progression de la CBP. Une surveillance étroite s’impose chez les patients atteints d’une cirrhose compensée, une affection hépatique concomitante (par ex. hépatite auto-immune, maladie du foie liée à l’alcoolisme) et/ou une maladie intercurrente grave avec de nouveaux signes d’hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombocytopénie chronique < 150 × 109/l) ou des augmentations au-dessus de la limite supérieure normale de la bilirubine totale, de la bilirubine conjuguée (directe) ou du temps de prothrombine. L’OCALIVA doit définitivement être arrêté chez des patients qui développent des signes biologiques ou cliniques d’une décompensation hépatique, sont atteints d’une cirrhose compensée et développent des signes d’hypertension portale, présentent des effets indésirables hépatiques cliniquement significatifs ou développent une obstruction totale des voies biliaires (voir « Contre-indications » et « Mises en garde et précautions »).
Prise en charge de patients avec prurit non tolérable sous OCALIVA
Une ou plusieurs des solutions suivantes doivent être envisagées chez les patients présentant une intolérance en raison d’un prurit non tolérable sous OCALIVA :
§Ajout de résines chélatrices des acides biliaires ou d’antihistaminiques.
§Réduction de la dose d’OCALIVA à :
o5 mg un jour sur deux, chez les patients intolérants à 5 mg une fois par jour.
o5 mg une fois par jour, chez les patients intolérants à 10 mg une fois par jour.
§Interruption provisoire de la dose d’OCALIVA pendant 2 semaines maximum puis reprise du traitement à une dose réduite.
Pour les patients dont la posologie est réduite ou le traitement suspendu, titrer la dose en fonction de la réponse biochimique et de la tolérance (voir « Posologie recommandée »).
Une interruption du traitement par OCALIVA doit être envisagée pour les patients qui continuent de présenter un prurit persistant intolérable.
Résines chélatrices des acides biliaires
Les patients prenant des résines chélatrices des acides biliaires doivent prendre l’acide obéticholique au moins 4 à 6 heures avant ou 4 à 6 heures après l’administration d’une résine chélatrice des acides biliaires, ou en respectant un intervalle aussi long que possible (voir « Interactions »).
Instructions posologiques particulières
Patients âgés (≥ 65 ans)
Les données disponibles à ce jour chez les patients âgés sont limitées. Aucun ajustement posologique n’est nécessaire chez les patients âgés (voir « Pharmacocinétique »).
Patients présentant des troubles de la fonction rénale
Aucune adaptation de la posologie n’est nécessaire chez les patients atteints d’insuffisance rénale (analyse d’une dose unique, voir « Pharmacocinétique »). La sécurité d’OCALIVA en traitement d’entretien chez des patients insuffisants rénaux n’a pas été étudiée.
Patients présentant des troubles de la fonction hépatique
Des cas de décompensation ou d’insuffisance hépatique, parfois fatale ou imposant une greffe de foie, ont été rapportés lors du traitement par OCALIVA de patients atteints de CBP avec une cirrhose, compensée ou décompensée (voir « Mises en garde et précautions »). OCALIVA est contre-indiqué chez les patients ayant une cirrhose décompensée (par ex. classe Child-Pugh B ou C), ayant connu un événement antérieur de décompensation ou présentant une cirrhose compensée associée à une hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombopénie chronique < 150 × 109/l) (voir « Contre-indications » et « Mises en garde et précautions »).
Lors d’essais cliniques sur la CBP, une relation dose-réponse a été observée pour la survenue d’effets indésirables hépatiques avec OCALIVA (voir « Mises en garde et précautions »). L’exposition plasmatique à l’acide obéticholique et ses conjugués actifs augmente significativement chez les patients atteints d’insuffisance hépatique modérée à sévère (voir « Pharmacocinétique »).
Les patients devront être surveillés durant le traitement par OCALIVA afin de détecter la survenue d’éventuels effets indésirables hépatiques (voir « Mises en garde et précautions »). OCALIVA doit être définitivement arrêté chez les patients chez lesquels des effets indésirables hépatiques cliniquement significatifs sont survenus (voir ci-dessus « Surveillance en vue d’évaluer la sécurité, nécessité de suspendre le traitement par OCALIVA » ainsi que « Contre-indications » et « Mises en garde et précautions »).
Enfants et adolescents
Il n’existe pas d’utilisation justifiée de l’acide obéticholique chez les enfants et les adolescents dans l’indication de la cholangite biliaire primitive (CBP).
Mode d’administration
Le comprimé doit être pris par voie orale, pendant ou en dehors des repas.
Contre-indications§Cirrhose décompensée (par ex. classe Child-Pugh B ou C) ou un événement antérieur de décompensation (voir « Mises en garde et précautions »)
§Cirrhose compensée avec signes d’hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombopénie chronique < 150 × 109/l) (voir « Mises en garde et précautions »).
§Obstruction complète des voies biliaires.
§Hypersensibilité au principe actif ou à l’un des excipients cités sous « Composition ».
Mises en garde et précautionsDécompensation et insuffisance hépatiques chez des patients atteints de CBP avec une cirrhose
Des cas de décompensation ou d’insuffisance hépatiques, parfois fatale ou imposant une greffe de foie, ont été rapportés lors du traitement par OCALIVA de patients atteints de CBP avec une cirrhose, compensée ou décompensée. Les cas rapportés après la commercialisation indiquent une durée médiane de 4 mois pour les patients avec une cirrhose compensée ; la durée médiane avant un nouvel événement de décompensation (par ex. encéphalopathie hépatique) était de 2,5 mois chez les patients avec une cirrhose décompensée.
Certains de ces cas se sont présentés chez des patients atteints d’une cirrhose décompensée lorsqu’ils étaient traités avec des doses supérieures à celles recommandées pour ce groupe de patients ; toutefois, même chez des patients avec une cirrhose décompensée ayant reçu la posologie recommandée, des cas de décompensation et d’insuffisance hépatiques ont été rapportés.
Une hépatotoxicité a été observée lors des essais cliniques avec OCALIVA. Une relation dose-réponse a été observée quant à la survenue d’effets indésirables hépatiques, y compris ictère, aggravation d’ascite, exacerbation de cholangite biliaire primitive, avec des posologies d’OCALIVA de 10 mg une fois/jour à 50 mg une fois/jour (jusqu’à 5 fois la dose maximale recommandée), déjà dans le mois ayant suivi le début du traitement avec OCALIVA lors de deux essais cliniques de 3 mois contre placebo, avec des patients atteints de CBP à un stade principalement précoce (voir « Surdosage »). Dans une analyse regroupée de trois essais cliniques contrôlés par placebo avec des patients atteints de CBP à un stade principalement précoce, les taux d’incidence ajustés à l’exposition pour tous les effets indésirables hépatiques graves ou autrement significatifs sur le plan clinique ainsi que des élévations isolées lors de tests biochimiques de la fonction hépatique, pour 100 patients-années d’exposition (PAE) étaient : 5,2 dans le groupe d’OCALIVA à 10 mg (dose maximale recommandée), 19,8 dans le groupe d’OCALIVA à 25 mg (2,5 fois la dose maximale recommandée) et 54,5 dans le groupe d’OCALIVA à 50 mg (5 fois la dose maximale recommandée) par rapport à 2,4 dans le groupe placebo.
Des élévations de l’alanine aminotransférase (ALAT) et de l’aspartate aminotransférase (ASAT) ont été observées chez les patients prenant de l’acide obéticholique. Des signes et symptômes de décompensation hépatique ont également été observés. Ces effets se sont produits parfois dès le premier mois de traitement.
Après l’initiation du traitement, tous les patients doivent être régulièrement surveillés par rapport à la progression de la CBP, y compris aux effets indésirables hépatiques, à l'aide d'analyses de laboratoire ainsi que d'examens cliniques afin de déterminer si le traitement par acide obéticholique doit être interrompu (voir « Posologie/Mode d’emploi »)
Une surveillance étroite s’impose chez les patients avec une cirrhose compensée, une maladie hépatique concomitante (par ex. hépatite auto-immune, maladie du foie liée à l’alcool) et/ou une maladie intercurrente grave avec de nouveaux signes d’hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombopénie chronique < 150 × 109/l) ou des augmentations au-dessus de la limite supérieure normale de la bilirubine totale, de la bilirubine conjuguée (directe) ou du temps de prothrombine, pour déterminer si le traitement doit être suspendu (voir « Posologie/Mode d’emploi »).
Le traitement par OCALIVA doit être définitivement arrêté chez les patients qui :
§développent des signes biologiques ou cliniques d’une décompensation hépatique (par ex. ascite, ictère, hémorragies de varices, encéphalopathie hépatique, classe Child-Pugh B ou C) (voir « Contre-indications »).
§présentent une cirrhose compensée et développent des signes d’hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombopénie chronique < 150 × 109/l) (voir « Contre-indications »).
§subissent des effets indésirables hépatiques cliniquement significatifs
§développent une obstruction totale des voies biliaires (voir « Contre-indications »).
En cas de maladie intercurrente grave, le traitement par acide obéticholique doit être interrompu et la fonction hépatique du patient surveillée. Avant de reprendre le traitement par acide obéticholique après guérison de la maladie intercurrente, il faut en évaluer les risques et bénéfices potentiels.
Prurit sévère
Dans l’étude de phase III sur OCALIVA (POISE), un prurit sévère a été signalé chez 23% des patients du bras OCALIVA 10 mg, chez 19% des patients du bras OCALIVA avec augmentation de dose et chez 7% des patients du bras placebo. Le délai médian d’apparition du prurit sévère était de respectivement 11, 158 et 75 jours pour les patients des bras OCALIVA 10 mg, OCALIVA avec augmentation de dose et placebo. Les stratégies de prise en charge incluent l’ajout de résines chélatrices des acides biliaires ou d’antihistaminiques, la réduction de la posologie, la réduction de la fréquence d’administration et/ou l’interruption temporaire du traitement (voir « Posologie/Mode d’emploi » et « Effets indésirables »).
InteractionsEffets d’autres médicaments sur l’acide obéticholique
Résines chélatrices des acides biliaires
Les résines chélatrices des acides biliaires, telles que la cholestyramine, le colestipol ou le colésévélam, absorbent et réduisent l’absorption des acides biliaires et peuvent réduire l’efficacité de l’acide obéticholique. En cas d’association avec des résines chélatrices des acides biliaires, l’acide obéticholique doit être pris au moins 4 à 6 heures avant ou 4 à 6 heures après l’administration des résines chélatrices des acides biliaires, ou en respectant un intervalle aussi long que possible.
Effets de l’acide obéticholique sur d’autres médicaments
Substrats du CYP1A2 à indice thérapeutique étroit
L’acide obéticholique peut augmenter l’exposition aux médicaments concomitants substrats du CYP1A2. Une surveillance clinique est recommandée lors de l’administration de médicaments substrats du CYP1A2 à indice thérapeutique étroit (par ex. : théophylline et tizanidine).
Grossesse, allaitementGrossesse
Il n’existe à ce jour aucune donnée sur l’utilisation de l’acide obéticholique chez la femme enceinte. Les études chez l’animal n’ont pas montré d’effets délétères directs sur la reproduction (voir « Données précliniques »). Par mesure de précaution, l’acide obéticholique ne doit pas être utilisé pendant la grossesse, à moins que l’état clinique de la femme enceinte ne nécessite un traitement.
Allaitement
On ne sait pas si l’acide obéticholique est excrété dans le lait maternel. Sur la base des études effectuées chez l’animal et des données pharmacologiques, l’acide obéticholique ne devrait pas interférer avec l’allaitement ni avec la croissance ou le développement d’un enfant allaité. Une décision doit être donc prise entre l’arrêt de l’allaitement ou le renoncement au traitement par l’acide obéticholique/l’interruption du traitement par l’acide obéticholique. Ce faisant, le bénéfice de l’allaitement pour l’enfant doit être considéré au regard du bénéfice du traitement pour la mère (voir « Données précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL’acide obéticholique n’a aucune influence ou une influence négligeable sur l’aptitude à la conduite ou l’utilisation de machines.
Effets indésirablesAu total, 432 patients souffrant de CBP ont été évalués dans trois études en double aveugle contrôlées contre placebo. Parmi eux, 290 ont été traités pendant au moins 6 mois par OCALIVA, 232 ont été traités pendant au moins 12 mois et 70 pendant au moins 2 ans. 131 patients prenaient OCALIVA 10 mg une fois par jour et 70 patients OCALIVA 5 mg une fois par jour.
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés étaient le prurit (63%) et la fatigue (22%). Le taux d’interruption de traitement était au total de 12% dans le bras OCALIVA 10 mg, de 10% dans le bras OCALIVA avec augmentation de dose, et de 4% dans le bras placebo de l’étude de phase III POISE sur OCALIVA. L’effet indésirable le plus fréquent ayant entraîné l’interruption du traitement était le prurit. La plupart des cas de prurit se sont produits durant le premier mois du traitement et avaient tendance à disparaître lors de la poursuite du traitement.
Liste des effets indésirables
Les effets indésirables rapportés avec OCALIVA au cours de l’étude clinique de phase III sont énumérés dans le tableau ci-dessous par classe de systèmes d’organes MedDRA et par fréquence. Les fréquences sont définies comme suit : très fréquents (≥ 1/10), fréquents (≥ 1/100, < 1/10), occasionnels (≥ 1/1000, < 1/100), rares (≥ 1/10 000, < 1/1000), très rares (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque catégorie de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 1 : Fréquence des effets indésirables chez les patients atteints de CBP
|
Classe de systèmes d’organes
|
Très fréquents
|
Fréquents
|
Fréquence inconnue
| |
Affections endocriniennes
|
|
Troubles de la fonction thyroïdienne*
|
| |
Affections du système nerveux
|
|
Vertiges*
|
| |
Affections cardiaques
|
|
Palpitations*
|
| |
Affections respiratoires, thoraciques et médiastinales
|
|
Douleur oropharyngée*
|
| |
Affections gastro-intestinales
|
Douleurs et gêne abdominales* (14%)
|
Constipation*
|
| |
Affections hépatiques et biliaires
|
|
|
Insuffisance hépatique#, augmentation du taux de bilirubine#, ictère#, cirrhose#
| |
Affections de la peau et du tissu sous-cutané
|
Prurit* (63%)
|
Eczéma*, rash cutané*
|
| |
Affections musculo-squelettiques et systémiques
|
|
Douleur articulaire*
|
| |
Troubles généraux et anomalies au site d’administration
|
Fatigue* (22%)
|
Œdème périphérique*, fièvre*
|
|
* Les effets indésirables sont définis comme des événements survenant à une fréquence de ≥ 5% dans le groupe de traitement par acide obéticholique et selon une fréquence de ≥ 1% par rapport au groupe de traitement par placebo.
# Relevés après la commercialisation, voir également ci-après sous « Effets indésirables après la commercialisation ».
Description de certains effets indésirables et informations complémentaires
Prurit
Environ 60% des patients présentaient des antécédents de prurit à l’inclusion dans l’étude de phase III. Le prurit lié au traitement est généralement apparu durant le premier mois suivant l’instauration du traitement.
Comparés aux patients ayant commencé par une dose de 10 mg d’OCALIVA une fois par jour, les patients inclus dans le bras OCALIVA avec augmentation de dose ont présenté une plus faible incidence de prurit (70% et 56% respectivement) et un taux inférieur d’interruption de traitement liée au prurit (10% et 1% respectivement).
Le nombre de patients avec un prurit nécessitant une intervention (c’est-à-dire un ajustement posologique, une interruption de traitement ou l’instauration d’antihistaminiques ou de résines chélatrices des acides biliaires) était de 30 sur 51 patients (59%) dans le bras OCALIVA 10 mg, de 24 sur 39 (62%) dans le bras OCALIVA avec augmentation de dose et de 14 sur 28 (50%) dans le bras placebo.
Effets indésirables après la commercialisation
Affections hépatiques et des voies biliaires occasionnellement avec issue fatale, augmentation du taux de bilirubine, ictère, cirrhose (voir « Mises en garde et précautions »).
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLors des essais cliniques, les patients atteints de CBP ayant reçu OCALIVA à raison de 25 mg une fois par jour (2,5 fois la dose maximale recommandée) ou 50 mg une fois par jour (5 fois la dose maximale recommandée), ont connu une augmentation dose-dépendante de l’incidence d’effets indésirables hépatiques, tels que des valeurs hépatiques élevées aux tests biochimiques, une ascite, un ictère, une hypertension portale et une poussée de cholangite biliaire primitive (voir « Mises en garde et précautions »).
En cas de surdosage, les patients doivent être placés sous observation étroite et un traitement de soutien doit être administré si nécessaire.
Propriétés/EffetsCode ATC
A05AA04
Classe pharmacothérapeutique : acides biliaires et dérivés
Mécanisme d’action
L’acide obéticholique est un agoniste sélectif et puissant du récepteur farnésoïde X (FXR), un récepteur nucléaire exprimé à des taux élevés dans le foie et l’intestin. Le récepteur FXR est considéré comme un régulateur clé des acides biliaires et des voies inflammatoires, fibrotiques et métaboliques. L’activation du FXR fait baisser la concentration intracellulaire d’acides biliaires à l’intérieur des hépatocytes en inhibant la synthèse de novo à partir du cholestérol et en augmentant le transport des acides biliaires en dehors des hépatocytes. Ces mécanismes limitent la quantité globale d’acides biliaires circulants, tout en stimulant la cholérèse, ce qui réduit l’exposition hépatique aux acides biliaires.
Pharmacodynamique
Titration de la dose
Dans une étude de phase III d’une durée de 12 mois, randomisée, en double aveugle, contrôlée contre placebo et avec groupes parallèles (étude POISE), le taux de PAL de la plupart des patients traités par OCALIVA 5 mg une fois par jour s’est stabilisé après environ 3 mois. Une augmentation de la dose d’OCALIVA à 10 mg une fois par jour en fonction de la tolérance et de la réponse a conduit chez la majorité des patients à une baisse supplémentaire du taux de PAL (voir « Posologie/Mode d’emploi »).
Marqueurs pharmacodynamiques
Dans l’étude POISE, l’administration d’OCALIVA 10 mg une fois par jour était associée à une hausse de 173% des concentrations de FGF19, une entérokine induite par le FXR qui participe à l’homéostasie des acides biliaires, entre le début du traitement et le mois 12. Les concentrations d’acide cholique et d’acide chénodésoxycholique ont baissé (1,4 μM et 2,7 μM, respectivement) entre l’initiation du traitement et le mois 12. La signification clinique de ces résultats est incertaine.
Électrophysiologie cardiaque
À une dose correspondant à 10 fois la dose maximale recommandée, OCALIVA n’a pas rallongé l’intervalle QT dans une mesure cliniquement significative.
Efficacité clinique
Une étude de phase III d’une durée de 12 mois, randomisée, en double aveugle, contrôlée par placebo et avec groupes parallèles (étude POISE) a évalué la sécurité d’emploi et l’efficacité d’OCALIVA chez 216 patients atteints de CBP qui ont pris de l’AUDC pendant au moins 12 mois (dose stable depuis ≥ 3 mois avant l’inclusion dans l’étude) ou qui ne toléraient pas l’AUDC et qui n’en avaient pas reçu dans les ≥ 3 mois avant l’inclusion dans l’étude. Les patients étaient inclus dans l’étude si le taux de phosphatases alcalines (PAL) était ≥ 1,67 × la limite supérieure de la normale (LSN) et/ou si le taux de bilirubine totale était > 1 × la LSN mais < 2 × la LSN.
Les patients ont été randomisés (1:1:1) pour recevoir une fois par jour le placebo, OCALIVA 10 mg ou un schéma de titration de dose d’OCALIVA (5 mg augmenté à 10 mg à 6 mois selon la réponse thérapeutique/la tolérance). La majorité des patients (93%) ont reçu un traitement en association avec l’AUDC et un petit nombre de patients (7%) qui ne toléraient pas l’AUDC ont reçu le placebo, OCALIVA (10 mg) ou un schéma d´augmentation de dose d’OCALIVA (5 mg augmenté à 10 mg) en monothérapie.
Les taux de phosphatases alcalines (PAL) et de bilirubine totale ont été évalués en tant que variables nominales du critère d’évaluation composite principal, ainsi qu’en tant que variables continues dans le temps.
La population de l’étude était essentiellement composée de femmes (91%) et de patients d’origine caucasienne (94%). L’âge moyen était de 56 ans et la majorité des patients étaient âgés de moins de 65 ans. Les taux moyens de PAL en situation initiale variaient de 316 U/l à 327 U/l. Les taux moyens de bilirubine totale en situation initiale variaient de 10 μmol/l à 12 μmol/l pour les différents groupes de traitement, 92% des patients présentant des valeurs dans les limites normales.
Le traitement par OCALIVA 10 mg ou par le schéma de titration d’OCALIVA (5 mg à 10 mg) a entraîné des augmentations cliniquement et statistiquement significatives (p < 0,0001), par rapport au placebo, du nombre de patients atteignant le critère d’évaluation composite principal à toutes les échéances de l’étude (voir Tableau 2). Ces réponses ont été observées en partie dès 2 semaines de traitement et étaient dépendantes de la dose (OCALIVA 5 mg vs 10 mg à 6 mois, p = 0,0358).
Tableau 2 : Pourcentage des patients atteints de CBP atteignant le critère d’évaluation composite principal au mois 6 et au mois 12 (avec ou sans AUDC)b
|
|
OCALIVA
10 mgc
(N = 73)
|
OCALIVA
Titrationc
(N = 70)
|
Placebo
(N = 73)
| |
Mois 6
|
|
|
| |
Répondeur, n (%)
IC à 95% correspondant
|
37 (51)
39%, 63%
|
24 (34)
23%, 47%
|
5 (7)
2%, 15%
| |
Valeur pd
|
< 0,0001
|
< 0,0001
|
S.O.
| |
Mois 12
|
|
|
| |
Répondeur, n (%)
IC à 95% correspondant
|
35 (48)
36%, 60%
|
32 (46)
34%, 58%
|
7 (10)
4%, 19%
| |
Valeur p d
|
< 0,0001
|
< 0,0001
|
S.O.
| |
Composants du critère d’évaluation principale
| |
Taux de PAL < 1,67 × LSN, n (%)
|
40 (55)
|
33 (47)
|
12 (16)
| |
Réduction du taux de PAL d’au moins 15%, n (%)
|
57 (78)
|
54 (77)
|
21 (29)
| |
Bilirubine totale de ≤ 1 × LSN, n (%)
|
60 (82)
|
62 (89)
|
57 (78)
|
a Pourcentage des patients atteignant une réponse, définie comme un taux de PAL inférieur à 1,67 fois la LSN, un taux de bilirubine totale dans les limites normales et une réduction du taux de PAL d’au moins 15%. Les valeurs manquantes étaient considérées comme des non-réponses. Le test exact de Fisher a été utilisé pour calculer les intervalles de confiance (IC) à 95%.
b Dans cette étude, 16 patients (7%) étaient intolérants et n’ont pas reçu l’AUDC en médicament concomitant : 6 patients (8%) dans le bras OCALIVA 10 mg, 5 patients (7%) dans le bras OCALIVA avec augmentation de dose et 5 patients (7%) dans le groupe placebo.
c Les patients ont été randomisés (1:1:1) pour recevoir OCALIVA 10 mg une fois par jour pendant les 12 mois complets de l’étude, ou le schéma de titration d’OCALIVA (5 mg une fois par jour pendant les 6 premiers mois, la dose pouvant être augmentée à 10 mg une fois par jour pendant les 6 derniers mois, si le patient tolérait OCALIVA, mais présentait un taux de PAL au moins 1,67 fois supérieur à la LSN, et/ou un taux de bilirubine totale supérieure à la LSN, ou une réduction du taux de PAL inférieure à 15%) ou le placebo.
d Titration d’OCALIVA et OCALIVA 10 mg par rapport au placebo. Les valeurs de p ont été obtenues à l’aide du test Cochran-Mantel-Haenszel d’Association Générale, stratifié selon l’intolérance à l’AUDC et les taux de PAL prétraitements supérieurs à 3 fois la LSN et/ou l’ASAT supérieur à 2 fois la LSN et/ou un taux de bilirubine totale supérieur à la LSN.
e Les taux de réponse ont été calculés sur la base de l’analyse des cas observés (c’est-à-dire [n= répondeur observé]/[N=population en intention de traiter (ITT)]); le pourcentage de patients présentant des valeurs au mois 12 est de 86%, 91% et 96% pour le bras OCALIVA 10 mg, le bras OCALIVA avec augmentation de dose et le bras placebo, respectivement.
f Les taux moyens de bilirubine totale en situation initiale variaient de 10 μmol/l à 12 μmol/l pour les différents bras de traitement, 92% des patients inclus présentant des valeurs dans les limites normales (c’est-à-dire ≤ LSN).
Réduction moyenne des taux de PAL
La figure 1 montre la réduction moyenne du taux de PAL chez les patients traités par OCALIVA par rapport au placebo. Une réduction a été observée dès la semaine 2. Elle s’est stabilisée au mois 3 et s’est maintenue jusqu’au mois 12 chez les patients qui avaient reçu la même dose sur les 12 mois complets. Bien qu’une titration ait été évaluée à 6 mois dans l’étude POISE, les données parlent en faveur d’une titration d’OCALIVA dès 3 mois. Une réduction supplémentaire du taux de PAL a été observée au mois 12 chez une majorité des patients du bras de titration d’OCALIVA, dont la dose d’OCALIVA était passée de 5 mg une fois par jour à 10 mg une fois par jour (voir « Pharmacocinétique »).
Figure 1 : Taux moyen de PAL sur 12 mois dans l’étude POISE, par bras de traitement, avec ou sans AUDCa
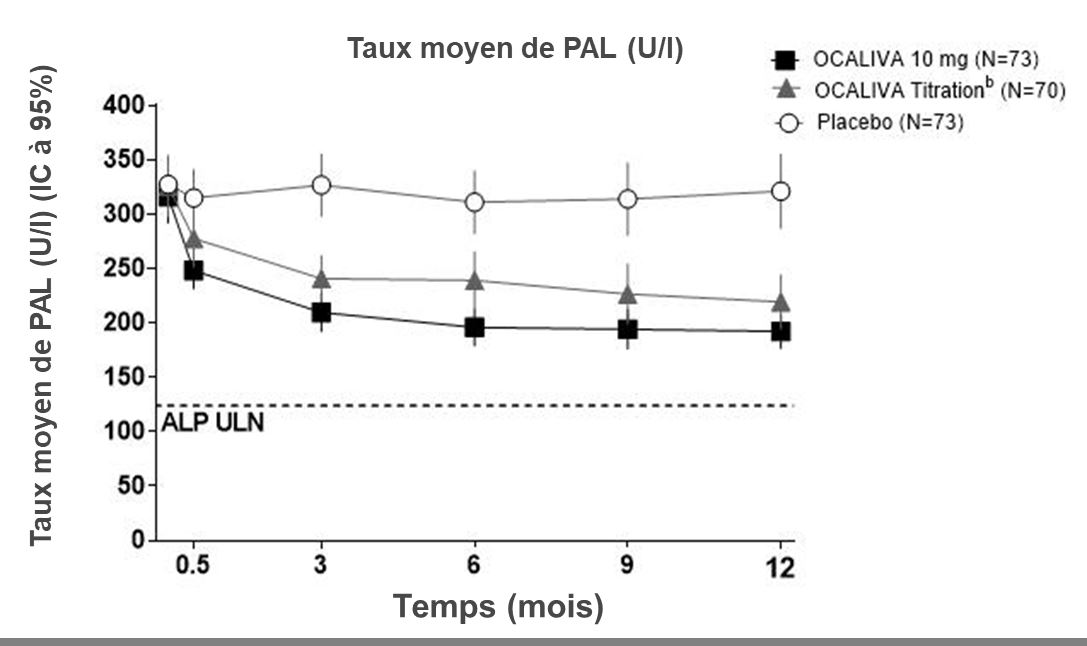
a Dans cette étude, 16 patients (7%) n’ont pas reçu l’AUDC en médicament concomitant en raison d’une intolérance : 6 patients (8%) dans le bras OCALIVA 10 mg, 5 patients (7%) dans le bras OCALIVA avec augmentation de dose et 5 patients (7%) dans le bras placebo.
b Les patients répartis aléatoirement dans le bras OCALIVA avec augmentation de dose ont reçu 5 mg d’OCALIVA une fois par jour pendant les 6 premiers mois. À partir du mois 6, les patients qui toléraient bien OCALIVA mais qui présentaient un taux de PAL au moins 1,67 fois supérieur à la limite supérieure de la normale (LSN) et/ou un taux de bilirubine totale supérieur à la LSN ou une diminution du taux de PAL de moins de 15% étaient jugés admissibles pour passer d’une dose de 5 mg une fois par jour à une dose de 10 mg une fois par jour pendant les 6 derniers mois de l’étude.
Réduction moyenne des taux de GGT
La réduction moyenne (IC à 95%) des taux de gamma-glutamyl transférase (GGT) était de 178 (137, 219) U/l dans le bras OCALIVA 10 mg, de 138 (102, 174) U/l dans le bras OCALIVA avec augmentation de dose et de 8 (-32, 48) U/l dans le bras placebo.
OCALIVA en monothérapie
La réponse biochimique à OCALIVA administré en monothérapie a été évaluée chez cinquante-et-un patients atteints de CBP présentant un taux de PAL en situation initiale 1,67 fois supérieur à la LSN ou plus, et/ou un taux de bilirubine totale supérieure à la LSN, (24 patients ont reçu OCALIVA 10 mg une fois par jour et 27 patients ont reçu un placebo) dans le cadre d’une méta-analyse des données émanant d’une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, d’une durée de 12 mois (étude POISE), ainsi que d’une étude clinique randomisée, en double aveugle, contrôlée contre placebo, d’une durée de 3 mois. Au mois 3, 9 (38%) des patients traités par OCALIVA ont obtenu une réponse en termes de critère d’évaluation composite, par rapport à 1 patient (4%) dans le groupe placebo. La réduction moyenne (IC à 95%) des taux de PAL chez les patients traités par OCALIVA était de 246 (165, 327) U/l, par rapport à une augmentation de 17 (−7, 42) U/l chez les patients traités par placebo.
PharmacocinétiqueAbsorption
L’acide obéticholique est absorbé avec des concentrations plasmatiques maximales (Cmax) atteintes après un délai médian (tmax) d’environ 1,5 heure. L’administration concomitante d’aliments ne modifie pas le niveau d’absorption de l’acide obéticholique.
Distribution
Chez l’homme, le taux de liaison aux protéines plasmatiques de l’acide obéticholique et de ses conjugués est supérieur à 99%. Le volume de distribution de l’acide obéticholique est de 618 L. Le volume de distribution des conjugués taurine et glycine de l’acide obéticholique n’a pas encore été déterminé.
Métabolisme
L’acide obéticholique est conjugué à la glycine ou à la taurine au niveau hépatique, puis est sécrété dans la bile. Ces conjugués glycine et taurine de l’acide obéticholique sont absorbés dans l’intestin grêle, entraînant ainsi une recirculation entérohépatique. Les conjugués peuvent être déconjugués dans l’iléon et le côlon par le microbiote intestinal, conduisant à la conversion en acide obéticholique, qui est susceptible d’être réabsorbé ou excrété dans les fèces, la voie fécale constituant la principale voie d’élimination.
Après administration quotidienne d’acide obéticholique, on observe une accumulation de ses conjugués glycine et taurine, dont l’activité pharmacologique in vitro est équivalente à celle de la molécule mère. Les rapports métabolites-molécule mère des conjugués glycine et taurine de l’acide obéticholique étaient respectivement de 13,8 et 12,3 après l’administration quotidienne. Un troisième métabolite supplémentaire de l’acide obéticholique, le 3-glucuronide, se forme, mais son activité pharmacologique est considérée comme minimale.
Élimination
Après administration d’acide obéticholique radiomarqué, le composé est excrété à plus de 87% par voie fécale. L’excrétion urinaire est inférieure à 3%.
Proportionnalité dose/temps
À la suite de l’administration de doses répétées de 5, 10 ou 25 mg une fois par jour pendant 14 jours, l’exposition systémique à l’acide obéticholique a augmenté proportionnellement à la dose. Les expositions aux conjugués glycine et taurine de l’acide obéticholique et à l’acide obéticholique total ont augmenté selon un rapport plus que proportionnel à la dose. L’état d’équilibre (Steady State) atteint le jour 14 (AUC0-24h) de l’acide obéticholique totale était respectivement de 4,2, 6,6 ou 7,8 fois de l’exposition systématique (AUC0-24h) observée le jour 1 après une administration unique de 5, 10 ou 25 mg.
Cinétique pour certains groupes de patients
Poids corporel
En se basant sur la pharmacocinétique par population, le poids corporel s’est révélé un prédicateur significatif pour la pharmacocinétique de l’acide obéticholique, avec l’expectation du moindre exposition à l’acide obéticholique dans le plasma avec un poids corporel plus élevé. Il paraît probable que l’effet du poids corporel sur l’efficacité est négligeable.
Patients âgés
Il existe uniquement des données pharmacocinétiques limitées chez les patients âgés (≥ 65 ans). L’analyse de la pharmacocinétique de population, réalisée sur la base de données provenant de patients âgés de 65 ans maximum, indique que l’âge ne devrait pas influencer considérablement l’élimination de l’acide obéticholique de la circulation.
Enfants et adolescents
Aucune étude de pharmacocinétique n’a été effectuée avec l’acide obéticholique chez les patients âgés de moins de 18 ans.
Sexe
L’analyse de la pharmacocinétique de population a révélé que le sexe n’influence pas les paramètres pharmacocinétiques de l’acide obéticholique.
Origine ethnique
L’analyse de la pharmacocinétique de population a révélé que l’origine ethnique ne devrait pas influencer les paramètres pharmacocinétiques de l’acide obéticholique.
Troubles de la fonction rénale
Une étude pharmacocinétique spécifique avec une dose unique de 25 mg d’acide obéticholique a révélé une augmentation de l’exposition plasmatique à l’acide obéticholique et ces conjugués chez des sujets avec une insuffisance rénale légère (Modification of Diet in Renal Disease [MDRD] eGFR ≥ 60 et < 90 ml/min/1,73 m2), modérée (MDRD eGFR ≥ 30 et < 60 ml/min/1,73 m2) et sévère (MDRD eGFR ≥ 15 et < 30 ml/min/1,73 m2) d’environ 1,4 à 1,6 fois par rapport à des sujets avec une fonction rénale normale. La sécurité d’Ocaliva en traitement d’entretien chez des patients insuffisants rénaux n’a pas été étudiée.
Troubles de la fonction hépatique
L’acide obéticholique est métabolisé par le foie et les intestins. L’exposition systémique à l’acide obéticholique, à ses conjugués actifs et aux acides biliaires endogènes augmente chez les patients présentant une insuffisance hépatique modérée à sévère, par rapport aux témoins sains. Le traitement par l’acide obéticholique est contre-indiqué chez des patients ayant une cirrhose décompensée (par ex. classe Child-Pugh B ou C), un événement antérieur de décompensation ou présentant une cirrhose compensée associée à une hypertension portale (par ex. ascite, varices gastro-œsophagiennes, thrombopénie chronique < 150 × 109/l) (voir « Contre-indications » et « Mises en garde et précautions »).
L’effet d’une insuffisance hépatique légère (classe A de Child-Pugh) sur la pharmacocinétique de l’acide obéticholique était négligeable.
Chez les sujets atteints d’insuffisance hépatique légère, modérée et sévère (classe A, B et C de Child-Pugh, respectivement), l’ASC moyenne (aire sous la courbe) de l’acide obéticholique total, la somme de l’acide obéticholique et de ses deux conjugués actifs, a augmenté d’un facteur de respectivement 1,13, 4 et 17 par rapport aux sujets présentant une fonction hépatique normale, à la suite de l’administration d’une dose unique de 10 mg d’acide obéticholique.
Données précliniquesL’administration orale d’acide obéticholique à une posologie supérieure à la dose sans effet nocif observé (NOAEL) à des souris, des rats et des chiens dans le cadre d’études pivotales de toxicologie en administration répétée a principalement induit des effets sur le système hépatobiliaire. Il s’agissait notamment d’une augmentation du poids du foie, de modifications des paramètres sériques (ALAT, ASAT, LDH, PAL, GGT et/ou bilirubine) et de modifications macroscopiques/microscopiques. Toutes ces modifications ont été réversibles après l’interruption du traitement, ont été cohérentes et ont fourni des informations sur la prédiction de la toxicité limitant la dose chez l’être humain.
Le potentiel cancérogène de l’acide obéticholique a été évalué dans des études de carcinogénicité d’une durée maximale de 2 ans menées chez la souris et le rat. Chez la souris, aucun effet néoplasique lié au médicament n’a été constaté aux doses maximales de 25 mg/kg/jour d’acide obéticholique. Ces doses produisaient chez la souris des expositions systémiques d’environ 12 fois l’exposition humaine, à la dose maximale recommandée chez l’homme de 10 mg. Chez le rat, l’acide obéticholique a été administré aux doses de 2, 7 et 20 mg/kg/jour. À la dose de 20 mg/kg/jour (environ 12 fois l’exposition humaine à la DMRH), l’acide obéticholique a entraîné une augmentation de l’incidence des tumeurs bénignes des cellules de la granulosa des ovaires et des tumeurs bénignes des cellules de la granulosa du vagin et du col de l’utérus chez les rates. Aucun effet néoplasique lié au médicament n’a été constaté chez les rats mâles.
L’acide obéticholique n’était pas génotoxique dans le test d’Ames, un test d’aberration chromosomique sur les lymphocytes du sang périphérique humain, et un test du micronoyau effectué chez la souris. De même, le conjugué de glycine de l’acide obéticholique n’était pas génotoxique dans le test d’Ames ni dans le test d’aberration chromosomique sur les lymphocytes du sang périphérique humain. Le conjugué de taurine de l’acide obéticholique n’était pas génotoxique dans le test d’Ames et était négatif dans un test d’aberration chromosomique sur les lymphocytes du sang périphérique humain en présence d’une activation métabolique ; les résultats du test d’aberration chromosomique en l’absence d’activation métabolique n’étaient pas concluants.
L’acide obéticholique a été administré à des doses orales de 5, 25 et 50 mg/kg/jour à des rats mâles pendant 28 jours avant l’accouplement et pendant la période d’accouplement, et à des rates à partir de 14 jours avant l’accouplement, pendant la période d’accouplement et jusqu’au jour 7 de la gestation. Quelle que soit la dose (la dose de 50 mg/kg/jour équivaut à environ 13 fois l’exposition humaine à la DMRH), ni la fertilité des mâles ou des femelles ni le développement embryonnaire précoce n’a été altéré.
Au cours d’une étude sur le développement embryonnaire chez le rat, de l’acide obéticholique a été administré oralement lors de l’organogenèse à des doses de 5, 25 et 75 mg/kg/jour. À 25 mg/kg/jour (une dose correspondant à une exposition systémique d’env. 13 fois celle chez l’homme à une MRHD de 10 mg), aucune toxicité maternelle ou sur le développement n’a été notée. À 75 mg/kg/jour (environ 40 fois l’exposition humaine avec la MRHD), le poids des embryons a été réduit et un nombre plus important de résorptions précoces ou tardives et d’embryons non viables a été noté. Chez les mères, une mortalité ainsi qu’une perte d’embryons, une réduction du poids corporel, une réduction de la prise de nourriture et une réduction de la prise de poids ont été observées à 75 mg/kg/jour. C’est pourquoi la toxicité sur le développement observée à cette dose pourrait être une manifestation secondaire de la toxicité maternelle. Chez le lapin, de l’acide obéticholique a été administré oralement pendant l’organogenèse à des doses de 3, 9 et 20 mg/kg/jour. À des doses ≤ 20 mg/kg/jour (environ 6 fois l’exposition humaine avec la MRHD), cette molécule n’a pas été tératogène et aucune indication de lésion chez les fœtus n’a été mise en évidence.
Au cours d’une étude sur le développement pré- et post-natal chez le rat, l’acide obéticholique a été administré à des doses de 5, 25 et 40 mg/kg/jour, de l’organogenèse à la lactation. Aucune dose n’a entraîné des conséquences sur la gestation, la mise-bas ou le développement post-natal (la dose de 40 mg/kg/jour correspond environ à 21 fois l’exposition humaine avec la MRHD). Lors de cette étude, la forme de l’acide obéticholique conjuguée à la taurine a été détectée chez les jeunes animaux allaités par une mère traitée par acide obéticholique.
Remarques particulièresStabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention « EXP » sur l’emballage.
Remarques particulières concernant le stockage
Tenir hors de portée des enfants.
Conserver dans l’emballage à température ambiante (15–25 °C).
Numéro d’autorisation66530 (Swissmedic)
PrésentationOCALIVA 5 mg : 30 comprimés pelliculés en flacon en PEHD avec fermeture de sécurité enfant (B)
OCALIVA 10 mg : 30 comprimés pelliculés en flacon en PEHD avec fermeture de sécurité enfant (B)
Titulaire de l’autorisationAdvanz Pharma Specialty Medicine Switzerland GmbH, Zurich
Mise à jour de l’informationMars 2022
|