CompositionPrincipes actifs
Durvalumab.
Excipients
L-histidine, chlorhydrate de L-histidine monohydraté, α,α-tréhalose dihydraté, polysorbate 80, eau pour préparations injectables.
Indications/Possibilités d’emploiIndications faisant l'objet d'une autorisation ordinaire
Cancer du poumon non à petites cellules (CPNPC)
Imfinzi est indiqué dans le traitement des patients adultes atteints d'un cancer du poumon non à petites cellules (CPNPC) localement avancé et non résécable, dont la maladie n'a pas progressé après chimioradiothérapie (CRT) définitive à base de platine.
Cancer du poumon à petites cellules (CPPC)
Imfinzi en monothérapie est indiqué dans le traitement des patients adultes atteints d'un cancer du poumon à petites cellules non opérable à un stade limité (LS-SCLC, limited-stage small cell lung cancer), dont la maladie n'a pas progressé après chimioradiothérapie (CRT) à base de platine.
Imfinzi, en association avec l'étoposide et soit le carboplatine soit le cisplatine, est indiqué dans le traitement de première intention des patients adultes atteints d'un cancer du poumon à petites cellules de stade avancé (ES-SCLC, extensive-stage small cell lung cancer).
Cancer de la vessie
Imfinzi, en association avec la gemcitabine et le cisplatine comme traitement néoadjuvant, suivis d'Imfinzi en monothérapie adjuvante après une cystectomie radicale, est indiqué dans le traitement des patients adultes atteints de cancer de la vessie infiltrant le muscle (TVIM).
L'étude NIAGARA n'était pas conçue pour analyser et évaluer séparément l'efficacité d'Imfinzi dans la phase de traitement néoadjuvant ou adjuvant (voir «Propriétés/Effets»). Le traitement adjuvant supplémentaire par Imfinzi par rapport à la phase de traitement néoadjuvant seul était lié à une toxicité supplémentaire (voir «Mises en garde et précautions»).
Indications faisant l'objet d'une autorisation à durée limitée
Cancer du poumon non à petites cellules (CPNPC)
Imfinzi, en association avec une chimiothérapie à base de platine comme traitement néoadjuvant, suivi d'Imfinzi en monothérapie après l'opération, est indiqué dans le traitement des patients adultes atteints d'un CPNPC résécable (tumeurs ≥4 cm et/ou envahissement ganglionnaire positif) en l'absence connus de mutations du récepteur du facteur de croissance épidermique (EGFR) ou de réarrangements de la kinase du lymphome anaplasique (ALK).
L'étude AEGEAN n'était pas conçue pour analyser et évaluer séparément l'efficacité d'Imfinzi dans la phase de traitement néoadjuvant ou adjuvant (voir «Propriétés/Effets»). Le traitement adjuvant supplémentaire par Imfinzi par rapport à la phase de traitement néoadjuvant seul était lié à une toxicité supplémentaire (voir «Mises en garde et précautions»).
Cancer des voies biliaires (CVB)
Imfinzi, en association avec la gemcitabine et le cisplatine, est indiqué dans le traitement de première intention des patients adultes atteints d'un cancer des voies biliaires localement avancé ou métastatique (voir rubrique «Efficacité clinique»).
En raison de données cliniques incomplètes au moment de l'examen de la demande d'autorisation, ces indications font l'objet d'une autorisation à durée limitée (art. 9a de la Loi sur les produits thérapeutiques). L'autorisation à durée limitée est impérativement liée à la satisfaction de charges en temps opportun. Une fois ces charges satisfaites, l'autorisation à durée limitée pourra être transformée en autorisation ordinaire.
Posologie/Mode d’emploiLe traitement par Imfinzi doit être instauré et surveillé par un oncologue expérimenté.
Pour garantir la traçabilité des médicaments fabriqués à partir des biotechnologies, il est recommandé de documenter la marque commerciale et le numéro de lot pour chaque traitement.
Mode d'administration
Utilisation intraveineuse.
Pour des instructions sur la dilution de ce médicament avant utilisation, voir la rubrique « Remarques particulières ».
Posologie usuelle
La dose recommandée d'Imfinzi dépend de l'indication. Imfinzi est administré en perfusion intraveineuse sur une période de 60 minutes.
CPNPC localement avancé
La dose recommandée d'Imfinzi est de 10 mg/kg toutes les 2 semaines ou de 1500 mg toutes les 4 semaines jusqu'à progression de la maladie ou toxicité inacceptable, pendant 12 mois au plus.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, la posologie doit se baser sur le poids et correspond à 10 mg/kg d'Imfinzi toutes les 2 semaines ou à 20 mg/kg toutes les 4 semaines en monothérapie jusqu'à l'obtention d'un poids supérieur à 30 kg.
Les recommandations posologiques pour une administration de 1500 mg toutes les 4 semaines et pour les patients d'un poids corporel inférieur ou égal à 30 kg sont basées sur les simulations de pharmacocinétique de population.
CPNPC résécable
La dose recommandée d'Imfinzi est de 1500 mg en association avec une chimiothérapie toutes les 3 semaines jusqu'à 4 cycles avant l'opération, suivis de 1500 mg toutes les 4 semaines en monothérapie après l'opération, jusqu'à ce que la maladie soit considérée comme étant non résécable, jusqu'à ce qu'elle récidive ou qu'une toxicité inacceptable survienne, ou pendant 12 cycles au maximum après l'opération.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, une posologie basée sur le poids de 20 mg/kg est nécessaire, à savoir 20 mg/kg en association avec une chimiothérapie toutes les 3 semaines (21 jours) avant l'opération, suivis d'une monothérapie à la posologie de 20 mg/kg toutes les 4 semaines après l'opération jusqu'à l'atteinte d'un poids supérieur à 30 kg.
Imfinzi doit être administré avant la chimiothérapie lorsque les deux sont administrés le même jour. Se référer à l'information professionnelle correspondante pour le dosage de l'agent chimiothérapeutique. Pour les recommandations posologiques de la chimiothérapie, voir rubrique «Efficacité clinique».
LS-SCLC
La dose recommandée d'Imfinzi est de 1500 mg toutes les 4 semaines jusqu'à progression de la maladie, toxicité inacceptable ou pendant 24 mois au plus.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, la posologie doit se baser sur le poids et correspond à 20 mg/kg toutes les 4 semaines en monothérapie jusqu'à l'obtention d'un poids supérieur à 30 kg. Ces recommandations posologiques sont basées sur les simulations de pharmacocinétique de population.
CPPC avancé (ES-SCLC)
La dose recommandée d'Imfinzi est de 1500 mg en association à une chimiothérapie toutes les 3 semaines (21 jours) pendant 4 cycles suivis de 1500 mg toutes les 4 semaines en monothérapie jusqu'à progression de la maladie ou toxicité inacceptable.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, une posologie d'Imfinzi basée sur le poids est nécessaire, à savoir 20 mg/kg d'Imfinzi en association à une chimiothérapie toutes les 3 semaines (21 jours) pendant 4 cycles suivis d'une monothérapie à la posologie de 20 mg/kg toutes les 4 semaines jusqu'à l'atteinte d'un poids supérieur à 30 kg. Ces recommandations posologiques sont basées sur les simulations de pharmacocinétique de population.
Imfinzi doit être administré avant la chimiothérapie lorsque les deux sont administrés le même jour. Se référer aux différentes informations professionnelles pour les dosages de l'étoposide et du carboplatine ou du cisplatine
CVB
La dose recommandée d'Imfinzi est de 1500 mg en association à une chimiothérapie toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis de 1500 mg toutes les 4 semaines en monothérapie jusqu'à progression de la maladie ou toxicité inacceptable.
Pour une chimiothérapie, 1000 mg/m2 de gemcitabine et 25 mg/m2 de cisplatine sont administrés (chacun étant administré les jours 1 et 8) toutes les 3 semaines (21 jours) jusqu'à 8 cycles.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, une posologie d'Imfinzi basée sur le poids est nécessaire, à savoir 20 mg/kg d'Imfinzi en association à une chimiothérapie toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis d'une monothérapie à la posologie de 20 mg/kg toutes les 4 semaines jusqu'à l'atteinte d'un poids supérieur à 30 kg.
Imfinzi doit être administré avant la chimiothérapie lorsque les deux sont administrés le même jour.
TVIM
La dose recommandée d'Imfinzi est de 1500 mg en association avec une chimiothérapie toutes les 3 semaines (21 jours) pendant 4 cycles avant l'opération, suivis de 1500 mg toutes les 4 semaines en monothérapie après l'opération, jusqu'à la progression de la maladie excluant une opération définitive, jusqu'à la récidive de la maladie, jusqu'à la survenue d'une toxicité inacceptable ou jusqu'à un nombre maximal de 8 cycles après l'opération.
Chez les patients d'un poids corporel inférieur ou égal à 30 kg, une posologie d'Imfinzi basée sur le poids de 20 mg/kg est nécessaire. Imfinzi, en association avec une chimiothérapie, est administré à raison de 20 mg/kg toutes les 3 semaines (21 jours) avant l'opération, suivis d'une monothérapie à la posologie de 20 mg/kg toutes les 4 semaines après l'opération jusqu'à l'atteinte d'un poids supérieur à 30 kg.
Imfinzi doit être administré avant la chimiothérapie lorsque les deux sont administrés le même jour.
Ajustement de la posologie du fait d'effets indésirables/d'interactions
Une diminution ou une augmentation de la dose d'Imfinzi n'est pas recommandée. En général, Imfinzi doit être arrêté en cas d'effets indésirables à médiation immunitaire sévères (grade 3). Imfinzi doit être définitivement arrêté en cas d'effets indésirables à médiation immunitaire menaçant le pronostic vital (grade 4), d'effets indésirables à médiation immunitaire sévères récurrents (grade 3) qui exigent un traitement immunosuppresseur systémique et si la dose de corticoïde ne peut pas être réduite dans les 12 semaines après initiation du corticoïde à 10 mg ou moins de prednisone par jour ou équivalent. Les effets indésirables à médiation immunitaire qui exigent une gestion spécifique sont répertoriés dans le Tableau 1.
Voir la rubrique « Mises en garde et précautions » pour de plus amples informations sur la surveillance et l'évaluation.
Tableau 1. Recommandations relatives à la modification du traitement par Imfinzi
|
Effets indésirables
|
Degré de gravitéa
|
Modification du traitement par Imfinzi
| |
Pneumonite à médiation immunitaire/pneumopathie interstitielle
|
Grade 2
|
Suspension de l'administrationb
| |
Grade 3 ou 4
|
Arrêt définitif
| |
Hépatite à médiation immunitaire
|
ALT ou AST > 3 à ≤5 x LSN ou bilirubine totale > 1,5 à ≤3 x LSN
|
Suspension de l'administrationb
| |
ALT ou AST > 5 – ≤10 x LSN
|
Suspension de l'administrationb
| |
À la fois, ALT ou AST > 3 x LSN et bilirubine totale > 2 x LSN
|
Arrêt définitif
| |
ALT ou AST > 10 x LSN ou bilirubine totale > 3 x LSN
|
Arrêt définitif
| |
Colite ou diarrhée à médiation immunitaire
|
Grade 2 ou 3
|
Suspension de l'administrationb
| |
Grade 4
|
Arrêt définitif
| |
Endocrinopathies à médiation immunitaire: hyperthyroïdie, thyroïdite
|
Grade 2 à 4
|
Suspension de l'administration jusqu'à stabilité clinique
| |
Endocrinopathies à médiation immunitaire:
hypothyroïdie
|
Grade 2 à 4
|
Aucune modification
| |
Endocrinopathies à médiation immunitaire:
insuffisance surrénalienne,
hypophysite/insuffisance hypophysaire
|
Grade 2 à 4
|
Suspension de l'administration jusqu'à stabilité clinique
| |
Endocrinopathies à médiation immunitaire:
diabète sucré de type 1
|
Grade 2 à 4
|
Suspension de l'administration jusqu'à stabilité clinique
| |
Néphrite à médiation immunitaire
|
Grade 2 avec créatinine sérique > 1,5 à 3 x LSN ou valeur de référence
|
Suspension de l'administrationb
| |
Grade 3 avec créatinine sérique > 3 x valeur de référence ou > 3 à 6 x LSN; grade 4 avec créatinine sérique > 6 x LSN
|
Arrêt définitif
| |
Éruption cutanée ou dermatite à médiation immunitaire (incluant pemphigoïde)
|
Grade 2> 1 semaine ou grade 3
|
Suspension de l'administrationb
| |
Grade 4
|
Arrêt définitif
| |
Myocardite à médiation immunitaire
|
Grade 2 à 4
|
Arrêt définitif
| |
Myosite/polymyosite à médiation immunitaire/rhabdomyolyse
|
Grade 2 ou 3
|
Suspension de l'administrationb,c
| |
Grade 4
|
Arrêt définitif
| |
Réactions liées à la perfusion
|
Grade 1 ou 2
|
Interruption de la perfusion ou ralentissement du débit de perfusion
| |
Grade 3 ou 4
|
Arrêt définitif
| |
Myasthénie grave à médiation immunitaire
|
Grade 2 à 4
|
Arrêt définitif
| |
Encéphalite à médiation immunitaire
|
Grade 2 à 4
|
Arrêt définitif
| |
Syndrome de Guillain-Barré à médiation immunitaire
|
Grade 2 à 4
|
Arrêt définitif
| |
Myélite transverse à médiation immunitaire
|
Tous les grades
|
Arrêt définitif
| |
Autres effets indésirables à médiation immunitaired
|
Grade 2 ou 3
|
Suspension de l'administrationb
| |
Grade 4
|
Arrêt définitif
| |
Effet indésirable récidivant de grade 3 ou 4
|
Effet indésirable récidivant de grade 3 ou 4 (grave ou potentiellement fatal)
|
Arrêt définitif
|
a Critères terminologiques généraux pour les effets indésirables, version 4.03. ALT: alanine aminotransférase, AST: aspartate aminotransférase, LSN: limite supérieure de la normale.
b Après une suspension, le traitement par Imfinzi peut être repris dans les 12 semaines si les effets indésirables ont régressé (≤ grade 1) et la dose de corticoïde a été réduite à ≤10 mg/j de prednisone ou équivalent. En cas d'effets indésirables récurrents de grade 3, Imfinzi doit être arrêté définitivement.
c Imfinzi doit être définitivement arrêté si l'effet indésirable ne régresse pas (≤ grade 1) dans un délai de 30 jours ou en cas de survenue de signes d'insuffisance respiratoire.
d Comprend immunothrombocytopénie, pancréatite, arthrite, uvéite et la polymyalgie rhumatismale.
En cas de réactions autres que des réactions à médiation immunitaire, le traitement par Imfinzi doit être suspendu si elles sont de grade 2 ou 3 et définitivement arrêté si elles sont de grade 4. Les effets indésirables doivent être traités conformément aux normes de l'établissement.
Instructions posologiques particulières
Un ajustement posologique en raison de l'âge, du poids corporel, du sexe ou du groupe ethnique du patient n'est pas nécessaire (voir « Pharmacocinétique »).
Patients présentant des troubles de la fonction rénale
La sécurité et l'efficacité d'Imfinzi n'ont pas été établies chez les patients souffrant d'insuffisance rénale. D'après une analyse pharmacocinétique de population, aucun ajustement de la dose d'Imfinzi n'est recommandé chez les patients souffrant d'insuffisance rénale légère ou modérée (voir « Pharmacocinétique »). Les données disponibles chez les patients souffrant d'insuffisance rénale sévère sont insuffisantes et ne permettent pas de tirer des conclusions concernant cette population (voir « Pharmacocinétique »).
Patients présentant des troubles de la fonction hépatique
La sécurité et l'efficacité d'Imfinzi n'ont pas été établies chez les patients souffrant d'insuffisance hépatique. D'après une analyse pharmacocinétique de population, aucun ajustement de la dose d'Imfinzi n'est recommandé chez les patients souffrant d'insuffisance hépatique légère ou modérée. Imfinzi n'a pas été étudié chez des patients souffrant d'insuffisance hépatique sévère (voir « Pharmacocinétique »).
Patients âgés
D'après une analyse pharmacocinétique de population, aucun ajustement posologique n'est nécessaire pour les patients âgés (≥65 ans) (voir « Propriétés/Effets »). Parmi les 476 patients atteints d'un CPNPC inopérable localement avancé (population principalement étudiée pour déterminer l'efficacité), qui ont été traités par Imfinzi, 215 avaient 65 ans ou plus. Globalement, aucune différence cliniquement significative en termes de sécurité n'a été rapportée chez les patients ≥65 ans comparés aux patients plus jeunes. Les données des patients de 75 ans et plus issues de l'étude PACIFIC (7,6%) sont trop limitées pour pouvoir tirer des conclusions concernant cette population.
Parmi les 401 patients atteints d'un CPNPC résécable traités dans l'étude AEGEAN par Imfinzi associé à une chimiothérapie, 209 patients (52 %) étaient âgés de 65 ans au moins et 49 patients (12 %) étaient âgés de 75 ans au moins. Concernant la sécurité ou l'efficacité, globalement aucune différence cliniquement pertinente n'a été rapportée entre les patients ≥65 ans et les plus jeunes.
Parmi les 262 patients avec LS-SCLC traités par Imfinzi, 103 patients (39,3 %) étaient âgés de 65 ans au moins. Concernant la sécurité ou l'efficacité, globalement aucune différence cliniquement pertinente n'a été rapportée entre les patients ≥65 ans et les plus jeunes.
Parmi les 265 patients avec CPPC avancé traités par Imfinzi associé à une chimiothérapie, 101 patients (38%) étaient âgés de 65 ans au moins. Concernant la sécurité ou l'efficacité, aucune différence cliniquement pertinente n'a été rapportée entre les patients ≥65 ans et les plus jeunes.
Parmi les 338 patients atteints de CVB traités par Imfinzi associé à une chimiothérapie, 158 patients (46,7%) étaient âgés de 65 ans au moins. Concernant la sécurité ou l'efficacité, aucune différence cliniquement pertinente n'a été rapportée entre les patients ≥65 ans et les plus jeunes.
Parmi les 533 patients atteints de TVIM traités par Imfinzi associé à une chimiothérapie, 275 patients (51,6 %) étaient âgés de 65 ans au moins. Concernant la sécurité ou l'efficacité, il n'y avait globalement aucune différence cliniquement pertinente entre les patients ≥65 ans et les plus jeunes.
Population pédiatrique
L'utilisation d'Imfinzi dans la population pédiatrique n'est pas autorisée. En dehors de l'indication autorisée, l'utilisation d'Imfinzi a été étudiée chez des enfants âgés de 1 à 17 ans atteints de neuroblastome, de tumeurs solides et de sarcomes.
Les résultats de cette étude n'ont pas démontré l'efficacité d'Imfinzi dans cette population.
Les données actuellement disponibles sont décrites à la rubrique «Effets indésirables».
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients selon la composition.
Mises en garde et précautionsVoir le Tableau 1 à la rubrique « Posologie/Mode d'emploi » pour connaître les recommandations relatives à la modification du traitement.
En cas de suspicion d'effets indésirables à médiation immunitaire, une évaluation appropriée doit être réalisée afin de confirmer l'étiologie ou d'exclure d'autres étiologies. En fonction du grade de sévérité de l'effet indésirable, le traitement par Imfinzi devra être interrompu ou arrêté définitivement. Une corticothérapie ou une thérapie endocrinienne devra être instaurée. En cas d'événements nécessitant une corticothérapie ou après amélioration jusqu'à un grade ≤1, la corticothérapie devra être réduite progressivement sur une période d'au moins 1 mois. L'augmentation de la dose de corticoïdes et/ou l'utilisation d'immunosuppresseurs systémiques supplémentaires sont à envisager en cas d'aggravation ou d'absence d'amélioration.
Si les corticoïdes ne peuvent pas être réduits progressivement et qu'une réduction à ≤10 mg de prednisone par jour (ou équivalent) n'est pas possible en l'espace de 12 semaines après la dernière dose d'Imfinzi, le traitement par Imfinzi doit être arrêté définitivement.
Pneumonite à médiation immunitaire
Après administration d'Imfinzi, une pneumonite et une pneumopathie interstitielle à médiation immunitaire, d'issue fatale dans certains cas, sont survenues sans autre étiologie identifiable et ont nécessité une corticothérapie systémique (voir « Effets indésirables »). En cas d'événements de grade de sévérité 2, un traitement par une dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement. En cas d'événements de grade 3 ou 4, un traitement par une dose initiale de 2 à 4 mg de méthylprednisolone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement.
Pneumonite et pneumonite radique
La pneumonite radique est souvent observée chez les patients traités par radiothérapie des poumons. Les tableaux cliniques de la pneumonite et de la pneumonite radique sont très similaires. Les patients doivent être surveillés pour déceler d'éventuels signes et symptômes d'une pneumonite ou d'une pneumonite radique. Une pneumonite suspectée doit être confirmée à l'aide d'un examen radiographique, en excluant autres causes infectieuses et liées à la maladie, et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ».
Hépatite à médiation immunitaire
Après administration d'Imfinzi, une hépatite à médiation immunitaire, d'issue fatale dans certains cas, est survenue sans autre étiologie identifiable et a nécessité une corticothérapie systémique (voir « Effets indésirables »). Il convient de tester régulièrement les patients avant et pendant le traitement par Imfinzi afin de déceler toute anomalie des valeurs hépatiques. L'hépatite à médiation immunitaire doit être traitée conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». Pour tous les grades de sévérité, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement.
Colite à médiation immunitaire
Après administration d'Imfinzi, une colite ou une diarrhée à médiation immunitaire est survenue chez certains patients et a nécessité une corticothérapie systémique. Aucune autre étiologie n'a été clairement établie (voir « Effets indésirables »). Les patients doivent être surveillés pour déceler d'éventuels signes et symptômes de colite ou de diarrhée et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'événements de grade de sévérité 2 à 4, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement. En cas de suspicion de perforation intestinale tous grades confondus, un chirurgien doit être consulté immédiatement.
Endocrinopathies à médiation immunitaire
Hypothyroïdie/hyperthyroïdie/thyroïdite à médiation immunitaire
Une hypothyroïdie à médiation immunitaire, hyperthyroïdie/thyroïdite est survenue chez des patients traités par Imfinzi (voir « Effets indésirables »). Les patients doivent être régulièrement testés avant et pendant le traitement afin de déceler toute anomalie de la fonction thyroïdienne et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'hypothyroïdie à médiation immunitaire de grade de sévérité 2 à 4, un traitement substitutif par hormones thyroïdiennes doit être instauré si celui-ci est cliniquement indiqué. En cas d'hyperthyroïdie à médiation immunitaire ou de thyroïdite de grade de sévérité 2 à 4, un traitement symptomatique peut être instauré.
Insuffisance surrénalienne à médiation immunitaire
Une insuffisance surrénalienne à médiation immunitaire est survenue chez des patients traités par Imfinzi (voir « Effets indésirables »). Les patients doivent être examinés pour déceler d'éventuels signes cliniques et symptômes d'une insuffisance surrénalienne. En cas d'insuffisance surrénalienne symptomatique, les patients doivent être traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'insuffisance surrénalienne à médiation immunitaire de grade de sévérité 2 à 4, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent, suivi d'une posologie réduite progressivement, et un traitement hormonal substitutif, pour autant qu'il soit cliniquement indiqué, doivent être instaurés.
Diabète sucré de type 1 à médiation immunitaire
Un diabète sucré de type 1 à médiation immunitaire, qui peut être accompagné d'une acidocétose diabétique, est survenu chez des patients traités par Imfinzi (voir « Effets indésirables »). Les patients doivent être examinés pour déceler d'éventuels signes cliniques et symptômes d'un diabète sucré de type 1. En cas de diabète sucré de type 1 symptomatique, les patients doivent être traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'événements de grade de sévérité 2 à 4, une insulinothérapie peut être instaurée si elle est cliniquement indiquée.
Hypophysite/insuffisance hypophysaire à médiation immunitaire
Une hypophysite ou une insuffisance hypophysaire à médiation immunitaire est survenue chez des patients traités par Imfinzi (voir « Effets indésirables »). Les patients doivent être examinés pour déceler d'éventuels signes cliniques et symptômes d'une hypophysite. En cas d'hypophysite ou d'insuffisance hypophysaire symptomatique, les patients doivent être traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'événements de grade de sévérité 2 à 4, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent, suivi d'une posologie réduite progressivement, et un traitement hormonal substitutif, pour autant qu'il soit cliniquement indiqué, doivent être instaurés.
Néphrite à médiation immunitaire
Après administration d'Imfinzi, une néphrite à médiation immunitaire est survenue chez certains patients sans autre étiologie identifiable et a nécessité une corticothérapie systémique (voir « Effets indésirables »). Les patients doivent être régulièrement testés avant et pendant le traitement par Imfinzi afin de déceler toute anomalie de la fonction rénale et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas de néphrite à médiation immunitaire de grade de sévérité 2 à 4, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement.
Exanthème à médiation immunitaire
Après administration d'Imfinzi, un exanthème ou une dermatite à médiation immunitaire (incluant pemphigoïde) sont survenus chez certains patients sans autre étiologie identifiable et ont nécessité une corticothérapie systémique (voir « Effets indésirables »). Les patients doivent être surveillés pour déceler d'éventuels signes et symptômes d'exanthème ou de dermatite et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas d'exanthème à médiation immunitaire de grade de sévérité 2 pendant > 1 semaine ou de grade de sévérité 3 ou 4, un traitement par corticoïdes à la dose initiale de 1 à 2 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement.
Myocardite à médiation immunitaire
Une myocardite à médiation immunitaire, qui peut être fatale, est survenue chez des patients ayant reçu Imfinzi (voir « Effets indésirables »). Les patients doivent être surveillés pour déceler d'éventuels signes et symptômes de myocardite à médiation immunitaire et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». En cas de myocardite à médiation immunitaire de grade de sévérité 2 à 4, un traitement par corticoïdes à la dose initiale de 2 à 4 mg de prednisone/kg/jour ou équivalent doit être instauré, suivi d'une posologie réduite progressivement. Si, malgré l'administration de corticoïdes, aucune amélioration ne survient dans les 2 à 3 jours, un traitement immunosuppresseur supplémentaire doit être instauré immédiatement. Après convalescence (grade 0), les corticoïdes doivent être réduits progressivement sur une période d'au moins 1 mois.
Lymphohistiocytose hémophagocytaire à médiation immunitaire (LHH)
Une LHH est survenue chez des patients traités par Imfinzi (voir « Effets indésirables »). La LHH est un syndrome potentiellement mortel s'accompagnant d'une activation pathologique des défenses immunitaires. En l'absence de diagnostic et de traitement précoces, la LHH a fréquemment une évolution létale. Cette maladie se caractérise par des signes et symptômes cliniques d'une inflammation systémique sévère, tels que fièvre, éruption cutanée, hépatosplénomégalie, cytopénie (surtout anémie et thrombocytopénie), lymphadénopathie, symptômes neurologiques, taux élevé de ferritine sérique, hypertriglycéridémie, ainsi que troubles de la fonction hépatique et de la coagulation. Les patients présentant de tels signes et symptômes doivent immédiatement être examinés et leur état doit être évalué en vue d'un éventuel diagnostic de LHH. L'administration d'Imfinzi doit être suspendue tant qu'une autre étiologie n'a pas pu être établie.
Autres effets indésirables à médiation immunitaire
En raison du mécanisme d'action d'Imfinzi, d'autres effets indésirables à médiation immunitaire peuvent survenir. Les patients doivent être surveillés pour déceler d'éventuels signes et symptômes et traités conformément aux recommandations figurant à la rubrique « Posologie/Mode d'emploi ». Chez les patients traités par Imfinzi en monothérapie dans le cadre d'études cliniques (n = 4045) les effets indésirables à médiation immunitaire cliniquement significatifs suivants sont survenus avec une fréquence de < 1%: méningite aseptique, anémie hémolytique, thrombocytopénie immunitaire, cystite non infectieuse, myosite, rhabdomyolyse, encéphalite, pancréatite, syndrome de Guillain-Barré, arthrite, uvéite et la polymyalgie rhumatismale (voir « Effets indésirables ») et maladies oculaires inflammatoires, y compris kératite. Une polymyosite d'issue fatale a été rapportée chez un patient inclus dans une étude clinique en cours. Dans de rares cas, la myasthénie grave peut apparaître comme un effet indésirable à médiation immunitaire.
Après commercialisation, des cas de myélite transverse ont été observés sous traitement par Imfinzi. Les patients doivent être surveillés à la recherche de signes et symptômes évocateurs d'une myélite.
Des cas de maladie cœliaque ont été observés sous traitement par Imfinzi.
Les effets indésirables à médiation immunitaire cliniquement significatifs suivants ont été rapportés pour d'autres produits de la même classe: syndrome de Stevens-Johnson (SSJ)/nécrolyse épidermique toxique (NET), pancréatite, insuffisance pancréatique exocrine, syndrome de réponse inflammatoire systémique, lymphadénite histiocytaire nécrosante, démyélinisation, vasculite, anémie hémolytique, anémie aplasique iritis, paralysie du nerf facial et du nerf oculomoteur externe, polymyalgie rhumatismale, neuropathie auto-immune, syndrome de Guillain-Barré et syndrome de Vogt-Koyanagi-Harada.
Patients porteurs d'une maladie auto-immune préexistante
Chez les patients porteurs d'une maladie auto-immune (MAI) préexistante, les données issues d'études observationnelles indiquent un risque accru d'effets indésirables immunomédiés après un traitement par un inhibiteur du point de contrôle immunitaire par rapport aux patients sans MAI préexistante. En outre, des poussées de la MAI sous-jacente sont souvent survenues, mais la plupart d'entre elles étaient légères et faciles à traiter.
Réactions liées à la perfusion
Les patients doivent être examinés pour déceler d'éventuels signes et symptômes de réactions liées à la perfusion. Chez des patients sous Imfinzi, des réactions sévères liées à la perfusion ont été observées (voir « Effets indésirables »). En cas de réactions de grade de sévérité 1 ou 2, une prémédication pour la prophylaxie des réactions liées à la perfusion suivantes peut être envisagée. Les réactions liées à la perfusion sévères de grade 3 ou 4 doivent être traitées selon les procédures standard de l'établissement, les lignes directrices applicables pour la pratique clinique et/ou les lignes directrices des sociétés spécialisées.
Événements cérébrovasculaires
Des événements cérébrovasculaires, d'issue fatale dans certains cas, ont été observés chez des patients atteints de CVB ayant reçu Imfinzi associé à une chimiothérapie dans le cadre de l'étude TOPAZ-1. La plupart de ces patients avaient des facteurs de risque cérébrovasculaire (voir « Effets indésirables »).
Réactions indésirables chez les patients transplantés
Chez des patients traités par des inhibiteurs de PD-1/PD-L1, un rejet des organes solides transplantés a été observé lors des études post-commercialisation. Chez ces patients, le bénéfice du traitement par des inhibiteurs de PD-1/PD-L1, y compris durvalumab, doit être mis en balance avec le risque d'un éventuel rejet d'organe.
Précautions spécifiques à la maladie
Cholangite et infections des voies biliaires chez les patients atteints de CVB
La cholangite et les infections des voies biliaires ne sont pas rares chez les patients atteints de CVB avancés. Des événements de type cholangite ont été rapportés dans l'étude TOPAZ-1 dans les deux bras de traitement (14,5 % [IMFINZI + chimiothérapie] vs 8,2 % [placebo + chimiothérapie]); ces cas étaient principalement associés à des prothèses biliaires et n'étaient pas d'étiologie à médiation immunitaire. Les patients atteints de CVB (en particulier ceux qui sont porteurs d'une prothèse biliaire) doivent être étroitement surveillés pour détecter l'apparition d'une cholangite ou d'une infection des voies biliaires avant l'instauration du traitement et régulièrement par la suite.
CPNPC au stade précoce
L'analyse finale de la SG de l'étude AEGEAN est en cours, un bénéfice en termes de survie avec le traitement périopératoire par le durvalumab par rapport au placebo n'ayant pas encore pu être démontré. Par comparaison avec la chimiothérapie seule, l'administration de durvalumab en plus de la chimiothérapie était associée à un taux accru d'événements indésirables à médiation immunitaire (EIMI) (voir rubrique «Effets indésirables»). Par comparaison avec le traitement adjuvant par placebo, le traitement adjuvant par le durvalumab après un traitement néoadjuvant était lié à une toxicité supplémentaire. Durant la phase néoadjuvante de l'étude AEGEAN (avec DCO du 14 août 2023), la fréquence des événements indésirables graves (EIG) était de 20,7 % vs 16,6 %, la fréquence des événements indésirables survenus pendant le traitement (EIST) de grade le plus élevé ≥3 de 34,4 % vs 37,4 % et la fréquence des EIST de grade 5 de 2,0 % vs 1,0 % des patients qui recevaient le durvalumab et une chimiothérapie, resp. le placebo et une chimiothérapie. Durant toute la phase de traitement, la fréquence des EIG était de 38,9 % vs 31,7 %, la fréquence des EIST de grade le plus élevé ≥3 de 49,1 % vs 47,0 % et la fréquence des EIST de grade 5 de 5,7 % vs 3,8 % des patients qui recevaient le durvalumab et une chimiothérapie, resp. le placebo et une chimiothérapie. Les données disponibles ne permettent pas de tirer une conclusion sur l'influence du durvalumab en cas de CPNPC au stade précoce sur un traitement systémique anti-PD-(L)1 ultérieur dans le cas d'une récidive avancée/métastatique, y compris la possibilité d'une résistance (acquise) à un traitement systémique anti-PD-(L)1.
Cancer de la vessie infiltrant le muscle
Par comparaison avec la chimiothérapie seule, l'administration de durvalumab en plus de la chimiothérapie était associée à un taux accru d'événements indésirables à médiation immunitaire (EIMI) (voir rubrique «Effets indésirables»). Par comparaison avec l'absence de traitement adjuvant, le traitement adjuvant par le durvalumab après un traitement néoadjuvant était lié à une toxicité supplémentaire. Durant la phase néoadjuvante de l'étude NIAGARA (DCO du 29 avril 2024), la fréquence des événements indésirables graves (EIG) était de 23,6 % vs 22,4 %, la fréquence des événements indésirables survenus pendant le traitement (EIST) de grade le plus élevé 3 ou 4 de 46,8 % vs 50,8 % et la fréquence des EIST de grade 5 de 1,1 % vs 1,9 % des patients qui recevaient le durvalumab et une chimiothérapie néoadjuvante, resp. une chimiothérapie néoadjuvante. Durant toute la phase de traitement, la fréquence des EIG était de 61,5 % vs 54,6 %, la fréquence des EIST de grade le plus élevé 3 ou 4 de 66,6 % vs 63,9 % et la fréquence des EIST de grade 5 de 5,1 % vs 5,5 % des patients qui recevaient le durvalumab et une chimiothérapie, resp. une chimiothérapie. Les données disponibles ne permettent pas de tirer une conclusion sur l'influence du durvalumab en cas de TVIM sur un traitement systémique anti-PD-(L)1 ultérieur dans le cas d'une récidive avancée/métastatique, y compris la possibilité d'une résistance (acquise) à un traitement systémique anti-PD-(L)1.
Populations de patients non évalués dans des études cliniques
Les groupes de patients suivants ont été exclus des études cliniques: patients avec un poids corporel < 30 kg, patients avec maladies auto-immunes ou inflammatoires actives, maladies intercurrentes non contrôlées incluant une pneumopathie interstitielle (Interstitial Lung Disease, ILD), infections actives incluant tuberculose, hépatite C ou VIH, patients ayant reçu un vaccin vivant atténué dans les 30 jours avant la première dose de durvalumab, patients avec utilisation actuelle ou préalable de médicaments immunosuppresseurs systémiques à haute dose dans les 14 jours avant la première dose de durvalumab et patients avec un statut de performance ECOG ≥2.
InteractionsLe durvalumab est un anticorps humanisé qui n'est pas excrété principalement par voie hépatique/rénale, mais qui est essentiellement éliminé par catabolisme protéique par le biais du système réticulo-endothélial ou par une élimination du complexe anticorps-protéine cible. Étant donné qu'aucune interaction pharmacocinétique avec d'autres médicaments n'est donc attendue, aucune étude formelle des interactions pharmacocinétiques n'a été effectuée.
Les interactions pharmacocinétiques entre le durvalumab et la chimiothérapie ont été analysées dans l'étude CASPIAN et ont montré que l'administration concomitante avec le durvalumab n'avait pas influencé la pharmacocinétique de l'étoposide, du carboplatine ou du cisplatine.
On ne s'attend pas non plus à ce que le durvalumab active ou inhibe le métabolisme d'autres médicaments par le cytochrome P450.
L'utilisation de corticoïdes systémiques ou d'immunosuppresseurs avant le début du traitement par durvalumab est à éviter, car ils peuvent entraver l'activité pharmacodynamique et diminuer l'efficacité de ce dernier. Des corticoïdes systémiques ou d'autres immunosuppresseurs peuvent cependant être utilisés après le début du traitement par durvalumab afin de traiter les effets indésirables à médiation immunitaire (voir rubrique « Mises en garde et précautions »).
Grossesse, allaitementGrossesse
Dans des études sur la reproduction animale menées chez des macaques de Java, l'administration de durvalumab pendant la période de gestation à des doses 6 à 20 fois ou 3 à 11 fois plus élevées que la dose clinique de 10 mg/kg de durvalumab toutes les 2 semaines ou 1500 mg de durvalumab toutes les 3 semaines (niveau d'exposition estimé sur la base de l'ASC) a été associée à des naissances prématurées, à des fausses couches (avortements spontanés et décès in utero) et à une augmentation des cas de décès néonataux (voir « Données précliniques »). Il n'existe pas de données cliniques concernant l'emploi du durvalumab chez la femme enceinte. En raison de son mécanisme d'action, le durvalumab peut avoir un effet sur le maintien de la grossesse et entraîner des anomalies fœtales s'il est administré à des femmes enceintes. Les IgG1 humaines sont capables de passer la barrière placentaire. Imfinzi ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue. Les femmes en âge de procréer devraient utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 3 mois après l'administration de la dernière dose.
Allaitement
On ne dispose d'aucune donnée concernant la présence du durvalumab dans le lait maternel, son absorption, ses effets sur le nourrisson allaité et son incidence sur la production lactique. L'IgG humaine est excrétée dans le lait maternel humain. Dans des études sur la reproduction menées chez des femelles macaques de Java gravides auxquelles le durvalumab a été administré, l'excrétion d'une faible quantité dose-dépendante de durvalumab dans le lait maternel a été observée. En raison des effets indésirables potentiels du durvalumab chez les nourrissons allaités, il est recommandé aux femmes qui allaitent d'interrompre l'allaitement pendant le traitement et pendant au moins 3 mois après la dernière dose.
Effet sur l’aptitude à la conduite et l’utilisation de machinesD'après les propriétés pharmacodynamiques, il est peu probable que le durvalumab ait une influence sur l'aptitude à la conduite ou l'utilisation de machines. Si des patients développent néanmoins des effets indésirables qui influent sur leur capacité de concentration et de réaction, la prudence est de mise en cas de conduite de véhicules ou d'utilisation de machines.
Effets indésirablesL'utilisation d'Imfinzi est associée à des effets indésirables à médiation immunitaire. La plupart, y compris les réactions sévères, ont régressé après instauration d'un traitement médical approprié ou arrêt d'Imfinzi (voir « Description de certains effets indésirables »).
Durvalumab en monothérapie
Résumé du profil de sécurité
Des données poolées de 4642 patients issues de 15 études sur différents types de tumeurs (Durvalumab-Pan-Tumor-Pool) sont disponibles sur la sécurité d'Imfinzi en monothérapie. Les effets indésirables le plus fréquents (≥10%) étaient la toux/la toux productive (18,1 %), la diarrhée (15,1 %), l'éruption cutanée (15,0 %), la fièvre (12,5 %), les douleurs abdominales (11,8 %), l'infection des voies respiratoires supérieures (11,8 %), l'hypothyroïdie (11,6 %) et le prurit (11,1 %). Les effets indésirables les plus fréquents (≥1 %) de grade 3 à 4 étaient la pneumonie (3,5 %), l'augmentation de l'aspartate aminotransférase ou de l'alanine aminotransférase (2,5 %) et les douleurs abdominales (1,6 %).
Durvalumab en association avec une chimiothérapie
Résumé du profil de sécurité
La sécurité d'Imfinzi administré en association avec une chimiothérapie repose sur les données poolées chez 1868 patients de 5 études (TOPAZ-1, CASPIAN, POSEIDON, AEGEAN et NIAGARA). Les effets indésirables les plus fréquents (≥10%) ont été la neutropénie (42,0 %), l'anémie (40,7 %), les nausées (39,3 %), la fatigue (38,0 %), la constipation (28,6 %), la diminution de l'appétit (22,6 %), la thrombocytopénie (20,3 %), l'éruption cutanée (18,4 %), la diarrhée (16,5 %), les vomissements (16,1 %), la leucopénie (14,8 %), les douleurs abdominales (14,3 %), l'alopécie (14,2 %), la fièvre (13,8 %), le prurit (12,0 %), l'augmentation de l'aspartate aminotransférase ou de l'alanine aminotransférase (11,3 %), la toux/toux productive (11,0 %) et la neuropathie périphérique (10,5 %). Les effets indésirables les plus fréquents (≥1 %) de grade 3 à 4 étaient la neutropénie (25,2 %), l'anémie (14,1 %), la thrombocytopénie (7,1 %), la leucopénie (4,6 %), la fatigue (3,2 %), la pneumonie (2,7 %), l'atteinte rénale aiguë (2,5 %), l'augmentation de l'aspartate aminotransférase ou de l'alanine aminotransférase (2,0 %), la neutropénie fébrile (2,0 %), l'hypokaliémie (1,9 %), l'embolie pulmonaire (1,9 %), l'hyponatrémie (1,8 %), la septicémie (1,6 %), l'augmentation de l'amylase (1,2 %), la diarrhée (1,2 %), l'augmentation de la gamma-glutamyl transférase (1,2 %) et la dyspnée (1,0 %).
Tableau récapitulatif des effets indésirables
Le Tableau 2 liste les incidences des effets indésirables de l'ensemble des données de sécurité en monothérapie et les effets indésirables survenant de manière avérée lors d'une administration d'Imfinzi seul ou dans le cadre d'une chimiothérapie ou lors d'un traitement par une association de ces médicaments, également lorsque ces effets n'ont pas été signalés au cours des études cliniques évaluant ce traitement en association. Les effets indésirables sont présentés par classe de systèmes d'organes, selon la terminologie du MedDRA. Au sein des classes de systèmes d'organes, les effets indésirables sont répertoriés par fréquence, puis par degré de gravité, dans l'ordre décroissant. La fréquence des effets indésirables est définie comme suit: Très fréquents (≥1/10), fréquents (< 1/10, ≥1/100), occasionnels (< 1/100, ≥1/1000), rares (< 1/1000, ≥1/10 000), très rares (< 1/10 000) et fréquence inconnue.
|
Tableau 2: Effets indésirables chez les patients traités par Imfinzi
| |
|
Monothérapie
|
En association avec une chimiothérapie
| |
Infections et infestations
| |
Très fréquents
|
Infections des voies respiratoires supérieuresa (11,8%)
|
| |
Fréquents
|
Pneumonieb,c, grippe, candidose buccale, infections dentaires et des tissus mous buccauxd
|
Pneumonieb,c, infections des voies respiratoires supérieuresa, grippe, infections dentaires et des tissus mous buccauxd, septicémiec
| |
Occasionnels
|
|
Candidose buccale
| |
Affections hématologiques et du système lymphatique
| |
Très fréquents
|
|
Neutropéniev (42,0%), anémie (40,7%), thrombocytopéniew (20,3%), leucopéniex (14,8%)
| |
Fréquents
|
|
Neutropénie fébrile, pancytopénie
| |
Occasionnels
|
Thrombocytopénie immunitairec
|
| |
Rares
|
|
Lymphohistiocytose hémophagocytairec
| |
Fréquence inconnue
|
Lymphohistiocytose hémophagocytaire
|
| |
Affections endocriniennes
| |
Très fréquents
|
Hypothyroïdie (11,6 %)e
|
| |
Fréquents
|
Hyperthyroïdief, augmentation de la TSH
|
Hyperthyroïdief, hypothyroïdiee, augmentation de la TSH
| |
Occasionnels
|
Réduction de la TSH,
thyroïditeg, insuffisance surrénalienne, hypophysite/insuffisance hypophysaire8, diabète sucré de type 1
|
Insuffisance surrénalienne, réduction de la TSH, hypophysite/insuffisance hypophysaire8, thyroïditeg, diabète sucré de type 1
| |
Rares
|
Diabète insipide
|
| |
Affections oculaires
| |
Rares
|
Uvéite
|
| |
Affections du métabolisme et de la nutrition
| |
Très fréquents
|
|
Diminution de l'appétitc (22,6%)
| |
Fréquents
|
|
Hypomagnésémie2, hypokaliémie, hyponatrémie3, déshydratationc, hypocalcémie9
| |
Occasionnels
|
|
Hypophosphatémie
| |
Affections psychiatriques
| |
Fréquents
|
|
Insomnie
| |
Affections du système nerveux
| |
Très fréquents
|
|
Neuropathie périphérique1 (10,5 %)
| |
Fréquents
|
|
Événements cérébrovasculairesc,u Céphalées
| |
Occasionnels
|
Encéphalitei, myasthénie graveh
|
Myasthénie graveh
| |
Rares
|
|
Encéphalitei
| |
Fréquence inconnue
|
Myélite transverse6, syndrome de Guillain-Barréc
|
| |
Affections de l'oreille et du labyrinthe
| |
Fréquents
|
|
Acouphènes
| |
Affections cardiaques
| |
Fréquents
|
|
Tachycardie
| |
Occasionnels
|
Myocardite
|
Myocarditec
| |
Affections vasculaires
| |
Fréquents
|
|
Hypotension
| |
Affections respiratoires, thoraciques et médiastinales
| |
Très fréquents
|
Toux/toux productive (18,1%)
|
Toux/toux productive (11,0%)
| |
Fréquents
|
Pneumonitec,10, dysphonie
|
Pneumonite c,10, dysphonie, dyspnée, embolie pulmonairec, hoquet
| |
Occasionnels
|
Pneumopathie interstitielle
|
Pneumopathie interstitiellec
| |
Affections gastro-intestinales
| |
Très fréquents
|
Diarrhée (15,1%), douleurs abdominalesj (11,8%)
|
Nausées (39,3%), constipation (28,6%), vomissements (16,1%), diarrhée (16,5%),douleurs abdominalesj (14,3%)
| |
Fréquents
|
|
Stomatitey, augmentation de l'amylase4, colitek
| |
Occasionnels
|
Colitek, pancréatitel
|
Pancréatitel
| |
Rares
|
Maladie cœliaque
|
Maladie cœliaque
| |
Affections hépatobiliaires
| |
Très fréquents
|
|
Augmentation de l'aspartate aminotransférase ou de l'alanine aminotransférasem (11,3%)
| |
Fréquents
|
Hépatitec,n, augmentation de l'aspartate aminotransférase ou de l'alanine aminotransférasec,m
|
Hépatitec,n, augmentation de la bilirubine dans le sang5, augmentation de la gamma-glutamyl transférase
| |
Affections de la peau et du tissu sous-cutané
| |
Très fréquents
|
Éruption cutanéeo (15,0%), prurit (11,1%)
|
Alopécie (14,2%), éruption cutanéeo (18,4%), prurit (12,0%)
| |
Fréquents
|
Sueurs nocturnes
|
Dermatite, prurit
| |
Occasionnels
|
Dermatite, psoriasis, pemphigoïdep
|
Pemphigoïdep, sueurs nocturnes, psoriasis
| |
Affections musculo-squelettiques et du tissu conjonctif
| |
Fréquents
|
Myalgie
|
Dorsalgie, myalgie, spasmes musculaires
| |
Occasionnels
|
Myosite7, arthrite à médiation immunitaire11
|
Arthrite à médiation immunitaire11, myosite7
| |
Rares
|
Polymyalgie rhumatismale
|
Polymyalgie rhumatismale12
| |
Fréquence inconnue
|
Polymyositeq
|
| |
Affections du rein et des voies urinaires
| |
Fréquents
|
Augmentation du taux de créatinine dans le sang, dysurie
|
Augmentation du taux de créatinine dans le sang , dysurie, atteinte rénale aiguëc, protéinurie
| |
Occasionnels
|
Néphriter, cystite non infectieuse
|
Néphriter, cystite non infectieuse
| |
Troubles généraux et anomalies au site d'administration
| |
Très fréquents
|
Fièvre (12,5%)
|
Fatiguez (38,0%), fièvre (13,8%)
| |
Fréquents
|
Œdème périphériques
|
Œdème périphériques, frissons, œdème, malaise
| |
Lésions, intoxications et complications liées aux procédures
| |
Fréquents
|
Réaction liée à la perfusiont
|
Réaction liée à la perfusiont
|
La fréquence des effets indésirables peut ne pas être entièrement attribuée au durvalumab seul, étant donné que bien plus la maladie sous-jacente ou d'autres médicaments utilisés en association peuvent y contribuer.
a Comprend laryngite, naso-pharyngite, abcès péritonsillaire, pharyngite, rhinite, sinusite, tonsillite, trachéobronchite et infections des voies respiratoires supérieures.
b Comprend pneumonie à Pneumocystis jirovecii, pneumonie à Candida, pneumonie à Legionella, pneumonie adénovirale, pneumonie bactérienne, pneumonie à cytomégalovirus, pneumonie à Haemophilus, pneumonie à pneumocoques et streptocoques et pneumonie à Klebsiella.
c Y compris d'issue fatale.
d Comprend gingivite, infection buccale, parodontite, pulpite, abcès dentaire et infection dentaire.
e Comprend hypothyroïdie, hypothyroïdie auto-immune et hypothyroïdie à médiation immunitaire.
f Comprend hyperthyroïdie, maladie de Basedow et hyperthyroïdie à médiation immunitaire.
g Comprend thyroïdite auto-immune, thyroïdite à médiation immunitaire, thyroïdite et thyroïdite subaiguë.
h La fréquence rapportée des études cliniques sponsorisées par AstraZeneca en dehors du pool des données est rare et il n'y avait aucun cas de grade > 2.
i Comprend encéphalite, encéphalite à médiation immunitaire, encéphalite auto-immune et encéphalite non infectieuse.
j Comprend les douleurs abdominales, de l'abdomen inférieur, de l'abdomen supérieur et des flancs.
k Comprend colite, entérite, entérocolite, entérocolite à médiation immunitaire et rectite.
l Comprend pancréatite à médiation immunitaire, pancréatite et pancréatite aiguë.
m Comprend augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase, augmentation des enzymes hépatiques et des transaminases.
n Comprend hépatite, hépatite auto-immune, hépatite toxique, hépatite aiguë, hépatotoxicité, cytolyse hépatique et hépatite à médiation immunitaire.
o Comprend exanthème érythémateux, exanthème maculeux, exanthème maculo-papuleux, exanthème papuleux, exanthème prurigineux, exanthème pustuleux, érythème, eczéma et exanthème.
p Comprend pemphigoïde, dermatite bulleuse et pemphigus.
q Une polymyosite d'évolution fatale a été rapportée chez un patient traité par Imfinzi dans une étude en cours en dehors des données poolées. Rare dans chaque grade, rare dans le grade 3 et plus selon CTCAE.
r Comprend néphrite auto-immune, néphrite tubulo-interstitielle, néphrite à médiation immunitaire, néphrite, glomérulonéphrite et glomérulonéphrite membraneuse.
s Comprend œdème périphérique et gonflement périphérique.
t Les réactions liées à la perfusion comprennent les réactions provoquées par la perfusion et l'urticaire au début de la médication ou un jour après l'administration du médicament.
u Comprend infarctus cérébrale, accident vasculaire cérébral ischémique, hémorragie cérébrale et attaque cérébrovasculaire.
v Comprend neutropénie et baisse du taux de neutrophiles.
w Comprend thrombocytopénie et baisse du taux de plaquettes.
x Comprend leucopénie et baisse du taux de globules blancs.
y Comprend stomatite et inflammation des muqueuses.
z Comprend fatigue et asthénie.
1 Comprend neuropathie périphérique, paresthésie et neuropathie sensorielle périphérique.
2 Comprend diminution du taux de magnésium dans le sang et hypomagnésémie.
3 Comprend diminution du taux de sodium dans le sang et hyponatrémie.
4 Comprend augmentation de l'amylase et hyperamylasémie.
5 Comprend augmentation de la bilirubine dans le sang et hyperbilirubinémie.
6 Rapportée après commercialisation.
7 Comprend myosite et rhabdomyolyse.
8 Comprend hypophysite et insuffisance hypophysaire.
9 Comprend diminution du taux de calcium dans le sang et hypocalcémie.
10 Comprend pneumopathie à médiation immunitaire et pneumonite.
11 Comprend arthrite auto-immune et polyarthrite.
11 Comprend arthrite auto-immune, arthrite à médiation immunitaire, polyarthrite et polyarthrite rhumatoïde.
12 Pas observé dans le groupe Imfinzi+chimiothérapie, mais observé dans d'autres études cliniques dont AstraZeneca était le promoteur.
Description de certains effets indésirables
Les données ci-dessous sur les effets indésirables significatifs concernent l'ensemble des données poolées de sécurité pour Imfinzi en monothérapie pour différents types de tumeurs (n = 4642, Durvalumab-Pan-Tumor-Pool) ou des données poolées de sécurité pour Imfinzi + chimiothérapie (n = 1868), sauf indication contraire. L'ensemble Imfinzi + pool chimiothérapie comprenait des patients de 5 études: étude TOPAZ-1 (durvalumab + gemcitabine/cisplatine (gem/cis); traitement de première intention en cas de CVB avancé), étude CASPIAN (durvalumab + étoposide et soit carboplatine ou cisplatine en association avec une chimiothérapie à base de platine; traitement de première intention du ES-SCLC), étude POSEIDON (durvalumab + chimiothérapie à base de platine; traitement de première intention en cas de CPNPC métastatique), étude AEGEAN (durvalumab + chimiothérapie avant l'opération, suivis de durvalumab après l'opération; CPNPC résécable) et NIAGARA (durvalumab + chimiothérapie à base de platine comme traitement néoadjuvant suivis de durvalumab en monothérapie adjuvante après la cystectomie radicale; TVIM).
Des détails sont présentés lorsque des différences cliniquement pertinentes étaient notées lors de l'utilisation d'Imfinzi en association avec une chimiothérapie.
Les directives relatives au traitement de ces effets indésirables sont décrites dans les rubriques «Posologie/Mode d'emploi» et «Mises en garde et précautions».
Pneumonite à médiation immunitaire
Sous Imfinzi en monothérapie, une pneumonite à médiation immunitaire est survenue chez 147 patients (3,2%); sont compris 37 patients (0,8%) de grade 3, 2 patients (<0,1%) de grade 4 et 10 patients (0,2%) de grade 5. Le délai médian de survenue était de 56 jours (extrêmes: 1-1308 jours). 114 patients sur 147 patients ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour), 4 patients ont également reçu d'autres immunosuppresseurs dont l'infliximab et la ciclosporine en traitement adjuvant. Le traitement par Imfinzi a été arrêté chez 60 patients. Quatre-vingt-cinq patients se sont rétablis. Par comparaison, la pneumonite à médiation immunitaire était plus fréquente chez les patients de l'étude PACIFIC qui avaient terminé une radiochimiothérapie concomitante dans les 1 à 42 jours avant le début de l'étude (9,9 %) par rapport aux autres patients enregistrés dans les données de sécurité combinées (1,8 %).
Sous Imfinzi + chimiothérapie, une pneumonite à médiation immunitaire est survenue chez 45 patients (2,4%), dont 12 cas de grade 3 (0,6%) 2 cas (0,1 %) de grade 4 et 5 cas (0,3 %) de grade 5. Le délai médian de survenue a été de 161 jours (fourchette: 11-529 jours). 22 patients sur 45 ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Deux patients ont reçu d'autres immunosuppresseurs. Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez 13 patients. 28 patients se sont rétablis.
Parmi les patients de l'étude PACIFIC atteints d'un CPNPC inopérable, localement avancé (n = 475 dans le groupe Imfinzi et n = 234 dans le groupe placebo) ayant subi un traitement par chimioradiothérapie 1 à 42 jours avant le début du traitement à l'étude, une pneumonite à médiation immunitaire est survenue chez 47 patients (9,9%) dans le groupe Imfinzi et chez 14 patients (6,0%) dans le groupe placebo. Sont inclus 9 cas de grade 3 (1,9%) dans le groupe Imfinzi versus 6 cas de grade 3 (2,6%) dans le groupe placebo, ainsi que 4 cas de grade 5 (0,8%) dans le groupe Imfinzi versus 3 cas de grade 5 (1,3%) dans le groupe placebo. Le délai médian de survenue était de 46 jours (extrêmes: 2-342 jours) dans le groupe Imfinzi et de 57 jours (extrêmes: 26-253 jours) dans le groupe placebo. Dans le groupe Imfinzi, 30 patients ont reçu une corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour), et 2 patients ont reçu l'infliximab en traitement adjuvant. Dans le groupe placebo, 12 patients ont été traités par corticothérapie systémique et un patient a également été traité par cyclophosphamide et tacrolimus. 29 patients se sont rétablis dans le groupe Imfinzi tandis qu'ils étaient 6 dans le groupe placebo.
Parmi les patients de l'étude ADRIATIC atteints d'un LS-SCLC (n = 262 dans le groupe Imfinzi et n = 265 dans le groupe placebo) ayant subi un traitement par chimioradiothérapie 1 à 42 jours avant le début du traitement à l'étude, une pneumonite à médiation immunitaire est survenue chez 31 patients (11,8 %) dans le groupe Imfinzi et chez 8 patients (3,0 %) dans le groupe placebo. Sont inclus 5 cas de grade 3 (1,9 %) dans le groupe Imfinzi versus 1 cas de grade 3 (0,4 %) dans le groupe placebo, ainsi que 1 cas de grade 5 (0,4 %) dans le groupe Imfinzi. Le délai médian de survenue était de 55 jours dans le groupe Imfinzi (extrêmes: 1-375 jours) et 65,5 jours (fourchette: 24-124 jours) dans le groupe placebo. Dans le groupe Imfinzi, 25 patients ont reçu une corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour), et 1 patient a reçu l'infliximab en traitement adjuvant. Dans le groupe placebo, 7 patients ont reçu une corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). 18 patients se sont rétablis dans le groupe Imfinzi contre 3 dans le groupe placebo.
La pneumonite radique est souvent observée chez les patients traités par radiothérapie des poumons. Les tableaux cliniques de la pneumonite et de la pneumonite radique sont très similaires. Dans l'étude PACIFIC, une pneumonite (pneumonite à médiation immunitaire et pneumonite radique) est survenue chez 161 patients (33,9%) dans le groupe traité par Imfinzi et chez 58 patients (24,8%) dans le groupe placebo, dont 3,4%/3,0% de grade 3 et 1,1%/1,7% de grade 5.
Dans l'étude ADRIATIC, une pneumonite ou une pneumonite radique est survenue chez 100 patients (38,2 %) dans le groupe Imfinzi et chez 80 patients (30,2 %) dans le groupe placebo, dont 8 cas (3,1 %) de grade 3 dans le groupe Imfinzi par comparaison avec 6 (2,3 %) dans le groupe placebo et 1 cas (0,4 %) de grade 5 dans le groupe Imfinzi par comparaison avec 0 dans le groupe placebo.
Hépatite à médiation immunitaire
Sous monothérapie par Imfinzi, une hépatite à médiation immunitaire est survenue chez 120 patients (2,6%), dont 70 cas de grade 3 (1,5%), 9 patients (0,2%) de grade 4 et 6 patients (0,1%) de grade 5. Le délai médian de survenue a été de 36 jours (extrêmes: 1-644 jours). Quatre-vingt-quatorze patients sur 120 ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Neuf patients ont reçu d'autres immunosuppresseurs en traitement adjuvant, dont le mycophénolate. Le traitement par Imfinzi a été arrêté chez 30 patients. Cinquante-six patients se sont rétablis.
Sous Imfinzi + chimiothérapie, une hépatite à médiation immunitaire est survenue chez 40 patients (2,1%), dont 20 cas de grade 3 (1,1%), 7 cas de grade 4 (0,4%) et 1 cas de grade 5 (0,1%). Le délai médian de survenue a été de 78 jours (fourchette: 6-455 jours). Vingt-sept des 40 patients ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Deux patients ont reçu d'autres immunosuppresseurs. Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez 15 patients. Trente-cinq patients se sont rétablis.
Colite ou diarrhée à médiation immunitaire
Sous monothérapie par Imfinzi, une colite ou une diarrhée à médiation immunitaire est survenue chez 79 patients (1,7%), dont 15 cas de grade 3 (0,3%), 2 cas de grade 4 (< 0,1%) et un cas (< 0,1 %) de grade 5. Le délai médian de survenue était de 72 jours (extrêmes: 1-920 jours). Cinquante-cinq des 79 patients ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Cinq ont reçu d'autres immunosuppresseurs en traitement adjuvant, dont l'infliximab et le mycophénolate. Le traitement par Imfinzi a été arrêté chez 15 patients. Cinquante-quatre patients se sont rétablis.
Sous Imfinzi + chimiothérapie, une colite ou une diarrhée à médiation immunitaire est survenue chez 24 patients (1,3%), dont 5 cas de grade 3 (0,3%). Le délai médian de survenue a été de 193 jours (fourchette: 6-657 jours). Quatorze patients sur 24 ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez 9 patients. 20 patients se sont rétablis.
Endocrinopathies à médiation immunitaire
Hypothyroïdie à médiation immunitaire
Sous monothérapie par Imfinzi, une hypothyroïdie à médiation immunitaire est survenue chez 384 patients (8,3%), dont 7 cas de grade 3 (<0,2%). Le délai médian de survenue était de 91 jours (extrêmes: 1-951 jours). Parmi les 384 patients, 379 ont reçu un traitement hormonal substitutif, 7 une corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour) suivie d'un traitement hormonal substitutif. Un patient a été contraint d'interrompre le traitement par Imfinzi en raison d'une hypothyroïdie à médiation immunitaire. L'hypothyroïdie à médiation immunitaire était précédée d'une hyperthyroïdie à médiation immunitaire chez 25 patients et d'une thyroïdite à médiation immunitaire chez 2 patients.
Sous Imfinzi + chimiothérapie, une hypothyroïdie à médiation immunitaire est survenue chez 163 patients (7,7%) et un cas de grade 3 a été rapporté. Le délai médian de survenue a été de 175 jours (fourchette: 1-659 jours). 121 des 163 patients ont reçu un traitement hormonal substitutif. Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez un patient en raison d'une hypothyroïdie à médiation immunitaire.
Hyperthyroïdie à médiation immunitaire
Sous monothérapie par Imfinzi, une hyperthyroïdie à médiation immunitaire est survenue chez 76 patients (1,6%), et aucun cas de grade 3 ou 4 n'a été rapporté. Le délai médian de survenue était de 43 jours (extrêmes: 1-253 jours). Soixante et onze patients sur 76 ont reçu un traitement médicamenteux (thiamazole, carbimazole, propylthiouracil, perchlorate, bloqueur des canaux calciques ou bêtabloquant), 15 patients ont été traités par corticothérapie systémique, dont 8 à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Un patient a été contraint d'interrompre le traitement par Imfinzi en raison d'une hyperthyroïdie à médiation immunitaire. Soixante-deux patients se sont rétablis. Une hypothyroïdie est survenue chez 31 patients à la suite d'une hyperthyroïdie.
Sous Imfinzi + chimiothérapie, une hyperthyroïdie à médiation immunitaire est survenue chez 40 patients (2,1%), dont 1 cas de grade 3 (0,1%). Le délai médian de survenue a été de 68 jours (fourchette: 21-372 jours). Trente-deux des 40 patients ont reçu un traitement hormonal substitutif, 4 patients ont été traités par corticothérapie systémique à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi et/ou par la chimiothérapie n'a été arrêté chez aucun patient en raison d'une hyperthyroïdie à médiation immunitaire. 27 patients se sont rétablis.
Thyroïdite à médiation immunitaire
Sous Imfinzi en monothérapie, une thyroïdite à médiation immunitaire est survenue chez 21 patients (0,5%), dont 2 patients (<0,1%) de grade 3. Le délai médian de survenue était de 57 jours (extrêmes: 14-217 jours). Dix-huit des 21 patients ont été traités par traitement hormonal de substitution, 3 patients ont reçu une corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Imfinzi a été arrêté chez un patient à cause d'une thyroïdite à médiation immunitaire. Huit patients se sont rétablis. Une hypothyroïdie est survenue chez 5 patients à la suite d'une thyroïdite.
Sous Imfinzi + chimiothérapie, une thyroïdite à médiation immunitaire est survenue chez 11 patients (0,6%) et aucun cas de grade 3 ou 4 n'a été rapporté. Le délai médian de survenue a été de 100 jours (fourchette: 43-260 jours). Dix des 11 patients ont reçu un traitement hormonal substitutif et 3 patients ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez deux patients en raison d'une thyroïdite à médiation immunitaire. 3 patients se sont rétablis.
Insuffisance surrénalienne à médiation immunitaire
Sous monothérapie par Imfinzi, une insuffisance surrénalienne à médiation immunitaire est survenue chez 24 patients (0,5%), dont 8 cas de grade 3 (< 0,2%). Le délai médian de survenue était de 158 jours (extrêmes: 20-547 jours). Les 24 patients ont été traités par corticothérapie systémique, dont 8 à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Un patient a été contraint d'interrompre le traitement par Imfinzi en raison d'une insuffisance surrénalienne. Six patients se sont rétablis.
Sous Imfinzi + chimiothérapie, une insuffisance surrénalienne à médiation immunitaire est survenue chez 15 patients (0,8%), dont 1 cas de grade 3 (0,1%). Le délai médian de survenue a été de 234 jours (fourchette: 86-739 jours). Douze patients sur 15 ont été traités par une corticothérapie systémique, 2 patients sur 15 ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez un patient en raison d'une insuffisance surrénalienne à médiation immunitaire. 2 patients se sont rétablis.
Diabète sucré de type 1 à médiation immunitaire
Sous Imfinzi en monothérapie, un diabète sucré de type 1 à médiation immunitaire de grade 3 est survenu chez 5 patients (0,1%), dont 3 cas de grade 3 (0,1%) et 1 cas de grade 4 (<0,1%). Le délai médian de survenue était de 43 jours (fourchette: 29-631 jours). L'ensemble des 5 patients ont reçu un traitement hormonal substitutif. Le traitement par Imfinzi a été arrêté chez un patient en raison d'un diabète sucré de type 1 à médiation immunitaire. 2 patients se sont rétablis.
Sous Imfinzi + chimiothérapie, un diabète sucré de type 1 à médiation immunitaire est survenu chez 4 patients (0,2%), dont 2 cas de grade 3 (0,1%) et 2 cas de grade 4 (0,1%). Le délai médian de survenue a été de 157 jours (fourchette: 68-316 jours). Un patient a été traité par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). L'ensemble des 4 patients ont reçu un traitement hormonal substitutif. Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté définitivement chez 1 patient en raison d'un diabète sucré de type 1 à médiation immunitaire.
Hypophysite/Insuffisance hypophysaire à médiation immunitaire
Sous monothérapie par Imfinzi, une hypophysite/insuffisance hypophysaire à médiation immunitaire est survenue chez 6 patients (0,1 %), dont 5 cas (0,1 %) de grade 3. Le délai médian de survenue était 85 jours (fourchette: 44-225 jours). L'ensemble des 6 patients ont été traités par corticothérapie systémique, et 3 patients sur 6 ont reçu une haute dose de corticostéroïdes (au moins 40 mg de prednisone ou l'équivalent par jour). Imfinzi a été arrêté chez trois patients à cause d'une hypophysite/insuffisance hypophysaire à médiation immunitaire.
Sous Imfinzi + chimiothérapie, une hypophysite/insuffisance hypophysaire à médiation immunitaire est survenue chez 6 patients (0,3%), dont 1 cas de grade 3 (0,1%). Le délai médian de survenue a été de 335 jours (fourchette: 53-420 jours). Cinq patients ont été traités par corticothérapie systémique et le traitement par Imfinzi et/ou par la chimiothérapie n'a été arrêté chez aucun patient en raison d'une hypophysite/insuffisance hypophysaire.
Néphrite à médiation immunitaire
Sous monothérapie par Imfinzi, une néphrite à médiation immunitaire est survenue chez 17 patients (0,4%), dont 4 cas de grade 3 (0,1%) et 1 cas de grade 4 (< 0,1%). Le délai médian de survenue était de 84 jours (extrêmes: 4-393 jours). Douze patients (0,3%) ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour) et un patient a également été traité par mycophénolate. Le traitement par Imfinzi a été arrêté chez 7 patients. Huit patients se sont rétablis.
Sous Imfinzi + chimiothérapie, une néphrite à médiation immunitaire est survenue chez 13 patients (0,7%), dont 4 cas de grade 3 (0,2%). Le délai médian de survenue a été de 152 jours (fourchette: 8-414 jours). Onze patients ont été traités par corticothérapie systémique, et l'ensemble des patients ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi et/ou par la chimiothérapie a été arrêté chez 8 patients. 7 patients se sont rétabli.
Exanthème à médiation immunitaire
Sous monothérapie par Imfinzi, un exanthème ou une dermatite à médiation immunitaire (incluant pemphigoïde) sont survenus chez 74 patients (1,6%), dont 20 cas de grade 3 (0,4%). Le délai médian de survenue était de 56 jours (extrêmes: 4-600 jours). L'ensemble des 74 patients ont été traités par corticothérapie systémique, et 37 patients sur 74 ont été traités par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par Imfinzi a été arrêté chez 5 patients. Quarante-six patients se sont rétablis.
Sous Imfinzi + chimiothérapie, un exanthème ou une dermatite à médiation immunitaire (incluant pemphigoïde) sont survenus chez 61 patients (3,3%), dont 12 cas de grade 3 (0,6%). Le délai médian de survenue était de 90 jours (fourchette: 1-856 jours). Trente-huit patients sur 61 ont été traités par une corticothérapie systémique et 11 de ces patients par corticothérapie à haute dose (au moins 40 mg de prednisone ou équivalent par jour). 1 patient a reçu d'autres immunosuppresseurs. Le traitement par Imfinzi et/ou la chimiothérapie a été arrêté chez 7 patients. 53 patients se sont rétablis.
Réactions liées à la perfusion
Sous monothérapie par Imfinzi, des réactions liées à la perfusion sont survenues chez 70 patients (1,5%), dont un grade de maximum 3 ou 4 chez 6 patients (0,1%). Aucun cas de grade 5 n'a été observé.
Sous Imfinzi + chimiothérapie, des réactions liées à la perfusion sont survenues chez 37 patients (2,0%) dont un grade de maximum 3 ou 4 chez un patient (< 0,1 %). Aucun cas de grade 5 n'a été observé.
Immunogénicité
Les résultats des tests d'immunogénicité dépendent directement de plusieurs facteurs, parmi lesquels la sensibilité et la spécificité du test, la méthodologie utilisée, la manipulation des échantillons, la date de prélèvement des échantillons, l'administration concomitante de médicaments ainsi que la maladie sous-jacente. En raison de la performance limitée du test, l'incidence du développement d'anticorps chez les patients sous Imfinzi peut être sous-estimée et une comparaison entre l'incidence des anticorps dirigés contre Imfinzi et l'incidence des anticorps dirigés contre d'autres produits peut être trompeuse.
Comme d'autres protéines thérapeutiques, le durvalumab possède également un potentiel immunogène. Parmi les 3511 patients du Durvalumab-Pan-Tumor-Pool ayant pu être évalués par un dosage des anticorps anti-médicament (Anti-Drug Antibodies, ADAs), 2,6 % (93/3511) ont présenté des résultats positifs au test de détection des anticorps anti-médicament. Des anticorps neutralisants dirigés contre le durvalumab ont été mis en évidence chez 0,5% (19/3511).
Dans l'étude ADRIATIC, 7 des 206 patients (3,4 %) traités par Imfinzi en monothérapie et évaluables en termes de présence d'ADA se sont révélés positifs aux ADA dus au traitement. Des anticorps neutralisants dirigés contre le durvalumab ont été mis en évidence chez 1 % (2/206) des patients. En raison du faible nombre de patients présentant des résultats positifs au test de détection des ADA pendant le traitement, on ne dispose pas de données suffisantes permettant d'évaluer si la présence d'ADA avait des effets sur la pharmacocinétique ou la sécurité du durvalumab.
Dans l'étude AEGEAN, parmi les 375 patients traités par Imfinzi 1500 mg en association avec une chimiothérapie toutes les 3 semaines avant l'opération, suivis d'Imfinzi 1500 mg toutes les 4 semaines après l'opération, et évaluables sur la présence d'ADA, 25 (6,7 %) ont présenté des résultats positifs au test de détection des ADA. Des anticorps neutralisants dirigés contre le durvalumab ont été mis en évidence chez 2 patients (0,5 %). En raison du faible nombre de patients présentant des résultats positifs au test de détection des ADA pendant le traitement, on ne dispose pas de données suffisantes permettant d'évaluer si la présence d'ADA avait des effets sur la pharmacocinétique ou la sécurité du durvalumab.
Dans l'étude CASPIAN, le résultat du test des anticorps anti-médicament était positif chez 0 (0%) des 201 patients évaluables sur la présence d'ADA recevant Imfinzi 1500 mg toutes les 3 semaines associé à une chimiothérapie. Les effets sur la pharmacocinétique et la sécurité clinique du durvalumab n'ont donc pas pu être évalués. Le développement d'ADA était associé à des niveaux d'exposition au durvalumab plus faibles, la réduction n'a toutefois pas été considérée comme cliniquement pertinente. Rien n'indique que les ADA aient une influence sur la sécurité du durvalumab. Les effets possibles des ADA sur l'efficacité du durvalumab n'ont pas pu être évalués puisque le nombre de patients positifs aux ADA dus au traitement était insuffisant.
Sur les 240 patients ayant été traités par Imfinzi 1500 mg toutes les 3 semaines associé à une chimiothérapie, suivis d'Imfinzi 1500 mg toutes les 4 semaines dans le cadre de l'étude TOPAZ-1 et étant évaluables sur la présence d'ADA, 2 patients (0,8%) ont eu un résultat positif au test des ADA apparus sous traitement. Le nombre de patients chez lesquels des ADA étaient survenus pendant le traitement ou présentant des anticorps neutralisants (2 patients dans chaque cas) était trop faible pour pouvoir déterminer si les ADA ont un impact sur la pharmacocinétique et l'efficacité clinique du durvalumab.
Sur les 453 patients ayant été traités par Imfinzi 1500 mg toutes les 3 semaines en association avec une chimiothérapie avant l'opération, puis par Imfinzi 1500 mg toutes les 4 semaines après l'opération dans le cadre de l'étude NIAGARA et étant évaluables sur la présence d'ADA, 8 patients (1,8 %) ont eu un résultat positif au test des ADA apparus sous traitement. Des anticorps neutralisants dirigés contre le durvalumab ont été mis en évidence chez 6 patients (1,3 %). La présence d'ADA n'a eu aucun effet apparent sur la pharmacocinétique ou la sécurité.
Enfants et adolescents
Dans l'étude clinique D419EC00001, dans laquelle 50 patients pédiatriques (< 18 ans) ont été exami-nés, les événements indésirables de vomissements, anémie, maux de tête, nausées, pyrexie, douleurs abdominales et augmentation de l'alanine-aminotransférase sont apparus dans toutes les phases de traitement chez plus de 20% des patients. Dans cette étude, des événements indésirables de grade 3 ou 4 ont été signalés chez 20 patients sur 50 (40%). Dans la population pédiatrique, aucun nouveau signal de sécurité n'a été observé par rapport aux profils de sécurité connus d'Imfinzi chez les adultes.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe aucun traitement spécifique en cas de surdosage au durvalumab, et les symptômes d'un surdosage ne sont pas connus. En cas de surdosage, prendre les mesures de secours générales et traiter les éventuels symptômes.
Propriétés/EffetsCode ATC
L01FF03
Mécanisme d'action/pharmacodynamique
L'expression du ligand de mort cellulaire programmée de type 1 (PD-L1) est une réponse immunitaire adaptative qui dissimule les tumeurs et les protège contre le système immunitaire. Le PD-L1 peut être activé par des signaux inflammatoires (p.ex. IFNγ) et exprimé sur les cellules tumorales comme sur les cellules immunitaires associées à la tumeur dans le micro-environnement tumoral. Le PD-L1 inhibe le fonctionnement des lymphocytes T et leur activation par le biais de son interaction avec le PD-1 et la protéine CD80 (B7-1). En se fixant à leurs récepteurs, le PD-L1 diminue l'activité et la multiplication des lymphocytes T, ainsi que la production de cytokine.
Le durvalumab est un anticorps monoclonal à forte affinité de liaison, entièrement humanisé, de la sous-classe d'immunoglobuline de type G1 kappa (IgG1κ) qui inhibe de manière sélective l'interaction du PD-L1 avec le PD-1 et la CD80 (B7-1), sans toutefois influer sur l'interaction PD-1/PD-L2. Le durvalumab ne provoque aucune cytotoxicité à médiation cellulaire dépendante des anticorps (ADCC). L'inhibition sélective des interactions PD-L1/PD-1 et PD-L1/CD80 accroît la réponse immunitaire antitumorale. Cette réponse antitumorale peut entraîner l'élimination des tumeurs.
Dans des études précliniques, l'inhibition du PD-L1 a induit une activation accrue des lymphocytes T et une réduction de la taille des tumeurs.
Efficacité clinique
Cancer du poumon non à petites cellules (CPNPC) – Étude PACIFIC
Sur la base de la relation modélisée entre l'exposition et l'efficacité/la sécurité, étayée par des données cliniques limitées, aucune différence clinique pertinente n'a été observée pour les posologies d'Imfinzi de 10 mg/kg toutes les 2 semaines et de 1500 mg toutes les 4 semaines chez des patients présentant un cancer du poumon non à petites cellules (CPNPC) localement avancé et non résécable, dont la maladie n'a pas progressé après chimioradiothérapie définitive à base de platine.
L'efficacité d'Imfinzi a été établie dans le cadre de l'étude multicentrique PACIFIC, contrôlée par placebo, randomisée et menée en double chez 713 patients présentant un CPNPC inopérable localement avancé, confirmé par des analyses histologiques ou cytologiques. Les patients avaient subi au moins deux cycles de chimioradiothérapie définitive à base de platine 1 à 42 jours avant le début du traitement à l'étude et présentaient un score de performance ECOG de 0 ou 1. 92% des patients ont reçu une dose de rayonnement totale de 54 à 66 Gy. Ont été exclus de cette étude les patients montrant une progression après une chimioradiothérapie, les patients souffrant d'une maladie auto-immune active ou d'un antécédent documenté de maladie auto-immune dans les 2 ans précédant le début de l'étude, les patients immunodéprimés, les patients ayant développé des réactions indésirables graves à médiation immunitaire, les patients atteints de maladies nécessitant une immunosuppression, sauf une dose physiologique de corticoïdes systémiques, les patients souffrant d'une tuberculose active ou d'une hépatite B ou C ou infectés par le VIH et les patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant ou suivant le début du traitement par Imfinzi. Les patients ont été randomisés selon un rapport 2:1 et ont reçu 10 mg/kg d'Imfinzi (n = 476) ou un placebo (n = 237) par perfusion intraveineuse toutes les 2 semaines pendant 12 mois, jusqu'à la survenue d'une toxicité inacceptable ou jusqu'à la progression confirmée de la maladie. La randomisation a été stratifiée selon le sexe, l'âge (< 65 ans vs ≥65 ans) et le statut tabagique (fumeur ou non-fumeur). Des évaluations de la réponse tumorale ont été réalisées toutes les 8 semaines pendant les 12 premiers mois, puis toutes les 12 semaines.
Les patients ont été inclus dans l'étude indépendamment du statut d'expression tumorale PD-L1. Lorsqu'ils étaient disponibles, des échantillons de tissus tumoraux d'archives prélevés avant la chimioradiothérapie ont été testés rétrospectivement pour l'expression de PD-L1 sur des cellules tumorales en utilisant le test IHC VENTANA PD-L1 (SP263).
Les caractéristiques démographiques et pathologiques à l'inclusion étaient équilibrées entre les groupes de l'étude. La composition de la population totale au début de l'étude était la suivante: hommes (70%), âge ≥65 ans (45%), caucasiens (69%), asiatiques (27%), autres (4%), fumeurs (16%), anciens fumeurs (75%) et non-fumeurs (9%), score OMS/ECOG de 0 (49%), score OMS/ECOG de 1 (51%). Les caractéristiques pathologiques étaient les suivantes: stade IIIA (53 %), stade IIIB (45%), sous-groupes histologiques de types squameux (46%), non squameux (54%).
Les principaux critères de jugement de l'étude ont été la survie globale (SG) et la survie sans progression (SSP) sous Imfinzi vs placebo.
L'étude a démontré une amélioration statistiquement significative de la SG dans le groupe Imfinzi comparativement au groupe placebo [risque relatif (RR) = 0,68 (IC à 95%: 0,53 à 0,87), p = 0,00251]. Au moment de l'analyse provisoire de la SG (22 mars 2018), la durée médiane de suivi était de 25,2 mois; 299 patients (183 (38,4%) dans le groupe durvalumab et 116 patients (48,9%) dans le groupe placebo) étaient décédés. La survie globale médiane n'avait pas encore été atteinte dans le groupe durvalumab; dans le groupe placebo, elle était de 28,7 mois selon l'estimation de Kaplan-Meier. La survie globale après 24 mois était de 66,3% dans le groupe Imfinzi et de 55,6% dans le groupe placebo.
Au moment de l'actualisation des données au bout du suivi de 5 ans, 23 nouveaux événements SG au total ont été rapportés depuis la deuxième analyse du suivi SG (au total 419 décès [264 dans le groupe Imfinzi et 155 dans le groupe placebo]). Le bénéfice de survie d'Imfinzi par rapport au placebo était cohérent avec l'analyse primaire SG avec une réduction du risque de mortalité de 28% par rapport au placebo [HR = 0,72 (IC à 95%: 0,59-0,89)]. L'estimation de Kaplan-Meier de la SG médiane était de 29,1 mois dans le groupe placebo par rapport à 47,5 mois dans le groupe Infimzi. Les taux de survie globale estimés étaient de 42,9% pour le groupe Imfinzi et 33,4% pour le groupe placebo.
À la date de l'analyse intermédiaire (13 février 2017) par un comité d'examen central indépendant agissant en aveugle conformément aux critères RECIST 1.1, l'étude a montré une amélioration statistiquement significative de la SSP dans le groupe Imfinzi comparé au groupe placebo [RR = 0,52 (IC à 95%: 0,42 à 0,65), p < 0,0001]. À cette date, 371 patients (214 (45,0%) dans le groupe durvalumab et 157 (66,2%) dans le groupe placebo présentaient une progression de la maladie ou étaient décédés1. La SSP médiane selon l'estimation de Kaplan-Meier a été de 16,8 mois dans le groupe Imfinzi et de 5,6 mois dans le groupe placebo. La SSP à 12 mois a été de 55,9% dans le groupe Imfinzi et de 35,5% dans le groupe placebo. La SSP à 18 mois a été de 44,2 % dans le groupe Imfinzi et de 27,0% dans le groupe placebo. Des améliorations de la SG et de la SSP au bénéfice des patients du groupe Imfinzi par rapport au groupe placebo ont été observées en permanence dans l'ensemble des sous-groupes prédéfinis analysés (groupe ethnique, âge, sexe, statut tabagique, statut de mutation de l'EGFR et histologie).
Au moment de l'analyse du suivi de 5 ans (11 janvier 2021) basée sur les évaluations de la SSP par un comité d'examen central indépendant conformément à RECIST 1.1, le bénéfice de SSP [HR: 0,55 (IC à 95%: 0,45-0,68)] constaté dans le groupe Imfinzi était cohérent avec l'analyse primaire de la SSP [HR: 0,52: (IC à 95%: 0,42-0,65)]. L'estimation de Kaplan-Meier de la SSP médiane s'élevait à 16,9 mois dans le groupe durvalumab (IC à 95%: 13,0-23,9) par comparaison avec 5,6 mois dans le groupe placebo (IC à 95%: 4,8-7,7).
Dans le groupe traité par Imfinzi, un effet indésirable d'issue fatale est survenu chez 21 patients (4,4%), et un effet indésirable d'issue fatale est survenu chez 14 patients (6,0%) du groupe placebo.
Analyse post-hoc exploratoire en sous-groupe en fonction de l'expression du PD-L1
Sur les 713 patients randomisés de l'étude PACIFIC, 451 (63%) ont fourni un échantillon de tissu d'une qualité et d'une quantité suffisantes pour déterminer l'expression du PD-L1. Parmi les 451 échantillons, 33% présentaient une expression du PD-L1 sur les cellules tumorales < 1%, 67% une expression du PD-L1 sur les cellules tumorales ≥1%, 32% une expression du PD-L1 sur les cellules tumorales 1 - < 25%, 35% une expression du PD-L1 sur les cellules tumorales ≥25%.
Les analyses post-hoc en sous-groupes ont été effectuées afin d'évaluer l'efficacité dans les cas supplémentaires présentant des niveaux d'expression tumorale PD-L1 (< 1%, ≥1%, 1 à < 25%) qui n'étaient pas prévus dans le plan d'analyse statistique.
SSP:
Dans le sous-groupe ne présentant pas d'expression du PD-L1 (expression du PD-L1 sur les cellules tumorales < 1%), le RR de la SSP était de 0,73; IC à 95%: 0,48 à 1,11.
Pour les sous-groupes analysés présentant une expression du PD-L1 positive (expression du PD-L1 sur les cellules tumorales ≥1%) et les patients dont le statut d'expression PD-L1 ne pouvait être établi (PD-L1 sur les cellules tumorales inconnu), les résultats suivants ont été observés: expression du PD-L1 sur les cellules tumorales ≥1% RR: 0,46; IC à 95%: 0,33 à 0,64; expression du PD-L1 sur les cellules tumorales jusqu'à < 25% RR: 0,49; IC à 95%: 0,30 à 0,80; expression du PD-L1 sur les cellules tumorales ≥25% RR: 0,41; IC à 95%: 0,26 à 0,65; expression du PD-L1 sur les cellules tumorales inconnue RR: 0,59; IC à 95%: 0,42 à 0,83).
SG:
Dans le sous-groupe ne présentant pas d'expression du PD-L1 (expression du PD-L1 sur les cellules tumorales <1%), le RR de la SG était de 1,36; IC à 95%: 0,79 à 2,34.
Pour les sous-groupes analysés présentant une expression du PD-L1 positive (expression du PD-L1 sur les cellules tumorales ≥1%) et le sous-groupe dont le statut d'expression PD-L1 ne pouvait être établi (expression du PD-L1 sur les cellules tumorales inconnue), les résultats suivants ont été observés: expression du PD-L1 sur les cellules tumorales ≥1% RR: 0,53; IC à 95%: 0,36 à 0,77; expression du PD-L1 sur les cellules tumorales 1 à < 25% RR: 0,60; IC à 95%: 0,35 à 1,03; expression du PD-L1 sur les cellules tumorales ≥25% RR: 0,46; IC à 95%: 0,27 à 0,78; expression du PD-L1 sur les cellules tumorales inconnue RR: 0,62; IC à 95%: 0,43 à 0,89.
CPNPC résécable – étude AEGEAN
AEGEAN est une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, multicentrique évaluant l'efficacité d'Imfinzi en association avec une chimiothérapie comme traitement néoadjuvant, suivis d'une monothérapie par Imfinzi après l'opération chez les patients présentant un CPNPC résécable (stade IIA à stade IIIB (N2) chez certains patients [AJCC, 8e édition]). Des patients naïfs de traitement atteints d'un CPNPC épidermoïde ou non épidermoïde documenté sans exposition antérieure à une immunothérapie, présentant un indice de performance OMS/ECOG de 0 ou 1 et au moins une lésion cible selon les critères RECIST 1.1. ont été inclus dans l'étude. Le niveau d'expression tumorale PD-L1 a été confirmé par le test Ventana PD-L1 (SP263) avant la randomisation.
Les patients présentant une maladie auto-immune active ou un antécédent documenté de maladie auto-immune ou ayant utilisé des médicaments immunosuppresseurs dans les 14 jours précédant la première dose de durvalumab ont été exclus de l'étude. Étaient exclus de l'étude les patients dont l'opération planifiée était une résection segmentaire, une résection cunéiforme ou (après une modification du protocole) une pneumonectomie. Les opérations réalisées pendant l'étude ont été effectuées dans le cadre du traitement standard et pouvaient comporter une pneumonectomie. Après une modification du protocole, aucun patient atteint de tumeurs T4 avec invasion du diaphragme, invasion médiastinale, invasion du cœur, invasion des gros vaisseaux, de la trachée, du nerf laryngé récurrent, de l'œsophage ou de la carène n'a plus été inclus ultérieurement dans l'étude. Les patients atteints de mutations de l'EGFR connues ou de réarrangements de l'ALK ont été exclus de la population de l'étude pour l'analyse de l'efficacité (analyse en intention de traiter modifiée [ITTm]).
La randomisation a été stratifiée en fonction du stade de la maladie (stade II vs stade III) et du niveau d'expression PD-L1 (TC < 1 % vs TC ≥1 %).
Dans l'étude AEGEAN, 802 patients ont été randomisés dans un rapport 1:1 pour recevoir le traitement périopératoire par Imfinzi (bras 1) ou le placebo (bras 2) en association avec une chimiothérapie néoadjuvante. Le crossover entre les bras de l'étude n'était pas autorisé. L'analyse de l'efficacité a été effectuée sur la base de 740 patients dans la population ITTm.
·Bras 1: Imfinzi 1500 mg + chimiothérapie toutes les 3 semaines pendant jusqu'à 4 cycles avant l'opération, suivis d'Imfinzi 1500 mg toutes les 4 semaines pendant jusqu'à 12 cycles après l'opération.
·Bras 2: placebo + chimiothérapie toutes les 3 semaines pendant jusqu'à 4 cycles avant l'opération, suivis du placebo toutes les 4 semaines pendant jusqu'à 12 cycles après l'opération.
Le choix de la chimiothérapie standard en tenant compte de l'histologie était à la discrétion de l'investigateur. En cas de tumeurs épidermoïdes: carboplatine (ASC 6) et paclitaxel (200 mg/m2) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum ou cisplatine (75 mg/m2) le jour 1 et gemcitabine (1250 mg/m2) les jours 1 et 8 de chaque cycle de 3 semaines pendant 4 cycles au maximum. L'utilisation de carboplatine (ASC 5) associé à la gemcitabine (au lieu du cisplatine) était autorisée chez les patients présentant des comorbidités ou qui, selon le jugement de l'investigateur, ne toléraient pas le cisplatine. En cas de tumeurs non épidermoïdes: pémétrexed (500 mg/m2) et cisplatine (75 mg/m2) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum ou pémétrexed (500 mg/m2) et carboplatine (ASC 5) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum. En cas de mauvaise tolérance, les patients ont pu passer du traitement par le cisplatine au traitement par le carboplatine à tout moment de l'étude (lorsqu'ils étaient éligibles à ce traitement).
Les patients qui avaient subi une opération avec des marges de résection R0/R1 ont pu poursuivre le traitement adjuvant.
Une évaluation de la tumeur selon les critères RECIST 1.1 a été effectuée à l'inclusion et au terme de la phase néoadjuvante (avant l'opération). Le premier examen postopératoire CT/IRM du thorax et de l'abdomen (y compris de la totalité du foie et des deux glandes surrénales) a été effectué 5 semaines ± 2 semaines après et avant l'opération, mais à une date aussi proche que possible de l'instauration du traitement adjuvant. Ensuite, des évaluations de la tumeur ont eu lieu toutes les 12 semaines (en fonction de la date de l'opération) jusqu'à la semaine 48, toutes les 24 semaines (en fonction de la date de l'opération) jusqu'à la semaine 192 (env. 4 ans) et ensuite toutes les 48 semaines (en fonction de la date de l'opération) jusqu'à la progression de la maladie (PM) confirmée radiologiquement selon les critères RECIST 1.1, jusqu'à la révocation du consentement ou jusqu'au décès.
Le statut de survie a été contrôlé 2, 3 et 4 mois après la fin du traitement, ensuite tous les 2 mois jusqu'au mois 12 puis tous les 3 mois.
Les critères d'évaluation primaires de l'étude étaient la réponse pathologique complète (RPC) conformément à un examen pathologique central en aveugle ainsi que la survie sans événements (SSE) conformément à une évaluation centrale indépendante en aveugle (BICR). Les critères d'évaluation secondaires principaux étaient la réponse pathologique majeure (RPM) conformément à un examen pathologique central en aveugle, la survie sans maladie (SSM) conformément à une BICR et la survie globale (SG). D'autres objectifs d'efficacité secondaires étaient la SSE (population de l'analyse PD-L1-TC ≥1 %) et la RPC (population de l'analyse PD-L1-TC ≥1 %).
Lors de l'analyse intermédiaire planifiée de la RPC, l'étude a atteint la limite prédéfinie pour la déclaration de la signification statistique pour la RPC et la RPM. Par la suite, lors de la première analyse intermédiaire planifiée de la SSE, l'étude a atteint sa limite prédéfinie pour la déclaration de la signification statistique pour la SSE.
Les caractéristiques démographiques et pathologiques au début de l'étude étaient équilibrées dans les deux bras de l'étude (366 patients dans le bras 1 et 374 patients dans le bras 2 de la population ITTm). Les caractéristiques initiales démographiques et spécifiques à la maladie de la population pour l'analyse de l'efficacité (ITTm) étaient les suivantes: hommes (71,6 %), femmes (28,4 %), âge ≥65 ans (51,6 %), âge médian 65 ans (fourchette: 30 à 88 ans), score OMS/ECOG-PS 0 (68,4 %), score OMS/ECOG-PS 1 (31,6 %), blancs (53,6 %), asiatiques (41,5 %), noirs ou afro-américains (0,9 %), amérindiens ou indigènes d'Alaska (1,4 %), autre origine (2,6 %), hispaniques ou latino-américains (16,1 %), non hispaniques ou non latino-américains (83,9 %), fumeurs actuels ou anciens fumeurs (85,5 %), non-fumeurs (14,5 %), histologie épidermoïde (48,6 %) et histologie non épidermoïde (50,7 %), stade II (28,4 %), stade III (71,6 %), niveau d'expression PD-L1 TC ≥1 % (66,6 %), niveau d'expression PD-L1 TC < 1 % (33,4 %). Les données démographiques et les caractéristiques initiales de la population ITTm étaient comparables à celles de la population ITT, à l'exception de l'absence de patients présentant des mutations de l'EGFR connues ou des réarrangements de l'ALK.
L'étude a montré une amélioration statistiquement significative de la SSE [HR = 0,68 (IC à 95 %: 0,53; 0,88), p = 0,003902] dans le bras Imfinzi par comparaison avec le bras placebo. En outre, l'étude a montré une amélioration statistiquement significative de la RPC [différence des proportions 12,96 % (IC à 95 %: 8,67, 17,57)] dans le bras Imfinzi par comparaison avec le bras placebo. Les données relatives à la survie globale (SG) n'étaient pas suffisamment matures au moment de l'analyse de la SSE. Voir Tableau 3.
Tableau 3: Résultats d'efficacité de l'étude AEGEAN (ITTm)
|
|
Imfinzi + chimiothérapie
(n = 366)
|
Placebo + chimiothérapie
(n = 374)
| |
SSEa
| |
Nombre d'événements, n (%)
|
98 (26,8)
|
138 (36,9)
| |
SSE médiane (IC à 95 %) (mois)
|
NR (31,9; NR)
|
25,9 (18,9; NR)
| |
SSE après 12 mois, % (IC à 95 %)
|
73,4 (67,9; 78,1)
|
64,5 (58,8; 69,6)
| |
SSE après 24 mois, % (IC à 95 %)
|
63,3 (56,1; 69,6)
|
52,4 (45,4; 59,0)
| |
Hazard Ratio (IC à 95 %)
|
0,68 (0,53; 0,88)
| |
Valeur p bilatéraled
|
0,003902
| |
RPCa,b,d
| |
Nombre de patients présentant une réponse
|
63
|
16
| |
Taux de réponse, % (IC à 95 %)
|
17,21 (13,49; 21,48)
|
4,28 (2,46; 6,85)
| |
Différence des proportions, % (IC à 95 %)
|
12,96 (8,67; 17,57)
| |
RPMa,c,d
| |
Nombre de patients présentant une réponse
|
122
|
46
| |
Taux de réponse, % (IC à 95 %)
|
33,33 (28,52; 38,42)
|
12,30 (9,15; 16,06)
| |
Différence des proportions, % (IC à 95 %)
|
21,03 (15,14; 26,93)
|
a Les résultats se basent sur l'analyse intermédiaire de la SSE préplanifiée et l'analyse finale de la RPC/RPM (DCO: 10 novembre 2022) qui a été effectuée 46,3 mois après le début de l'étude.
b Basée sur une analyse intermédiaire de la RPC prédéfinie (DCO: 14 janvier 2022), chez n = 402, le taux deRPC était statistiquement significatif (p = 0,000036) par comparaison avec le niveau de signification de 0,0082 %.
c Basée sur une analyse intermédiaire de la RPM prédéfinie (DCO: 14 janvier 2022), chez n = 402, le taux de RPM était statistiquement significatif (p = 0,000002) par comparaison avec le niveau de signification de 0,0082 %.
d La valeur p bilatérale pour la RPC et la RPM a été calculée sur la base d'un test CMH stratifié. La valeur p bilatérale pour la SSE a été calculée sur la base d'un test du log-rank stratifié. Les facteurs de stratification étaient le PD-L1 et le stade de la maladie.
La limite pour la déclaration de la signification statistique pour chacun des critères d'évaluation de l'efficacité a été déterminée par une fonction de dépense alpha selon la méthode de Lan et DeMets voisine d'une approche de O'Brien et Flemming (SSE = 0,9899 %, RPC = 0,0082 %, RPM = 0,0082 %, bilatéral).
Au moment de la DCO le 14 août 2023, la maturité concernant la survie globale (SG) était de 28,6 % (212 événements de SG). Dans le sous-groupe PD-L1 TC <1 % de la population ITTm, 27 patients (22,1 %) dans le bras Imfinzi + chimiothérapie et 39 patients (31,2 %) dans le bras placebo + chimiothérapie étaient décédés. Dans le sous-groupe PD-L1 TC ≥1 %, 72 patients (29,5 %) et 74 patients (29,7 %) étaient décédés dans le bras de l'étude respectif.
Cancer du poumon à petites cellules à un stade limité (LS-SCLC) – Étude ADRIATIC
Dans l'étude ADRIATIC, l'efficacité d'Imfinzi a été évaluée avec ou sans trémélimumab. L'étude ADRIATIC était une étude randomisée, en double aveugle, contrôlée contre placebo, multicentrique sur 730 patients atteints de LS-SCLC confirmé par des analyses histologiques ou cytologiques (stade I à III conformément à la 8e édition de l'AJCC), dont la maladie n'a pas progressé après une chimioradiothérapie concomitante. Les patients de stade I ou II devaient être inopérables, comme constaté par l'investigateur. Les patients ont subi 3 à 4 cycles de chimioradiothérapie définitive à base de platine, soit avec 60 à 66 Gy une fois par jour (QD) pendant 6 semaines, soit avec 45 Gy deux fois par jour (BID) pendant 3 semaines, 1 à 42 jours avant la première dose du traitement à l'étude. Une irradiation crânienne prophylactique (PCI) a pu être réalisée à la discrétion de l'investigateur après la fin de la chimioradiothérapie 1 à 42 jours avant la première dose du traitement à l'étude.
Les patients exclus étaient les patients présentant une maladie auto-immune active ou un antécédent documenté d'une maladie auto-immune dans les 5 années précédant le début de l'étude, des antécédents d'immunodéficience primaire active, de pneumonite de grade ≥2, de tuberculose active, d'hépatite B ou C ou d'infection par le VIH, les patients présentant une pneumopathie interstitielle, les patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant le début du traitement par Imfinzi ainsi que les patients avec métastases cérébrales, compression de la colonne vertébrale ou antécédent de carcinomatose leptoméningée. Les patients présentant une histologie mixte de CPPC et de CPNPC ont également été exclus de l'étude.
La randomisation a été stratifiée en fonction du stade de la maladie (I/II ou III) et PCI (oui ou non). Les patients ont été randomisés selon un rapport 1:1:1 et répartis dans les groupes de traitement suivants:
·Bras 1: 1500 mg d'Imfinzi + placebo toutes les 4 semaines pendant 4 cycles, suivis de 1500 mg d'Imfinzi toutes les 4 semaines.
·Bras 2: placebo + deuxième placebo toutes les 4 semaines pendant 4 cycles, suivis d'un placebo toutes les 4 semaines.
·Bras 3: 1500 mg d'Imfinzi + 75 mg de trémélimumab toutes les 4 semaines pendant 4 cycles, suivis de 1500 mg d'Imfinzi toutes les 4 semaines.
Après que 600 patients ont été randomisés au total dans les trois bras de l'étude, les patients suivants ont été randomisés selon un rapport 1:1 et répartis dans les bras 1 et 2. Ils ont reçu soit 1500 mg d'Imfinzi toutes les 4 semaines soit un placebo toutes les 4 semaines.
Le traitement a été poursuivi jusqu'à progression de la maladie, toxicité inacceptable ou pendant 24 mois au plus. Des évaluations de la tumeur ont eu lieu toutes les 8 semaines jusqu'à la semaine 72, puis toutes les 12 semaines jusqu'à la semaine 96 et ensuite toutes les 24 semaines.
Les caractéristiques démographiques et pathologiques à l'inclusion étaient bien équilibrées entre les bras de l'étude. Les données démographiques initiales et les caractéristiques pathologiques à l'inclusion dans les groupes Imfinzi et placebo étaient: hommes (69,1 %), âge ≥65 ans (39,2 %), blancs (50,4 %), noirs ou afro-américains (0,8 %), asiatiques (47,5 %), autre origine (1,3 %), hispaniques ou latino-américains (4,2 %), fumeurs actuels (22,3 %), anciens fumeurs (68,5 %), non-fumeurs (9,2 %), score OMS/ECOG-PS 0 (48,7 %), score OMS/ECOG-PS 1 (51,3 %), stade I (3,6 %), stade II (9,1 %), stade III (87,4 %).
Avant la randomisation, tous les patients ont reçu une chimiothérapie à base de platine (66,2 % cisplatine + étoposide, 33,8 % carboplatine + étoposide); 72,1 % des patients ont reçu une RT QD (dont 92,4 % avec ≥60 - ≤66 Gy QD); 27,9 % ont reçu une RT BID (dont 96,6 % avec 45 Gy BID) et 53,8 % des patients ont reçu une PCI. La réponse à la CRT était comme suit: réponse complète (12,3 %), réponse partielle (73,8 %), maladie stable (14,0 %).
Les deux critères d'évaluation primaires de l'étude ont été la SG et la SSP sous Imfinzi vs placebo. Les critères d'évaluation secondaires ont été l'ORR sous Imfinzi vs placebo. L'évaluation de la SSP et de l'ORR était effectuée par BICR selon RECIST v1.1.
Lors d'une analyse intermédiaire prévue, l'étude a montré une amélioration statistiquement significative et cliniquement pertinente de la SG pour Imfinzi par comparaison avec le placebo [HR = 0,73 (IC à 95 %: 0,569, 0,928), p=0,01042]. L'étude a en outre montré une amélioration statistiquement significative et cliniquement pertinente de la SSP pour Imfinzi par comparaison avec le placebo [HR = 0,76 (IC à 95 %: 0,606, 0,950), p=0,01608]. Voir Tableau 4.
Tableau 4. Données d'efficacité de l'étude ADRIATIC
|
|
Bras 1: Imfinzi (n = 264)
|
Bras 2: Placebo (n = 266)
| |
SGa
| |
Nombre de décès (%)
|
115 (43,6)
|
146 (54,9)
| |
SG médiane (mois) (IC à 95 %)b
|
55,9 (37,3, NR)
|
33,4 (25,5, 39,9)
| |
HR (IC à 95 %)c
|
0,73 (0,569, 0,928)
| |
Valeur pd
|
0,01042
| |
SG au bout de 24 mois (%) (IC à 95 %)b
|
68,0 (61,9, 73,3)
|
58,5 (52,3, 64,3)
| |
SG au bout de 36 mois (%) (IC à 95 %)b
|
56,5 (50,0, 62,5)
|
47,6 (41,3, 53,7)
| |
SSPe
| |
Nombre d'événements (%)
|
139 (52,7)
|
169 (63,5)
| |
SSP médiane (mois) (IC à 95 %)b
|
16,6 (10,2, 28,2)
|
9,2 (7,4, 12,9)
| |
HR (IC à 95 %)f
|
0,76 (0,606, 0,950)
| |
Valeur pd
|
0,01608
| |
SSP au bout de 18 mois (%) (IC à 95 %)b
|
48,8 (42,2, 55,0)
|
36,1 (29,9, 42,2)
| |
SSP au bout de 24 mois (%) (IC à 95 %)b
|
46,2 (39,6, 52,5)
|
34,2 (28,2, 40,3)
| |
ORRe
| |
ORRg n (%)
|
53/175 (30,3)
|
54/169 (32,0)
| |
Réponse complète n (%)
|
5 (2,9)
|
4 (2,4)
| |
Réponse partielle n (%)
|
48 (27,4)
|
50 (29,6)
| |
Odds Ratio (IC à 95 %)
|
-1,2 (-11,0, 8,5)
| |
DdRb,e médiane (mois)
(IC à 95 %)
|
33,0 (22,4, NR)
|
27,7 (9,6, NR)
| |
Pourcentage de patients en rémission après 12 moisb,e (%) (IC à 95 %)
|
73,7 (59,0, 83,8)
|
60,3 (44,5, 72,9)
| |
Pourcentage de patients en rémission après 18 moisb,e (%) (IC à 95 %)
|
71,5 (56,6, 82,0)
|
55,2 (39,4, 68,5)
|
a La durée médiane du suivi de SG chez les patients censurés a été de 37,19 mois dans le groupe Imfinzi et de 37,24 mois dans le groupe placebo.
b Calculé en utilisant la méthode de Kaplan-Meier. IC de la médiane généré sur la base de la méthode de Brookmeyer-Crowley.
c L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié; tous deux ajustés pour le traitement par PCI.
d Valeur p se basant sur les résultats de l'analyse intermédiaire préplanifiée. Sur la base d'une fonction de dépense de l'alpha de Lan-DeMets avec une limite de type O'Brien Fleming et le nombre réel d'événements observés, la limite pour la déclaration de la signification statistique pour la SG était de 0,01679 pour un alpha global de 4,5 % et pour la SSP de 0,02805 pour un alpha global de 5 % (Lan et DeMets, 1983).
e Évalué par un BICR conformément à RECIST v1.1.
f L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié; tous deux ajustés en fonction du statut TNM et pour le traitement par PCI.
g Sur la base d'un sous-groupe de l'ensemble d'analyse global avec une maladie mesurable à l'inclusion conformément à RECIST v1.1; Imfinzi (n = 175), placebo (n = 169).
Les améliorations de la SG et de la SSP au bénéfice des patients traités par Imfinzi par rapport aux patients ayant reçu le placebo étaient globalement cohérentes sur l'ensemble des sous-groupes prédéfinis analysés.
Cancer du poumon à petites cellules avancé (ES-SCLC) – Étude CASPIAN
L'efficacité d'Imfinzi associé à l'étoposide et soit au carboplatine soit au cisplatine chez les patients ES-SCLC naïfs de traitement a été évaluée dans CASPIAN, une étude en ouvert, multicentrique, randomisée, avec contrôle actif. L'état de santé général selon WHO/ECOG des patients admissibles était de 0 ou 1 avec un poids corporel >30 kg; ils étaient éligibles à une chimiothérapie à base de platine comme traitement de première ligne du SCLC et avaient une espérance de vie ≥12 semaines. Les patients avec métastases cérébrales asymptomatiques ou traitées, les patients avec au moins une lésion cible selon RECIST 1.1 et une fonction organique et médullaire suffisante étaient également éligibles. Le choix des sels de platine était laissé à la discrétion de l'investigateur compte tenu de la clairance de la créatinine calculée. Les patients exclus étaient les patients avec antécédents de radiothérapie thoracique ou une radiothérapie thoracique de consolidation planifiée; intervention chirurgicale majeure dans les 28 jours avant la première dose; maladie intercurrente non contrôlée; carcinomatose leptoméningée; antécédents d'immunodéficience primaire active; maladie auto-immune y compris syndrome paranéoplasique (SNP); maladie auto-immune active ou documentée ou maladies inflammatoires; utilisation d'immunosuppresseurs systémiques dans les 14 jours avant la première administration du traitement, à l'exception de doses physiologiques de corticostéroïdes systémiques; infection active, y compris la tuberculose, l'hépatite B ou C, ou le VIH et patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant le début du traitement par Imfinzi ou devant le recevoir.
La randomisation était stratifiée en fonction du traitement à base de platine prévu au cycle 1 (carboplatine ou cisplatine).
L'évaluation de l'efficacité en cas d'ES-SCLC était basée sur une comparaison entre les groupes:
·Imfinzi 1500 mg et, selon le choix de l'investigateur, soit carboplatine (AUC 5 ou 6 mg/ml/min) ou cisplatine (75-80 mg/m2) le jour 1 et étoposide (80-100 mg/m2) en intraveineux les jours 1, 2 et 3 de chaque cycle de 21 jours pendant 4 cycles, suivis d'Imfinzi 1500 mg toutes les 4 semaines jusqu'à progression de la maladie ou toxicité inacceptable.
·Selon le choix de l'investigateur, soit carboplatine (AUC 5 ou 6 mg/ml/min) ou cisplatine (75-80 mg/m2) le jour 1 et étoposide (80-100 mg/m2) en intraveineux les jours 1, 2 et 3 de chaque cycle de 21 jours pendant jusqu'à 6 cycles. Irradiation crânienne prophylactique (Prophylactic Cranial Irradiation, PCI) après la fin de la chimiothérapie, à la discrétion de l'investigateur.
L'administration d'Imfinzi en monothérapie était également autorisée après une progression de la maladie dans la mesure où le patient était cliniquement stable et qu'un bénéfice clinique pouvait être attendu de l'avis de l'investigateur.
Les évaluations des tumeurs ont eu lieu 6 et 12 semaines après la randomisation et toutes les 8 semaines ensuite jusqu'à constatation objective et confirmée d'une progression de la maladie. Le statut de survie était contrôlé tous les 2 mois après la fin du traitement.
Le critère d'évaluation primaire de l'étude était la survie globale (OS) sous Imfinzi + chimiothérapie vs chimiothérapie seule. Le critère d'évaluation secondaire principal était la survie sans progression (PFS). Les autres critères d'évaluation secondaires étaient le taux de réponse objective (ORR), les jalons OS et PFS ainsi que les Patient Reported Outcomes (PRO). L'évaluation de la PFS et de l'ORR était effectuée par l'investigateur selon RECIST v1.1.
Une analyse intermédiaire (primaire) planifiée a montré qu'Imfinzi + chimiothérapie vs chimiothérapie avait atteint le critère principal de l'OS. Les résultats sont résumés ci-dessous.
Les caractéristiques démographiques et pathologiques à l'inclusion étaient équilibrées dans les deux groupes (268 patients dans le groupe Imfinzi + chimiothérapie et 269 patients dans le groupe chimiothérapie). Au début de l'étude, la population totale présentait les caractéristiques suivantes: homme (69,6%), âge ≥65 ans (39,6%), âge médian 63 ans (extrêmes: 28 à 82 ans), caucasiens (83,8%), asiatiques (14,5%), noirs ou afro-américains (0,9%), autres (0,6%), non hispaniques ou latino-américains (96,1%), fumeur actuel ou ancien fumeur (93,1%), non fumeur (6,9%), score 0 WHO/ECOG-PS (35,2%), score 1 WHO/ECOG-PS (64,8%), stade IV 90,3%, 24,6% des patients ont reçu du cisplatine et 74,1% du carboplatine. Dans le groupe chimiothérapie, 56,8% des patients ont reçu 6 cycles de chimiothérapie et 7,8% une PCI.
L'analyse primaire de l'étude a montré une amélioration statistiquement significative de l'OS sous Imfinzi + chimiothérapie vs chimiothérapie seule [HR=0,73 [IC à 95%: 0,591, 0,909], p=0,0047]. La survie globale médiane était de 13,0 mois dans le groupe Imfinzi + chimiothérapie (IC à 95%: 11,5, 14,8) et de 10,3 mois dans le groupe chimiothérapie (IC à 95%: 9,3, 11,2). 336 patients sont décédés (155 (57,8%] dans le groupe Imfinzi + chimiothérapie et 181 [67,3%] dans le groupe chimiothérapie). La survie globale après 12 resp. 18 mois était de 53,7% resp. 33,9% dans le groupe Imfinzi + chimiothérapie et de 39,8% resp. 24,7% dans le groupe chimiothérapie.
Dans l'analyse du suivi au long cours avec une période de suivi médiane de 39,3 mois, une amélioration durable de la SG [HR=0,71 (IC à 95%: 0,595; 0,858)] sous Imfinzi + chimiothérapie vs chimiothérapie seule a également été mise en évidence. La SG médiane s'élevait à 12,9 mois (IC à 95%: 11,3; 14,7) dans le groupe Imfinzi + chimiothérapie et à 10,5 mois (IC à 95%: 9,3; 11,2) dans le groupe chimiothérapie. 469 patients sont décédés (221 [82,5%] dans le groupe Imfinzi + chimiothérapie et 248 [92,2%] sous chimiothérapie seule). La SG au bout de 24 mois et 36 mois était de 22,9% et 17,6% respectivement dans le groupe Imfinzi + chimiothérapie et de 13,9% et 5,8% respectivement dans le groupe chimiothérapie.
Une amélioration de la PFS par rapport à la chimiothérapie seule a été constatée sous Imfinzi + chimiothérapie [HR=0,78 (IC à 95%: 0,645, 0,936) valeur de p nominale = 0,0078]. 459 patients ont présenté une progression ou sont décédés (226 (84,3%] dans le groupe Imfinzi + chimiothérapie et 233 (86,6%) dans le groupe chimiothérapie). La PFS médiane était de 5,1 mois dans le groupe Imfinzi + chimiothérapie et de 5,4 mois dans le groupe chimiothérapie. La PFS après 6 mois était de 45,4% dans le groupe Imfinzi + chimiothérapie et de 45,6% dans le groupe chimiothérapie. La PFS après 12 mois était de 17,5% dans le groupe Imfinzi + chimiothérapie et de 4,7% dans le groupe chimiothérapie.
L'ORR était de 67,9% (182/268) dans le groupe Imfinzi + chimiothérapie et de 57,6% (155/269) dans le groupe chimiothérapie. Il y a eu 6 cas de réponse complète dans le groupe Imfinzi + chimiothérapie et de 2 cas dans le groupe chimiothérapie.
Analyse des sous-groupes
L'amélioration de l'OS chez les patients traités par Imfinzi + chimiothérapie par rapport à la chimiothérapie seule était observée de manière cohérente pour les différents sous-groupes en fonction de la démographie, de la région géographique, de l'utilisation du carboplatine ou du cisplatine et des sous-groupes préspécifiés de caractéristiques de la maladie.
Analyse exploratoire des sous-groupes en fonction de l'expression de PD-L1
Des analyses exploratoires limitées relatives à l'expression de PD-L1 ont été réalisées rétrospectivement dans le tissu tumoral avec le test VENTANA PD-L1 (SP263) IHC et comprenaient les données des cellules immunitaires (IC) et tumorales (TC) (52,3% de l'ensemble d'analyse). Le statut PD-L1 des TC a été déterminé par coloration membranaire sur fond tandis que le statut de PD-L1 des IC était déterminé par coloration sur fond des IC associés à la tumeur. La prévalence de l'expression de PD-L1 est faible, tandis qu'une expression positive de PD-L1 (≥1%) n'a été observée que chez 25,8% des IC et 5,7% des TC.
Dans le sous-groupe IC, les résultats de l'analyse finale ont montré une OS avec un HR de 0,64 (IC à 95%: 0,473, 0,855) chez les patients PD-L1 négatifs (< 1% IC) et une OS avec un HR de 0,59 (IC à 95%: 0,342, 1,019) chez les patients PD-L1 positifs (≥1% IC). Dans le sous-groupe TC, les résultats de l'analyse finale ont montré une OS avec un HR de 0,62 (IC à 95%: 0,475, 0,810) chez les patients PD-L1 négatifs (< 1% TC) et une OS avec un HR de 0,75 (IC à 95%: 0,238, 2,269) chez les patients PD-L1 positifs (≥1% TC). Dans l'ensemble, ces données n'ont pas montré de relation claire entre le statut de PD-L1 et l'efficacité relative à l'OS.
Cancer des voies biliaires – Étude TOPAZ-1
TOPAZ-1 était une étude conçue pour évaluer l'efficacité d'Imfinzi en association avec la gemcitabine et le cisplatine. TOPAZ-1 était une étude randomisée, en double aveugle, contrôlée contre placebo, multicentrique incluant 685 patients atteints d'un cancer des voies biliaires localement avancé ou métastatique confirmé par examen histologique et présentant un indice de performance ECOG de 0 ou 1. Les patients ayant développé une récidive plus de 6 mois après la chirurgie et/ou la fin d'un traitement adjuvant étaient également inclus. Les patients devaient présenter au moins une lésion cible selon les critères RECIST v1.1 et une fonction organique et médullaire adéquate.
L'étude excluait les patients ayant un carcinome de l'ampoule de Vater (carcinome papillaire), des affections auto-immunes ou inflammatoires documentées actives ou antérieures, une infection par le VIH ou d'autres infections actives (y compris une tuberculose ou une hépatite C) ou les patients recevant actuellement ou ayant reçu un médicament immunosuppresseur dans les 14 jours précédant la première dose d'Imfinzi.
La randomisation a été stratifiée selon le statut de la maladie et la localisation de la tumeur primitive.
Les patients ont été randomisés selon un rapport 1:1 et répartis dans les groupes de traitement suivants:
·bras 1: Imfinzi 1500 mg administrés par voie intraveineuse le jour 1 + gemcitabine 1000 mg/m² et cisplatine 25 mg/m² (chacun administré les jours 1 et 8) toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis d'Imfinzi 1500 mg toutes les 4 semaines aussi longtemps qu'un bénéfice clinique est observé ou jusqu'à la survenue d'une toxicité inacceptable ou
·bras 2: placebo administré par voie intraveineuse le jour 1 + gemcitabine 1000 mg/m² et cisplatine 25 mg/m² (chacun administré les jours 1 et 8) toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis d'un placebo toutes les 4 semaines aussi longtemps qu'un bénéfice clinique est observé ou jusqu'à la survenue d'une toxicité inacceptable.
Les évaluations tumorales ont eu lieu toutes les 6 semaines pendant les 24 premières semaines après la date de randomisation, puis toutes les 8 semaines jusqu'à progression objective confirmée de la maladie.
Le critère d'évaluation primaire de l'étude était la SG, le critère d'évaluation secondaire principal était la SSP. Les autres critères d'évaluation secondaires étaient l'ORR, la durée de réponse (DdR) et les données issues des questionnaires d'autoévaluation du patient (Patient Reported Outcomes (PRO)). La SSP, l'ORR et la DdR ont été évalués par l'investigateur selon les critères RECIST v1.1.
Les caractéristiques démographiques et de la maladie au début de l'étude étaient équilibrées entre les deux bras de l'étude (341 patients dans le bras 1 et 344 patients dans le bras 2). Les caractéristiques démographiques initiales de la population globale de l'étude étaient les suivantes: hommes (50,4%), âge <65 ans (53,3%), blancs (37,2%), asiatiques (56,4%), noirs ou afro-américains (2,0%), autres (4,2%), non hispaniques ou latino-américains (93,1%), ECOG PS 0 (49,1%), vs PS 1 (50,9%), localisation de la tumeur primitive: cholangiocarcinome intrahépatique (55,9%), cholangiocarcinome extrahépatique (19,1%) et carcinome de la vésicule biliaire (25,0%), statut de la maladie: récidive (19,1%) vs non résécable initialement (80,7%), métastatique (86,0%) vs localement avancée (13,9%).
L'étude a démontré une amélioration statistiquement significative de la SG et la SSP lors d'une analyse intermédiaire préplanifiée sur la base d'une fonction de dépense de l'alpha de Lan-DeMets avec une limite de type O'Brien Fleming et le nombre réel d'événements observés (Lan et DeMets, 1983) [HR = 0,80 (IC à 95%: 0,66, 0,97), p = 0,021 pour la SG et HR = 0,75 (IC à 95%: 0,63, 0,89), p = 0,001 pour la SSP]. La maturité pour la SG était de 61,9% et la maturité pour la SSP était de 83,6%. Les résultats de cette analyse pour la SSP sont présentés dans le Tableau 5.
Une analyse de suivi supplémentaire de la SG a été effectuée 6,5 mois après l'analyse intermédiaire avec une maturité de SG de 76,9%. Le HR pour la SG était de 0,76 (IC à 95%: 0,64, 0,91) et la survie médiane était de 12,9 mois (IC à 95%: 11,6, 14,1). Les résultats de cette analyse pour la SG sont présentés dans le Tableau 5.
Tableau 5. Résultats d'efficacité de l'étude TOPAZ-1
|
|
Imfinzi + gemcitabine et cisplatine
(n = 341)
|
Placebo + gemcitabine et cisplatine
(n = 344)
| |
SG (DCO: 25 févr. 2022)
|
| |
Nombre de décès (%)
|
248 (72,7)
|
279 (81,1)
| |
SG médiane (mois) (IC à 95%)a
|
12,9
(11,6; 14,1)
|
11,3
(10,1; 12,5)
| |
HR (IC à 95%)b
|
0,76 (0,64; 0,91)
| |
SG au bout de 24 mois (%) (IC à 95%)a
|
23,624,9
(18,7; 28,9) (17,9; 32,5)
|
11,510,4
(7,6; 16,2) (4,7; 18,8)
| |
SSP (DCO: 11 août 2021)
|
| |
Nombre d'événements (%)
|
276 (80,9)
|
297 (86,3)
| |
SSP médiane (mois)
(IC à 95%)a
|
7,2
(6,7; 7,4)
|
5,7
(5,6; 6,7)
| |
HR (IC à 95%)b
|
0,75 (0,63; 0,89)
| |
Valeur pb,c
|
0,001
|
a Calculé en utilisant la méthode de Kaplan-Meier. IC de la médiane généré sur la base de la méthode de Brookmeyer-Crowley.
b L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié, tous deux ajustés en fonction du statut de la maladie et de la localisation de la tumeur primitive.
c Valeur p se basant sur les résultats de l'analyse intermédiaire préplanifiée. Sur la base d'une fonction de dépense alpha de Lan-DeMets avec une limite de type Pocock et le nombre réel d'événements observés.
Une analyse a été réalisée en sous-groupes selon la région géographique, se basant sur les patients qui ont été randomisés dans des centres d'essais cliniques en Asie (178 patients pour durvalumab + gem/cis et 196 patients pour placebo + gem/cis), mais aussi sur des patients randomisés dans des centres d'essai cliniques répartis dans le monde entier (163 patients, resp. 148 patients). Le hazard ratio pour la SG (IC à 95%) était de 0,68 (0,54, 0,85) en Asie et de 0,91 (IC à 95%: 0,70, 1,18) dans le reste du monde (voir Figures 1 et 2).
Figure 1: Courbe de Kaplan-Meier pour la SG en Asie (DCO: 25 févr. 2022)
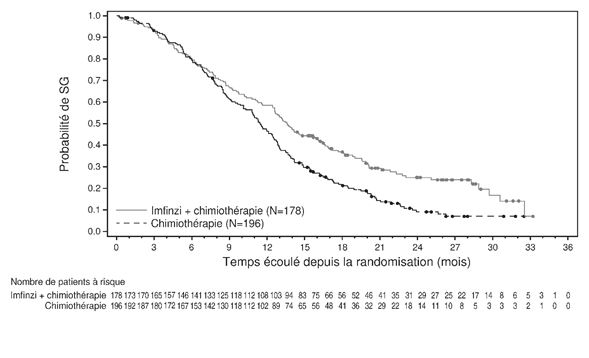
Figure 2: Courbe de Kaplan-Meier pour la SG dans le reste du monde (DCO: 25 févr. 2022)
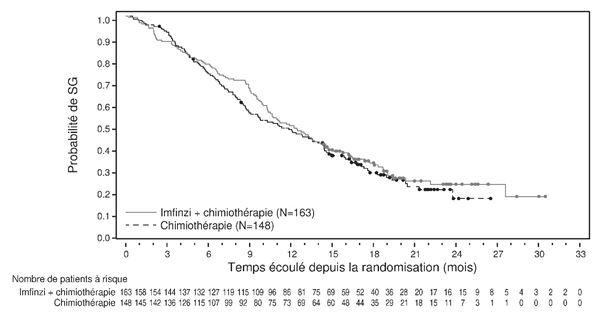
Cancer de la vessie infiltrant le muscle (TVIM) – Étude NIAGARA
NIAGARA était une étude de phase III randomisée, en ouvert, multicentrique évaluant l'efficacité d'Imfinzi comme traitement néoadjuvant en association avec la gemcitabine et le cisplatine, suivis d'Imfinzi en monothérapie adjuvante chez des patients atteints de TVIM. 1063 patients présentant un stade clinique de la tumeur T2-T4aN0/1M0, éligibles à un traitement au cisplatine et à une cystectomie radicale et qui n'avaient pas reçu auparavant de chimiothérapie systémique ni de traitement à médiation immunitaire pour traiter le TVIM, ont été randomisés dans l'étude. Les patients ayant une fonction rénale adéquate (clairance de la créatinine [ClCr] ≥60 ml/min) ou limite (ClCr ≥40 ml/min à < 60 ml/min) pouvaient participer à l'étude. Les patients présentant exclusivement une histologie non urothéliale, toute histologie à petites cellules et un cancer urothélial primitif autre qu'un cancer de la vessie (c.-à-d. cancer de l'uretère, de l'urètre ou du bassinet), les patients présentant une maladie auto-immune active ou un antécédent documenté de maladie auto-immune, les patients présentant une tuberculose active, une hépatite B ou C ou une infection par le VIH ou les patients ayant pris des immunosuppresseurs dans les 14 jours précédant la première dose de durvalumab, à l'exception des corticostéroïdes systémiques utilisés à des doses physiologiques ou en prémédication, étaient exclus de l'étude.
La randomisation a été stratifiée selon le stade clinique de la tumeur T2N0 vs > T2N0 (y compris T2N1, T3 et T4a), de la fonction rénale (fonction rénale adéquate: ClCr ≥60 ml/min vs fonction rénale limite: ClCr ≥40 ml/min et < 60 ml/min), et du statut d'expression PD-L1 (élevé vs faible/négatif).
Les patients ont été randomisés selon un rapport 1:1 pour recevoir Imfinzi en périopératoire associé à une chimiothérapie néoadjuvante (bras 1) ou une chimiothérapie néoadjuvante seule (bras 2).
·Bras 1 (Imfinzi + chimiothérapie): Imfinzi 1500 mg + gemcitabine 1000 mg/m² et cisplatine 70 mg/m² toutes les 3 semaines pendant 4 cycles avant l'opération, suivis d'Imfinzi 1500 mg toutes les 4 semaines jusqu'à 8 cycles après l'opération ou
·Bras 2 (chimiothérapie): gemcitabine 1000 mg/m² et cisplatine 70 mg/m² toutes les 3 semaines pendant 4 cycles avant l'opération, sans traitement postopératoire.
Les patients présentant une fonction rénale limite ont reçu une dose fractionnée de cisplatine de 35 mg/m² les jours 1 et 8 de chaque cycle.
Une évaluation de la tumeur selon les critères RECIST-1-1 a été effectuée au début de l'étude et après la fin du traitement néoadjuvant (avant l'opération). Après l'opération, des évaluations de la tumeur selon les critères RECIST-1.1 ont eu lieu toutes les 12 semaines pendant les 24 premiers mois, toutes les 24 semaines au cours des 36 mois suivants, puis toutes les 52 semaines jusqu'à la progression de la maladie, jusqu'à la fin de l'étude ou jusqu'au décès.
Les critères d'évaluation primaires étaient la réponse pathologique complète ( RPC) conformément à un examen pathologique central en aveugle et la survie sans événement ( SSE), comprenant une évaluation centrale indépendante en aveugle (BICR). Le critère d'évaluation secondaire principal était la survie globale (SG).
Les données démographiques et les caractéristiques pathologiques au début de l'étude étaient globalement bien équilibrées entre les 533 patients du bras 1 et les 530 patients du bras 2. Les données démographiques au début de l'étude étaient comme suit: hommes (81,8 %), âge < 65 ans (46,9 %), caucasiens (67 %), asiatiques (27,9 %), hispaniques ou latino-américains (8,0 %), noirs ou afro-américains (0,9 %), autre origine (0,8 %), et score ECOG PS 0 (78,4 %) vs score ECOG PS 1 (21,6 %). Les caractéristiques pathologiques étaient comme suit: stade tumoral T2N0 (40,3 %) et > T2N0a (59,7 %), ganglions lymphatiques régionaux N0 (94,5 %) et N1 (5,5 %), fonction rénale adéquate (81,1 %) et fonction rénale limite (18,9 %), et statut d'expression PD-L1 élevé (73,1 %) et faible/négatif (26,9 %). Les sous-types histologiques comprenaient le carcinome urothélial (84,5 %), le carcinome urothélial avec une différenciation épidermoïde (8,2 %), le carcinome urothélial avec une variante histologique (5,0 %) et le carcinome urothélial avec une différenciation glandulaire (2,4 %).
Lors de la deuxième analyse intermédiaire préspécifiée, l'étude a montré une amélioration de la survie statistiquement significative dans le bras Imfinzi + chimiothérapie en comparaison avec le bras chimiothérapie [HR = 0,68 (IC à 95 %: 0,56; 0,82), p < 0,0001]. L'étude a aussi montré une amélioration de la SG statistiquement significative dans le bras Imfinzi + chimiothérapie en comparaison avec le bras chimiothérapie [HR = 0,75 (IC à 95 %: 0,59; 0,93), p = 0,0106]. Une amélioration numérique des taux de RPC, qui s'est cependant avérée être statistiquement non significative, a été observée dans le bras Imfinzi + chimiothérapie [taux de réponse = 37,3 % (IC à 95 %: 33,2; 41,6)] en comparaison avec le bras chimiothérapie [taux de réponse = 27,5 % (IC à 95 %: 23,8, 31,6)]. Voir Tableau 6.
Tableau 6: Résultats d'efficacité de l'étude NIAGARA
|
|
Imfinzi + chimiothérapie
(n = 533)
|
Chimiothérapie
(n = 530)
| |
SSEa
| |
Nombre d'événements (%)
|
187 (35,1)
|
246 (46,4)
| |
SSE médiane (mois) (IC à 95 %)b
|
NR (NR; NR)
|
46,1 (32,2; NR)
| |
HR (IC à 95 %)c
|
0,68 (0,56; 0,82)
| |
Valeur p bilatéraled,e
|
< 0,0001
| |
SSE après 24 mois (%) (IC à 95 %)b
|
67,8 (63,6; 71,7)
|
59,8 (55,4; 64,0)
| |
RPCf
| |
Nombre de patients présentant une réponse
|
199
|
146
| |
Taux de réponse, % (IC à 95 %)g
|
37,3 (33,2; 41,6)
|
27,5 (23,8; 31,6)
| |
Odds Ratio (IC à 95 %)h
|
1,60 (1,23; 2,09)
| |
SGa
| |
Nombre d'événements (%)
|
136 (25,5)
|
169 (31,9)
| |
SG médiane (mois) (IC à 95 %)b
|
NR (NR; NR)
|
NR (NR; NR)
| |
HR (IC à 95 %)c
|
0,75 (0,59; 0,93)
| |
Valeur p bilatéraled,e
|
0,0106
| |
SG après 24 mois (%) (IC à 95 %)b
|
82,2 (78,7; 85,2)
|
75,2 (71,3; 78,8)
|
a Les résultats se basent sur une analyse intermédiaire prédéfinie (DCO: 29 avril 2024) qui a été effectuée 68 mois après le début de l'étude.
b Calculée en utilisant la méthode de Kaplan-Meier.
c Basé sur un modèle à risques proportionnels de Cox stratifié selon les facteurs suivants: stade tumoral [T2N0 vs > T2N0], fonction rénale [adéquate vs limite] et statut PD-L1 [élevé vs faible/négatif].
d Basée sur un test du log-rank stratifié selon les facteurs suivants: stade tumoral [T2N0 vs > T2N0], fonction rénale [adéquate vs limite] et statut PD-L1 [élevé vs faible/négatif].
e La limite pour la déclaration de la signification statistique pour les critères d'efficacité primaires (taux de RPC, SSE) et le critère d'évaluation secondaire principal (SG) a été déterminée par une procédure de tests multiples utilisant une approche exhaustive de recyclage de l'alpha. L'alpha attribué lors de l'analyse intermédiaire pour la SSE et pour la SG se basait sur une fonction de dépense de l'alpha de Lan-DeMets avec une approche de O'Brien Fleming. (RPC = 0,001, SSE = 0,0412, SG = 0,0154, bilatérale).
f Basée sur une analyse descriptive actualisée du critère d'évaluation primaire. Lors de l'analyse finale antérieure de la RPC préspécifiée (DCO: 14 janvier 2022), une amélioration numérique des taux de RPC a été observée dans le bras Imfinzi + chimiothérapie [taux de réponse = 33,8 % (IC à 95 %: 29,8; 38,0)] en comparaison avec le bras chimiothérapie [taux de réponse = 25,8 % (IC à 95 %: 22,2; 29,8)] [Odds Ratio 1,49 (IC à 95 %: 1,14; 1,96], p = 0,0038).
g L'IC a été calculé avec la méthode de Clopper-Pearson.
h Établi par régression logistique, ajusté en fonction des facteurs de stratification (fonction rénale [adéquate vs limite], stade tumoral [T2N0 vs > T2N0] et statut PD-L1 [élevé vs faible/négatif] par SVI).
IC = intervalle de confiance, HR = Hazard Ratio, NR = Non atteint
PharmacocinétiqueLa pharmacocinétique (PK) du durvalumab a été évaluée pour Imfinzi en monothérapie et en association avec une chimiothérapie. La pharmacocinétique (PK) du durvalumab a été testée chez 2903 patients présentant des tumeurs solides à des doses comprises entre 0,1 et 20 mg/kg, administrées toutes les 2, 3 ou 4 semaines. ll n'a pas été observé de différence cliniquement significative entre la PK du durvalumab administré en monothérapie et en association avec une chimiothérapie.
Absorption
L'exposition PK a augmenté plus fortement que proportionnellement à la dose (PK non linéaire) à des doses < 3 mg/kg et proportionnellement à la dose (PK linéaire) à des doses ≥3 mg/kg.
Distribution
L'équilibre a été atteint à 16 semaines environ. D'après l'analyse de la population PK, qui a inclus 1'878 patients dans la plage posologique de 10 mg/kg toutes les 2 semaines, de 15 mg/kg toutes les 3 semaines et de 20 mg/kg toutes les 4 semaines, la moyenne géométrique du volume de distribution à l'état d'équilibre (Vss) a été de 5,64 l.
Métabolisme
Le métabolisme du durvalumab n'a pas été déterminé. Le durvalumab étant un anticorps IgG1 monoclonal entièrement humanisé, on s'attend à ce qu'à l'instar de l'IgG endogène il soit catabolisé par de petits peptides et acides aminés.
Élimination
La clairance du durvalumab (Cl) a diminué progressivement. Au jour 365, la moyenne géométrique de la clairance à l'état d'équilibre (Clss) a atteint 8,16 ml/h; la baisse de la Clss n'a pas été considérée comme cliniquement significative. La demi-vie d'élimination (t1/2), calculée à partir de la clairance de référence, a été de 18 jours environ. L'élimination s'effectue principalement par catabolisme protéique par le biais du système réticulo-endothélial ou par une élimination du complexe anticorps-protéine cible.
Cinétique pour certains groupes de patients
Une analyse pharmacocinétique de population a montré que l'âge (19–96 ans), le poids (31–149 kg), le sexe, la présence d'anticorps anti-médicament (anti-drug antibodies, ADAs), les taux d'albumine, de LDH et de créatinine, le PD-L1 soluble, le type de tumeur, le groupe ethnique et le score OMS/ECOG n'avaient aucune incidence cliniquement significative sur la pharmacocinétique du durvalumab.
Patients âgés:
Une analyse pharmacocinétique de population a montré que l'âge (19–96 ans) n'avait aucune incidence cliniquement significative sur la pharmacocinétique du durvalumab.
Troubles de la fonction rénale:
Une analyse pharmacocinétique de population a montré qu'une insuffisance rénale légère (clairance de la créatinine [ClCr] de 60 à 89 ml/min) et une insuffisance rénale modérée (ClCr de 30 à 59 ml/min) n'avaient aucune incidence cliniquement significative sur la pharmacocinétique du durvalumab. L'incidence de l'insuffisance rénale sévère (ClCr de 15 à 29 ml/min) sur la pharmacocinétique du durvalumab n'est pas connue.
Troubles de la fonction hépatique:
Une analyse pharmacocinétique de population a montré qu'une insuffisance hépatique légère (bilirubine ≤ LSN et AST > LSN ou bilirubine > 1,0 à 1,5 x LSN et tout AST) ou qu'une insuffisance hépatique modérée (bilirubine > 1,5 à 3 x LSN et tout AST) n'avaient aucune incidence cliniquement significative sur la pharmacocinétique du durvalumab. L'incidence de l'insuffisance hépatique sévère (bilirubine > 3,0 x LSN et tout AST) sur la pharmacocinétique du durvalumab n'est pas connue.
Population pédiatrique
La pharmacocinétique du durvalumab a été analysée dans une étude réalisée auprès de 50 patients pédiatriques (dont 42 ont été inclus dans les données de pharmacocinétique de population) âgés de 1 à 17 ans (étude D419EC00001). Les patients ont reçu soit du durvalumab 20 mg/kg en association avec du trémélimumab 1 mg/kg, soit du durvalumab 30 mg/kg en association avec du trémélimumab 1 mg/kg toutes les 4 semaines pendant 4 cycles, suivis du durvalumab en monothérapie toutes les 4 semaines. D'après une analyse pharmacocinétique de population, l'exposition systémique au durvalumab chez les patients pédiatriques ≥35 kg recevant du durvalumab 20 mg/kg toutes les 4 semaines était comparable à l'exposition chez les adultes recevant du durvalumab 20 mg/kg toutes les 4 semaines, alors que chez les patients pédiatriques ≥35 kg recevant du durvalumab 30 mg/kg toutes les 4 semaines, l'exposition était environ 1,5 fois plus élevée par rapport à l'exposition chez les adultes recevant du durvalumab 20 mg/kg toutes les 4 semaines. Chez les patients pédiatriques < 35 kg recevant du durvalumab 30 mg/kg toutes les 4 semaines, l'exposition systémique était comparable à l'exposition chez les adultes recevant du durvalumab 20 mg/kg toutes les 4 semaines.
Données précliniquesCarcinogénicité et mutagénicité
Le potentiel carcinogène et génotoxique du durvalumab n'a pas été établi.
Toxicité sur la reproduction
Il n'existe aucune donnée relative aux effets potentiels du durvalumab sur la fertilité humaine. Comme l'indiquent les données de la littérature, la voie de signalisation PD-1/PD-L1 joue un rôle central dans le maintien de la grossesse, car elle permet de maintenir la tolérance immunitaire de la mère vis-à-vis du fœtus. Il a été démontré à l'aide de modèles murins gravides allogéniques qu'une interruption des signaux PD-L1 entraînait une augmentation de l'incidence de mort fœtale. Dans des études sur la reproduction animale menées chez des macaques gestantes de Java, l'administration de durvalumab pendant la période de gestation à des doses 6 à 20 fois ou 3 à 11 fois plus élevées que la dose clinique de 10 mg/kg de durvalumab toutes les 2 semaines ou de 1500 mg toutes les 3 semaines (niveau d'exposition estimé sur la base de l'ASC) a été associée à des naissances prématurées, à des fausses couches (avortements spontanés et décès in utero) et à une augmentation des cas de décès néonataux (voir «Grossesse, Allaitement»).
Toxicologie et pharmacologie animales
Des études de toxicité avec administration répétée de durvalumab à des macaques de Javas sexuellement matures pendant une durée maximale de 3 mois n'ont pas mis en évidence d'effets indésirables d'importance notable pour l'être humain.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention « EXP » sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Conserver dans l'emballage d'origine et protéger de la lumière.
Ne pas congeler.
Ne pas agiter.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Imfinzi ne contient aucun conservateur. Administrer la solution pour perfusion immédiatement après préparation. Si la solution pour perfusion n'est pas administrée immédiatement, mais conservée, il faut tenir compte des recommandations suivantes:
·La stabilité chimique et physique de la solution pour perfusion reconstituée dans la poche de perfusion a été démontrée pour une période allant jusqu'à 30 jours entre 2 et 8°C et pour une période allant jusqu'à 24 heures à température ambiante (ne dépassant pas 25°C) à partir du moment de la reconstitution.
D'un point de vue microbiologique, la solution pour perfusion reconstituée dans la poche pour perfusion doit être utilisée immédiatement. Si elle n'est pas utilisée immédiatement, les délais et conditions de conservation avant utilisation sont sous la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures à une température entre 2 et 8°C ou 12 heures à température ambiante (ne dépassant pas 25°C), à moins que la dilution n'ait eu lieu dans des conditions aseptiques contrôlées et validées.
Préparation de la solution
Imfinzi est conditionné en flacon perforable monodose et ne contient aucun conservateur. Respectez les règles d'asepsie.
·Inspectez visuellement le médicament pour déceler les éventuelles particules et colorations. Imfinzi est une solution transparente à opalescente et incolore à jaunâtre. Jetez les flacons perforables lorsque la solution est trouble ou colorée ou contient des particules en suspension. Ne pas agiter le flacon perforable.
·Prélevez la quantité de solution requise dans le flacon perforable d'Imfinzi et ajoutez-la à une poche pour perfusion intraveineuse contenant une solution de chlorure de sodium à 0,9 % ou de dextrose à 5 %. Mélangez cette solution diluée par inversion avec précaution. La concentration finale de la solution diluée doit se situer entre 1 et 15 mg/ml. Ne pas congeler et ne pas agiter la solution.
·Veillez à préserver la stérilité des solutions préparées.
·Ne pas réutiliser le flacon perforable après avoir prélevé le médicament; n'administrez toujours qu'une seule dose par flacon perforable.
·Jetez tout le liquide résiduel du flacon perforable.
Administration
·La solution pour perfusion est administrée sur une période de 60 minutes par une voie veineuse munie d'un filtre en ligne stérile à faible absorption protéique d'une épaisseur de 0,2 ou 0,22 µm.
·N'administrer aucun autre médicament par la même ligne de perfusion intraveineuse.
Tout médicament inutilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation66548 (Swissmedic)
PrésentationImfinzi 120 mg/2,4 ml: 1 flacon perforable [A]
Imfinzi 500 mg/10 ml: 1 flacon perforable [A]
Titulaire de l’autorisationAstraZeneca AG, 6340 Baar
Mise à jour de l’informationAvril 2025
|