CompositionPrincipes actifs
Benralizumab.
Benralizumab est un anticorps monoclonal humanisé produit dans des cellules ovariennes de hamster chinois par la technologie de l'ADN recombinant.
Excipients
L-histidine, chlorhydrate de L-histidine monohydraté, tréhalose dihydraté, polysorbate 20, eau pour préparations injectables.
Indications/Possibilités d’emploiAsthme
Fasenra est indiqué chez l'adulte à partir de 18 ans en complément d'un traitement d'entretien dans l'asthme sévère à éosinophiles caractérisé par les critères suivants:
·au moins 2 exacerbations au cours des 12 derniers mois sous traitement standard actuel (corticostéroïdes à inhaler à haute dose et bronchodilatateurs à longue durée d'action) et/ou nécessitant un traitement par des corticostéroïdes systémiques.
·Taux sanguin d'éosinophiles ≥0,3 G/litre (soit ≥300 cellules/μl).
Pour des informations plus détaillées sur les populations de patients ayant pris part aux études, voir «Efficacité clinique».
Granulomatose éosinophilique avec polyangéite (GEPA)
Fasenra est indiqué chez l'adulte à partir de 18 ans en tant que traitement d'appoint de la granulomatose éosinophilique avec polyangéite (GEPA) récidivante ou résistante au traitement caractérisée par les critères suivants:
·Absence de GEPA active menaçant un organe ou le pronostic vital
·Stabilisation préalable de la pathologie avec des corticostéroïdes systémiques
·Nécessité d'un traitement d'entretien par corticostéroïdes systémiques et éventuellement immunosuppresseurs d'épargne stéroïdienne
Pour de plus amples informations relatives aux populations de patients examinées dans les études, voir «Efficacité clinique».
Posologie/Mode d’emploiFasenra doit être prescrit par un médecin expérimenté dans le diagnostic et le traitement des pathologies pour lesquelles le benralizumab est indiqué (voir «Indications/Possibilités d'emploi»).
En cas de réponse, Fasenra est destiné à un traitement à long terme. La nécessité de poursuivre le traitement doit être réévaluée au moins une fois par an, selon un rythme déterminé par le degré de sévérité de la pathologie, le degré de contrôle de la pathologie et la numération des éosinophiles dans le sang.
Posologie usuelle
Asthme
La dose recommandée est de 30 mg par voie sous-cutanée à administrer toutes les 4 semaines pour les trois premières doses, puis toutes les 8 semaines.
Le succès thérapeutique doit être évalué au plus tard après 5 administrations de Fasenra afin de décider si le traitement doit être poursuivi ou non. L'évaluation de la réponse au traitement additionnel inclut une évaluation soigneuse du contrôle de l'asthme, du besoin de corticostéroïdes systémiques et de la fréquence des exacerbations avant et pendant le traitement.
GEPA
Aucune étude clinique de recherche de dose spécifique à la GEPA n'a été menée.
La dose recommandée est de 30 mg en injection sous-cutanée toutes les 4 semaines.
Chez les patients qui développent des manifestations de GEPA menaçant le pronostic vital, il convient de réévaluer la nécessité de poursuivre le traitement, car Fasenra n'a pas été étudié dans cette population de patients.
Traçabilité
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale (voir «Pharmacocinétique»).
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés (voir «Pharmacocinétique»).
Enfants et adolescents
Les données disponibles concernant les adolescents de moins de 18 ans atteints d'asthme sévère à éosinophiles sont limitées. Par conséquent, l'utilisation de ce médicament dans cette catégorie d'âge n'est pas recommandée.
La sécurité et l'efficacité de Fasenra chez les enfants et les adolescents (âgés de moins de 18 ans) atteints de GEPA n'ont pas encore été établies.
Mode d'administration
Fasenra doit être administré en injection sous-cutanée, au départ par un médecin ou un professionnel de santé. L'injection est effectuée dans la cuisse ou l'abdomen.
Si le médecin le juge adapté, les patients qui ont déjà bien toléré Fasenra lors d'une exposition répétée peuvent s'auto-administrer le médicament après avoir reçu une formation adéquate ou se le faire administrer par un proche aidant dûment formé (voir le mode d'emploi à la fin des informations destinées aux patients). Lorsque l'injection est administrée par un tiers, elle peut également être effectuée dans la partie supérieure du bras.
Fasenra ne doit pas être administré dans des zones où la peau est sensible ou indurée ou en présence d'un érythème ou d'un hématome (voir «Remarques particulières»).
Conformément à la pratique clinique courante, il est recommandé de surveiller les patients après l'administration d'un principe actif d'origine biologique (voir rubrique «Mises en garde et précautions»).
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsFasenra ne doit pas être utilisé pour traiter les exacerbations aiguës de l'asthme.
Les patients doivent être instruits de consulter un médecin si les symptômes d'asthme restent mal contrôlés ou s'aggravent après le début du traitement.
Il est déconseillé d'arrêter brutalement les corticostéroïdes après l'instauration du traitement par Fasenra. Les doses de corticoïdes doivent être réduites progressivement sous surveillance médicale.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité (p.ex. anaphylaxie, angio-œdème, urticaire, urticaire papuleuse, éruption cutanée) ont été observées après l'administration de Fasenra. Ces réactions se produisent généralement dans les premières heures suivant l'administration, mais parfois aussi plus tardivement (après plusieurs jours). Les patients doivent être informés de ce risque et des options de traitement de réactions allergiques graves.
En présence d'une réaction d'hypersensibilité, le traitement par Fasenra doit être arrêté et un traitement adapté doit être instauré.
En cas d'auto-administration, les patients doivent recevoir pour consigne de consulter immédiatement un médecin en cas de réactions allergiques systémiques graves, p.ex. urticaire, angio-œdème, difficultés respiratoires ou troubles cardio-vasculaires.
Infections parasitaires (helminthes)
Les éosinophiles peuvent être impliqués dans la réponse immunitaire à certaines helminthiases. Les patients présentant une helminthiase connue ont été exclus des études cliniques. On ignore si Fasenra influence la réaction du patient aux helminthiases.
Les patients présentant une helminthiase doivent être traités avant de commencer le traitement par Fasenra. Si une helminthiase survient pendant le traitement par Fasenra et qu'elle ne répond pas à un traitement antihelminthique, il faut envisager une interruption temporaire du traitement par Fasenra jusqu'à la guérison de l'infestation parasitaire.
GEPA menaçant un organe ou le pronostic vital
Fasenra n'a pas été étudié chez les patients présentant des manifestations actives de GEPA menaçant un organe ou le pronostic vital (voir «Posologie/Mode d'emploi»).
InteractionsAucune étude formelle n'a été effectuée pour examiner les interactions du benralizumab avec d'autres médicaments.
Dans une étude randomisée en groupes parallèles et en double aveugle réalisée auprès de 103 patients atteints d'asthme sévère, âgés de 12 à 21 ans, la réaction humorale aux anticorps mesurée suite à la vaccination contre la grippe saisonnière variait en fonction de la souche vaccinale et du critère d'évaluation de la mesure. En présence d'une grande variabilité de réaction, aucune différence cohérente entre le placebo et le benralizumab n'a pu être mise en évidence.
Grossesse, allaitementGrossesse
On ne dispose pas de données pertinentes concernant l'utilisation du benralizumab pendant la grossesse.
Les études effectuées chez l'animal n'ont fourni aucun indice de toxicité pour la reproduction (voir «Données précliniques»).
Au cours de la grossesse, les anticorps monoclonaux, comme p.ex. le benralizumab, traversent le placenta. Ce transport se fait de façon linéaire. C'est pourquoi une exposition potentielle de l'enfant à naître est probablement plus importante lors du deuxième et troisième trimestres de la grossesse.
Fasenra ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
On ne dispose pas de données concernant le passage du benralizumab dans le lait maternel humain ou animal. Par conséquent, un risque pour le nourrisson ne peut pas être exclu.
Il faut donc cesser soit l'allaitement, soit le traitement par benralizumab après avoir évalué les avantages de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère.
Fertilité
Des études portant sur la fertilité chez l'être humain n'ont pas été réalisées. Les études effectuées chez l'animal n'ont pas mis en évidence d'effet indésirable d'un traitement par benralizumab sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesRésumé du profil de sécurité
Le profil de sécurité du benralizumab est similaire dans l'asthme et la GEPA.
Dans les études cliniques réalisées auprès de patients atteints d'asthme sévère non contrôlé à éosinophiles, les effets indésirables le plus souvent rapportés au cours du traitement étaient des céphalées et une pharyngite.
Les effets indésirables les plus fréquemment rapportés en cas de GEPA étaient les céphalées.
Liste des effets indésirables médicamenteux
Asthme
Un nombre total de 1'663 patients atteints d'asthme sévère non contrôlé à éosinophiles ont reçu le benralizumab dans le cadre d'études cliniques d'une durée de 48 à 56 semaines. Les effets indésirables médicamenteux suivants ont été observés dans 2 études contrôlées versus placebo réalisées auprès de patients ayant reçu les 3 premières doses de 30 mg de benralizumab toutes les 4 semaines, puis 30 mg de benralizumab toutes les 8 semaines.
Granulomatose éosinophilique avec polyangéite (GEPA)
Le profil de sécurité chez un total de 70 patients atteints de GEPA ayant reçu du benralizumab à raison de 30 mg toutes les 4 semaines dans une étude clinique de phase 3 contrôlée contre traitement actif d'une durée de 52 semaines était comparable au profil de sécurité établi du benralizumab. L'incidence des effets indésirables était comparable à celle observée dans l'asthme, à l'exception des céphalées et des infections des voies urinaires qui sont survenues chez respectivement 17% et 7,1% des patients traités par benralizumab. Aucun effet indésirable supplémentaire n'a été constaté.
Les fréquences sont définies comme suit: très fréquents (≥1/10), fréquents (≥1/100 et < 1/10), occasionnels (≥1/1'000 et < 1/100), rares (≥1/10'000 et < 1/1'000), très rares (< 1/10'000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Affections du système nerveux
Très fréquent: céphalées
Infections et infestations
Fréquent: pharyngite*, infections des voies urinaires
Troubles généraux et anomalies au site d'administration
Fréquent: fièvre, réactions au site d'injection
Affections du système immunitaire
Fréquent: réaction d'hypersensibilité**
Fréquence indéterminée: anaphylaxie (y compris réaction anaphylactique, angio-œdème)***
* pharyngite inclut les termes normalisés suivants: pharyngite, pharyngite bactérienne, pharyngite virale, pharyngite due à des streptocoques
** réaction d'hypersensibilité inclut les termes normalisés suivants: urticaire, urticaire papuleuse et éruption cutanée. Voir rubrique «Mises en garde et précautions», exemples des manifestations associées ayant été rapportées et leur délai de survenue.
*** Il s'agit d'un effet indésirable lié à l'utilisation de Fasenra qui a été observé après l'autorisation de mise sur le marché. Il n'est généralement pas possible de déterminer la fréquence de ces effets, étant donné qu'il s'agit de rapports spontanés issus d'une population dont la taille exacte n'est pas connue avec précision. La fréquence de ces effets indésirables est par conséquent indiquée avec la mention‚ fréquence indéterminée' (non estimable sur la base des données disponibles).
Description de certains effets indésirables médicamenteux
Réactions au site d'injection
Dans les études sur l'asthme contrôlées versus placebo, des réactions au site d'injection (p.ex. douleurs, érythème, prurit, papules) sont apparues chez 2,2% des patients traités par benralizumab contre 1,9% des patients traités par placebo.
Sécurité à long terme
Dans le cadre de l'étude BORA (étude 4), une étude d'extension de 56 semaines, randomisée et en double aveugle qui incluait des patients asthmatiques préalablement inclus dans les études 1, 2 et 3, 842 patients ont été traités par Fasenra à la posologie autorisée. Le profil d'effets indésirables correspondait au profil d'effets indésirables décrit dans les études sur l'asthme précitées.
L'étude 5 (MELTEMI) était une étude d'extension, en ouvert, examinant la sécurité qui incluait des patients adultes traités pendant au moins 16 semaines dans le cadre de l'étude 4. 226 patients au total ont été traités par Fasenra pendant une durée maximale de 43 mois à la posologie recommandée. Associé à la durée de traitement des études précédentes, cela correspond à un suivi médian de 3,4 ans (fourchette: 8,5 mois – 5,3 ans). Le profil de sécurité durant cette période de suivi était cohérent avec le profil de sécurité connu de Fasenra.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
Immunogénicité
Pendant l'intervalle de traitement de la phase 3, d'une durée de 48 à 56 semaines et contrôlé contre placebo, durant lequel les patients asthmatiques ont été traités par Fasenra selon le schéma posologique recommandé, 107 patients sur 809 (13%) ont développé une réponse immunitaire liée au traitement en produisant des anticorps anti-médicaments (ADA). La majorité des participants ayant développé une réponse ADA présentaient des anticorps neutralisants in vitro. Par rapport aux participants de l'étude sans anticorps, les patients présentant un titre élevé d'ADA ont été associés à des concentrations sériques réduites de benralizumab et à des taux accrus d'éosinophiles dans le sang. Jusqu'à ce jour, aucun lien n'a été observé entre les ADA et des variations en termes d'efficacité et de sécurité.
Après la deuxième année de traitement par Fasenra au schéma posologique recommandé, des anticorps sont apparus chez 18 autres patients asthmatiques des 510 (4%) traités auparavant par Fasenra et chez 29 des 279 (10,4%) patients traités auparavant par placebo. La prévalence globale des anticorps (indépendamment du traitement antérieur) était de 11,7% et celle des anticorps neutralisants de 9,5%.
Les titres chez les patients qui étaient positifs aux ADA dans les études précédentes restaient en moyenne stables durant la première année et montraient plutôt une tendance à la baisse durant la deuxième année de traitement. Toutefois, chez certains patients, les titres demeuraient toujours élevés. Les effets indésirables rapportés chez les patients positifs aux ADA, y compris les patients avec des titres d'ADA qui demeuraient élevés, étaient similaires à ceux des patients négatifs aux ADA et étaient conformes à la maladie sous-jacente du groupe de patients étudié.
Conformément aux résultats des études précédentes, aucun lien n'a été mis en évidence entre les anticorps anti-benralizumab et l'efficacité ou la sécurité.
Chez les patients atteints de GEPA, une réponse des ADA liée au traitement s'est développée chez 6 des 67 (9%) patients sous benralizumab pendant la phase de traitement de 52 semaines de l'étude de phase 3 contrôlée contre traitement actif. Une activité des anticorps neutralisants a été détectée chez un des patients ADA-positifs.
Ces données reflètent la part de patients ayant présenté des résultats positifs aux tests spécifiques de détection d'anticorps contre le benralizumab. L'incidence observée de la réponse anticorps dépend fortement de différents facteurs tels que la sensibilité et la spécificité du test, la méthode du test, la prise en charge des échantillons, le timing du prélèvement des échantillons, la médication concomitante et la présence d'une maladie sous-jacente. C'est pourquoi il peut s'avérer trompeur de comparer l'incidence d'anticorps anti-benralizumab avec l'incidence d'anticorps contre d'autres produits.
SurdosageLors des études cliniques, des patients atteints d'asthme à éosinophiles ont reçu des doses unitaires allant jusqu'à 200 mg par voie sous-cutanée sans aucun indice de toxicité dose-dépendante.
Il n'existe aucun traitement spécifique en cas de surdosage de benralizumab. En cas de surdosage, le patient doit recevoir le traitement symptomatique approprié à son cas et être surveillé en conséquence.
Propriétés/EffetsCode ATC
R03DX10
Mécanisme d'action
Le benralizumab est un anticorps monoclonal humanisé (IgG1, kappa) non fucosylé qui se lie à la sous-unité alpha du récepteur de l'interleukine-5 (IL-5Rα) humaine avec une haute affinité (16 pM) et spécificité. Le récepteur d'IL-5 s'exprime en particulier à la surface des granulocytes éosinophiles et basophiles. L'absence de fucose dans la partie Fc du benralizumab entraîne une liaison d'une forte affinité (45,5 nM) aux récepteurs FcγRIII sur les cellules immunitaires effectrices, comme p.ex. les cellules tueuses naturelles (NK). Ce lien conduit à un renforcement de la cytotoxicité cellulaire dépendante des anticorps (ADCC) et ainsi à l'apoptose des éosinophiles et des basophiles.
L'inflammation éosinophilique contribue de manière essentielle à la pathogenèse de la maladie dans les formes d'asthme à éosinophiles et dans la GEPA.
Pharmacodynamique
Effet sur les éosinophiles dans le sang
Le traitement par benralizumab chez les patients asthmatiques conduit dans les 24 heures après la première dose à une déplétion presque totale des éosinophiles dans le sang qui se maintient pendant toute la durée du traitement. La déplétion des éosinophiles dans le sang s'accompagne d'une réduction des protéines granulaires éosinophiles dans le sérum, d'une réduction de la neurotoxine dérivée de l'éosinophile (EDN) et de la protéine cationique éosinophile (ECP), ainsi que d'une réduction des basophiles dans le sang.
Dans les études cliniques 1 (SIROCCO) et 2 (CALIMA), le nombre d'éosinophiles dans le sang a baissé jusqu'à une valeur absolue médiane de 0 cellules/μl, ce qui correspond à une baisse médiane de 100%, après l'administration sous-cutanée de la dose recommandée de benralizumab (voir «Efficacité clinique»). L'ampleur de la baisse a été constatée au premier moment de l'observation ainsi qu'après 4 semaines de traitement et s'est maintenue pendant toute la durée du traitement.
On a observé le maintien de la déplétion des éosinophiles sur les 56 semaines de la phase d'extension (étude 4, BORA), ce qui concorde avec les résultats des études précédentes.
Chez les patients atteints de GEPA, la déplétion des éosinophiles dans le sang était cohérente avec l'effet observé dans les études sur l'asthme. La déplétion des éosinophiles dans le sang a été constatée au premier point d'observation une semaine après le traitement et s'est maintenue pendant toute la phase de traitement de 52 semaines.
Effet sur les éosinophiles dans la muqueuse des voies respiratoires
L'effet du benralizumab sur les éosinophiles de la muqueuse des voies respiratoires a été évalué chez des patients asthmatiques avec un nombre accru d'éosinophiles dans les expectorations (≥2,5%) dans une étude clinique de phase 1 de 12 semaines, randomisée, en double aveugle, contrôlée versus placebo avec l'administration sous-cutanée de 100 ou 200 mg de benralizumab. Cette étude a révélé une diminution médiane des éosinophiles dans la muqueuse des voies respiratoires de 96% au total par rapport à la valeur initiale dans le groupe traité par benralizumab, en comparaison à 47% dans le groupe placebo (p=0,039).
Efficacité clinique
Asthme
L'efficacité de Fasenra a été examinée dans 3 études cliniques randomisées, en double aveugle, contrôlées versus placebo, d'une durée de 28 à 56 semaines chez des patients dès l'âge de 12 ans.
Dans ces études, Fasenra a été administré en complément du traitement standard à la dose de 30 mg une fois toutes les 4 semaines pour les 3 premières doses, puis toutes les 4 ou 8 semaines et examiné par comparaison au traitement standard plus placebo.
Les deux études contrôlées versus placebo ayant évalué l'effet sur les exacerbations, étude 1 (SIROCCO) et étude 2 (CALIMA), d'une durée de resp. 48 et 56 semaines, ont inclus un total de 2'510 patients (adultes ou adolescents à partir de 12 ans) atteints d'asthme non contrôlé; 822 d'entre eux ont été traités à la dose proposée. Au moins 2 épisodes d'exacerbation de l'asthme ayant exigé un traitement corticoïde par voie orale ou systémique devaient figurer dans l'anamnèse des patients au cours des 12 mois précédents. De plus, le résultat du test de contrôle de l'asthme ACQ-6 devait être de 1,5 ou plus au dépistage, et la fonction pulmonaire altérée au départ (volume expiratoire maximal par seconde (VEMS) avant bronchodilatation < 80% chez les adultes et < 90% chez les adolescents). Les patients de l'étude 1 recevaient un traitement régulier par des corticostéroïdes inhalés (CSI) à hautes doses et les patients de l'étude 2 recevaient un traitement par CSI à moyennes ou hautes doses en association à des bêta-agonistes à longue durée d'action (LABA). Le nombre moyen d'exacerbations rapportées au cours de l'année précédente était de 3 et le VEMS moyen prédit «avant bronchodilatation» de 57,5%. La stratification des patients a été effectuée selon leur lieu de résidence géographique, leur âge et le taux d'éosinophiles dans le sang périphérique (≥300 cellules/μl ou < 300 cellules/μl).
Au total 220 patients asthmatiques (61% de femmes; âge moyen de 51 ans) ont participé à l'étude 3 contrôlée versus placebo (ZONDA) ayant examiné la diminution de l'utilisation des corticostéroïdes oraux (CSO); 73 d'entre eux ont été traités par la dose proposée. Les patients inclus dans l'étude étaient traités quotidiennement par des CSO (7,5–40 mg par jour) et en plus, régulièrement, par des CSI à hautes doses et des LABA, avec ou sans autres médicaments complémentaires nécessaires au maintien du contrôle de l'asthme. L'étude comprenait une phase de préinclusion de 8 semaines permettant de réduire la dose de CSO par titration à la dose efficace minimale sans perte du contrôle de l'asthme. La dose moyenne de CSO au stade initial était à un niveau similaire dans tous les groupes de traitement. Le taux d'éosinophiles dans le sang périphérique devait être d'au moins 150 cellules/μl et les patients devaient avoir eu au moins une exacerbation au cours des 12 mois précédents. La dose moyenne de CSO était de 10 mg (fourchette 8-40 mg) au stade initial dans les 3 groupes de traitement. Au total, 598 patients (64% de femmes; âge moyen de 53 ans) ont participé à l'étude 6 (PONENTE) à un bras portant sur la diminution des CSO; ils ont tous été traités par la dose proposée. La dose moyenne de CSO était de 10 mg (fourchette 5–60 mg) au stade initial.
Alors que les études 1, 2 et 3 examinaient deux schémas posologiques, le schéma posologique recommandé de Fasenra prévoit d'administrer une dose toutes les 4 semaines pour les 3 premières doses, puis une dose toutes les 8 semaines (voir «Posologie/Mode d'emploi»).
Exacerbations
Le critère d'évaluation primaire de l'étude 1 (SIROCCO) et de l'étude 2 (CALIMA) était la fréquence annuelle d'exacerbations cliniquement significatives de l'asthme chez des patients présentant au stade initial un taux d'éosinophiles dans le sang d'au moins 300 cellules/μl et utilisant des CSI à hautes doses et des LABA. Conformément à la définition utilisée, une exacerbation cliniquement significative de l'asthme était une péjoration de l'asthme nécessitant l'utilisation de corticostéroïdes par voie orale ou systémique pendant au moins 3 jours et/ou une hospitalisation dans le cadre de laquelle le traitement requis comprenait des corticostéroïdes par voie orale/systémique et/ou une admission dans un service d'urgence. Chez les patients prenant des corticostéroïdes par voie orale en traitement d'entretien, une exacerbation cliniquement significative de l'asthme était définie comme suit: augmentation à court terme pendant au moins 3 jours des corticostéroïdes oraux ou systémiques utilisés jusque-là à une dose stable ou administration d'une injection unique de corticostéroïde à effet retard.
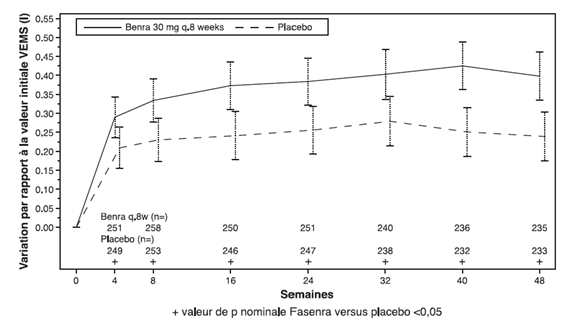
Dans les études 1 et 2, les patients avec au moins 300 cellules/μl d'éosinophiles dans le sang ont bénéficié sous Fasenra d'une diminution significative du taux annuel d'exacerbations par rapport au placebo (tableau 1). La variation du VEMS moyen par rapport à la valeur initiale a été mesurée dans les deux études et a au moins montré des avantages numériques à tout moment à partir de la semaine 4 (voir Figure 1). L'effet s'est maintenu jusqu'à la fin du traitement, comme le montre le tableau 1.
Tableau 1. Résultats concernant le taux annuel d'exacerbations et la fonction pulmonaire au terme du traitement dans les études 1 (SIROCCO) et 2 (CALIMA) selon le nombre d'éosinophiles.
|
|
Étude 1
|
Étude 2
| |
Fasenra
|
Placebo
|
Fasenra
|
Placebo
| |
Taux d'éosinophiles dans le sang ≥300 cellules/μla
|
n=267
|
n=267
|
n=239
|
n=248
| |
Exacerbations cliniquement significatives
| |
Taux
|
0,74
|
1,52
|
0,73
|
1,01
| |
Différence
|
-0,78
|
-0,29
| |
Ratio des taux (IC à 95%)
|
0,49 (0,37; 0,64)
|
0,72 (0,54; 0,95)
| |
Valeur de p
|
< 0,001
|
0,019
| |
VEMS avant bronchodilatation (litre)
| |
Valeur initiale moyenne
|
1,660
|
1,654
|
1,758
|
1,815
| |
Amélioration versus valeur initiale
|
0,398
|
0,239
|
0,330
|
0,215
| |
Différence (IC à 95%)
|
0,159 (0,068; 0,249)
|
0,116 (0,028; 0,204)
| |
Valeur de p
|
0,001
|
0,010
|
a population en intention de traiter (ITT) (patients traités par des CSI à haute dose et avec un taux d'éosinophiles dans le sang ≥300 cellules/μl).
Une analyse combinée des études 1 et 2 a mis en évidence une plus forte diminution du taux d'exacerbations en présence d'un taux plus élevé d'éosinophiles dans le sang au stade initial. De même, une analyse combinée a montré une amélioration plus importante du VEMS chez les patients avec un taux croissant d'éosinophiles dans le sang.
Dans l'étude 1 (SIROCCO), le taux d'exacerbations ayant nécessité une hospitalisation et/ou une consultation au service des urgences était de 0,09 chez les patients sous Fasenra versus 0,25 sous placebo (ratio des taux 0,37, IC à 95%: 0,20, 0,67, p ≤0,001). Dans l'étude 2 (CALIMA), ce taux était de 0,12 versus 0,10 (ratio des taux 1,23; IC à 95%: 0,64; 2,35; p=0,538). L'étude 2 n'a pas révélé d'avantages numériques pour Fasenra dans ce domaine. Toutefois, le taux d'événements était trop bas pour pouvoir tirer des conclusions sur l'effet de Fasenra sur les exacerbations sévères exigeant une hospitalisation et/ou une consultation au service des urgences.
Figure 1. Variation moyenne versus valeur initiale du VEMS avant bronchodilatation (litre), études 1 (SIROCCO) et 2 (CALIMA)
Étude 1
Étude 2
Le nombre de patients ayant souffert d'au moins une exacerbation était plus bas sous Fasenra que sous placebo (35% vs 51% dans l'étude 1 et 40% vs 51% dans l'étude 2), et le temps écoulé jusqu'à la première exacerbation était plus long sous Fasenra dans les deux études (Hazard ratio 0,60; IC à 95%: 0,46; 0,78 et Hazard ratio 0,73; IC à 95%: 0,55; 0,95, resp. dans les études 1 et 2).
De plus, au terme du traitement, on a observé une amélioration de la variation moyenne du débit expiratoire de pointe (PEF) matinal et vespéral par rapport au stade initial chez les patients sous Fasenra par rapport au placebo.
Dans les études 1 et 2, les patients ayant reçu Fasenra ont bénéficié d'une réduction statistiquement significative des symptômes d'asthme (score total de l'asthme) par rapport aux patients ayant reçu un placebo. Une amélioration cohérente en lien avec Fasenra a été observée au moyen du questionnaire de contrôle de l'asthme (Asthma Control Questionnaire-6: ACQ-6) et du questionnaire standardisé portant sur la qualité de vie des personnes asthmatiques âgées de plus de 12 ans (Standardised Asthma Quality of Life Questionnaire for 12 Years and Older: AQLQ(S)+12) (Tableau 2).
Tableau 2. Différence thérapeutique à la fin du traitement exprimée par la variation moyenne par rapport aux valeurs initiales du score des symptômes d'asthme, de l'ACQ-6 et de l'AQLQ(s)+12a
|
Variables d'efficacité
|
Étude 1
(SIROCCO)
|
Étude 2
(CALIMA)
| |
Fasenra
na=267
|
Placebo
na=267
|
Fasenra
na=239
|
Placebo
na=248
| |
Score total des symptômes d'asthmeb
| |
Valeur initiale moyenne
|
2,68
|
2,74
|
2,76
|
2,71
| |
Amélioration par rapport à la valeur initialeb
|
- 1,30
|
- 1,04
|
-1,40
|
-1,16
| |
Différence (IC à 95%)
|
-0,25 (-0,45; -0,06)
|
-0,23 (-0,43; -0,04)
| |
Valeur de p
|
0,012
|
0,019
| |
ACQ-6
| |
Valeur initiale moyenne
|
2,81
|
2,90
|
2,80
|
2,75
| |
Amélioration par rapport à la valeur initiale
|
-1,46
|
-1,17
|
-1,44
|
-1,19
| |
Différence (IC à 95%)
|
-0,29 (-0,48; -0,10)
|
-0,25 (-0,44; -0,07)
| |
AQLQ(S)+12
| |
Valeur initiale moyenne
|
3,93
|
3,87
|
3,87
|
3,93
| |
Amélioration par rapport à la valeur initiale
|
1,56
|
1,26
|
1,56
|
1,31
| |
Différence (IC à 95%)
|
0,30 (0,10, 0,50)
|
0,24 (0,04; 0,45)
|
a Le nombre de patients (n) varie légèrement en raison de la disponibilité des données relatives aux différentes variables. Les résultats indiqués font référence aux dernières données disponibles pour la variable en question.
b Score des symptômes d'asthme: score total allant de 0 (le plus bas) à 6 (le plus élevé); score des symptômes d'asthme diurne et nocturne allant de 0 (le plus bas) à 3 (le plus élevé) symptômes. Pris individuellement, les scores diurnes et nocturnes étaient similaires.
Analyses en sous-groupes selon les antécédents d'exacerbations
Une analyse en sous-groupes à partir des études 1 et 2 a identifié un taux accru d'exacerbations dans les antécédents comme un prédicteur potentiel d'une meilleure réponse au traitement par le benralizumab. Seul ou combiné avec un taux accru d'éosinophiles dans le sang au stade initial, le taux d'exacerbations peut être utilisé pour identifier les patients susceptibles de bénéficier d'une meilleure réponse au traitement par le benralizumab.
Dans les deux études, les patients ayant eu 3 exacerbations ou plus au cours des 12 mois de la randomisation Fasenra présentaient une diminution du taux d'exacerbation plus importante que les patients ayant eu moins de 3 exacerbations antérieures (voir Tableau 3).
Tableau 3. Taux d'exacerbations et fonction pulmonaire (VEMS) au terme du traitement selon le nombre d'exacerbations au cours de l'année précédente, études 1 (SIROCCO) et 2 (CALIMA) (population en intention de traiter)
|
|
Étude 1
|
Étude 2
| |
|
Fasenra (N=267)
|
Placebo (N=267)
|
Fasenra (N=239)
|
Placebo (N=248)
| |
Valeur initiale avec 2 exacerbations
| |
n
|
164
|
149
|
144
|
151
| |
Taux d'exacerbations
|
0,57
|
1,04
|
0,63
|
0,62
| |
Différence
|
-0,47
|
0,01
| |
Taux d'incidence
(IC à 95%)
|
0,55
(0,37; 0,80)
|
1,01
(0,70;1,46)
| |
Variation moyenne du VEMS avant bronchodilatation
|
0,343
|
0,230
|
0,266
|
0,236
| |
Taux d'incidence
(IC à 95%)
|
0,113
(-0,002; 0,228)
|
0,029
(-0,079; 0,137)
| |
Valeur initiale avec plus de 3 exacerbations
| |
n
|
103
|
118
|
95
|
97
| |
Taux d'exacerbations
|
0,95
|
2,23
|
0,82
|
1,65
| |
Différence
|
-1,28
|
-0,84
| |
Taux d'incidence
(IC à 95%)
|
0,43
(0,29; 0,63)
|
0,49
(0,33; 0,74)
| |
Variation moyenne du VEMS avant bronchodilatation
|
0,486
|
0,251
|
0,440
|
0,174
| |
Taux d'incidence
(IC à 95%)
|
0,235
(0,088; 0,382)
|
0,265
(0,115; 0,415)
|
S'agissant des symptômes d'asthme, les patients avec 3 exacerbations ou plus au cours des 12 mois précédant la randomisation avaient au terme du traitement par Fasenra des différences de score moyennes dans les études 1 et 2 de resp. -0,32 et -0,41 par rapport au stade initial (étude 1 IC à 95%: -0,62, -0,01; étude 2 IC à 95%: -0,73, -0,09). S'agissant des symptômes d'asthme, les patients avec 2 exacerbations au cours des 12 mois précédant la randomisation avaient au terme du traitement par Fasenra des différences de score moyennes dans les études 1 et 2 de resp. -0,22 et -0,12 par rapport au stade initial (étude 1: IC à 95%: -0,49; -0,04; étude 2: IC à 95%: -0,37; -0,13).
Études portant sur la diminution de l'utilisation des corticostéroïdes oraux (CSO)
L'étude ZONDA (étude 3), une étude contrôlée versus placebo et l'étude PONENTE (étude 6), une étude en ouvert, ont évalué l'effet de Fasenra sur la diminution de l'utilisation des CSO en traitement d'entretien.
Dans l'étude 3, le critère d'évaluation primaire était la diminution en pour cent de la dose de CSO au terme de l'étude dans les semaines 24 à 28 par rapport à la valeur initiale tout en maintenant le contrôle de l'asthme. Par rapport au placebo, les patients traités par Fasenra ont pu diminuer plus fortement la dose quotidienne d'entretien des corticostéroïdes oraux tout en maintenant le contrôle de l'asthme. Une diminution de la dose de CSO de 50% ou plus a été observée chez 48 patients (66%) recevant Fasenra par rapport à 28 patients (37%) recevant le placebo. Le tableau 4 résume les résultats de l'étude 3.
Tableau 4. Effet de Fasenra sur la diminution de la dose de CSO, étude 3 (ZONDA)
|
|
Fasenra
N=73
|
Placebo
N=75
| |
Test de la somme des rangs de Wilcoxon (méthode d'analyse primaire)
| |
Diminution médiane en pour cent de la dose quotidienne de CSO par rapport à la valeur initiale (IC à 95%)
|
75 (60; 88)
|
25 (0; 33)
| |
Test de la somme des rangs de Wilcoxon, valeur de p
|
< 0,001
|
| |
Proportional Odds Model (analyse de sensibilité)
| |
Diminution en pour cent des CSO par rapport à la valeur initiale jusqu'à la semaine 28
| |
≥90%
|
27 (37%)
|
9 (12%)
| |
≥75%
|
37 (51%)
|
15 (20%)
| |
≥50%
|
48 (66%)
|
28 (37%)
| |
> 0%
|
58 (79%)
|
40 (53%)
| |
Pas de variation ou pas de diminution des CSO
|
15 (21%)
|
35 (47%)
| |
Odds Ratio (IC à 95%)
|
4,12 (2,22; 7,63)
|
| |
Diminution de la dose quotidienne de CSO à
0 mg/jour*
|
22 (52%)
|
8 (19%)
| |
Odds Ratio (IC à 95%)
|
4,19 (1,58; 11,12)
|
| |
Diminution de la dose quotidienne de CSO à
≤5 mg/jour
|
43 (59%)
|
25 (33%)
| |
Odds Ratio (IC à 95%)
|
2,74 (1,41; 5,31)
|
| |
Taux d'exacerbations
|
0,54
|
1,83
| |
Ratio des taux (IC à 95%)
|
0,30 (0,17; 0,53)
|
| |
Taux d'exacerbations ayant exigé une hospitalisation/consultation au service des urgences
|
0,02
|
0,32
| |
Ratio des taux (IC à 95%)
|
0,07 (0,01; 0,63)
|
|
* Seuls les patients recevant au stade initial une dose optimisée de CSO de 12,5 mg ou moins ont pu obtenir une diminution de 100% de la dose de CSO durant l'étude
Par ailleurs, la fonction pulmonaire, le score des symptômes d'asthme, l'ACQ-6 et l'AQLQ(S)+12 ont été examinés. Ces résultats étaient comparables à ceux des études 1 et 2.
L'étude 6 incluait 598 patients adultes présentant un asthme sévère corticodépendant (taux d'éosinophiles dans le sang ≥150 cellules/μl à l'inclusion dans l'étude ou ≥300 cellules/μl au cours des 12 derniers mois si le taux à l'inclusion dans l'étude était < 150 cellules/μl). Les critères d'évaluation primaire étaient la proportion de patients ayant pu arrêter les CSO tout en maintenant le contrôle de l'asthme et la proportion de patients dont la dose finale de CSO était inférieure ou égale à 5 mg tout en maintenant le contrôle de l'asthme et en tenant compte de la fonction surrénalienne. La proportion de patients qui ont pu arrêter le traitement d'entretien par les CSO était de 62,9%. La proportion de patients pour qui la dose finale de CSO était inférieure ou égale à 5 mg (tout en maintenant le contrôle de l'asthme et non limité par la fonction surrénalienne) était de 81,9%. Les effets en termes de diminution des CSO étaient comparables indépendamment du taux d'éosinophiles dans le sang à l'inclusion dans l'étude (y compris pour les patients présentant un taux d'éosinophiles dans le sang < 150 cellules/μl) et ils se sont maintenus pendant une période de 24 à 32 semaines. Le taux annuel d'exacerbations dans l'étude 6 était comparable à celui rapporté dans les études précédentes.
Étude d'extension à long terme
La sécurité à long terme de Fasenra a été évaluée dans le cadre d'une étude d'extension de phase 3 de 56 semaines, randomisée, en double aveugle et en groupes parallèles (étude 4, BORA). La sécurité à long terme de Fasenra a été, en outre, examinée dans le cadre de l'étude d'extension de sécurité à long terme en ouvert MELTEMI (étude 5) (voir «Effets indésirables»). L'étude 4 (BORA) incluait 2123 patients adultes et adolescents (âgés de 12 ans et plus) préalablement inclus dans les études 1 (SIROCCO), 2 (CALIMA) et 3 (ZONDA). Parmi eux, 842 patients ont été traités par Fasenra à la posologie autorisée.
L'étude 4 (BORA) visait principalement à évaluer la sécurité et la tolérance à long terme de Fasenra. L'évolution du taux annuel d'exacerbations, la fonction pulmonaire, l'ACQ-6, l'AQLQ(S)+12 et la dose de CSO ont également été observés. Les schémas posologiques évalués dans les deux études précédentes ont pour cela été appliqués.
La diminution du taux d'exacerbations et de la dose de CSO constatée dans les études 1 et 2 précédentes comparativement au placebo a également été maintenue pendant la deuxième année d'application du schéma posologique autorisé. L'amélioration de la fonction pulmonaire, de l'ACQ-6 et de l'AQLQ(S)+12 constatée dans les études 1 et 2 précédentes était également maintenue pendant la deuxième année d'application du schéma posologique autorisé.
La sécurité à long terme a été examinée dans le cadre de l'étude 5 chez 226 patients qui avaient reçu au moins une dose de Fasenra au schéma posologique recommandé. Le taux annuel d'exacerbations dans l'étude 5 (0,47) était comparable à celui des études 1 (0,74), 2 (0,73) et 4 (0,46).
Granulomatose éosinophilique avec polyangéite (GEPA)
Chez les patients atteints de GEPA âgés de 18 ans et plus, l'efficacité et la sécurité de Fasenra ont été évaluées dans le cadre d'une étude clinique de non-infériorité randomisée, en double aveugle et contrôlée contre traitement actif avec une durée de traitement de 52 semaines. Au total, 140 patients présentant une affection récidivante ou réfractaire ont été randomisés. Fasenra 30 mg a été comparé au mépolizumab 300 mg par voie sous-cutanée toutes les 4 semaines en ajout au traitement de base par prednisolone/prednisone avec ou sans traitement immunosuppresseur. La dose de CSO a été réduite progressivement selon l'appréciation du médecin investigateur. Les patients qui étaient traités activement par le cyclophosphamide ou le rituximab au début de l'étude ont été exclus.
Les données démographiques et les caractéristiques initiales figurent dans le Tableau 5.
Tableau 5. Données démographiques et caractéristiques initiales dans l'étude portant sur la GEPA
|
|
Population globale (N=140)
| |
Âge moyen (ans)
|
52
| |
Sexe féminin (%)
|
60
| |
Origine caucasienne (%)
|
79
| |
Temps écoulé depuis le diagnostic de GEPA, années, moyenne (ET)
|
5,2 (5,64)
| |
≥1 récidive confirmée au cours des 2 dernières années (%)
|
79
| |
Affection réfractaire (%)
|
60
| |
Dose quotidienne initiale de corticostéroïdes orauxa, mg, médiane (étendue)
|
10 (5–40)
| |
Maintien du traitement immunosuppresseurb (%)
|
36
| |
Positivité ANCAc (%)
|
29
|
ET = écart type.
a Équivalent prednisone ou prednisolone.
b Azathioprine, méthotrexate, mycophénolate.
c Antécédents de positivité ou positivité à la sélection pour les anticorps anti-cytoplasme des polynucléaires neutrophiles (ANCA).
Rémission
Le critère d'évaluation principal était le pourcentage de patients en rémission, définie comme un score BVAS (Birmingham Vasculitis Activity Score) = 0 (absence de vascularite active) plus dose de prednisolone/prednisone ≤4 mg/jour, aussi bien à la Semaine 36 qu'à la Semaine 48. Comme le montre le Tableau 6, Fasenra présentait une non-infériorité par rapport au mépolizumab en ce qui concerne le critère d'évaluation principal. Les résultats pour la durée cumulée de la rémission et les composantes de la rémission sont également présentés dans le Tableau 6.
Tableau 6. Rémission et composantes de la rémission dans la GEPA
|
|
Rémission
(CSO ≤4 mg/jour +
BVAS=0)
|
CSO ≤4 mg/jour
|
BVAS=0
| |
Fasenraa
N=70
|
Mépob
N=70
|
Fasenraa
N=70
|
Mépob
N=70
|
Fasenraa
N=70
|
Mépob
N=70
| |
Patients en rémission après 36 et 48 semaines
| |
Patients, n (%)c
|
40 (58)
|
40 (57)
|
42 (61)
|
41 (58)
|
58 (83)
|
59 (84)
| |
Différences dans le taux de rémission, (%)c (IC à 95%)
|
1,21
(-14,11; 16,53)
|
--
--
|
2,64
(-12,67; 17,95)
|
--
--
|
-1,17
(-13,27; 10,94)
|
--
--
| |
Durée cumulée sur 52 semaines, n (%)
| |
0 semained
|
9 (13)
|
15 (21)
|
9 (13)
|
12 (17)
|
0
|
0
| |
> 0 à < 12 semaines
|
13 (19)
|
10 (14)
|
11 (16)
|
12 (17)
|
0
|
2 (3)
| |
12 à < 24 semaines
|
8 (11)
|
8 (11)
|
9 (13)
|
8 (11)
|
2 (3)
|
2 (3)
| |
24 à < 36 semaines
|
20 (29)
|
19 (27)
|
19 (27)
|
18 (26)
|
6 (9)
|
7 (10)
| |
≥36 semaines
|
20 (29)
|
18 (26)
|
22 (31)
|
20 (29)
|
62 (89)
|
59 (84)
| |
Odds Ratioe
(IC à 95%)
|
1,32
(0,72; 2,40)
|
--
--
|
1,27
(0,70; 2,31)
|
--
--
|
1,50
(0,54; 4,15)
|
--
--
|
N = nombre de patients dans l'analyse.
a Fasenra 30 mg toutes les 4 semaines.
b Mépolizumab (mépo) 300 mg toutes les 4 semaines.
c Pourcentages ajustés selon le modèle.
d Aucune rémission obtenue, quel que soit le moment.
e Un odds ratio > 1 est en faveur de Fasenra.
Le pourcentage de patients ayant obtenu une rémission au cours des 24 premières semaines de traitement et ayant maintenu cette rémission jusqu'à la Semaine 52 atteignait 42% pour Fasenra et 37% pour le mépolizumab (différence des taux de réponse 5,54%, IC à 95%: -9,30; 20,37).
En cas d'utilisation d'une définition alternative de la rémission, à savoir un score BVAS = 0 plus prednisolone/prednisone ≤7,5 mg/jour, une efficacité cohérente entre les groupes a été observée pour ces critères d'évaluation.
Les patients ont atteint les critères principaux de rémission dans tous les sous-groupes prédéterminés de données démographiques et de caractéristiques initiales.
Récidives
Le hazard ratio pour le délai jusqu'à la première récidive (définie comme une détérioration de la vascularite, de l'asthme ou des symptômes sinusaux nécessitant une augmentation de la dose de corticostéroïdes ou du traitement immunosuppresseur) atteignait 0,98 (IC à 95%: 0,53; 1,82). Des récidives ont été rapportées chez 30% des patients sous Fasenra et chez 30% des patients sous mépolizumab. Le taux de récidive annualisé atteignait 0,50 pour les patients sous Fasenra contre 0,49 pour les patients sous mépolizumab (ratio des taux 1,03, IC à 95%: 0,56; 1,90). Les types de récidives étaient comparables chez les patients sous Fasenra et mépolizumab.
Réduction des corticostéroïdes oraux
La dose de CSO quotidienne moyenne au cours des Semaines 48 à 52 est présentée dans le Tableau 7. Une réduction de la dose de CSO de 100% a été observée chez 41% des patients sous Fasenra, par comparaison avec 26% des patients qui ont reçu le mépolizumab (différence 15,69%, IC à 95%: 0,67; 30,71). Une réduction de 50% ou plus a été observée chez 85% des patients sous Fasenra, par comparaison avec 74% des patients qui ont reçu le mépolizumab (différence 10,79%, IC à 95%: -2,25; 23,83).
Tableau 7. Dose quotidienne moyenne de corticostéroïdes oraux au cours des Semaines 48 à 52 dans la GEPA
|
|
Nombre de patients (%)
| |
Fasenraa (N=70)
|
Mépolizumabb (N=70)
| |
0 mg
> 0 à ≤4,0 mg
> 4,0 à ≤7,5 mg
> 7,5 mg
|
29 (41)
19 (27)
15 (21)
7 (10)
|
19 (27)
30 (43)
13 (19)
8 (11)
| |
Odds Ratioc (IC à 95%)
|
1,38 (0,75; 2,54)
|
--
|
N = nombre de patients dans l'analyse.
a Fasenra 30 mg toutes les 4 semaines.
b Mépolizumab 300 mg toutes les 4 semaines.
c Un odds ratio > 1 est en faveur de Fasenra.
PharmacocinétiqueLes propriétés pharmacocinétiques du benralizumab ci-dessous reposent sur les analyses pharmacocinétiques de population issues des études portant sur l'asthme.
Administré par voie sous-cutanée à des patients atteints d'asthme, le benralizumab a présenté à des doses allant de 2 à 200 mg une pharmacocinétique proportionnelle à la dose.
Absorption
Après l'administration sous-cutanée à des patients asthmatiques, la demi-vie d'absorption s'élevait à 3,5 jours. Selon l'analyse pharmacocinétique de population, la biodisponibilité absolue estimée se situait à environ 59% et aucune différence cliniquement pertinente n'est apparue s'agissant de la biodisponibilité relative lors d'une administration dans l'abdomen, la cuisse ou la partie supérieure du bras.
Distribution
Selon l'analyse pharmacocinétique de population, le volume de distribution central et périphérique est estimé à resp. 3,1 litres et 2,5 litres pour une personne de 70 kg.
Biotransformation
Le benralizumab est un anticorps monoclonal humanisé de type IgG1. Il est dégradé par des enzymes protéolytiques présentes non seulement dans le foie mais dans le corps entier.
Élimination
L'analyse pharmacocinétique de population du benralizumab suggère une pharmacocinétique linéaire sans voie de clairance médiée par le récepteur cible. La clairance systémique estimée (CL) du benralizumab était de 0,29 litre par jour. Chez les patients atteints de GEPA, la clairance systémique estimée d'après le modèle était de 0,22 litre par jour. Après l'administration sous-cutanée, la demi-vie d'élimination s'élevait à env. 15,5 jours.
Interactions avec d'autres médicaments et autres formes d'interactions
Aucune étude formelle d'interaction avec d'autres médicaments n'a été effectuée.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude clinique n'a été effectuée pour évaluer l'influence d'une insuffisance hépatique sur la pharmacocinétique du benralizumab. Étant donné que les anticorps monoclonaux IgG ne sont pas essentiellement dégradés par le foie, la clairance du benralizumab ne devrait guère être influencée par une insuffisance hépatique. Selon les analyses pharmacocinétiques de population, les biomarqueurs de la fonction hépatique mesurés au stade initial (ALAT, ASAT et bilirubine) n'avaient pas d'effet cliniquement pertinent sur la clairance du benralizumab.
Troubles de la fonction rénale
Aucune étude clinique formelle n'a été effectuée pour évaluer l'influence d'une insuffisance rénale sur la pharmacocinétique du benralizumab. Sur la base des analyses pharmacocinétiques de population, la clairance du benralizumab chez les patients dont la clairance de la créatinine est comprise entre 30 et 80 ml/min est comparable à celle de patients à la fonction rénale normale. Les données disponibles sur les patients présentant une clairance de la créatinine inférieure à 30 ml/min sont limitées, mais le benralizumab n'est pas éliminé par voie rénale.
Patients âgés
L'analyse pharmacocinétique de population n'a pas fourni d'indices d'une influence de l'âge sur la clairance du benralizumab.
Sexe
L'analyse pharmacocinétique de population a montré que le sexe n'a pas d'influence significative sur la clairance du benralizumab.
Données précliniquesPharmacologie de sécurité
Les données précliniques issues des études conventionnelles de pharmacologie de sécurité ou de toxicité en administration répétée réalisées sur des singes n'ont pas révélé de risque particulier pour l'être humain. Le benralizumab étant un anticorps monoclonal, aucune étude de génotoxicité ou de carcinogénicité n'a été effectuée.
Une diminution du nombre d'éosinophiles dans le sang périphérique et dans la moelle osseuse a été observée chez la majorité des singes cynomolgus après administration intraveineuse et sous-cutanée de benralizumab. L'absence de réponse chez certains singes n'a pu être élucidée de façon concluante. La réduction du nombre d'éosinophiles n'a pas entraîné d'anomalie toxicologique pertinente.
Toxicité sur la reproduction
Dans une étude portant sur le développement pré- et postnatal chez des singes cynomolgus, l'administration de 10 ou 30 mg de benralizumab/kg n'a pas eu d'impact sur le développement maternel, embryo-fœtal ou postnatal. Chez les singes qui étaient exposés au benralizumab pendant la gestation, une réduction des éosinophiles a été mise en évidence chez la progéniture.
Fertilité
Dans les études en administration répétée, l'administration de doses de benralizumab allant jusqu'à 30 mg/kg n'a pas influencé la fertilité des singes cynomolgus mâles et femelles.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Fasenra peut être conservé jusqu'à 25°C pendant un maximum de 14 jours. Fasenra doit être utilisé ou jeté dans les 14 jours suivant sa sortie du réfrigérateur. Ne pas exposer à la chaleur. Ne pas congeler.
Tenir la seringue préremplie dans l'emballage originale à l'abri de la lumière et hors de la portée des enfants.
Ne pas agiter.
Remarques concernant la manipulation
Ce médicament est destiné exclusivement à un usage unique.
La solution injectable Fasenra est fournie dans une seringue préremplie stérile contenant une dose unique pour un usage individuel. Ne pas agiter. Ne plus utiliser si le produit a été congelé.
Avant l'administration, la boîte de Fasenra doit être conservée à température ambiante (jusqu'à 25°C) pendant en règle générale 30 minutes afin de tempérer le médicament. Inspecter visuellement Fasenra à la recherche de particules ou d'un changement de couleur avant l'administration.
Fasenra est une solution claire, incolore à jaune, et peut contenir des particules translucides ou blanches à blanc cassé. Ne pas utiliser Fasenra si le liquide est trouble, présente un changement de coloration ou s'il contient des grosses particules ou des composants étrangers.
La notice d'emballage contient un mode d'emploi détaillé.
Numéro d’autorisation66582, 67581 (Swissmedic)
PrésentationFasenra: emballage contenant 1 seringue préremplie à 30 mg/1 ml. [B]
Fasenra Pen: emballage contenant 1 stylo prérempli à 30 mg/1 ml. [B]
Titulaire de l’autorisationAstraZeneca AG, 6340 Baar
Mise à jour de l’informationAoût 2024
|