Propriétés/EffetsCode ATC
J05AX18
Mécanisme d'action
Prevymis (létermovir) est un médicament antiviral contre le CMV.
Le létermovir inhibe le complexe terminase de l'ADN du CMV qui est indispensable à la réplication virale. Les analyses biochimiques et électromiscroscopiques ont montré que le létermovir affecte la formation d'unités génomiques de longueur adéquate et interfère avec la maturation des virions.
Pharmacodynamique
Activité antivirale
Dans un modèle d'infection en culture cellulaire, la CE50 médiane du létermovir contre un ensemble d'isolats du CMV obtenus en clinique a été de 2,1 nM (intervalle de 0,7 nM à 6,1 nM; n = 74).
Résistance virale
En culture cellulaire
Les gènes UL51, UL56 et UL89 du CMV codent des sous-unités de l'ADN terminase du CMV. Des mutants CMV présentant une sensibilité réduite au létermovir ont été sélectionnés en culture cellulaire, et les mutations correspondent à pUL51 (P91S, A95V), pUL56 (C25F, S229F, V231A, V231L, N232Y, V236A, V236L, V236M, E237D, L241P, T244K, T244R, L254F, L257F, L257I, K258E, F261C, F261L, F261S, Y321C, C325F, C325R, C325W, C325Y, L328V, M329T, A365S, N368D, R369G, R369M, R369S) et pUL89 (N320H, D344E). Les valeurs de CE50 pour les mutants CMV recombinants exprimant ces substitutions sont 1,6 à 9300 fois plus élevées que celles pour les virus de référence de type sauvage.
Dans les études cliniques
Lors d'une étude de phase 2b évaluant des doses de létermovir de 60, 120 ou 240 mg/jour vs placebo sur une période allant jusqu'à 84 jours chez 131 receveurs de GCSH, une analyse de la séquence d'ADN d'une région choisie du gène UL56 (acides aminés 231 à 369) a été réalisée sur des échantillons obtenus auprès de 12 patients traités par létermovir ayant présenté un échec de la prophylaxie et pour lesquels des échantillons étaient disponibles pour analyse. Un participant de l'étude (qui avait reçu 60 mg/jour) a présenté un variant génotypique (VG) résistant au létermovir (V236M).
Lors d'une étude de phase 3 (P001), une analyse de la séquence d'ADN de l'ensemble des régions codantes des gènes UL56 et UL89 a été réalisée sur des échantillons obtenus auprès de 40 patients traités par le létermovir, dans la population totale d'analyse (FAS, Full Analysis Set) ayant présenté un échec de la prophylaxie et pour lesquels des échantillons étaient disponibles pour analyse. Au total, deux substitutions associées à une résistance au létermovir ont été détectées chez deux patients, toutes les deux correspondant à pUL56. Un patient a présenté la substitution V236M, l'autre E237G. Un patient supplémentaire, qui avait un ADN du CMV détectable à l'inclusion (et ne faisait donc pas partie de la population totale d'analyse), a présenté des substitutions C325W et R369T de pUL56, détectées après arrêt du létermovir.
Lors d'une étude de phase 3 (P040), une analyse de la séquence d'ADN de l'ensemble des régions codantes des gènes UL51, UL56 et UL89 a été réalisée sur des échantillons obtenus auprès de 32 sujets (quel que soit le groupe de traitement) ayant présenté un échec de prophylaxie ou qui l'ont arrêtée prématurément en raison d'une virémie à CMV. Aucune substitution associée à une résistance au létermovir n'a été détectée au-dessus de la limite validée du test de 5%.
Lors d'une étude de phase 3 (P002), une analyse de la séquence d'ADN de l'ensemble des régions codantes des gènes UL51, UL56 et UL89 a été réalisée sur des échantillons obtenus auprès de 52 patients traités avec le létermovir qui ont présenté une maladie à CMV ou qui ont arrêté prématurément le traitement en raison d'une virémie à CMV. Aucune substitution associée à une résistance au létermovir n'a été détectée au-dessus de la limite validée du test de 5%.
Résistance croisée
Une résistance croisée avec un médicament d'une autre classe est peu probable. Le létermovir est pleinement actif contre les populations virales porteuses de substitutions conférant une résistance aux inhibiteurs de la polymérase de l'ADN du CMV (ganciclovir, cidofovir et foscarnet). Un panel de souches de CMV recombinantes porteuses de substitutions conférant une résistance au létermovir était totalement sensible au cidofovir, au foscarnet et au ganciclovir – à l'exception d'une souche recombinante porteuse de la substitution E237G de pUL56 qui confère une réduction de la sensibilité au ganciclovir de 2,1 fois par rapport au type sauvage.
Pharmacogénomique
L'impact de mutations affectant le gène OATP1B1 sur la pharmacocinétique du létermovir a été étudié sur 299 participants des études cliniques. Il s'agit des variants SLCO1B1 (rs4149056, rs2306283, rs4149032) et UGT1A1 (rs4148323 et mutation des répétitions du TA dans le promoteur). Ces variants n'ont pas donné lieu à des effets cliniquement significatifs en termes d'exposition au létermovir.
Électrophysiologie cardiaque
L'effet du létermovir sur l'intervalle QTc à des doses allant jusqu'à 960 mg administrées par voie IV a été évalué dans une étude de durée de QT randomisée, en dose unique, contrôlée versus placebo et comparateur actif (moxifloxacine 400 mg par voie orale), en cross-over de quatre périodes, menée chez 38 sujets sains. Le létermovir ne provoque pas d'allongement cliniquement significatif de l'intervalle QTc après l'administration d'une dose de 960 mg par voie IV, ce qui correspond à des concentrations plasmatiques environ 2 fois supérieures à celles de la dose de 480 mg par voie IV.
Efficacité clinique
Adultes séropositifs au CMV receveurs [R+] d'une greffe allogénique de cellules souches hématopoïétiques
Prophylaxie jusqu'à la semaine 14 (~100 jours) post-GCSH
Afin d'évaluer la capacité prophylactique du létermovir comme stratégie préventive contre l'infection ou la maladie à CMV, l'efficacité du létermovir a été évaluée lors d'une étude de phase 3 (P001) multicentrique, en double aveugle, contrôlée versus placebo chez des adultes séropositifs au CMV receveurs [R+] d'une GCSH allogénique. Les sujets ont été randomisés (2:1) pour recevoir soit le létermovir soit le placebo. La randomisation a été stratifiée en fonction du centre d'étude et du risque (élevé versus faible) de réactivation du CMV à l'inclusion. Le létermovir a été initié après la GCSH (jours 0-28 post-GCSH) et poursuivi jusqu'à la semaine 14 post-GCSH. Le létermovir a été administré soit par voie orale soit par voie IV. Les patients étaient suivis jusqu'à la semaine 24 post-GCSH pour le critère principal d'efficacité, avec une surveillance qui se poursuivait jusqu'à la semaine 48 post-GCSH.
Parmi les 565 patients traités, 373 ont reçu le létermovir (dont 99 qui ont reçu au moins une dose IV) et 192 ont reçu le placebo (dont 48 qui ont reçu au moins une dose IV). Le délai médian avant instauration du létermovir a été de 9 jours après la greffe. Au début de l'étude, 37% des patients présentaient une prise de la greffe (engraftment). L'âge médian était de 54 ans (intervalle de 18 à 78 ans). Au début de l'étude, 50% des patients recevaient un traitement myéloablatif, 52% de la ciclosporine et 42% du tacrolimus. Les motifs principaux les plus fréquents de la greffe étaient la leucémie myéloïde aiguë (38%), le syndrome myéloblastique (15%) et le lymphome (13%). Au début de l'étude, 12% des patients étaient positifs à l'ADN du CMV.
Au début de l'étude, 31% des patients étaient à haut risque de réactivation tel que défini par un ou plusieurs des critères suivants: donneur HLA (Human Leukocyte Antigen) apparenté (frère/sœur) avec au moins une incompatibilité sur l'un des trois loci suivants des gènes HLA: donneur HLA-A, B ou DR; donneur haplo-identique; donneur non apparenté avec au moins une incompatibilité sur l'un des quatre loci suivants des gènes HLA: HLA-A, B, C et DRB1; utilisation de sang de cordon ombilical comme source de cellules souches; utilisation de greffons avec cellules T déplétées ex vivo; réaction greffon contre l'hôte (GVHD) de grade 2 ou supérieur nécessitant des corticoïdes systémiques.
Infection à CMV cliniquement significative
Le critère principal d'efficacité de l'étude P001 était l'apparition d'une infection à CMV cliniquement significative jusqu'à la semaine 24 post-GCSH. L'infection à CMV cliniquement significative a été définie comme étant soit la survenue d'une maladie à CMV touchant un organe cible soit l'instauration d'un traitement préemptif (PET) en raison d'une virémie à CMV documentée (utilisation du Roche COBAS AmpliPrep/COBAS TaqMan Assay).
Dans l'analyse du critère d'évaluation principal, le létermovir a démontré une efficacité supérieure au placebo (voir Tableau 4). La différence estimée de traitement de -23,5% était statistiquement significative (valeur p unilatérale <0,0001).
Tableau 4: P001: Résultats d'efficacité chez les receveurs de GCSH (approche NC=F, population FAS)
|
Paramètres
|
Létermovir
(N=325)
n (%)
|
Placebo
(N=170)
n (%)
| |
Critère d'évaluation principal
(Pourcentage de patients en échec de prophylaxie)
|
122
(37,5)
|
103
(60,6)
| |
Raisons des échecs†
|
|
| |
Infection au CMV cliniquement significative jusqu'à la semaine 24‡
|
57 (17,5)
|
71 (41,8)
| |
Instauration d'un PET basé sur une virémie à CMV documentée
|
52 (16,0)
|
68 (40,0)
| |
Maladie à CMV d'un organe cible
|
5 (1,5)
|
3 (1,8)
| |
Sortie de l'étude avant la semaine 24
|
56 (17,2)
|
27 (15,9)
| |
Résultat manquant jusqu'à la fenêtre de consultation de la semaine 24
|
9 (2,8)
|
5 (2,9)
| |
Différence de traitement ajustée en fonction de la stratification (létermovir-placebo)§
|
|
| |
Différence (IC à 95%)
|
-23,5 (-32,5, -14,6)
| |
Valeur p
|
<0,0001
| |
†
Les catégories d'échec sont mutuellement exclusives et se basent sur la hiérarchie des catégories dans l'ordre indiqué.
‡ Une infection à CMV cliniquement significative a été définie comme étant soit la survenue d'une maladie à CMV touchant un organe cible soit l'instauration d'un PET en raison d'une virémie à CMV documentée et de l'état clinique du patient.
§ Les IC à 95% et la valeur p pour les différences entre traitements en pourcentage de réponse ont été calculés à l'aide de la méthode de Mantel-Haenszel ajustée en fonction des strates, en pondérant la différence en fonction de la moyenne harmonique de la taille de l'échantillon par bras pour chaque strate (risque élevé ou faible). Une valeur p unilatérale ≤0,0249 a été utilisée pour déterminer la significativité
statistique.
Remarque: FAS = population totale d'analyse (Full Analysis Set); cette population inclut les patients randomisés qui ont reçu au moins une dose de médicament à l'étude, et exclut les patients présentant un ADN du CMV détectable en début d'étude. Approche suivie pour le traitement des valeurs manquantes: approche abandon = échec (Non-Completer = Failure; NC = F). Dans l'approche NC = F, l'échec a été défini comme l'ensemble des patients qui ont développé une infection au CMV cliniquement significative ou qui sont sortis prématurément de l'étude, ou ceux pour lesquels aucun résultat n'était disponible jusqu'à la fenêtre de consultation de la semaine 24 post-GCSH.
N = nombre de patients dans chaque groupe de traitement.
n (%) = nombre (pourcentage) de patients dans chaque sous-catégorie.
|
À la semaine 24 post-GCSH, le taux d'événements Kaplan-Meier (KM) pour une infection à CMV cliniquement significative était de 18,9% dans le groupe létermovir vs 44,3% dans le groupe placebo (valeur p nominale du log-rank stratifié bilatéral <0,0001) (voir Fig. 1). Chez les patients traités par le létermovir, les facteurs liés à une infection à CMV cliniquement significative entre les semaines 14 et 24 post-GCSH ont été les suivants:
·risque élevé de réactivation du CMV en début d'étude,
·présence d'une GVHD et
·utilisation d'un stéroïde à un moment quelconque après randomisation.
Fig. 1: P001: Courbe de Kaplan-Meier du délai de survenue d'une infection à CMV cliniquement significative jusqu'à la semaine 24 post-greffe chez les receveurs de GCSH (population FAS)
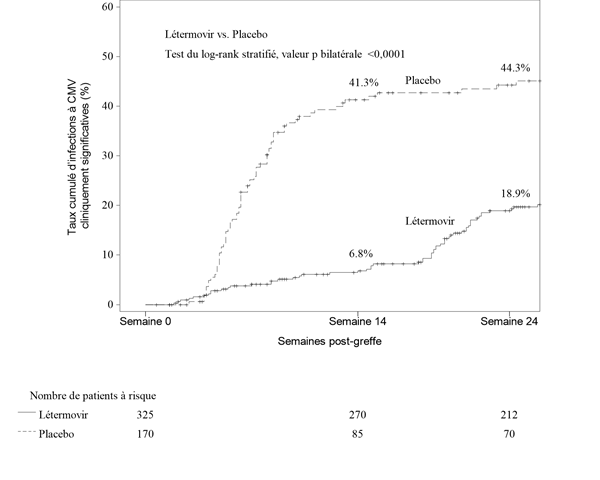
L'efficacité a été nettement supérieure dans tous les sous-groupes du bras létermovir, y compris les suivants:
·risque faible ou élevé de réactivation du CMV,
·traitements de conditionnement, et
·traitements immunosuppresseurs concomitants.
Mortalité
Le taux d'événements KM pour la mortalité toutes causes confondues dans le groupe létermovir vs placebo était de 12,1% vs 17,2% à la semaine 24 post-GCSH (valeur p nominale du log-rank stratifié bilatéral = 0,0401), respectivement et de 23,8% vs 27,6% à la semaine 48 post-GCSH (valeur p nominale du log-rank stratifié bilatéral = 0,2117). Le taux d'événements KM pour la mortalité toutes causes confondues par sexe (hommes vs femmes) à la semaine 24 post-GCSH était de 16,4% vs 6,6% dans le groupe létermovir et de 14,2% vs 25,4% dans le groupe placebo; ces taux d'événements décomposés par sexe sont à interpréter avec précaution; en effet, la randomisation n'a pas été stratifiée par sexe, ce qui a donné lieu en baseline à des déséquilibres entre groupes de traitement en termes de risque de mortalité spécifiquement liés au sexe.
Prophylaxie de la semaine 14 (~100 jours) à la semaine 28 (~200 jours) post-GCSH
L'efficacité de la prolongation de la prophylaxie par le létermovir de la semaine 14 (~100 jours) jusqu'à la semaine 28 (~200 jours) post-GCSH chez les patients à risque d'infection et de maladie à CMV tardives a été évaluée dans une étude de phase 3 (P040) multicentrique, en double aveugle, contrôlée par placebo chez des receveurs adultes séropositifs pour CMV [R+] d'une GCSH allogénique. Les sujets éligibles qui avaient terminé la prophylaxie de létermovir jusqu'à 100 jours environ post-GCSH, ont été randomisés (2:1) pour recevoir du létermovir ou un placebo de la semaine 14 jusqu'à la semaine 28 post-GCSH. Les sujets ont reçu une dose journalière de 480 mg de létermovir, qui a été ajustée à 240 mg en cas d'administration concomitante de ciclosporine. Le médicament à l'étude a été administré soit par voie orale soit par voie IV. Un sujet a reçu le létermovir par voie IV pour 2 jours. Les sujets ont été surveillés jusqu'à la semaine 28 post-GCSH pour le critère d'évaluation principal, avec un suivi continu hors traitement jusqu'à la semaine 48 post-GCSH.
Parmi les 218 sujets traités, 144 ont reçu le létermovir et 74 ont reçu un placebo. L'âge médian était de 55 ans (intervalle de 20 à 74 ans). Les raisons les plus fréquentes de greffe étaient la leucémie myéloïde aiguë (42%), la leucémie lymphoïde aiguë (15%) et le syndrome myélodysplasique (11%).
Lors de leur inclusion dans l'étude, tous les sujets présentaient des facteurs de risque d'une infection et d'une maladie à CMV tardives, 64% d'entre eux présentaient deux facteurs de risque ou plus. Les facteurs de risque comprenaient: un donneur HLA apparenté (frère/sœur) avec au moins une incompatibilité sur l'un des trois loci suivants des gènes HLA: HLA-A, -B ou -DR; donneur haplo-identique; donneur non apparenté avec au moins une incompatibilité sur l'un des quatre loci suivants du gène HLA: HLA-A, -B, -C et -DRB1; utilisation de sang de cordon ombilical comme source de cellules souches; utilisation de greffons avec cellules T déplétées ex-vivo; administration de globuline anti-thymocytes; administration d'alemtuzumab; utilisation de prednisone (ou équivalent) par voie systémique à une dose ≥1 mg/kg de poids corporel par jour.
Infection à CMV cliniquement significative
Le principal critère d'évaluation de l'efficacité de P040 était l'apparition d'une infection à CMV cliniquement significative jusqu'à la semaine 28 post-GCSH. Une infection à CMV cliniquement significative a été définie comme la survenue soit d'une maladie à CMV d'un organe cible, soit de l'initiation d'une PET sur la base d'une virémie à CMV documentée et de l'état clinique du patient. L'approche d'échec observé (Observed Failure, OF) a été utilisée, selon laquelle les sujets ayant arrêté prématurément l'étude sans virémie quelle que soit la raison ou en cas de données manquantes à la date de suivi, n'étaient pas considérés comme des échecs.
Le nombre des sujets qui avaient quitté l'étude sans virémie avant la semaine 28 était de 14 (9,7%) dans le groupe sous Prevymis et de 0 dans le groupe du placebo. Le nombre de sujets pour lesquels, lors de la fenêtre de consultation de la semaine 28, aucun résultat n'était disponible, était de 3 (2,1%) dans le groupe sous Prevymis et de 4 (5,4%) dans le groupe sous placebo. Aucun n'avait développé auparavant une virémie.
Les résultats d'efficacité de l'étude P040 sont présentés dans le Tableau 5. L'efficacité a été systématiquement en faveur du létermovir par rapport au placebo dans les sous-groupes de patients selon leurs caractéristiques (âge, sexe, appartenance ethnique) et selon les facteurs de risque d'infection et de maladie à CMV tardives lorsque l'approche OF a été utilisée.
Tableau 5: P040 Résultats d'efficacité chez les receveurs d'une GCSH présentant un risque d'infection et de maladie à CMV tardives (approche OF, population FAS)
|
Paramètre
|
Létermovir
(~200 jours de létermovir)
(N=144)
n (%)
|
Placebo
(~100 jours de létermovir)
(N=74)
n (%)
| |
Échecs†
|
4 (2,8)
|
14 (18,9)
| |
Infection à CMV cliniquement significative jusqu'à la semaine 28‡
|
2 (1,4)
|
13 (17,6)
| |
Initiation d'un PET sur la base d'une virémie à CMV documentée
|
1 (0,7)
|
11 (14,9)
| |
Maladie à CMV d'un organe cible
|
1 (0,7)
|
2 (2,7)
| |
Sortie de l'étude avec une virémie à CMV avant la semaine 28
|
2 (1,4)
|
1 (1,4)
| |
Différence de traitement ajustée en fonction de la stratification (létermovir (~200 jours de létermovir) - placebo (~100 jours de létermovir)) §
|
|
| |
Différence (IC à 95%)
|
-16,1 (-25,8; -6,5)
| |
Valeur p
|
0,0005
| |
†
Les catégories d'échec sont mutuellement exclusives et basées sur la hiérarchie des catégories dans l'ordre indiqué.
‡ Une infection à CMV cliniquement significative a été définie comme une maladie d'un organe cible à CMV (prouvée ou probable) ou l'initiation d'un PET sur la base d'une virémie à CMV documentée et de l'état clinique du patient.
§ Les IC à 95% et la valeur p pour les différences entre traitements en pourcentage de réponses ont été calculés à l'aide de la méthode de Mantel-Haenszel ajustée en fonction des strates, en pondérant la différence en fonction de la moyenne harmonique de la taille de l'échantillon par bras pour chaque strate (donneur haplo-identique oui ou non). Une valeur p unilatérale de ≤0,0249 a été utilisée pour déterminer la significativité statistique.
Approche suivie pour le traitement des valeurs manquantes: approche d'échec observé (Observed Failure, OF). Dans l'approche OF, l'échec a été défini comme l'ensemble des patients ayant développé une infection à CMV cliniquement significative ou qui sont sortis prématurément de l'étude avec une virémie à CMV de la semaine 14 (~100 jours) jusqu'à la semaine 28 (~200 jours) post-GCSH.
N = nombre de patients dans chaque groupe de traitement.
n (%) = nombre (pourcentage) de patients dans chaque sous-catégorie.
|
Le délai avant une infection à CMV cliniquement significative est présenté dans la Figure 2.
Figure 2: P040 Courbe de Kaplan-Meier du délai de survenue d'une infection à CMV cliniquement significative, de la semaine 14 jusqu'à la semaine 48 post-greffe, chez des receveurs d'une GCSH présentant un risque d'infection et de maladie à CMV tardives (population FAS)

LET = létermovir; PBO = placebo
Mortalité
Le taux de mortalité toutes causes confondues dans le bras sous létermovir et dans le bras sous placebo était respectivement de 2,1% et de 1,4% à la semaine 28 post-GCSH (valeur p nominale, unilatérale = 0,6244), et de 8,3% resp. 8,1% à la semaine 48 post-GCSH (valeur p nominale, unilatérale = 0,5264).
Adultes séronégatifs pour le CMV receveurs d'une greffe rénale provenant d'un donneur séropositif pour le CMV [D+/R-]
Pour évaluer la prophylaxie par létermovir comme stratégie préventive contre les maladies à CMV chez les receveurs d'une greffe rénale, l'efficacité du létermovir a été évaluée dans une étude de phase 3 (P002) de non-infériorité multicentrique, en double aveugle, contrôlée contre comparateur actif chez des adultes receveurs d'une greffe rénale à haut risque [D+/R-]. Les sujets ont été randomisés (1:1) pour recevoir soit du létermovir, soit du valganciclovir. Le létermovir a été administré à une dose de 480 mg une fois par jour (ajustée à 240 mg en cas d'administration concomitante de ciclosporine). Le létermovir a été administré en même temps que de l'aciclovir. Le valganciclovir a été administré en concomitance avec un placebo à l'aciclovir. La randomisation a été stratifiée selon l'utilisation ou non d'une immunothérapie anti-lymphocytaire hautement cytolytique pendant l'induction. Le médicament à l'étude a été initié entre le jour 0 et le jour 7 post-greffe rénale et a été poursuivi jusqu'à la semaine 28 (~200 jours) post-greffe. Le médicament à l'étude a été administré soit par voie orale, soit par voie IV. Trois sujets ont reçu le létermovir par voie IV pour une durée moyenne de 1,7 jour. Les sujets ont été surveillés jusqu'à la semaine 52 post-greffe.
Parmi les 589 sujets traités, 292 ont reçu le létermovir et 297 le valganciclovir. L'âge médian était de 51 ans (intervalle de 18 à 82 ans); 72% étaient des hommes; 84% étaient de type caucasien; 2% étaient d'origine asiatique; 9% étaient noirs; 17% étaient de type hispanique ou latino-américain; et 60% ont reçu un rein d'un donneur décédé. Les raisons principales les plus fréquentes de la greffe étaient une maladie rénale kystique congénitale (17%), une hypertension (16%) et un diabète/une néphropathie diabétique (14%).
Maladie à CMV
Le critère principal d'évaluation de l'efficacité de P002 a été la survenue d'une maladie à CMV (définie comme une maladie à CMV d'un organe cible ou syndrome à CMV, confirmé par un comité d'arbitrage indépendant) jusqu'à la semaine 52 post-greffe. L'approche d'échec observé OF a été utilisée, selon laquelle les patients ayant arrêté prématurément l'étude quelle que soit la raison ou en cas de données manquantes à la date de suivi n'étaient pas considérés comme des échecs.
Le nombre de sujets ayant quitté l'étude avant la semaine 52 étaient de 32 (11%) dans le groupe sous Prevymis et de 28 (9%) dans le groupe sous valganciclovir. Le nombre de sujets pour lesquels aucun résultat n'était disponible durant la fenêtre de consultation de la semaine 52 était de 24 (8%) dans le groupe sous Prevymis et de 25 (8%) dans le groupe sous valganciclovir.
Les résultats d'efficacité de l'étude P002 sont présentés dans le Tableau 6.
Tableau 6: P002 Résultats d'efficacité chez les receveurs d'une greffe rénale (approche OF, population FAS)
|
Paramètre
|
Létermovir
(N=289)
n (%)
|
Valganciclovir
(N=297)
n (%)
| |
Maladie à CMV† jusqu'à la semaine 52
Syndrome à CMV‡
Maladie à CMV d'un organe cible
|
30 (10,4)
24 (8,3)
6 (2,1)
|
35 (11,8)
34 (11,4)
1 (0,3)
| |
Différence de traitement ajustée en fonction de la stratification (létermovir-valganciclovir) §
|
| |
Différence (IC à 95%)
|
-1,4 (-6,5, 3,8) ¶
| |
†
Cas de maladies à CMV confirmés par un comité d'arbitrage indépendant.
‡ Défini comme la preuve de la présence de CMV dans le sang par isolation du virus, culture rapide, antigénémie ou tests faisant appel à la technologie des acides nucléiques, et deux des symptômes suivants ou plus: 1) fièvre ≥38 °C pendant au moins 2 jours, 2) apparition ou renforcement d'un malaise/d'une fatigue, 3) leucopénie ou neutropénie lors de deux mesures séparées distantes de 24 h au minimum, 4) ≥5% de lymphocytes atypiques, 5) thrombocytopénie, 6) élévation d'ALAT ou d'ASAT à des valeurs égales au double de la limite supérieure de la norme.
§ Les IC à 95% pour les différences entre les traitements en pourcentage de réponse ont été calculés à l'aide de la méthode de Mantel-Haenszel ajustée en fonction des strates, en pondérant la différence en fonction de la moyenne harmonique de la taille de l'échantillon par bras pour chaque strate (utilisation/non-utilisation d'une immunothérapie anti-lymphocytaire hautement cytolytique pendant l'induction).
¶ Sur la base d'une marge de non-infériorité de 10%.
Approche suivie pour le traitement des valeurs manquantes: approche d'échec observé (Observed Failure, OF).
N = nombre de patients dans chaque groupe de traitement.
n (%) = nombre (pourcentage) de patients dans chaque sous-catégorie.
|
L'efficacité était comparable dans tous les sous-groupes, y compris selon le sexe, l'âge, l'origine éthnique, la région et l'utilisation/non-utilisation d'une immunothérapie anti-lymphocytaire hautement cytolytique pendant l'induction.
|