Propriétés/EffetsCode ATC
A05AA04
Classe pharmacothérapeutique : acides biliaires et dérivés
Mécanisme d’action
L’acide obéticholique est un agoniste sélectif et puissant du récepteur farnésoïde X (FXR), un récepteur nucléaire exprimé à des taux élevés dans le foie et l’intestin. Le récepteur FXR est considéré comme un régulateur clé des acides biliaires et des voies inflammatoires, fibrotiques et métaboliques. L’activation du FXR fait baisser la concentration intracellulaire d’acides biliaires à l’intérieur des hépatocytes en inhibant la synthèse de novo à partir du cholestérol et en augmentant le transport des acides biliaires en dehors des hépatocytes. Ces mécanismes limitent la quantité globale d’acides biliaires circulants, tout en stimulant la cholérèse, ce qui réduit l’exposition hépatique aux acides biliaires.
Pharmacodynamique
Titration de la dose
Dans une étude de phase III d’une durée de 12 mois, randomisée, en double aveugle, contrôlée contre placebo et avec groupes parallèles (étude POISE), le taux de PAL de la plupart des patients traités par OCALIVA 5 mg une fois par jour s’est stabilisé après environ 3 mois. Une augmentation de la dose d’OCALIVA à 10 mg une fois par jour en fonction de la tolérance et de la réponse a conduit chez la majorité des patients à une baisse supplémentaire du taux de PAL (voir « Posologie/Mode d’emploi »).
Marqueurs pharmacodynamiques
Dans l’étude POISE, l’administration d’OCALIVA 10 mg une fois par jour était associée à une hausse de 173% des concentrations de FGF19, une entérokine induite par le FXR qui participe à l’homéostasie des acides biliaires, entre le début du traitement et le mois 12. Les concentrations d’acide cholique et d’acide chénodésoxycholique ont baissé (1,4 μM et 2,7 μM, respectivement) entre l’initiation du traitement et le mois 12. La signification clinique de ces résultats est incertaine.
Électrophysiologie cardiaque
À une dose correspondant à 10 fois la dose maximale recommandée, OCALIVA n’a pas rallongé l’intervalle QT dans une mesure cliniquement significative.
Efficacité clinique
Une étude de phase III d’une durée de 12 mois, randomisée, en double aveugle, contrôlée par placebo et avec groupes parallèles (étude POISE) a évalué la sécurité d’emploi et l’efficacité d’OCALIVA chez 216 patients atteints de CBP qui ont pris de l’AUDC pendant au moins 12 mois (dose stable depuis ≥ 3 mois avant l’inclusion dans l’étude) ou qui ne toléraient pas l’AUDC et qui n’en avaient pas reçu dans les ≥ 3 mois avant l’inclusion dans l’étude. Les patients étaient inclus dans l’étude si le taux de phosphatases alcalines (PAL) était ≥ 1,67 × la limite supérieure de la normale (LSN) et/ou si le taux de bilirubine totale était > 1 × la LSN mais < 2 × la LSN.
Les patients ont été randomisés (1:1:1) pour recevoir une fois par jour le placebo, OCALIVA 10 mg ou un schéma de titration de dose d’OCALIVA (5 mg augmenté à 10 mg à 6 mois selon la réponse thérapeutique/la tolérance). La majorité des patients (93%) ont reçu un traitement en association avec l’AUDC et un petit nombre de patients (7%) qui ne toléraient pas l’AUDC ont reçu le placebo, OCALIVA (10 mg) ou un schéma d´augmentation de dose d’OCALIVA (5 mg augmenté à 10 mg) en monothérapie.
Les taux de phosphatases alcalines (PAL) et de bilirubine totale ont été évalués en tant que variables nominales du critère d’évaluation composite principal, ainsi qu’en tant que variables continues dans le temps.
La population de l’étude était essentiellement composée de femmes (91%) et de patients d’origine caucasienne (94%). L’âge moyen était de 56 ans et la majorité des patients étaient âgés de moins de 65 ans. Les taux moyens de PAL en situation initiale variaient de 316 U/l à 327 U/l. Les taux moyens de bilirubine totale en situation initiale variaient de 10 μmol/l à 12 μmol/l pour les différents groupes de traitement, 92% des patients présentant des valeurs dans les limites normales.
Le traitement par OCALIVA 10 mg ou par le schéma de titration d’OCALIVA (5 mg à 10 mg) a entraîné des augmentations cliniquement et statistiquement significatives (p < 0,0001), par rapport au placebo, du nombre de patients atteignant le critère d’évaluation composite principal à toutes les échéances de l’étude (voir Tableau 2). Ces réponses ont été observées en partie dès 2 semaines de traitement et étaient dépendantes de la dose (OCALIVA 5 mg vs 10 mg à 6 mois, p = 0,0358).
Tableau 2 : Pourcentage des patients atteints de CBP atteignant le critère d’évaluation composite principal au mois 6 et au mois 12 (avec ou sans AUDC)b
|
|
OCALIVA
10 mgc
(N = 73)
|
OCALIVA
Titrationc
(N = 70)
|
Placebo
(N = 73)
| |
Mois 6
|
|
|
| |
Répondeur, n (%)
IC à 95% correspondant
|
37 (51)
39%, 63%
|
24 (34)
23%, 47%
|
5 (7)
2%, 15%
| |
Valeur pd
|
< 0,0001
|
< 0,0001
|
S.O.
| |
Mois 12
|
|
|
| |
Répondeur, n (%)
IC à 95% correspondant
|
35 (48)
36%, 60%
|
32 (46)
34%, 58%
|
7 (10)
4%, 19%
| |
Valeur p d
|
< 0,0001
|
< 0,0001
|
S.O.
| |
Composants du critère d’évaluation principale
| |
Taux de PAL < 1,67 × LSN, n (%)
|
40 (55)
|
33 (47)
|
12 (16)
| |
Réduction du taux de PAL d’au moins 15%, n (%)
|
57 (78)
|
54 (77)
|
21 (29)
| |
Bilirubine totale de ≤ 1 × LSN, n (%)
|
60 (82)
|
62 (89)
|
57 (78)
|
a Pourcentage des patients atteignant une réponse, définie comme un taux de PAL inférieur à 1,67 fois la LSN, un taux de bilirubine totale dans les limites normales et une réduction du taux de PAL d’au moins 15%. Les valeurs manquantes étaient considérées comme des non-réponses. Le test exact de Fisher a été utilisé pour calculer les intervalles de confiance (IC) à 95%.
b Dans cette étude, 16 patients (7%) étaient intolérants et n’ont pas reçu l’AUDC en médicament concomitant : 6 patients (8%) dans le bras OCALIVA 10 mg, 5 patients (7%) dans le bras OCALIVA avec augmentation de dose et 5 patients (7%) dans le groupe placebo.
c Les patients ont été randomisés (1:1:1) pour recevoir OCALIVA 10 mg une fois par jour pendant les 12 mois complets de l’étude, ou le schéma de titration d’OCALIVA (5 mg une fois par jour pendant les 6 premiers mois, la dose pouvant être augmentée à 10 mg une fois par jour pendant les 6 derniers mois, si le patient tolérait OCALIVA, mais présentait un taux de PAL au moins 1,67 fois supérieur à la LSN, et/ou un taux de bilirubine totale supérieure à la LSN, ou une réduction du taux de PAL inférieure à 15%) ou le placebo.
d Titration d’OCALIVA et OCALIVA 10 mg par rapport au placebo. Les valeurs de p ont été obtenues à l’aide du test Cochran-Mantel-Haenszel d’Association Générale, stratifié selon l’intolérance à l’AUDC et les taux de PAL prétraitements supérieurs à 3 fois la LSN et/ou l’ASAT supérieur à 2 fois la LSN et/ou un taux de bilirubine totale supérieur à la LSN.
e Les taux de réponse ont été calculés sur la base de l’analyse des cas observés (c’est-à-dire [n= répondeur observé]/[N=population en intention de traiter (ITT)]); le pourcentage de patients présentant des valeurs au mois 12 est de 86%, 91% et 96% pour le bras OCALIVA 10 mg, le bras OCALIVA avec augmentation de dose et le bras placebo, respectivement.
f Les taux moyens de bilirubine totale en situation initiale variaient de 10 μmol/l à 12 μmol/l pour les différents bras de traitement, 92% des patients inclus présentant des valeurs dans les limites normales (c’est-à-dire ≤ LSN).
Réduction moyenne des taux de PAL
La figure 1 montre la réduction moyenne du taux de PAL chez les patients traités par OCALIVA par rapport au placebo. Une réduction a été observée dès la semaine 2. Elle s’est stabilisée au mois 3 et s’est maintenue jusqu’au mois 12 chez les patients qui avaient reçu la même dose sur les 12 mois complets. Bien qu’une titration ait été évaluée à 6 mois dans l’étude POISE, les données parlent en faveur d’une titration d’OCALIVA dès 3 mois. Une réduction supplémentaire du taux de PAL a été observée au mois 12 chez une majorité des patients du bras de titration d’OCALIVA, dont la dose d’OCALIVA était passée de 5 mg une fois par jour à 10 mg une fois par jour (voir « Pharmacocinétique »).
Figure 1 : Taux moyen de PAL sur 12 mois dans l’étude POISE, par bras de traitement, avec ou sans AUDCa
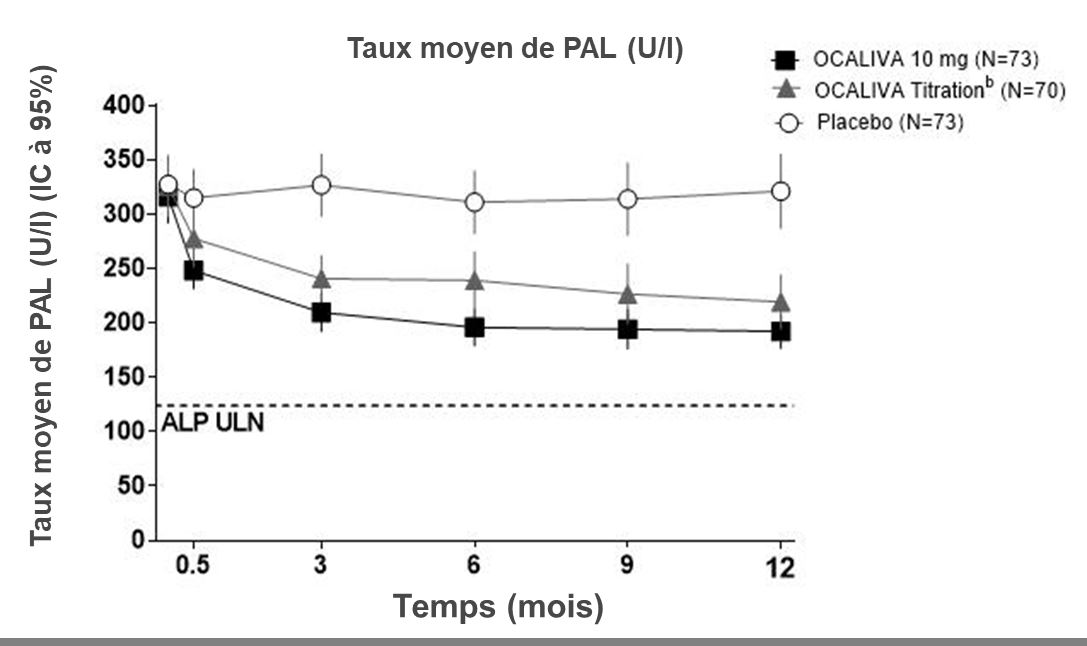
a Dans cette étude, 16 patients (7%) n’ont pas reçu l’AUDC en médicament concomitant en raison d’une intolérance : 6 patients (8%) dans le bras OCALIVA 10 mg, 5 patients (7%) dans le bras OCALIVA avec augmentation de dose et 5 patients (7%) dans le bras placebo.
b Les patients répartis aléatoirement dans le bras OCALIVA avec augmentation de dose ont reçu 5 mg d’OCALIVA une fois par jour pendant les 6 premiers mois. À partir du mois 6, les patients qui toléraient bien OCALIVA mais qui présentaient un taux de PAL au moins 1,67 fois supérieur à la limite supérieure de la normale (LSN) et/ou un taux de bilirubine totale supérieur à la LSN ou une diminution du taux de PAL de moins de 15% étaient jugés admissibles pour passer d’une dose de 5 mg une fois par jour à une dose de 10 mg une fois par jour pendant les 6 derniers mois de l’étude.
Réduction moyenne des taux de GGT
La réduction moyenne (IC à 95%) des taux de gamma-glutamyl transférase (GGT) était de 178 (137, 219) U/l dans le bras OCALIVA 10 mg, de 138 (102, 174) U/l dans le bras OCALIVA avec augmentation de dose et de 8 (-32, 48) U/l dans le bras placebo.
OCALIVA en monothérapie
La réponse biochimique à OCALIVA administré en monothérapie a été évaluée chez cinquante-et-un patients atteints de CBP présentant un taux de PAL en situation initiale 1,67 fois supérieur à la LSN ou plus, et/ou un taux de bilirubine totale supérieure à la LSN, (24 patients ont reçu OCALIVA 10 mg une fois par jour et 27 patients ont reçu un placebo) dans le cadre d’une méta-analyse des données émanant d’une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, d’une durée de 12 mois (étude POISE), ainsi que d’une étude clinique randomisée, en double aveugle, contrôlée contre placebo, d’une durée de 3 mois. Au mois 3, 9 (38%) des patients traités par OCALIVA ont obtenu une réponse en termes de critère d’évaluation composite, par rapport à 1 patient (4%) dans le groupe placebo. La réduction moyenne (IC à 95%) des taux de PAL chez les patients traités par OCALIVA était de 246 (165, 327) U/l, par rapport à une augmentation de 17 (−7, 42) U/l chez les patients traités par placebo.
|