Propriétés/EffetsCode ATC
L01FF03
Mécanisme d'action/pharmacodynamique
L'expression du ligand de mort cellulaire programmée de type 1 (PD-L1) est une réponse immunitaire adaptative qui dissimule les tumeurs et les protège contre le système immunitaire. Le PD-L1 peut être activé par des signaux inflammatoires (p.ex. IFNγ) et exprimé sur les cellules tumorales comme sur les cellules immunitaires associées à la tumeur dans le micro-environnement tumoral. Le PD-L1 inhibe le fonctionnement des lymphocytes T et leur activation par le biais de son interaction avec le PD-1 et la protéine CD80 (B7-1). En se fixant à leurs récepteurs, le PD-L1 diminue l'activité et la multiplication des lymphocytes T, ainsi que la production de cytokine.
Le durvalumab est un anticorps monoclonal à forte affinité de liaison, entièrement humanisé, de la sous-classe d'immunoglobuline de type G1 kappa (IgG1κ) qui inhibe de manière sélective l'interaction du PD-L1 avec le PD-1 et la CD80 (B7-1), sans toutefois influer sur l'interaction PD-1/PD-L2. Le durvalumab ne provoque aucune cytotoxicité à médiation cellulaire dépendante des anticorps (ADCC). L'inhibition sélective des interactions PD-L1/PD-1 et PD-L1/CD80 accroît la réponse immunitaire antitumorale. Cette réponse antitumorale peut entraîner l'élimination des tumeurs.
Dans des études précliniques, l'inhibition du PD-L1 a induit une activation accrue des lymphocytes T et une réduction de la taille des tumeurs.
Efficacité clinique
Cancer du poumon non à petites cellules (CPNPC) – Étude PACIFIC
L'efficacité d'Imfinzi a été établie dans le cadre de l'étude multicentrique PACIFIC, contrôlée par placebo, randomisée et menée en double chez 713 patients présentant un CPNPC inopérable localement avancé, confirmé par des analyses histologiques ou cytologiques. Les patients avaient subi au moins deux cycles de chimioradiothérapie définitive à base de platine 1 à 42 jours avant le début du traitement à l'étude et présentaient un score de performance ECOG de 0 ou 1. 92% des patients ont reçu une dose de rayonnement totale de 54 à 66 Gy. Ont été exclus de cette étude les patients montrant une progression après une chimioradiothérapie, les patients souffrant d'une maladie auto-immune active ou d'un antécédent documenté de maladie auto-immune dans les 2 ans précédant le début de l'étude, les patients immunodéprimés, les patients ayant développé des réactions indésirables graves à médiation immunitaire, les patients atteints de maladies nécessitant une immunosuppression, sauf une dose physiologique de corticoïdes systémiques, les patients souffrant d'une tuberculose active ou d'une hépatite B ou C ou infectés par le VIH et les patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant ou suivant le début du traitement par Imfinzi. Les patients ont été randomisés selon un rapport 2:1 et ont reçu 10 mg/kg d'Imfinzi (n = 476) ou un placebo (n = 237) par perfusion intraveineuse toutes les 2 semaines pendant 12 mois, jusqu'à la survenue d'une toxicité inacceptable ou jusqu'à la progression confirmée de la maladie. La randomisation a été stratifiée selon le sexe, l'âge (< 65 ans vs ≥65 ans) et le statut tabagique (fumeur ou non-fumeur). Des évaluations de la réponse tumorale ont été réalisées toutes les 8 semaines pendant les 12 premiers mois, puis toutes les 12 semaines.
Les patients ont été inclus dans l'étude indépendamment du statut d'expression tumorale PD-L1. Lorsqu'ils étaient disponibles, des échantillons de tissus tumoraux d'archives prélevés avant la chimioradiothérapie ont été testés rétrospectivement pour l'expression de PD-L1 sur des cellules tumorales en utilisant le test IHC VENTANA PD-L1 (SP263).
Les caractéristiques démographiques et pathologiques à l'inclusion étaient équilibrées entre les groupes de l'étude. La composition de la population totale au début de l'étude était la suivante: hommes (70%), âge ≥65 ans (45%), caucasiens (69%), asiatiques (27%), autres (4%), fumeurs (16%), anciens fumeurs (75%) et non-fumeurs (9%), score OMS/ECOG de 0 (49%), score OMS/ECOG de 1 (51%). Les caractéristiques pathologiques étaient les suivantes: stade IIIA (53 %), stade IIIB (45%), sous-groupes histologiques de types squameux (46%), non squameux (54%).
Les principaux critères de jugement de l'étude ont été la survie globale (SG) et la survie sans progression (SSP) sous Imfinzi vs placebo.
L'étude a démontré une amélioration statistiquement significative de la SG dans le groupe Imfinzi comparativement au groupe placebo [risque relatif (RR) = 0,68 (IC à 95%: 0,53 à 0,87), p = 0,00251]. Au moment de l'analyse provisoire de la SG (22 mars 2018), la durée médiane de suivi était de 25,2 mois; 299 patients (183 (38,4%) dans le groupe durvalumab et 116 patients (48,9%) dans le groupe placebo) étaient décédés. La survie globale médiane n'avait pas encore été atteinte dans le groupe durvalumab; dans le groupe placebo, elle était de 28,7 mois selon l'estimation de Kaplan-Meier. La survie globale après 24 mois était de 66,3% dans le groupe Imfinzi et de 55,6% dans le groupe placebo.
Au moment de l'actualisation des données au bout du suivi de 5 ans, 23 nouveaux événements SG au total ont été rapportés depuis la deuxième analyse du suivi SG (au total 419 décès [264 dans le groupe Imfinzi et 155 dans le groupe placebo]). Le bénéfice de survie d'Imfinzi par rapport au placebo était cohérent avec l'analyse primaire SG avec une réduction du risque de mortalité de 28% par rapport au placebo [HR = 0,72 (IC à 95%: 0,59-0,89)]. L'estimation de Kaplan-Meier de la SG médiane était de 29,1 mois dans le groupe placebo par rapport à 47,5 mois dans le groupe Infimzi. Les taux de survie globale estimés étaient de 42,9% pour le groupe Imfinzi et 33,4% pour le groupe placebo.
À la date de l'analyse intermédiaire (13 février 2017) par un comité d'examen central indépendant agissant en aveugle conformément aux critères RECIST 1.1, l'étude a montré une amélioration statistiquement significative de la SSP dans le groupe Imfinzi comparé au groupe placebo [RR = 0,52 (IC à 95%: 0,42 à 0,65), p < 0,0001]. À cette date, 371 patients (214 (45,0%) dans le groupe durvalumab et 157 (66,2%) dans le groupe placebo présentaient une progression de la maladie ou étaient décédés1. La SSP médiane selon l'estimation de Kaplan-Meier a été de 16,8 mois dans le groupe Imfinzi et de 5,6 mois dans le groupe placebo. La SSP à 12 mois a été de 55,9% dans le groupe Imfinzi et de 35,5% dans le groupe placebo. La SSP à 18 mois a été de 44,2 % dans le groupe Imfinzi et de 27,0% dans le groupe placebo. Des améliorations de la SG et de la SSP au bénéfice des patients du groupe Imfinzi par rapport au groupe placebo ont été observées en permanence dans l'ensemble des sous-groupes prédéfinis analysés (groupe ethnique, âge, sexe, statut tabagique, statut de mutation de l'EGFR et histologie).
Au moment de l'analyse du suivi de 5 ans (11 janvier 2021) basée sur les évaluations de la SSP par un comité d'examen central indépendant conformément à RECIST 1.1, le bénéfice de SSP [HR: 0,55 (IC à 95%: 0,45-0,68)] constaté dans le groupe Imfinzi était cohérent avec l'analyse primaire de la SSP [HR: 0,52: (IC à 95%: 0,42-0,65)]. L'estimation de Kaplan-Meier de la SSP médiane s'élevait à 16,9 mois dans le groupe durvalumab (IC à 95%: 13,0-23,9) par comparaison avec 5,6 mois dans le groupe placebo (IC à 95%: 4,8-7,7).
Dans le groupe traité par Imfinzi, un effet indésirable d'issue fatale est survenu chez 21 patients (4,4%), et un effet indésirable d'issue fatale est survenu chez 14 patients (6,0%) du groupe placebo.
Analyse post-hoc exploratoire en sous-groupe en fonction de l'expression du PD-L1
Sur les 713 patients randomisés de l'étude PACIFIC, 451 (63%) ont fourni un échantillon de tissu d'une qualité et d'une quantité suffisantes pour déterminer l'expression du PD-L1. Parmi les 451 échantillons, 33% présentaient une expression du PD-L1 sur les cellules tumorales < 1%, 67% une expression du PD-L1 sur les cellules tumorales ≥1%, 32% une expression du PD-L1 sur les cellules tumorales 1 - < 25%, 35% une expression du PD-L1 sur les cellules tumorales ≥25%.
Les analyses post-hoc en sous-groupes ont été effectuées afin d'évaluer l'efficacité dans les cas supplémentaires présentant des niveaux d'expression tumorale PD-L1 (< 1%, ≥1%, 1 à < 25%) qui n'étaient pas prévus dans le plan d'analyse statistique.
SSP:
Dans le sous-groupe ne présentant pas d'expression du PD-L1 (expression du PD-L1 sur les cellules tumorales < 1%), le RR de la SSP était de 0,73; IC à 95%: 0,48 à 1,11.
Pour les sous-groupes analysés présentant une expression du PD-L1 positive (expression du PD-L1 sur les cellules tumorales ≥1%) et les patients dont le statut d'expression PD-L1 ne pouvait être établi (PD-L1 sur les cellules tumorales inconnu), les résultats suivants ont été observés: expression du PD-L1 sur les cellules tumorales ≥1% RR: 0,46; IC à 95%: 0,33 à 0,64; expression du PD-L1 sur les cellules tumorales jusqu'à < 25% RR: 0,49; IC à 95%: 0,30 à 0,80; expression du PD-L1 sur les cellules tumorales ≥25% RR: 0,41; IC à 95%: 0,26 à 0,65; expression du PD-L1 sur les cellules tumorales inconnue RR: 0,59; IC à 95%: 0,42 à 0,83).
SG:
Dans le sous-groupe ne présentant pas d'expression du PD-L1 (expression du PD-L1 sur les cellules tumorales <1%), le RR de la SG était de 1,36; IC à 95%: 0,79 à 2,34.
Pour les sous-groupes analysés présentant une expression du PD-L1 positive (expression du PD-L1 sur les cellules tumorales ≥1%) et le sous-groupe dont le statut d'expression PD-L1 ne pouvait être établi (expression du PD-L1 sur les cellules tumorales inconnue), les résultats suivants ont été observés: expression du PD-L1 sur les cellules tumorales ≥1% RR: 0,53; IC à 95%: 0,36 à 0,77; expression du PD-L1 sur les cellules tumorales 1 à < 25% RR: 0,60; IC à 95%: 0,35 à 1,03; expression du PD-L1 sur les cellules tumorales ≥25% RR: 0,46; IC à 95%: 0,27 à 0,78; expression du PD-L1 sur les cellules tumorales inconnue RR: 0,62; IC à 95%: 0,43 à 0,89.
CPNPC résécable – étude AEGEAN
AEGEAN est une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, multicentrique évaluant l'efficacité d'Imfinzi en association avec une chimiothérapie comme traitement néoadjuvant, suivis d'une monothérapie par Imfinzi après l'opération chez les patients présentant un CPNPC résécable (stade IIA à stade IIIB (N2) chez certains patients [AJCC, 8e édition]). Des patients naïfs de traitement atteints d'un CPNPC épidermoïde ou non épidermoïde documenté sans exposition antérieure à une immunothérapie, présentant un indice de performance OMS/ECOG de 0 ou 1 et au moins une lésion cible selon les critères RECIST 1.1. ont été inclus dans l'étude. Le niveau d'expression tumorale PD-L1 a été confirmé par le test Ventana PD-L1 (SP263) avant la randomisation.
Les patients présentant une maladie auto-immune active ou un antécédent documenté de maladie auto-immune ou ayant utilisé des médicaments immunosuppresseurs dans les 14 jours précédant la première dose de durvalumab ont été exclus de l'étude. Étaient exclus de l'étude les patients dont l'opération planifiée était une résection segmentaire, une résection cunéiforme ou (après une modification du protocole) une pneumonectomie. Les opérations réalisées pendant l'étude ont été effectuées dans le cadre du traitement standard et pouvaient comporter une pneumonectomie. Après une modification du protocole, aucun patient atteint de tumeurs T4 avec invasion du diaphragme, invasion médiastinale, invasion du cœur, invasion des gros vaisseaux, de la trachée, du nerf laryngé récurrent, de l'œsophage ou de la carène n'a plus été inclus ultérieurement dans l'étude. Les patients atteints de mutations de l'EGFR connues ou de réarrangements de l'ALK ont été exclus de la population de l'étude pour l'analyse de l'efficacité (analyse en intention de traiter modifiée [ITTm]).
La randomisation a été stratifiée en fonction du stade de la maladie (stade II vs stade III) et du niveau d'expression PD-L1 (TC < 1 % vs TC ≥1 %).
Dans l'étude AEGEAN, 802 patients ont été randomisés dans un rapport 1:1 pour recevoir le traitement périopératoire par Imfinzi (bras 1) ou le placebo (bras 2) en association avec une chimiothérapie néoadjuvante. Le crossover entre les bras de l'étude n'était pas autorisé. L'analyse de l'efficacité a été effectuée sur la base de 740 patients dans la population ITTm.
·Bras 1: Imfinzi 1500 mg + chimiothérapie toutes les 3 semaines pendant jusqu'à 4 cycles avant l'opération, suivis d'Imfinzi 1500 mg toutes les 4 semaines pendant jusqu'à 12 cycles après l'opération.
·Bras 2: placebo + chimiothérapie toutes les 3 semaines pendant jusqu'à 4 cycles avant l'opération, suivis du placebo toutes les 4 semaines pendant jusqu'à 12 cycles après l'opération.
Le choix de la chimiothérapie standard en tenant compte de l'histologie était à la discrétion de l'investigateur. En cas de tumeurs épidermoïdes: carboplatine (ASC 6) et paclitaxel (200 mg/m2) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum ou cisplatine (75 mg/m2) le jour 1 et gemcitabine (1250 mg/m2) les jours 1 et 8 de chaque cycle de 3 semaines pendant 4 cycles au maximum. L'utilisation de carboplatine (ASC 5) associé à la gemcitabine (au lieu du cisplatine) était autorisée chez les patients présentant des comorbidités ou qui, selon le jugement de l'investigateur, ne toléraient pas le cisplatine. En cas de tumeurs non épidermoïdes: pémétrexed (500 mg/m2) et cisplatine (75 mg/m2) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum ou pémétrexed (500 mg/m2) et carboplatine (ASC 5) le jour 1 de chaque cycle de 3 semaines pendant 4 cycles au maximum. En cas de mauvaise tolérance, les patients ont pu passer du traitement par le cisplatine au traitement par le carboplatine à tout moment de l'étude (lorsqu'ils étaient éligibles à ce traitement).
Les patients qui avaient subi une opération avec des marges de résection R0/R1 ont pu poursuivre le traitement adjuvant.
Une évaluation de la tumeur selon les critères RECIST 1.1 a été effectuée à l'inclusion et au terme de la phase néoadjuvante (avant l'opération). Le premier examen postopératoire CT/IRM du thorax et de l'abdomen (y compris de la totalité du foie et des deux glandes surrénales) a été effectué 5 semaines ± 2 semaines après et avant l'opération, mais à une date aussi proche que possible de l'instauration du traitement adjuvant. Ensuite, des évaluations de la tumeur ont eu lieu toutes les 12 semaines (en fonction de la date de l'opération) jusqu'à la semaine 48, toutes les 24 semaines (en fonction de la date de l'opération) jusqu'à la semaine 192 (env. 4 ans) et ensuite toutes les 48 semaines (en fonction de la date de l'opération) jusqu'à la progression de la maladie (PM) confirmée radiologiquement selon les critères RECIST 1.1, jusqu'à la révocation du consentement ou jusqu'au décès.
Le statut de survie a été contrôlé 2, 3 et 4 mois après la fin du traitement, ensuite tous les 2 mois jusqu'au mois 12 puis tous les 3 mois.
Les critères d'évaluation primaires de l'étude étaient la réponse pathologique complète (RPC) conformément à un examen pathologique central en aveugle ainsi que la survie sans événements (SSE) conformément à une évaluation centrale indépendante en aveugle (BICR). Les critères d'évaluation secondaires principaux étaient la réponse pathologique majeure (RPM) conformément à un examen pathologique central en aveugle, la survie sans maladie (SSM) conformément à une BICR et la survie globale (SG). D'autres objectifs d'efficacité secondaires étaient la SSE (population de l'analyse PD-L1-TC ≥1 %) et la RPC (population de l'analyse PD-L1-TC ≥1 %).
Lors de l'analyse intermédiaire planifiée de la RPC, l'étude a atteint la limite prédéfinie pour la déclaration de la signification statistique pour la RPC et la RPM. Par la suite, lors de la première analyse intermédiaire planifiée de la SSE, l'étude a atteint sa limite prédéfinie pour la déclaration de la signification statistique pour la SSE.
Les caractéristiques démographiques et pathologiques au début de l'étude étaient équilibrées dans les deux bras de l'étude (366 patients dans le bras 1 et 374 patients dans le bras 2 de la population ITTm). Les caractéristiques initiales démographiques et spécifiques à la maladie de la population pour l'analyse de l'efficacité (ITTm) étaient les suivantes: hommes (71,6 %), femmes (28,4 %), âge ≥65 ans (51,6 %), âge médian 65 ans (fourchette: 30 à 88 ans), score OMS/ECOG-PS 0 (68,4 %), score OMS/ECOG-PS 1 (31,6 %), blancs (53,6 %), asiatiques (41,5 %), noirs ou afro-américains (0,9 %), amérindiens ou indigènes d'Alaska (1,4 %), autre origine (2,6 %), hispaniques ou latino-américains (16,1 %), non hispaniques ou non latino-américains (83,9 %), fumeurs actuels ou anciens fumeurs (85,5 %), non-fumeurs (14,5 %), histologie épidermoïde (48,6 %) et histologie non épidermoïde (50,7 %), stade II (28,4 %), stade III (71,6 %), niveau d'expression PD-L1 TC ≥1 % (66,6 %), niveau d'expression PD-L1 TC < 1 % (33,4 %). Les données démographiques et les caractéristiques initiales de la population ITTm étaient comparables à celles de la population ITT, à l'exception de l'absence de patients présentant des mutations de l'EGFR connues ou des réarrangements de l'ALK.
L'étude a montré une amélioration statistiquement significative de la SSE [HR = 0,68 (IC à 95 %: 0,53; 0,88), p = 0,003902] dans le bras Imfinzi par comparaison avec le bras placebo. En outre, l'étude a montré une amélioration statistiquement significative de la RPC [différence des proportions 12,96 % (IC à 95 %: 8,67, 17,57)] dans le bras Imfinzi par comparaison avec le bras placebo. Les données relatives à la survie globale (SG) n'étaient pas suffisamment matures au moment de l'analyse de la SSE. Voir Tableau 4.
Tableau 4: Résultats d'efficacité de l'étude AEGEAN (ITTm)
|
|
Imfinzi + chimiothérapie
(n = 366)
|
Placebo + chimiothérapie
(n = 374)
| |
SSEa
| |
Nombre d'événements, n (%)
|
98 (26,8)
|
138 (36,9)
| |
SSE médiane (IC à 95 %) (mois)
|
NR (31,9; NR)
|
25,9 (18,9; NR)
| |
SSE après 12 mois, % (IC à 95 %)
|
73,4 (67,9; 78,1)
|
64,5 (58,8; 69,6)
| |
SSE après 24 mois, % (IC à 95 %)
|
63,3 (56,1; 69,6)
|
52,4 (45,4; 59,0)
| |
Hazard Ratio (IC à 95 %)
|
0,68 (0,53; 0,88)
| |
Valeur p bilatéraled
|
0,003902
| |
RPCa,b,d
| |
Nombre de patients présentant une réponse
|
63
|
16
| |
Taux de réponse, % (IC à 95 %)
|
17,21 (13,49; 21,48)
|
4,28 (2,46; 6,85)
| |
Différence des proportions, % (IC à 95 %)
|
12,96 (8,67; 17,57)
| |
RPMa,c,d
| |
Nombre de patients présentant une réponse
|
122
|
46
| |
Taux de réponse, % (IC à 95 %)
|
33,33 (28,52; 38,42)
|
12,30 (9,15; 16,06)
| |
Différence des proportions, % (IC à 95 %)
|
21,03 (15,14; 26,93)
|
a Les résultats se basent sur l'analyse intermédiaire de la SSE préplanifiée et l'analyse finale de la RPC/RPM (DCO: 10 novembre 2022) qui a été effectuée 46,3 mois après le début de l'étude.
b Basée sur une analyse intermédiaire de la RPC prédéfinie (DCO: 14 janvier 2022), chez n = 402, le taux deRPC était statistiquement significatif (p = 0,000036) par comparaison avec le niveau de signification de 0,0082 %.
c Basée sur une analyse intermédiaire de la RPM prédéfinie (DCO: 14 janvier 2022), chez n = 402, le taux de RPM était statistiquement significatif (p = 0,000002) par comparaison avec le niveau de signification de 0,0082 %.
d La valeur p bilatérale pour la RPC et la RPM a été calculée sur la base d'un test CMH stratifié. La valeur p bilatérale pour la SSE a été calculée sur la base d'un test du log-rank stratifié. Les facteurs de stratification étaient le PD-L1 et le stade de la maladie.
La limite pour la déclaration de la signification statistique pour chacun des critères d'évaluation de l'efficacité a été déterminée par une fonction de dépense alpha selon la méthode de Lan et DeMets voisine d'une approche de O'Brien et Flemming (SSE = 0,9899 %, RPC = 0,0082 %, RPM = 0,0082 %, bilatéral).
Au moment de la DCO le 14 août 2023, la maturité concernant la survie globale (SG) était de 28,6 % (212 événements de SG). Dans le sous-groupe PD-L1 TC <1 % de la population ITTm, 27 patients (22,1 %) dans le bras Imfinzi + chimiothérapie et 39 patients (31,2 %) dans le bras placebo + chimiothérapie étaient décédés. Dans le sous-groupe PD-L1 TC ≥1 %, 72 patients (29,5 %) et 74 patients (29,7 %) étaient décédés dans le bras de l'étude respectif.
Cancer du poumon à petites cellules à un stade limité (LS-SCLC) – Étude ADRIATIC
Dans l'étude ADRIATIC, l'efficacité d'Imfinzi a été évaluée avec ou sans trémélimumab. L'étude ADRIATIC était une étude randomisée, en double aveugle, contrôlée contre placebo, multicentrique sur 730 patients atteints de LS-SCLC confirmé par des analyses histologiques ou cytologiques (stade I à III conformément à la 8e édition de l'AJCC), dont la maladie n'a pas progressé après une chimioradiothérapie concomitante. Les patients de stade I ou II devaient être inopérables, comme constaté par l'investigateur. Les patients ont subi 3 à 4 cycles de chimioradiothérapie définitive à base de platine, soit avec 60 à 66 Gy une fois par jour (QD) pendant 6 semaines, soit avec 45 Gy deux fois par jour (BID) pendant 3 semaines, 1 à 42 jours avant la première dose du traitement à l'étude. Une irradiation crânienne prophylactique (PCI) a pu être réalisée à la discrétion de l'investigateur après la fin de la chimioradiothérapie 1 à 42 jours avant la première dose du traitement à l'étude.
Les patients exclus étaient les patients présentant une maladie auto-immune active ou un antécédent documenté d'une maladie auto-immune dans les 5 années précédant le début de l'étude, des antécédents d'immunodéficience primaire active, de pneumonite de grade ≥2, de tuberculose active, d'hépatite B ou C ou d'infection par le VIH, les patients présentant une pneumopathie interstitielle, les patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant le début du traitement par Imfinzi ainsi que les patients avec métastases cérébrales, compression de la colonne vertébrale ou antécédent de carcinomatose leptoméningée. Les patients présentant une histologie mixte de CPPC et de CPNPC ont également été exclus de l'étude.
La randomisation a été stratifiée en fonction du stade de la maladie (I/II ou III) et PCI (oui ou non). Les patients ont été randomisés selon un rapport 1:1:1 et répartis dans les groupes de traitement suivants:
·Bras 1: 1500 mg d'Imfinzi + placebo toutes les 4 semaines pendant 4 cycles, suivis de 1500 mg d'Imfinzi toutes les 4 semaines.
·Bras 2: placebo + deuxième placebo toutes les 4 semaines pendant 4 cycles, suivis d'un placebo toutes les 4 semaines.
·Bras 3: 1500 mg d'Imfinzi + 75 mg de trémélimumab toutes les 4 semaines pendant 4 cycles, suivis de 1500 mg d'Imfinzi toutes les 4 semaines.
Après que 600 patients ont été randomisés au total dans les trois bras de l'étude, les patients suivants ont été randomisés selon un rapport 1:1 et répartis dans les bras 1 et 2. Ils ont reçu soit 1500 mg d'Imfinzi toutes les 4 semaines soit un placebo toutes les 4 semaines.
Le traitement a été poursuivi jusqu'à progression de la maladie, toxicité inacceptable ou pendant 24 mois au plus. Des évaluations de la tumeur ont eu lieu toutes les 8 semaines jusqu'à la semaine 72, puis toutes les 12 semaines jusqu'à la semaine 96 et ensuite toutes les 24 semaines.
Les caractéristiques démographiques et pathologiques à l'inclusion étaient bien équilibrées entre les bras de l'étude. Les données démographiques initiales et les caractéristiques pathologiques à l'inclusion dans les groupes Imfinzi et placebo étaient: hommes (69,1 %), âge ≥65 ans (39,2 %), blancs (50,4 %), noirs ou afro-américains (0,8 %), asiatiques (47,5 %), autre origine (1,3 %), hispaniques ou latino-américains (4,2 %), fumeurs actuels (22,3 %), anciens fumeurs (68,5 %), non-fumeurs (9,2 %), score OMS/ECOG-PS 0 (48,7 %), score OMS/ECOG-PS 1 (51,3 %), stade I (3,6 %), stade II (9,1 %), stade III (87,4 %).
Avant la randomisation, tous les patients ont reçu une chimiothérapie à base de platine (66,2 % cisplatine + étoposide, 33,8 % carboplatine + étoposide); 72,1 % des patients ont reçu une RT QD (dont 92,4 % avec ≥60 - ≤66 Gy QD); 27,9 % ont reçu une RT BID (dont 96,6 % avec 45 Gy BID) et 53,8 % des patients ont reçu une PCI. La réponse à la CRT était comme suit: réponse complète (12,3 %), réponse partielle (73,8 %), maladie stable (14,0 %).
Les deux critères d'évaluation primaires de l'étude ont été la SG et la SSP sous Imfinzi vs placebo. Les critères d'évaluation secondaires ont été l'ORR sous Imfinzi vs placebo. L'évaluation de la SSP et de l'ORR était effectuée par BICR selon RECIST v1.1.
Lors d'une analyse intermédiaire prévue, l'étude a montré une amélioration statistiquement significative et cliniquement pertinente de la SG pour Imfinzi par comparaison avec le placebo [HR = 0,73 (IC à 95 %: 0,569, 0,928), p=0,01042]. L'étude a en outre montré une amélioration statistiquement significative et cliniquement pertinente de la SSP pour Imfinzi par comparaison avec le placebo [HR = 0,76 (IC à 95 %: 0,606, 0,950), p=0,01608]. Voir Tableau 5.
Tableau 5: Données d'efficacité de l'étude ADRIATIC
|
|
Bras 1: Imfinzi (n = 264)
|
Bras 2: Placebo (n = 266)
| |
SGa
| |
Nombre de décès (%)
|
115 (43,6)
|
146 (54,9)
| |
SG médiane (mois) (IC à 95 %)b
|
55,9 (37,3, NR)
|
33,4 (25,5, 39,9)
| |
HR (IC à 95 %)c
|
0,73 (0,569, 0,928)
| |
Valeur pd
|
0,01042
| |
SG au bout de 24 mois (%) (IC à 95 %)b
|
68,0 (61,9, 73,3)
|
58,5 (52,3, 64,3)
| |
SG au bout de 36 mois (%) (IC à 95 %)b
|
56,5 (50,0, 62,5)
|
47,6 (41,3, 53,7)
| |
SSPe
| |
Nombre d'événements (%)
|
139 (52,7)
|
169 (63,5)
| |
SSP médiane (mois) (IC à 95 %)b
|
16,6 (10,2, 28,2)
|
9,2 (7,4, 12,9)
| |
HR (IC à 95 %)f
|
0,76 (0,606, 0,950)
| |
Valeur pd
|
0,01608
| |
SSP au bout de 18 mois (%) (IC à 95 %)b
|
48,8 (42,2, 55,0)
|
36,1 (29,9, 42,2)
| |
SSP au bout de 24 mois (%) (IC à 95 %)b
|
46,2 (39,6, 52,5)
|
34,2 (28,2, 40,3)
| |
ORRe
| |
ORRg n (%)
|
53/175 (30,3)
|
54/169 (32,0)
| |
Réponse complète n (%)
|
5 (2,9)
|
4 (2,4)
| |
Réponse partielle n (%)
|
48 (27,4)
|
50 (29,6)
| |
Odds Ratio (IC à 95 %)
|
-1,2 (-11,0, 8,5)
| |
DdRb,e médiane (mois)
(IC à 95 %)
|
33,0 (22,4, NR)
|
27,7 (9,6, NR)
| |
Pourcentage de patients en rémission après 12 moisb,e (%) (IC à 95 %)
|
73,7 (59,0, 83,8)
|
60,3 (44,5, 72,9)
| |
Pourcentage de patients en rémission après 18 moisb,e (%) (IC à 95 %)
|
71,5 (56,6, 82,0)
|
55,2 (39,4, 68,5)
|
a La durée médiane du suivi de SG chez les patients censurés a été de 37,19 mois dans le groupe Imfinzi et de 37,24 mois dans le groupe placebo.
b Calculé en utilisant la méthode de Kaplan-Meier. IC de la médiane généré sur la base de la méthode de Brookmeyer-Crowley.
c L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié; tous deux ajustés pour le traitement par PCI.
d Valeur p se basant sur les résultats de l'analyse intermédiaire préplanifiée. Sur la base d'une fonction de dépense de l'alpha de Lan-DeMets avec une limite de type O'Brien Fleming et le nombre réel d'événements observés, la limite pour la déclaration de la signification statistique pour la SG était de 0,01679 pour un alpha global de 4,5 % et pour la SSP de 0,02805 pour un alpha global de 5 % (Lan et DeMets, 1983).
e Évalué par un BICR conformément à RECIST v1.1.
f L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié; tous deux ajustés en fonction du statut TNM et pour le traitement par PCI.
g Sur la base d'un sous-groupe de l'ensemble d'analyse global avec une maladie mesurable à l'inclusion conformément à RECIST v1.1; Imfinzi (n = 175), placebo (n = 169).
Les améliorations de la SG et de la SSP au bénéfice des patients traités par Imfinzi par rapport aux patients ayant reçu le placebo étaient globalement cohérentes sur l'ensemble des sous-groupes prédéfinis analysés.
Cancer du poumon à petites cellules avancé (ES-SCLC) – Étude CASPIAN
L'efficacité d'Imfinzi associé à l'étoposide et soit au carboplatine soit au cisplatine chez les patients ES-SCLC naïfs de traitement a été évaluée dans CASPIAN, une étude en ouvert, multicentrique, randomisée, avec contrôle actif. L'état de santé général selon WHO/ECOG des patients admissibles était de 0 ou 1 avec un poids corporel >30 kg; ils étaient éligibles à une chimiothérapie à base de platine comme traitement de première ligne du SCLC et avaient une espérance de vie ≥12 semaines. Les patients avec métastases cérébrales asymptomatiques ou traitées, les patients avec au moins une lésion cible selon RECIST 1.1 et une fonction organique et médullaire suffisante étaient également éligibles. Le choix des sels de platine était laissé à la discrétion de l'investigateur compte tenu de la clairance de la créatinine calculée. Les patients exclus étaient les patients avec antécédents de radiothérapie thoracique ou une radiothérapie thoracique de consolidation planifiée; intervention chirurgicale majeure dans les 28 jours avant la première dose; maladie intercurrente non contrôlée; carcinomatose leptoméningée; antécédents d'immunodéficience primaire active; maladie auto-immune y compris syndrome paranéoplasique (SNP); maladie auto-immune active ou documentée ou maladies inflammatoires; utilisation d'immunosuppresseurs systémiques dans les 14 jours avant la première administration du traitement, à l'exception de doses physiologiques de corticostéroïdes systémiques; infection active, y compris la tuberculose, l'hépatite B ou C, ou le VIH et patients ayant reçu un vaccin vivant atténué dans les 30 jours précédant le début du traitement par Imfinzi ou devant le recevoir.
La randomisation était stratifiée en fonction du traitement à base de platine prévu au cycle 1 (carboplatine ou cisplatine).
L'évaluation de l'efficacité en cas d'ES-SCLC était basée sur une comparaison entre les groupes:
·Imfinzi 1500 mg et, selon le choix de l'investigateur, soit carboplatine (AUC 5 ou 6 mg/ml/min) ou cisplatine (75-80 mg/m2) le jour 1 et étoposide (80-100 mg/m2) en intraveineux les jours 1, 2 et 3 de chaque cycle de 21 jours pendant 4 cycles, suivis d'Imfinzi 1500 mg toutes les 4 semaines jusqu'à progression de la maladie ou toxicité inacceptable.
·Selon le choix de l'investigateur, soit carboplatine (AUC 5 ou 6 mg/ml/min) ou cisplatine (75-80 mg/m2) le jour 1 et étoposide (80-100 mg/m2) en intraveineux les jours 1, 2 et 3 de chaque cycle de 21 jours pendant jusqu'à 6 cycles. Irradiation crânienne prophylactique (Prophylactic Cranial Irradiation, PCI) après la fin de la chimiothérapie, à la discrétion de l'investigateur.
L'administration d'Imfinzi en monothérapie était également autorisée après une progression de la maladie dans la mesure où le patient était cliniquement stable et qu'un bénéfice clinique pouvait être attendu de l'avis de l'investigateur.
Les évaluations des tumeurs ont eu lieu 6 et 12 semaines après la randomisation et toutes les 8 semaines ensuite jusqu'à constatation objective et confirmée d'une progression de la maladie. Le statut de survie était contrôlé tous les 2 mois après la fin du traitement.
Le critère d'évaluation primaire de l'étude était la survie globale (OS) sous Imfinzi + chimiothérapie vs chimiothérapie seule. Le critère d'évaluation secondaire principal était la survie sans progression (PFS). Les autres critères d'évaluation secondaires étaient le taux de réponse objective (ORR), les jalons OS et PFS ainsi que les Patient Reported Outcomes (PRO). L'évaluation de la PFS et de l'ORR était effectuée par l'investigateur selon RECIST v1.1.
Une analyse intermédiaire (primaire) planifiée a montré qu'Imfinzi + chimiothérapie vs chimiothérapie avait atteint le critère principal de l'OS. Les résultats sont résumés ci-dessous.
Les caractéristiques démographiques et pathologiques à l'inclusion étaient équilibrées dans les deux groupes (268 patients dans le groupe Imfinzi + chimiothérapie et 269 patients dans le groupe chimiothérapie). Au début de l'étude, la population totale présentait les caractéristiques suivantes: homme (69,6%), âge ≥65 ans (39,6%), âge médian 63 ans (extrêmes: 28 à 82 ans), caucasiens (83,8%), asiatiques (14,5%), noirs ou afro-américains (0,9%), autres (0,6%), non hispaniques ou latino-américains (96,1%), fumeur actuel ou ancien fumeur (93,1%), non fumeur (6,9%), score 0 WHO/ECOG-PS (35,2%), score 1 WHO/ECOG-PS (64,8%), stade IV 90,3%, 24,6% des patients ont reçu du cisplatine et 74,1% du carboplatine. Dans le groupe chimiothérapie, 56,8% des patients ont reçu 6 cycles de chimiothérapie et 7,8% une PCI.
L'analyse primaire de l'étude a montré une amélioration statistiquement significative de l'OS sous Imfinzi + chimiothérapie vs chimiothérapie seule [HR=0,73 [IC à 95%: 0,591, 0,909], p=0,0047]. La survie globale médiane était de 13,0 mois dans le groupe Imfinzi + chimiothérapie (IC à 95%: 11,5, 14,8) et de 10,3 mois dans le groupe chimiothérapie (IC à 95%: 9,3, 11,2). 336 patients sont décédés (155 (57,8%] dans le groupe Imfinzi + chimiothérapie et 181 [67,3%] dans le groupe chimiothérapie). La survie globale après 12 resp. 18 mois était de 53,7% resp. 33,9% dans le groupe Imfinzi + chimiothérapie et de 39,8% resp. 24,7% dans le groupe chimiothérapie.
Dans l'analyse du suivi au long cours avec une période de suivi médiane de 39,3 mois, une amélioration durable de la SG [HR=0,71 (IC à 95%: 0,595; 0,858)] sous Imfinzi + chimiothérapie vs chimiothérapie seule a également été mise en évidence. La SG médiane s'élevait à 12,9 mois (IC à 95%: 11,3; 14,7) dans le groupe Imfinzi + chimiothérapie et à 10,5 mois (IC à 95%: 9,3; 11,2) dans le groupe chimiothérapie. 469 patients sont décédés (221 [82,5%] dans le groupe Imfinzi + chimiothérapie et 248 [92,2%] sous chimiothérapie seule). La SG au bout de 24 mois et 36 mois était de 22,9% et 17,6% respectivement dans le groupe Imfinzi + chimiothérapie et de 13,9% et 5,8% respectivement dans le groupe chimiothérapie.
Une amélioration de la PFS par rapport à la chimiothérapie seule a été constatée sous Imfinzi + chimiothérapie [HR=0,78 (IC à 95%: 0,645, 0,936) valeur de p nominale = 0,0078]. 459 patients ont présenté une progression ou sont décédés (226 (84,3%] dans le groupe Imfinzi + chimiothérapie et 233 (86,6%) dans le groupe chimiothérapie). La PFS médiane était de 5,1 mois dans le groupe Imfinzi + chimiothérapie et de 5,4 mois dans le groupe chimiothérapie. La PFS après 6 mois était de 45,4% dans le groupe Imfinzi + chimiothérapie et de 45,6% dans le groupe chimiothérapie. La PFS après 12 mois était de 17,5% dans le groupe Imfinzi + chimiothérapie et de 4,7% dans le groupe chimiothérapie.
L'ORR était de 67,9% (182/268) dans le groupe Imfinzi + chimiothérapie et de 57,6% (155/269) dans le groupe chimiothérapie. Il y a eu 6 cas de réponse complète dans le groupe Imfinzi + chimiothérapie et de 2 cas dans le groupe chimiothérapie.
Analyse des sous-groupes
L'amélioration de l'OS chez les patients traités par Imfinzi + chimiothérapie par rapport à la chimiothérapie seule était observée de manière cohérente pour les différents sous-groupes en fonction de la démographie, de la région géographique, de l'utilisation du carboplatine ou du cisplatine et des sous-groupes préspécifiés de caractéristiques de la maladie.
Analyse exploratoire des sous-groupes en fonction de l'expression de PD-L1
Des analyses exploratoires limitées relatives à l'expression de PD-L1 ont été réalisées rétrospectivement dans le tissu tumoral avec le test VENTANA PD-L1 (SP263) IHC et comprenaient les données des cellules immunitaires (IC) et tumorales (TC) (52,3% de l'ensemble d'analyse). Le statut PD-L1 des TC a été déterminé par coloration membranaire sur fond tandis que le statut de PD-L1 des IC était déterminé par coloration sur fond des IC associés à la tumeur. La prévalence de l'expression de PD-L1 est faible, tandis qu'une expression positive de PD-L1 (≥1%) n'a été observée que chez 25,8% des IC et 5,7% des TC.
Dans le sous-groupe IC, les résultats de l'analyse finale ont montré une OS avec un HR de 0,64 (IC à 95%: 0,473, 0,855) chez les patients PD-L1 négatifs (< 1% IC) et une OS avec un HR de 0,59 (IC à 95%: 0,342, 1,019) chez les patients PD-L1 positifs (≥1% IC). Dans le sous-groupe TC, les résultats de l'analyse finale ont montré une OS avec un HR de 0,62 (IC à 95%: 0,475, 0,810) chez les patients PD-L1 négatifs (< 1% TC) et une OS avec un HR de 0,75 (IC à 95%: 0,238, 2,269) chez les patients PD-L1 positifs (≥1% TC). Dans l'ensemble, ces données n'ont pas montré de relation claire entre le statut de PD-L1 et l'efficacité relative à l'OS.
Cancer des voies biliaires – Étude TOPAZ-1
TOPAZ-1 était une étude conçue pour évaluer l'efficacité d'Imfinzi en association avec la gemcitabine et le cisplatine. TOPAZ-1 était une étude randomisée, en double aveugle, contrôlée contre placebo, multicentrique incluant 685 patients atteints d'un cancer des voies biliaires localement avancé ou métastatique confirmé par examen histologique et présentant un indice de performance ECOG de 0 ou 1. Les patients ayant développé une récidive plus de 6 mois après la chirurgie et/ou la fin d'un traitement adjuvant étaient également inclus. Les patients devaient présenter au moins une lésion cible selon les critères RECIST v1.1 et une fonction organique et médullaire adéquate.
L'étude excluait les patients ayant un carcinome de l'ampoule de Vater (carcinome papillaire), des affections auto-immunes ou inflammatoires documentées actives ou antérieures, une infection par le VIH ou d'autres infections actives (y compris une tuberculose ou une hépatite C) ou les patients recevant actuellement ou ayant reçu un médicament immunosuppresseur dans les 14 jours précédant la première dose d'Imfinzi.
La randomisation a été stratifiée selon le statut de la maladie et la localisation de la tumeur primitive.
Les patients ont été randomisés selon un rapport 1:1 et répartis dans les groupes de traitement suivants:
·bras 1: Imfinzi 1500 mg administrés par voie intraveineuse le jour 1 + gemcitabine 1000 mg/m² et cisplatine 25 mg/m² (chacun administré les jours 1 et 8) toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis d'Imfinzi 1500 mg toutes les 4 semaines aussi longtemps qu'un bénéfice clinique est observé ou jusqu'à la survenue d'une toxicité inacceptable ou
·bras 2: placebo administré par voie intraveineuse le jour 1 + gemcitabine 1000 mg/m² et cisplatine 25 mg/m² (chacun administré les jours 1 et 8) toutes les 3 semaines (21 jours) jusqu'à 8 cycles, suivis d'un placebo toutes les 4 semaines aussi longtemps qu'un bénéfice clinique est observé ou jusqu'à la survenue d'une toxicité inacceptable.
Les évaluations tumorales ont eu lieu toutes les 6 semaines pendant les 24 premières semaines après la date de randomisation, puis toutes les 8 semaines jusqu'à progression objective confirmée de la maladie.
Le critère d'évaluation primaire de l'étude était la SG, le critère d'évaluation secondaire principal était la SSP. Les autres critères d'évaluation secondaires étaient l'ORR, la durée de réponse (DdR) et les données issues des questionnaires d'autoévaluation du patient (Patient Reported Outcomes (PRO)). La SSP, l'ORR et la DdR ont été évalués par l'investigateur selon les critères RECIST v1.1.
Les caractéristiques démographiques et de la maladie au début de l'étude étaient équilibrées entre les deux bras de l'étude (341 patients dans le bras 1 et 344 patients dans le bras 2). Les caractéristiques démographiques initiales de la population globale de l'étude étaient les suivantes: hommes (50,4%), âge <65 ans (53,3%), blancs (37,2%), asiatiques (56,4%), noirs ou afro-américains (2,0%), autres (4,2%), non hispaniques ou latino-américains (93,1%), ECOG PS 0 (49,1%), vs PS 1 (50,9%), localisation de la tumeur primitive: cholangiocarcinome intrahépatique (55,9%), cholangiocarcinome extrahépatique (19,1%) et carcinome de la vésicule biliaire (25,0%), statut de la maladie: récidive (19,1%) vs non résécable initialement (80,7%), métastatique (86,0%) vs localement avancée (13,9%).
L'étude a démontré une amélioration statistiquement significative de la SG et la SSP lors d'une analyse intermédiaire préplanifiée sur la base d'une fonction de dépense de l'alpha de Lan-DeMets avec une limite de type O'Brien Fleming et le nombre réel d'événements observés (Lan et DeMets, 1983) [HR = 0,80 (IC à 95%: 0,66, 0,97), p = 0,021 pour la SG et HR = 0,75 (IC à 95%: 0,63, 0,89), p = 0,001 pour la SSP]. La maturité pour la SG était de 61,9% et la maturité pour la SSP était de 83,6%. Les résultats de cette analyse pour la SSP sont présentés dans le Tableau 6.
Une analyse de suivi supplémentaire de la SG a été effectuée 6,5 mois après l'analyse intermédiaire avec une maturité de SG de 76,9%. Le HR pour la SG était de 0,76 (IC à 95%: 0,64, 0,91) et la survie médiane était de 12,9 mois (IC à 95%: 11,6, 14,1).
Une analyse exploratoire de la SG a été effectuée avec environ 3 ans de suivi à partir de la randomisation du dernier patient de la cohorte globale (mise à jour à 3 ans; DCO: 23 oct. 2023). Le HR pour la SG était de 0,74 (IC à 95 %: 0,63; 0,87) et était cohérente avec l'analyse intermédiaire à 6,5 mois. Les résultats de cette analyse de la SG sont présentés dans le Tableau 6.
Tableau 6: Résultats d'efficacité de l'étude TOPAZ-1
|
|
Imfinzi + gemcitabine et cisplatine
(n = 341)
|
Placebo + gemcitabine et cisplatine
(n = 344)
| |
SG (DCO: 23 oct. 2023)
|
| |
Nombre de décès (%)
|
293 (85,5)
|
319 (92,7)
| |
SG médiane (mois) (IC à 95%)a
|
12,9
(11,6; 14,1)
|
11,3
(10,1; 12,5)
| |
HR (IC à 95%)b
|
0,74 (0,63; 0,87)
| |
SG au bout de 24 mois (%) (IC à 95%)a
|
22,9 (18,5; 27,5)
|
13,1 (9,8; 17,0)
| |
SSP (DCO: 11 août 2021)
|
| |
Nombre d'événements (%)
|
276 (80,9)
|
297 (86,3)
| |
SSP médiane (mois)
(IC à 95%)a
|
7,2
(6,7; 7,4)
|
5,7
(5,6; 6,7)
| |
HR (IC à 95%)b
|
0,75 (0,63; 0,89)
| |
Valeur pb,c
|
0,001
|
a Calculé en utilisant la méthode de Kaplan-Meier. IC de la médiane généré sur la base de la méthode de Brookmeyer-Crowley.
b L'analyse du HR a été réalisée en utilisant un modèle à risques proportionnels de Cox stratifié et la valeur de p bilatérale repose sur un test du log-rank stratifié, tous deux ajustés en fonction du statut de la maladie et de la localisation de la tumeur primitive.
c Valeur p se basant sur les résultats de l'analyse intermédiaire préplanifiée. Sur la base d'une fonction de dépense alpha de Lan-DeMets avec une limite de type Pocock et le nombre réel d'événements observés.
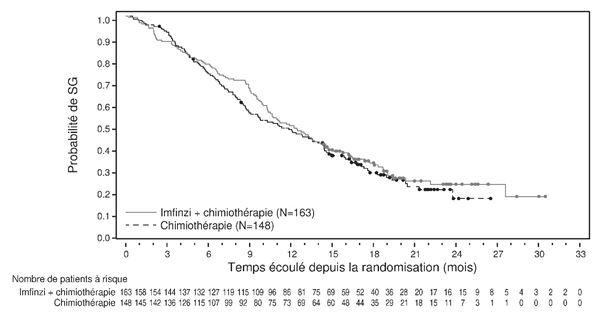
Pour la mise à jour à 3 ans, une analyse a été réalisée en sous-groupes selon la région géographique, se basant sur les patients qui ont été randomisés dans des centres d'essais cliniques en Asie (178 patients pour durvalumab + gem/cis et 196 patients pour placebo + gem/cis), mais aussi sur des patients randomisés dans des centres d'essai cliniques répartis dans le monde entier (163 patients, resp. 148 patients). Le hazard ratio pour la SG était de 0,62 (95% KI: 0,50; 0,78) en Asie et de 0.88 (95% KI: 0,69; 1,12) dans le reste du monde (voir Figures 1 et 2).
Figure 1: Courbe de Kaplan-Meier pour la SG en Asie (DCO: 23 oct. 2023)
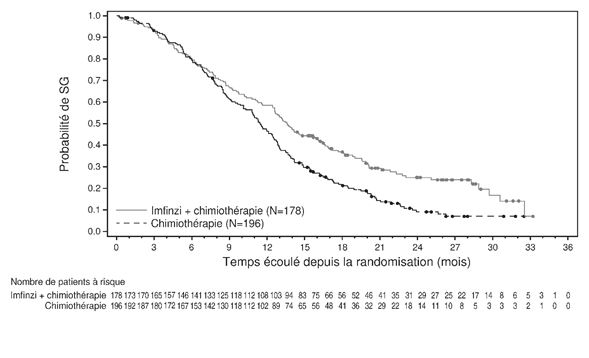
Figure 2: Courbe de Kaplan-Meier pour la SG dans le reste du monde DCO:23 oct. 2023)
Cancer de la vessie infiltrant le muscle (TVIM) – Étude NIAGARA
NIAGARA était une étude de phase III randomisée, en ouvert, multicentrique évaluant l'efficacité d'Imfinzi comme traitement néoadjuvant en association avec la gemcitabine et le cisplatine, suivis d'Imfinzi en monothérapie adjuvante chez des patients atteints de TVIM. 1063 patients présentant un stade clinique de la tumeur T2-T4aN0/1M0, éligibles à un traitement au cisplatine et à une cystectomie radicale et qui n'avaient pas reçu auparavant de chimiothérapie systémique ni de traitement à médiation immunitaire pour traiter le TVIM, ont été randomisés dans l'étude. Les patients ayant une fonction rénale adéquate (clairance de la créatinine [ClCr] ≥60 ml/min) ou limite (ClCr ≥40 ml/min à < 60 ml/min) pouvaient participer à l'étude. Les patients présentant exclusivement une histologie non urothéliale, toute histologie à petites cellules et un cancer urothélial primitif autre qu'un cancer de la vessie (c.-à-d. cancer de l'uretère, de l'urètre ou du bassinet), les patients présentant une maladie auto-immune active ou un antécédent documenté de maladie auto-immune, les patients présentant une tuberculose active, une hépatite B ou C ou une infection par le VIH ou les patients ayant pris des immunosuppresseurs dans les 14 jours précédant la première dose de durvalumab, à l'exception des corticostéroïdes systémiques utilisés à des doses physiologiques ou en prémédication, étaient exclus de l'étude.
La randomisation a été stratifiée selon le stade clinique de la tumeur T2N0 vs > T2N0 (y compris T2N1, T3 et T4a), de la fonction rénale (fonction rénale adéquate: ClCr ≥60 ml/min vs fonction rénale limite: ClCr ≥40 ml/min et < 60 ml/min), et du statut d'expression PD-L1 (élevé vs faible/négatif).
Les patients ont été randomisés selon un rapport 1:1 pour recevoir Imfinzi en périopératoire associé à une chimiothérapie néoadjuvante (bras 1) ou une chimiothérapie néoadjuvante seule (bras 2).
·Bras 1 (Imfinzi + chimiothérapie): Imfinzi 1500 mg + gemcitabine 1000 mg/m² et cisplatine 70 mg/m² toutes les 3 semaines pendant 4 cycles avant l'opération, suivis d'Imfinzi 1500 mg toutes les 4 semaines jusqu'à 8 cycles après l'opération ou
·Bras 2 (chimiothérapie): gemcitabine 1000 mg/m² et cisplatine 70 mg/m² toutes les 3 semaines pendant 4 cycles avant l'opération, sans traitement postopératoire.
Les patients présentant une fonction rénale limite ont reçu une dose fractionnée de cisplatine de 35 mg/m² les jours 1 et 8 de chaque cycle.
Une évaluation de la tumeur selon les critères RECIST-1-1 a été effectuée au début de l'étude et après la fin du traitement néoadjuvant (avant l'opération). Après l'opération, des évaluations de la tumeur selon les critères RECIST-1.1 ont eu lieu toutes les 12 semaines pendant les 24 premiers mois, toutes les 24 semaines au cours des 36 mois suivants, puis toutes les 52 semaines jusqu'à la progression de la maladie, jusqu'à la fin de l'étude ou jusqu'au décès.
Les critères d'évaluation primaires étaient la réponse pathologique complète ( RPC) conformément à un examen pathologique central en aveugle et la survie sans événement ( SSE), comprenant une évaluation centrale indépendante en aveugle (BICR). Le critère d'évaluation secondaire principal était la survie globale (SG).
Les données démographiques et les caractéristiques pathologiques au début de l'étude étaient globalement bien équilibrées entre les 533 patients du bras 1 et les 530 patients du bras 2. Les données démographiques au début de l'étude étaient comme suit: hommes (81,8 %), âge < 65 ans (46,9 %), caucasiens (67 %), asiatiques (27,9 %), hispaniques ou latino-américains (8,0 %), noirs ou afro-américains (0,9 %), autre origine (0,8 %), et score ECOG PS 0 (78,4 %) vs score ECOG PS 1 (21,6 %). Les caractéristiques pathologiques étaient comme suit: stade tumoral T2N0 (40,3 %) et > T2N0a (59,7 %), ganglions lymphatiques régionaux N0 (94,5 %) et N1 (5,5 %), fonction rénale adéquate (81,1 %) et fonction rénale limite (18,9 %), et statut d'expression PD-L1 élevé (73,1 %) et faible/négatif (26,9 %). Les sous-types histologiques comprenaient le carcinome urothélial (84,5 %), le carcinome urothélial avec une différenciation épidermoïde (8,2 %), le carcinome urothélial avec une variante histologique (5,0 %) et le carcinome urothélial avec une différenciation glandulaire (2,4 %).
Lors de la deuxième analyse intermédiaire préspécifiée, l'étude a montré une amélioration de la survie statistiquement significative dans le bras Imfinzi + chimiothérapie en comparaison avec le bras chimiothérapie [HR = 0,68 (IC à 95 %: 0,56; 0,82), p < 0,0001]. L'étude a aussi montré une amélioration de la SG statistiquement significative dans le bras Imfinzi + chimiothérapie en comparaison avec le bras chimiothérapie [HR = 0,75 (IC à 95 %: 0,59; 0,93), p = 0,0106]. Une amélioration numérique des taux de RPC, qui s'est cependant avérée être statistiquement non significative, a été observée dans le bras Imfinzi + chimiothérapie [taux de réponse = 37,3 % (IC à 95 %: 33,2; 41,6)] en comparaison avec le bras chimiothérapie [taux de réponse = 27,5 % (IC à 95 %: 23,8, 31,6)]. Voir Tableau 7.
Tableau 7: Résultats d'efficacité de l'étude NIAGARA
|
|
Imfinzi + chimiothérapie
(n = 533)
|
Chimiothérapie
(n = 530)
| |
SSEa
| |
Nombre d'événements (%)
|
187 (35,1)
|
246 (46,4)
| |
SSE médiane (mois) (IC à 95 %)b
|
NR (NR; NR)
|
46,1 (32,2; NR)
| |
HR (IC à 95 %)c
|
0,68 (0,56; 0,82)
| |
Valeur p bilatéraled,e
|
< 0,0001
| |
SSE après 24 mois (%) (IC à 95 %)b
|
67,8 (63,6; 71,7)
|
59,8 (55,4; 64,0)
| |
RPCf
| |
Nombre de patients présentant une réponse
|
199
|
146
| |
Taux de réponse, % (IC à 95 %)g
|
37,3 (33,2; 41,6)
|
27,5 (23,8; 31,6)
| |
Odds Ratio (IC à 95 %)h
|
1,60 (1,23; 2,09)
| |
SGa
| |
Nombre d'événements (%)
|
136 (25,5)
|
169 (31,9)
| |
SG médiane (mois) (IC à 95 %)b
|
NR (NR; NR)
|
NR (NR; NR)
| |
HR (IC à 95 %)c
|
0,75 (0,59; 0,93)
| |
Valeur p bilatéraled,e
|
0,0106
| |
SG après 24 mois (%) (IC à 95 %)b
|
82,2 (78,7; 85,2)
|
75,2 (71,3; 78,8)
|
a Les résultats se basent sur une analyse intermédiaire prédéfinie (DCO: 29 avril 2024) qui a été effectuée 68 mois après le début de l'étude.
b Calculée en utilisant la méthode de Kaplan-Meier.
c Basé sur un modèle à risques proportionnels de Cox stratifié selon les facteurs suivants: stade tumoral [T2N0 vs > T2N0], fonction rénale [adéquate vs limite] et statut PD-L1 [élevé vs faible/négatif].
d Basée sur un test du log-rank stratifié selon les facteurs suivants: stade tumoral [T2N0 vs > T2N0], fonction rénale [adéquate vs limite] et statut PD-L1 [élevé vs faible/négatif].
e La limite pour la déclaration de la signification statistique pour les critères d'efficacité primaires (taux de RPC, SSE) et le critère d'évaluation secondaire principal (SG) a été déterminée par une procédure de tests multiples utilisant une approche exhaustive de recyclage de l'alpha. L'alpha attribué lors de l'analyse intermédiaire pour la SSE et pour la SG se basait sur une fonction de dépense de l'alpha de Lan-DeMets avec une approche de O'Brien Fleming. (RPC = 0,001, SSE = 0,0412, SG = 0,0154, bilatérale).
f Basée sur une analyse descriptive actualisée du critère d'évaluation primaire. Lors de l'analyse finale antérieure de la RPC préspécifiée (DCO: 14 janvier 2022), une amélioration numérique des taux de RPC a été observée dans le bras Imfinzi + chimiothérapie [taux de réponse = 33,8 % (IC à 95 %: 29,8; 38,0)] en comparaison avec le bras chimiothérapie [taux de réponse = 25,8 % (IC à 95 %: 22,2; 29,8)] [Odds Ratio 1,49 (IC à 95 %: 1,14; 1,96], p = 0,0038).
g L'IC a été calculé avec la méthode de Clopper-Pearson.
h Établi par régression logistique, ajusté en fonction des facteurs de stratification (fonction rénale [adéquate vs limite], stade tumoral [T2N0 vs > T2N0] et statut PD-L1 [élevé vs faible/négatif] par SVI).
IC = intervalle de confiance, HR = Hazard Ratio, NR = Non atteint
Cancer de l'endomètre – étude DUO-E
DUO-E était une étude de phase III multicentrique, randomisée, contrôlée contre placebo, en double aveugle portant sur un traitement de première intention par chimiothérapie à base de platine en association avec Imfinzi, suivis d'un traitement d'entretien par Imfinzi avec ou sans olaparib chez des patientes atteintes d'un cancer de l'endomètre avancé ou récidivant. Les patientes devaient présenter un cancer de l'endomètre de l'une des catégories suivantes: maladie nouvellement diagnostiquée de stade III (maladie mesurable selon les critères RECIST 1.1 après l'opération ou la biopsie diagnostique), maladie nouvellement diagnostiquée de stade IV (avec ou sans maladie après l'opération ou la biopsie diagnostique) ou maladie récidivante (maladie mesurable ou non mesurable selon les critères RECIST 1.1), où le potentiel de guérison par chirurgie seule ou en association était faible. Chez les patientes présentant une maladie récidivante, un traitement antérieur par chimiothérapie était seulement autorisé si celui-ci avait été administré en tant que traitement adjuvant et si au moins 12 mois s'étaient écoulés entre la date de l'administration de la dernière dose de chimiothérapie et la récidive. L'étude a inclus des patientes atteintes de carcinomes épithéliaux de l'endomètre de tout sous-type histologique, y compris les carcinosarcomes. Les patientes présentant un sarcome de l'endomètre étaient exclues.
La randomisation a été stratifiée selon le statut MMR (réparation des mésappariements) du tissu tumoral (compétent [pMMR] versus déficient [dMMR]), le statut de la maladie (récidive versus nouveau diagnostic) et la région géographique (Asie versus reste du monde). Les patientes ont été randomisées selon un rapport 1:1:1 dans l'un des bras de traitement suivants:
·Bras 1 (chimiothérapie à base de platine): chimiothérapie à base de platine (paclitaxel et carboplatine) toutes les 3 semaines pendant 6 cycles au maximum avec un placebo de durvalumab toutes les 3 semaines. Après la fin de la chimiothérapie, les patientes sans progression objective de la maladie ont reçu en traitement d'entretien un placebo de durvalumab toutes les 4 semaines et des comprimés de placebo d'olaparib deux fois par jour jusqu'à la progression de la maladie.
·Bras 2 (chimiothérapie à base de platine + Imfinzi): chimiothérapie à base de platine (paclitaxel et carboplatine) toutes les 3 semaines pendant 6 cycles au maximum avec 1120 mg d'Imfinzi toutes les 3 semaines. Après la fin de la chimiothérapie, les patientes sans progression objective de la maladie ont reçu en traitement d'entretien 1500 mg de durvalumab toutes les 4 semaines avec des comprimés de placebo d'olaparib deux fois par jour jusqu'à la progression de la maladie.
·Bras 3 (chimiothérapie à base de platine + Imfinzi + olaparib): chimiothérapie à base de platine (paclitaxel et carboplatine) toutes les 3 semaines pendant 6 cycles au maximum avec 1120 mg d'Imfinzi toutes les 3 semaines. Après la fin de la chimiothérapie, les patientes sans progression objective de la maladie ont reçu en traitement d'entretien 1500 mg de durvalumab toutes les 4 semaines avec des comprimés de 300 mg d'olaparib deux fois par jour jusqu'à la progression de la maladie.
Le traitement a été poursuivi jusqu'à la progression de la maladie définie selon les critères RECIST 1.1 ou la survenue d'une toxicité inacceptable. Le statut tumoral a été évalué toutes les 9 semaines pendant les 18 premières semaines suivant la randomisation, puis toutes les 12 semaines.
Le critère d'évaluation primaire était la SSP évaluée par les investigateurs selon les critères RECIST 1.1. Les critères d'évaluation secondaires de l'efficacité incluaient la SG.
Les caractéristiques de la maladie à l'inclusion étaient dans l'ensemble équilibrées dans les trois bras de l'étude (241 patientes dans le bras 1, 238 patientes dans le bras 2 et 239 patientes dans le bras 3). Les caractéristiques démographiques à l'inclusion étaient: âge ≥65 ans (47 %), âge médian de 64 ans (fourchette: 22 à 86 ans), blanches (57 %), asiatiques (30 %), noires ou afro-américaines (5 %) et autre origine (4 %). Les caractéristiques de la maladie étaient: score OMS/ECOG PS 0 (67 %) vs PS 1 (33 %), 47 % de nouveaux diagnostics et 53 % de récidives; 67 % PD-L1 positifs et 30 % PD-L1 négatifs; 26 % présentaient une mutation HRR, 60 % ne présentaient pas de mutation HRR et 14 % avaient un statut mutationnel HRR inconnu; 11 % présentaient une mutation BRCA. Les sous-types histologiques étaient endométrioïde (60 %), séreux (21 %), carcinosarcome (7 %), épithélial mixte (4 %), à cellules claires (3 %), non différencié (2 %), mucineux (< 1 %) et autre (3 %).
La durée d'observation médiane de la SSP chez les patientes censurées était de 15,4 mois dans le bras de traitement chimiothérapie à base de platine + Imfinzi et de 12,6 mois dans le bras de traitement chimiothérapie à base de platine. Au moment de l'analyse primaire de la SSP, la maturité des données de SG de l'analyse intermédiaire était de 31 %, et chez 147 des 479 patientes ayant été traitées par chimiothérapie à base de platine + Imfinzi et par chimiothérapie à base de platine, des événements étaient survenus.
Tableau 8: Résultats d'efficacité de l'étude DUO-E
|
|
Chimiothérapie à base de platine + Imfinzi
n = 238
|
Chimiothérapie à base de platine
n = 241
| |
SSPa (analyse primaire, DCO: 12 avril 2023)
| |
Nombre d'événements (%)
|
139 (58,4)
|
173 (71,8)
| |
SSP médianeb (mois) (IC à 95 %)
|
10,2 (9,7-14,7)
|
9,6 (9,0-9,9)
| |
HRc (IC à 95 %)
|
0,71 (0,57-0,89)
| |
Valeur pd
|
0,003
|
a Évaluée par les investigateurs.
b Calculée par la méthode de Kaplan-Meier.
c Un HR inférieur à 1 est en faveur du bras de traitement chimiothérapie à base de platine + Imfinzi vs chimiothérapie à base de platine.
d La valeur p a été calculée en utilisant un test du log-rank.
Au moment d'une analyse descriptive ultérieure (DCO: 18 octobre 2023), 34,4 % des événements de SG étaient survenus. La SG médiane n'a été obtenue dans aucun des bras. Le HR de la SG était de 0,84 (0,62– 1,12), en faveur des patientes ayant été traitées par chimiothérapie à base de platine + Imfinzi par rapport à la chimiothérapie à base de platine.
Le statut de réparation des mésappariements (MMR) a été déterminé de manière centralisée au moyen d'un test de panel d'immunohistochimie spécifique au MMR. Sur un total de 718 patientes randomisées dans l'étude, 575 (80 %) présentaient un statut tumoral MMR compétent (pMMR) et 143 patientes (20 %) un statut tumoral MMR déficient (dMMR).
Patientes atteintes d'un carcinome de l'endomètre MMR déficient (dMMR)
Parmi les patientes ayant un statut tumoral dMMR, les caractéristiques démographiques et les caractéristiques à l'inclusion étaient de manière générale équilibrées entre les bras de traitement. Les caractéristiques démographiques à l'inclusion dans les trois bras de l'étude étaient: âge médian de 62 ans (fourchette: 34 à 85 ans), 41 % étaient âgées de 65 ans ou plus et 1,5 % de 75 ans ou plus, 62 % étaient blanches, 29 % asiatiques et 2 % étaient noires ou afro-américaines. Les caractéristiques de la maladie étaient: score ECOG PS 0 (58 %) ou 1 (42 %), 46 % de nouveaux diagnostics et 54 % de récidives, 80 % PD-L1 positives et 18 % PD-L1 négatives; 32 % présentaient une mutation HRR, 43 % ne présentaient pas de mutation HRR et 26 % avaient un statut mutationnel HRR inconnu; 13 % présentaient une mutation BRCA. Les sous-types histologiques étaient endométrioïde (83 %), épithélial mixte (5 %), séreux (3 %), carcinosarcome (3 %), non différencié (2 %) et autre (3 %).
Les résultats chez les patientes présentant un statut tumoral dMMR sont résumés dans le tableau 9. La durée médiane de suivi de la SSP chez les patientes censurées présentant un statut tumoral dMMR était de 15,5 mois dans le bras de traitement chimiothérapie à base de platine + Imfinzi et de 10,2 mois dans le bras de traitement chimiothérapie à base de platine. Au moment de l'analyse de la SSP, la maturité des données de SG de l'analyse intermédiaire était de 26 %, et chez 25 des 95 patientes ayant été traitées par chimiothérapie à base de platine + Imfinzi et par chimiothérapie à base de platine, des événements étaient survenus.
Tableau 9: Résultats d'efficacité de l'étude DUO-E (patientes présentant un statut tumoral dMMR)
|
|
Chimiothérapie à base de platine + Imfinzi
N = 46
|
Chimiothérapie à base de platine
N = 49
| |
SSP*,† (analyse primaire, DCO: 12 avril 2023)
| |
Nombre d'événements (%)
|
15 (32,6)
|
25 (51,0)
| |
SSP médiane, mois (IC à 95 %)‡
|
NA (NA; NA)
|
7,0 (6,7; 14,8)
| |
RR (IC à 95 %)
|
0,42 (0,22; 0,80)
|
-
|
* Évaluée par les investigateurs.
† Résultats de l'analyse primaire (SSP) (DCO: 12 avril 2023).
‡ Calculée par la méthode de Kaplan-Meier.
IC = intervalle de confiance; HR = hazard ratio; NA = non atteinte
Au moment d'une analyse descriptive ultérieure (DCO: 18 octobre 2023), 25,2 % des événements de SG étaient survenus. La SG médiane n'a été obtenue dans aucun des bras. Le HR de la SG était de 0,40 (0,17–0,86), en faveur des patientes ayant été traitées par chimiothérapie à base de platine + Imfinzi par rapport à la chimiothérapie à base de platine.
Adénocarcinome résécable de l'estomac ou de la jonction œsogastrique (GC/GEJC) – étude MATTERHORN
MATTERHORN était une étude de phase III multicentrique, randomisée, en double aveugle et contrôlée contre placebo évaluant l'efficacité d'Imfinzi en association avec une chimiothérapie FLOT comme traitement néoadjuvant et adjuvant, suivi d'Imfinzi en monothérapie adjuvante, chez des patients présentant un adénocarcinome résécable de l'estomac ou de la jonction œsogastrique (stade clinique II à IVA [cTNM, > T2 N0-3 M0 ou T0-4 N1-3 M0, AJCC, 8e édition]). Des patients naïfs de traitement atteints d'un GC/GEJC résécable, sans exposition antérieure à une immunothérapie et présentant un indice de performance OMS/ECOG de 0 ou 1 ont été inclus dans l'étude. Les patients ont été inclus dans l'étude indépendamment du statut d'expression de PD-L1 sur les cellules tumorales, qui a été confirmé par le test Ventana PD-L1 (SP263) avant la randomisation. Les patients présentant des métastases péritonéales, un carcinome adénosquameux, un carcinome épidermoïde ou des tumeurs stromales gastro-intestinales (GIST), une maladie auto-immune ou inflammatoire active ou un antécédent documenté de maladie auto-immune ou inflammatoire, ou ayant pris un médicament immunosuppresseur dans les 14 jours précédant la première dose d'Imfinzi ont été exclus de l'étude.
La randomisation a été stratifiée selon la région géographique (Asie vs autres régions), le statut clinique ganglionnaire (positif vs négatif) et le statut d'expression de PD-L1 (TAP < 1 % vs TAP ≥1 %).
Les patients ont été randomisés selon un rapport 1:1 dans l'un des bras de traitement suivants. Le crossover entre les bras de l'étude n'était pas autorisé.
·Bras 1: Imfinzi 1500 mg le jour 1 + chimiothérapie FLOT les jours 1 et 15 toutes les 4 semaines sur 4 cycles (1 dose d'Imfinzi et 2 doses de FLOT par cycle; 2 cycles pendant la phase de traitement néoadjuvant + 2 cycles pendant la phase de traitement adjuvant), puis Imfinzi 1500 mg au jour 1 toutes les 4 semaines pendant encore 10 cycles maximum après l'opération, soit 12 cycles au total (1 dose par cycle)
·Bras 2: placebo le jour 1 + chimiothérapie FLOT les jours 1 et 15 toutes les 4 semaines sur 4 cycles (1 dose de placebo et 2 doses de FLOT par cycle; 2 cycles pendant la phase de traitement néoadjuvant + 2 cycles pendant la phase de traitement adjuvant), puis placebo le jour 1 toutes les 4 semaines pendant encore 10 cycles maximum après l'opération, soit 12 cycles au total (1 dose par cycle)
Pendant la phase de traitement néoadjuvant ou adjuvant, les patients qui ont arrêté la chimiothérapie FLOT pour d'autres raisons qu'une progression de la maladie ou une récidive ont pu poursuivre le traitement d'Imfinzi en monothérapie de la manière décrite plus haut, à la discrétion de l'investigateur.
Une évaluation de référence de la tumeur selon les critères RECIST 1.1 a été effectuée avant le début du traitement néoadjuvant, et un examen de suivi avant l'opération a été réalisé dans les 4 semaines suivant la dernière dose de chimiothérapie. Un examen de référence de la phase de traitement adjuvant a été réalisé au plus tôt 4 semaines après l'opération et avant le début du traitement adjuvant. La tumeur a été évaluée (par rapport à l'examen de référence de la phase de traitement adjuvant) toutes les 12 semaines pendant 2 ans, puis toutes les 24 semaines jusqu'à la progression de la maladie confirmée par imagerie selon les critères RECIST 1.1, jusqu'à la révocation du consentement ou jusqu'au décès du patient.
Le critère d'évaluation primaire de l'étude était la SSE conformément à une évaluation centrale indépendante en aveugle (BICR). Les principaux critères d'évaluation secondaires étaient la SG et le taux de RPC conformément à un examen pathologique central en aveugle. Les PRO constituaient un autre critère d'efficacité secondaire.
Les caractéristiques démographiques et pathologiques à l'inclusions étaient globalement équilibrées entre les deux bras de l'étude (474 patients dans le bras 1 et 474 patients dans le bras 2). À l'incluson, la population globale présentait les caractéristiques suivantes: hommes (71,9 %), âge ≥65 ans (41,4 %), âge médian 62 ans (fourchette: 26 à 84 ans), blancs (67,8 %), asiatiques (20,4 %), noirs ou afro-américains (1,1 %), amérindiens ou indigènes d'Alaska (4,0 %), autre origine (1,7 %), non hispaniques ou non latino-américains (80,0 %), score OMS/ECOG PS 0 (74,2 %) versus PS 1 (25,8 %). Les caractéristiques pathologiques étaient les suivantes: stade clinique II (29,4 %), stade III (61,7 %), stade IVA (8,8 %); adénocarcinome de l'estomac (67,5 %), adénocarcinome de la jonction œsogastrique (32,5 %); type Siewert 1 (10,4 %), type Siewert 2 (14,8 %), type Siewert 3 (7,3 %); type intestinal (50,9 %), type diffus (26,3 %), type indéterminé (22,8 %); statut clinique ganglionnaire positif (70,4 %), statut clinique ganglionnaire négatif (29,2 %); statut d'expression de PD-L1 TAP ≥1 % (90,0 %), statut d'expression de PD-L1 TAP < 1 % (10,0 %).
Une chirurgie à visée curative a été tentée chez 431 patients (90,9 %) du bras 1 versus 428 patients (90,3 %) du bras 2. Une chirurgie à visée curative a été pratiquée chez 412 patients (86,9 %) du bras 1 versus 400 patients (84,4 %) du bras 2.
Lors d'une analyse intermédiaire prédéfinie, l'étude a montré une amélioration statistiquement et cliniquement significative de la SSE (HR = 0,71 [IC à 95 %: 0,58; 0,86]; p = 0,001) dans le bras Imfinzi en comparaison avec le bras placebo. L'étude a montré en outre une amélioration statistiquement significative du taux de RPC dans le bras Imfinzi (taux de réponse 19,2 % [IC à 95 %: 15,7, 23,0]) en comparaison avec le bras placebo (taux de réponse 7,2 % [IC à 95 %: 5,0, 9,9]). Lors d'une analyse finale de la SG, l'étude a montré une amélioration statistiquement significative et cliniquement pertinente de la SG (HR = 0,78 [IC à 95 %: 0,63, 0,96]; p = 0,021) dans le bras Imfinzi en comparaison avec le bras placebo, voir Tableau 10.
Tableau 10: Résultats d'efficacité de l'étude MATTERHORN
|
|
Imfinzi + chimiothérapie FLOT
(n = 474)
|
Placebo + chimiothérapie FLOT
(n = 474)
| |
SSEa
| |
Nombre d'événements, n (%)
|
167 (35,2 %)
|
218 (46,0 %)
| |
SSE médiane (IC à 95 %) (mois)b
|
NC (40,7, NR)
|
32,8 (27,9, NR)
| |
HR (IC à 95 %)c
|
0,71 (0,58, 0,86)
| |
Valeur p bilatéraled,e
|
< 0,001
| |
SSE après 24 mois, % (IC à 95 %)b
|
67,4 (62,9, 71,6)
|
58,5 (53,8; 63,0)
| |
SGf
| |
Nombre de décès (%)
|
160 (33,8 %)
|
192 (40,5 %)
| |
SG médiane (IC à 95 %) (mois)b
|
NR (NR, NR)
|
NR (NR, NR)
| |
HR (IC à 95 %)c
|
0,78 (0,63, 0,96)
| |
Valeur p bilatéraled,g
|
0,021
| |
SG après 24 mois, % (IC à 95 %)b
|
75,5 (71,3, 79,1)
|
70,4 (66,0, 74,3)
| |
SG après 36 mois, % (IC à 95 %)b
|
68,6 (64,2, 72,6)
|
61,9 (57,3, 66,2)
| |
RPCh
| |
Nombre de patients présentant une réponse
|
91
|
34
| |
Taux de réponse, % (IC à 95 %)g
|
19,2 (15,7, 23,0)
|
7,2 (5,0, 9,9)
| |
Odds Ratio, % (IC à 95 %)
|
3,08 (2,0, 4,7)
| |
Valeur bilatéralek,j
|
< 0,001
|
a Les résultats se basent sur une analyse intermédiaire prédéfinie de la SSE (DCO: 20 décembre 2024).
b Calculée en utilisant la méthode de Kaplan-Meier.
c Sur la base d'un modèle à risques proportionnels de Cox, stratifié selon la région géographique, le statut clinique ganglionnaire et le statut d'expression de PD-L1 au moment de la randomisation. Un HR < 1 est favorable à Imfinzi. L'IC a été calculé par la méthode de vraisemblance profilée.
d Sur la base d'un test du log-rank stratifié avec ajustement selon la région géographique, le statut clinique ganglionnaire et le statut d'expression de PD-L1 au moment de la randomisation.
e Sur la base d'une fonction de consommation du risque alpha selon la méthode de Lan et DeMets avec la limite d'O'Brien et Fleming, calculée à l'aide du nombre réel de patients à la DCO, la limite de déclaration de la signification statistique de la SSE était de 0,0239 pour un alpha total de 5 % (bilatéral).
f Les résultats se basent sur une analyse finale de la SG (DCO: 1er septembre 2025).
g Un alpha de 4,99 % (bilatéral) a été attribué à l'analyse finale de la SG, ce qui correspond à une limite pour la déclaration de signification statistique de 0,0499.
h Les résultats se basent sur l'analyse finale de la RPC (DCO: 1er février 2023).
i L'IC a été calculé par la méthode de Clopper et Pearson.
j Un alpha fixe de 0,1 % (bilatéral) a été attribué à l'analyse intermédiaire du taux de RPC, ce qui correspond à une limite de la valeur p pour la déclaration de signification statistique de 0,001.
k Sur la base d'un test de Cochran-Mantel-Haenszel stratifié avec ajustement selon la région géographique, le statut clinique ganglionnaire et le statut d'expression de PD-L1 au moment de la randomisation.
IC = intervalle de confiance, HR = Hazard Ratio, NR = non atteint
Pour le sous-groupe de patients présentant un adénocarcinome diffus qui a fait l'objet d'une analyse post hoc exploratoire le HR pour la SSE était de 0,93 (IC à 95 %: 0,66-1,32), le HR pour la SG était de 0,98 (IC à 95 %:0,68- 1,40), et le taux de RPC était de 6,9 % dans le bras D + FLOT contre 4,2 % dans le bras placebo.
|