Propriétés/EffetsCode ATC
L04AG04
Mécanisme d'action
Le stimulateur de lymphocytes B (BLyS, également appelé BAFF et TNFSF13) appartient à la famille des ligands du facteur de nécrose tumorale (TNF). C'est la molécule cible du bélimumab. Le BLyS inhibe l'apoptose des lymphocytes B et stimule la prolifération et la différenciation des lymphocytes B en plasmocytes producteurs d'immunoglobulines.
Le BLyS est surexprimé chez les patients atteints de LED. Il existe une forte corrélation entre l'activité du LED (évaluée à l'aide du «Safety of Estrogen in Lupus Erythematosus National Assessment-Systemic Lupus Erythematosus Disease Activity Index» [SELENA-SLEDAI]) et les taux plasmatiques de BLyS.
Le bélimumab est un anticorps monoclonal humain de type IgG1λ d'environ 147 kDa. Il se fixe spécifiquement au BLyS soluble humain et inhibe son activité biologique. Le bélimumab ne se fixe pas directement aux lymphocytes B, mais inhibe la survie des lymphocytes B – y compris des lymphocytes B auto-réactifs – et réduit la différenciation des lymphocytes B en plasmocytes producteurs d'immunoglobulines par une liaison et une neutralisation du BLyS.
Pharmacodynamique
Dans des études avec administration intraveineuse ou sous-cutanée, les concentrations médianes d'IgG ont diminué de 11 à 15% jusqu'à la 52e semaine chez les patients atteints de LED sous bélimumab et de 2,5 à 0,7% chez les patients sous placebo.
Parmi les patients atteints de LED ayant présenté des anticorps anti-ADN double brin au début de l'étude, une réduction a été observée sous bélimumab dès la semaine 4. À 52 semaines, les anticorps anti-ADN double brin étaient indétectables chez 16 à 18% des patients sous bélimumab, par rapport à 7 à 15% des patients sous placebo.
Chez les patients atteints de LED ayant présenté de faibles taux de complément au début de l'étude, le traitement par bélimumab a provoqué une augmentation des taux de complément (C3 et C4). Cette augmentation a été observée dès la semaine 4 et s'est maintenue par la suite. À 52 semaines, les taux de C3 et de C4 étaient normalisés chez 38 à 42% et 44 à 53% des patients sous bélimumab, par rapport à 17 à 21% et 19 à 20% des patients sous placebo.
Sous bélimumab, une diminution des lymphocytes B circulants, qu'il s'agisse des lymphocytes B transitionnels, naïfs et activés, des plasmocytes et des lymphocytes B auto-réactifs du LED, a été observée à la semaine 52. Les réductions de lymphocytes naïfs, de plasmocytes, de plasmocytes à courte durée de vie et de lymphocytes B auto-réactifs du LED étaient constatables dès la semaine 8. Le taux de lymphocytes B à mémoire a augmenté au début, puis baissé lentement aux valeurs initiales jusqu'à la semaine 52.
Dans une étude d'extension à long terme non contrôlée avec administration intraveineuse, menée chez des patients atteints de LED, des lymphocytes B (entre autres des lymphocytes naïfs, activés, des plasmocytes et le sous-groupe de lymphocytes B auto-réactifs du LED) ainsi que les concentrations d'IgG ont été observés sur plus de 7 ans sous traitement. Une régression marquée, durable et progressive de différents sous-groupes de lymphocytes B a été constatée, qui a entraîné une diminution médiane des lymphocytes B naïfs de 87%, des lymphocytes B à mémoire de 67%, des lymphocytes B activés de 99% et des plasmocytes de 92% après un traitement de plus de 7 ans. Après environ 7 ans, une diminution médiane des concentrations d'IgG de 28% a été observée, 1,6% des patients ayant montré une régression des concentrations d'IgG au-dessous de 400 mg/dl. L'incidence des effets indésirables signalés est restée globalement stable ou a légèrement reculé au cours de l'étude.
Après un traitement par Benlysta (10 mg/kg par voie intraveineuse) ou par placebo chez des patients atteints de néphrite lupique, une élévation des taux sériques d'IgG, associée à une diminution de la protéinurie, a été observée. Par rapport au placebo, on a observé une plus faible élévation du taux sérique d'IgG dans le groupe sous Benlysta, comme l'on pouvait s'y attendre au vu du mécanisme connu du bélimumab. À la semaine 104, le pourcentage médian d'élévation des taux d'IgG par rapport à la valeur initiale était de 17% pour Benlysta et de 37% pour le placebo. Les réductions observées des auto-anticorps, les élévations du complément et les réductions des lymphocytes B circulants totaux et des sous-types de lymphocytes B étaient conformes aux résultats des études sur le LED.
Efficacité clinique
Injection sous-cutanée dans le LED
L'efficacité du bélimumab administré par voie sous-cutanée a été examinée dans le cadre d'une étude de phase III randomisée, en double aveugle, contrôlée par placebo, d'une durée de 52 semaines (HGS1006-C1115; BEL112341), menée auprès de 836 patients atteints de LED diagnostiqué cliniquement selon les critères de classification de l'American College of Rheumatology. Les patients remplissant les critères de l'étude avaient un LED actif, défini par un score SELENA-SLEDAI de 8 au minimum, avec des résultats positifs de tests pour les anticorps antinucléaires (AAN ou anti-ADNdb; titre d'AAN de 1:80 au minimum et/ou anticorps anti-ADNdb de 30 unités/mL au minimum) lors de la sélection. Les patients étaient sous un régime thérapeutique stable pour leur LED (traitement standard) incluant les composants suivants (seuls ou en association): corticostéroïdes, antipaludiques, AINS ou autres immunosuppresseurs. Étaient exclus les patients souffrant d'un lupus sévère et actif avec atteinte du système nerveux central ou de néphrite lupique active sévère, les patients qui avaient reçu un autre médicament biologique encore en phase expérimentale au cours des 3 mois précédents, les patients qui avaient été testés positifs pour des anticorps anti-VIH, les antigènes de surface de l'hépatite B ou les anticorps dirigés contre l'hépatite C, ainsi que les patients transplantés et les patients souffrant d'hypogammaglobulinémie ou de déficit en IgA.
Cette étude a été menée aux États-Unis, en Amérique du Sud, en Europe et en Asie, Les patients avaient un âge médian de 37 ans (fourchette: de 18 à 77 ans) et étaient en majorité (94%) de sexe féminin. Les patients ont été randomisés dans un rapport de 2:1 pour recevoir en sous-cutané, pendant 52 semaines, soit le bélimumab 200 mg soit le placebo une fois par semaine.
Le critère primaire d'efficacité était un critère composite (SLE-Responder-Index, SRI), à l'aide duquel ont été définis comme répondeurs les patients qui, au bout de 52 semaines, satisfaisaient à tous les critères suivants par rapport au début de l'étude:
·régression du score SELENA-SLEDAI de 4 points au minimum et
·aucune nouvelle implication de systèmes d'organes BILAG-A ou absence de 2 nouvelles implications de systèmes d'organes BILAG-B (BILAG=British Isles Lupus Assessment Group), ainsi que
·aucune aggravation (élévation de <0,30 point) du score PGA (PGA=Physician's Global Assessment).
L'index SLE-Responder se base sur le score SELENA-SLEDAI comme mesure objective de la diminution de l'activité globale de la maladie, sur l'index BILAG pour exclure une aggravation significative dans un système d'organes spécifique et sur le score PGA pour vérifier que des améliorations de l'activité de la maladie ne s'obtiennent pas aux dépens de l'état général du patient.
Les critères secondaires d'efficacité étaient notamment le temps écoulé jusqu'à la première poussée sévère (obtenu à l'aide de l'index SELENA-SLEDAI-SLE-Flare modifié) et le pourcentage de patients avec une réduction de la dose moyenne de prednisone de ≥25% à ≤7,5 mg/jour pendant la période de la semaine 40 à 52, par rapport au début de l'étude. Un critère pour le succès du traitement portait sur la variation moyenne du score sur l'échelle FACIT-Fatigue (FACIT=Functional Assessment of Chronic Illness Therapy) au bout de 52 semaines.
Le bélimumab a entraîné des améliorations significatives de l'index SLE-Responder et du score SELENA-SLEDAI ainsi que de ses composantes individuelles (voir Tableau 1).
Tableau 1: Taux de réponse à la semaine 52
|
Réponse1
|
Placebo
(n=279)
|
Bélimumab
200 mg s.c. par semaine
(n=554)
| |
Index SLE-Responder
|
48,4%
|
61,4%
(p=0,0006).
| |
Composantes de l'index SLE-Responder
| |
Pourcentage de patients présentant une régression de SELENA-SLEDAI ≥4 (%)
|
49,1%
|
62,3%
(p=0,0005).
| |
Pourcentage de patients sans aggravation selon l'index BILAG (%)
|
74,2%
|
80,9%
(p=0,0305).
| |
Pourcentage de patients sans aggravation selon PGA (%)
|
72,8%
|
81,2%
(p=0,0061).
|
¹ Dans les analyses, tous les sujets pour lesquels une valeur initiale de l'un des composants manquait ont été exclus (1 dans le groupe placebo; 2 dans le groupe bélimumab).
Les différences entre les groupes de traitement étaient statistiquement significatives à partir de la semaine 16 et le sont restées jusqu'à la semaine 52 (Figure 1).
Figure 1: Pourcentage de SRI-Responder après visite

Les poussées sévères de LED ont été déterminées au moyen de l'index SELENA-SLEDAI-SLE-Flare modifié, la modification permettant d'exclure la saisie de poussées sévères exclusivement sur la base d'une valeur supérieure à 12 du score SELENA-SLEDAI. Le risque de poussées sévères a chuté de 49% (Hazard-Ratio=0,51; p=0,0004) dans le groupe sous bélimumab pendant l'observation de 52 semaines, par rapport au groupe sous placebo.
Au début de l'étude, 60% des patients recevaient de la prednisone (ou un équivalent de la prednisone) à des dosages de >7,5 mg/jour. Parmi ces patients, 18,2% ont réduit sous bélimumab leur dose moyenne de prednisone de 25% au moins à ≤7,5 mg/jour pendant les semaines 40 à 52, par rapport à 11,9% des patients sous placebo. Cette différence n'était cependant pas statistiquement significative (p=0,0732).
Le bélimumab, administré aussi bien par voie intraveineuse que par voie sous-cutanée a amélioré la fatigue par rapport au placebo, selon le score FACIT-Fatigue.
L'analyse par sous-groupes du critère primaire des études pivots sur l'administration intraveineuse et sous-cutanée a montré que ce sont les patients qui présentent une activité plus élevée de la maladie au début qui profitent le plus du traitement, notamment aussi les patients qui ont des scores SELENA-SLEDAI de 10 ou davantage, les patients qui ont des taux de complément abaissés, les patients qui ont besoin de stéroïdes pour contrôler la maladie ainsi que les patients qui ont à la fois des anticorps anti-ADNdb positifs et des taux de complément abaissés.
L'efficacité et la sécurité de Benlysta en association avec un seul cycle de rituximab ont été évaluées dans une étude de phase III randomisée, en double aveugle, contrôlée contre placebo, de 104 semaines, réalisée chez 292 patients (BLISS-BELIEVE). Le critère d'évaluation principal était la proportion de patients présentant le statut d'une maladie contrôlée, défini par un score SLEDAI-2K ≤2, qui était atteint à la semaine 52 sans immunosuppresseurs et avec des corticostéroïdes à une dose équivalente à ≤5 mg de prednisone par jour. Ceci a été atteint chez 19,4% (n = 28/144) des patients traités par Benlysta en association avec le rituximab et chez 16,7% (n = 12/72) des patients traités par Benlysta en association avec le placebo (Odds-Ratio 1,27; IC à 95%: 0,60; 2,71; p = 0,5342). Chez les patients traités par Benlysta en association avec le rituximab, des effets indésirables (91,7% vs 87,5%), des effets indésirables sévères (22,2% vs 13,9%) et des infections sévères (9,0% vs 2,8%) ont été observés plus fréquemment que chez les patients traités par Benlysta en association avec le placebo.
Perfusion intraveineuse dans la néphrite lupique
L'efficacité et la sécurité d'emploi du bélimumab 10 mg/kg, administré par voie intraveineuse sur une période d'une heure aux jours 0, 14 et 28 et ensuite tous les 28 jours, ont été évaluées dans une étude de phase III (BEL114054) randomisée (1:1), contrôlée contre placebo, en double aveugle, d'une durée de 104 semaines et réalisée chez 448 patients atteints de néphrite lupique active.
Au moment de la sélection, les patients présentaient un diagnostic clinique de LED posé selon les critères de classification de l'ACR, une néphrite lupique de classe III, IV et/ou V confirmée par une biopsie ainsi qu'une maladie rénale active qui rendait nécessaire un traitement standard (corticostéroïdes avec mycophénolate-mofétil pour l'induction et l'entretien ou cyclophosphamide pour le traitement d'induction, suivi d'azathioprine pour le traitement d'entretien). L'étude a été menée en Asie, en Amérique du Nord, en Amérique du Sud et en Europe. L'âge médian des patients était de 31 ans (fourchette: de 18 à 77 ans). Les participants étaient majoritairement de sexe féminin (88%).
Le critère principal d'efficacité était la réponse rénale primaire (Primary Efficacy Renal Response, PERR) à la semaine 104, définie comme une réponse à la semaine 100 confirmée par la répétition de la mesure des paramètres suivants à la semaine 104: rapport protéines urinaires/créatinine (uPCR) ≤0,7 et débit de filtration glomérulaire estimé (DFGe) ≥60 ml/min/1,73 m2 ou absence d'une chute du DFGe de >20% par rapport à la valeur mesurée avant la poussée.
Les principaux critères secondaires comprenaient:
·une réponse rénale complète (Complete Renal Response, CRR), définie comme une réponse à la semaine 100 confirmée par la répétition de la mesure des paramètres suivants à la semaine 104: uPCR <0,5 et DFGe ≥90 ml/min/1,73 m2 ou absence d'une chute du DFGe de >10% par rapport à la valeur mesurée avant la poussée
·une PERR à la semaine 52
·le temps écoulé jusqu'à un événement rénal ou jusqu'au décès (événement rénal défini comme le premier événement d'une insuffisance rénale terminale, doublement de la valeur de la créatinine sérique, détérioration de la fonction rénale [définie comme une augmentation de la protéinurie et/ou une altération de la fonction rénale] ou administration d'une thérapie non autorisée pour le traitement de maladies rénales)
En ce qui concerne les critères de PERR et CRR, les patients qui avaient quitté prématurément l'étude ou avaient reçu des médicaments non autorisés ont été considérés comme des non-répondeurs. Pour que le patient soit considéré comme répondeur en ce qui concerne les critères pertinents, le traitement par stéroïdes devait être réduit à ≤10 mg/jour à partir de la semaine 24.
Le pourcentage de patients qui ont atteint une PERR à la semaine 104 était significativement plus élevé dans le groupe sous bélimumab que dans le groupe sous placebo. De même, en ce qui concerne les principaux critères secondaires, le bélimumab a permis d'obtenir des résultats significativement meilleurs par rapport au placebo (Tableau 2).
Tableau 2: Résultats d'efficacité chez des patients adultes atteints de néphrite lupique
|
Critère d'efficacité
|
Placebo
n=223
|
Bélimumab
10 mg/kg
n=223
|
Différence observée vs placebo
|
Odds/Hazard Ratio vs placebo
(IC à 95%)
|
Valeur P
| |
PERR à la semaine 1041
Répondeurs
|
32,3%
|
43,0%
|
10,8%
|
OR 1,55
(1,04, 2,32)
|
0,0311
| |
Composantes de la PERR
| |
Rapport protéines urinaires/créatinine
≤0,7
|
33,6%
|
44,4%
|
10,8%
|
OR 1,54
(1,04, 2,29)
|
0,0320
| |
DGFe ≥60 ml/min/1,73 m2
ou absence de chute du DGFe de >20% par rapport à la valeur avant la poussée
|
50,2%
|
57,4%
|
7,2%
|
OR 1,32
(0,90, 1,94)
|
0,1599
| |
Pas d'échec du traitement
|
74,4%
|
83,0%
|
8,5%
|
OR 1,65
(1,03, 2,63)
|
0,0364
| |
CRR à la semaine 1041
Répondeurs
|
19,7%
|
30,0%
|
10,3%
|
OR 1,74
(1,11, 2,74)
|
0,0167
| |
Composantes de la CRR
| |
Rapport protéines urinaires/créatinine
<0,5
|
28,7%
|
39,5%
|
10,8%
|
OR 1,58
(1,05, 2,38)
|
0,0268
| |
DFGe ≥90 ml/min/1,73 m2
ou absence de chute du DFGe de >10% par rapport à la valeur avant la poussée
|
39,9%
|
46,6%
|
6,7%
|
OR 1,33
(0,90, 1,96)
|
0,1539
| |
Pas d'échec du traitement
|
74,4%
|
83,0%
|
8,5%
|
OR 1,65
(1,03, 2,63)
|
0,0364
| |
PERR à la semaine 521
Répondeurs
|
35,4%
|
46,6%
|
11,2%
|
OR 1,59
(1,06, 2,38)
|
0,0245
| |
Temps écoulé jusqu'à un événement rénal ou jusqu'au décès.1
Pourcentage de patients ayant subi l'événement2
|
28,3%
|
15,7%
|
-
|
|
| |
Temps écoulé jusqu'à l'événement [Hazard Ratio (IC à 95%)]
|
|
|
-
|
0,51
(0,34, 0,77)
|
0,0014
| |
1
La PERR à la semaine 104 était l'analyse d'efficacité primaire; la CRR à la semaine 104, la PERR à la semaine 52 et le temps écoulé jusqu'à l'événement rénal ou le décès faisaient partie de la hiérarchie des tests préalablement définis.
2 Si l'on exclut de l'analyse les cas de décès (un sous bélimumab, deux sous placebo), le pourcentage de patients ayant présenté un événement rénal était de 15,2% sous bélimumab et de 27,4% sous placebo (HR=0,51; IC à 95%: 0,34, 0,78).
|
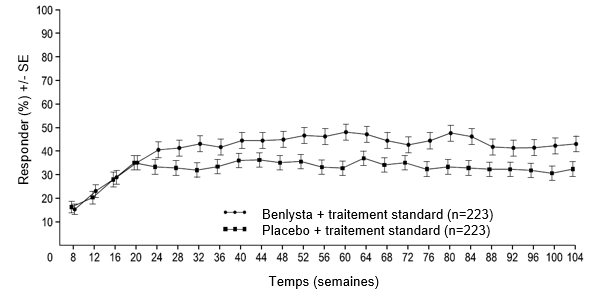
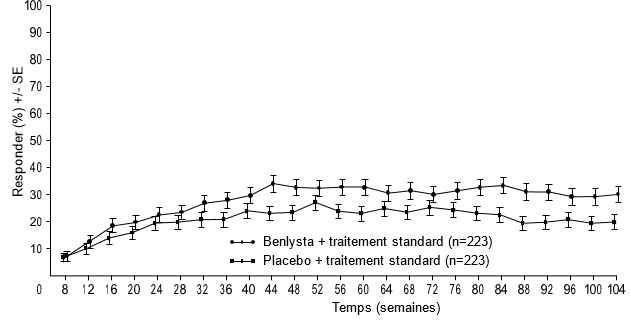
À partir de la semaine 24, un pourcentage numériquement plus élevé de patients sous bélimumab a atteint la PERR par rapport aux patients sous placebo. Cette différence entre traitements s'est maintenue jusqu'à la semaine 104. À partir de la semaine 12, un pourcentage numériquement plus élevé de patients sous bélimumab a atteint la CRR par rapport aux patients sous placebo. Cette différence numérique s'est maintenue jusqu'à la semaine 104 (Figure 2).
Figure 2: Taux de réponse des adultes atteints de néphrite lupique par visite
Réponse rénale primaire (Primary Efficacy Renal Response, PERR)
Réponse rénale complète (Complete Renal Response, CRR)
Dans des analyses descriptives de sous-groupes, les taux de PERR et de CRR ont été examinés par régime d'induction (mycophénolate ou cyclophosphamide), par classe de biopsie (classe III ou IV, classe III + V ou classe IV + V ou classe V) et selon les valeurs initiales de l'uPCR (<3 g/g ou ≥3 g/g; analyse post hoc) ont été examinés (Figure 3).
Figure 3: Odds Ratio pour la PERR et la CRR à la semaine 104 dans les divers sous-groupes
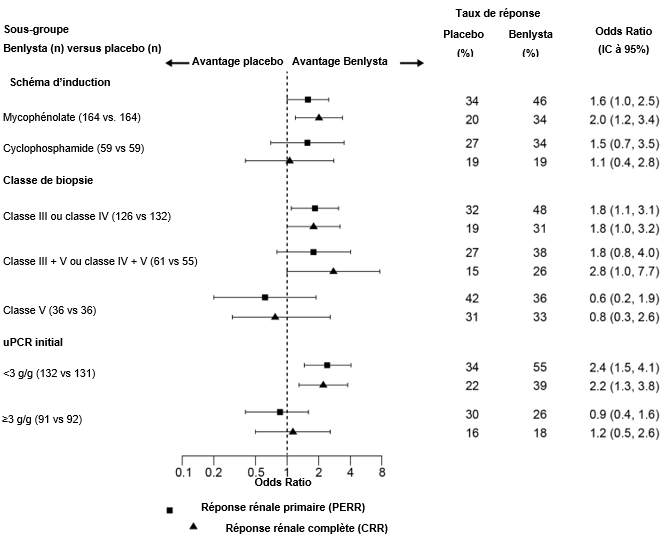
¹ Classe III=néphrite lupique focale proliférative; classe IV=néphrite lupique diffuse proliférative; classe V=néphrite lupique membraneuse; classe III + V=néphrite lupique mixte membraneuse – focale proliférative; classe IV + V=néphrite lupique mixte membraneuse – diffuse proliférative.
² Le rapport protéines urinaires initiales:créatinine (uPCR) était une analyse post hoc.
|