CompositionPrincipe actif: letermovir.
Excipients des comprimés pelliculés: cellulose microcristalline, croscarmellose sodique, povidone 25, silice colloïdale anhydre, stéarate de magnésium et les excipients suivants pour le pelliculage: lactose monohydraté, hypromellose 2910, dioxyde de titane, triacétine, oxyde de fer jaune, oxyde de fer rouge (uniquement pour les comprimés à 480 mg), cire de carnauba comme agent de polissage.
1 ml de solution pour perfusion contient les excipients suivants: hydroxypropylbetadex (150 mg), chlorure de sodium (3,1 mg), hydroxyde de sodium (1,2 mg), eau pour préparations injectables. La quantité d'hydroxyde de sodium peut être ajustée afin d'obtenir un pH de 7,5 environ.
Forme galénique et quantité de principe actif par unitéChaque comprimé pelliculé de Prevymis contient 240 mg ou 480 mg de letermovir.
Prevymis solution à diluer pour perfusion est une solution limpide et stérile dénuée d'agent conservateur en flacons à dose unique de 240 mg ou 480 mg de letermovir par flacon. 1 ml de solution contient 20 mg de letermovir. Prevymis solution à diluer pour perfusion doit être dilué avant administration par perfusion. Administration par perfusion intraveineuse (i.v.) uniquement.
Indications/Possibilités d’emploiPrevymis est indiqué dans la prophylaxie de l'infection à cytomégalovirus (CMV) et de la maladie à CMV chez les adultes séropositifs au CMV receveurs [R+] d'une greffe allogénique de cellules souches hématopoïétiques (GCSH).
Posologie/Mode d’emploiLa posologie recommandée de Prevymis est de 480 mg une fois par jour. Prevymis doit être initié après la GCSH. Prevymis peut être débuté le jour de la greffe et au plus tard 28 jours après celle-ci. Prevymis peut être débuté avant ou après la prise de la greffe (engraftment). Le traitement par Prevymis doit être poursuivi jusqu'à 100 jours après la greffe. La solution à perfuser Prevymis contient de l'hydroxypropylbetadex et doit être utilisée uniquement chez les patients qui ne peuvent pas prendre de traitement par voie orale. Les patients doivent passer aux comprimés pelliculés Prevymis dès qu'ils sont en mesure de prendre des médicaments par voie orale. Voir section «Pharmacocinétique» du letermovir après administration par voies i.v. et orale.
Prevymis comprimé pelliculé et solution à diluer pour perfusion peuvent être utilisés de manière interchangeable, à la discrétion du médecin; aucun ajustement posologique n'est requis.
Le comprimé doit être avalé entier et peut être pris avec ou sans nourriture. Le comprimé ne doit pas être divisé, écrasé ou croqué.
Prevymis solution à diluer pour perfusion doit être dilué avant administration par perfusion (voir section «Remarques particulières»). Administration par perfusion intraveineuse (i.v.) uniquement. Ne pas administrer en injection rapide ou en bolus. Après dilution, administrez Prevymis en perfusion intraveineuse sur une durée totale de 60 minutes environ à l'aide d'un cathéter veineux périphérique ou central. La totalité du contenu de la poche de perfusion doit être administrée.
Si Prevymis est co-administré avec de la ciclosporine, la posologie de Prevymis doit être réduite à 240 mg une fois par jour (voire sections «Mises en garde et précautions» et «Interactions»).
·Si la ciclosporine est initiée après le début du traitement par Prevymis, la dose suivante de Prevymis doit être réduite à 240 mg une fois par jour.
·Si la ciclosporine est arrêtée après le début du traitement par Prevymis, la dose suivante de Prevymis doit être augmentée à 480 mg une fois par jour.
·Si l'administration de ciclosporine est temporairement interrompue en raison de taux élevés de ciclosporine, aucun ajustement posologique de Prevymis n'est requis.
En cas d'oubli d'une dose
Les patients doivent être informés que s'ils oublient une dose de Prevymis, ils doivent la prendre dès qu'ils s'en souviennent. S'ils ne s'en aperçoivent qu'au moment de prendre la dose suivante, la dose oubliée ne doit pas être prise et ils doivent reprendre leur schéma d'administration habituel. Les patients doivent être informés qu'ils ne doivent pas doubler la dose suivante ni dépasser la dose prescrite.
Instructions spéciales pour la posologie
Prevymis n'est pas recommandé chez les patients présentant une insuffisance hépatique modérée associée à une insuffisance rénale modérée ou sévère (voir section «Interactions»).
Patients en insuffisance rénale
Aucun ajustement posologique de Prevymis n'est requis chez les patients présentant une clairance de la créatinine >10 ml/min (Voir section «Pharmacocinétique» ). On ne dispose pas de données concernant les patients en insuffisance rénale terminale (ClCr <10 ml/min) et les patients dialysés, si bien qu'aucune recommandation posologique ne peut être formulée. La solution concentrée Prevymis à diluer pour perfusion contient de l'hydroxypropylbetadex. L'exposition clinique attendue à l'hydroxypropylbetadex est approximativement de 3600 mg/jour lorsque le letermovir est administré par voie intraveineuse à la dose de 480 mg. Une accumulation de l'hydroxypropylbetadex peut survenir chez les patients en insuffisance rénale (clairance de la créatinine <50 ml/min) recevant une perfusion de Prevymis. Les taux de créatinine sérique doivent être étroitement surveillés chez ces patients.
Patients en insuffisance hépatique
Aucun ajustement posologique de Prevymis n'est requis en cas d'insuffisance hépatique légère (Child-Pugh classe A) à modérée (Child-Pugh classe B). Prevymis n'est pas recommandé chez les patients en insuffisance hépatique sévère (Child-Pugh classe C).
Prevymis n'est pas recommandé chez les patients présentant une insuffisance hépatique modérée associée à une insuffisance rénale modérée ou sévère (voir section «Pharmacocinétique»).
Patients âgés
Sur les 373 patients traités par Prevymis dans le cadre d'une étude de phase 3 randomisée et menée en double aveugle contre placebo (P001), 56 (15,0%) étaient âgés de 65 ans ou plus. Les patients âgés et jeunes n'ont pas présenté de différences en termes de sécurité et d'efficacité. Aucune adaptation posologique de Prevymis n'est requise en fonction de l'âge.
Enfants et adolescents
La sécurité et l'efficacité de Prevymis ne sont pas démontrées chez les patients de moins de 18 ans. Aucune donnée disponible (voir section «Pharmacocinétique»).
Contre-indicationsPrevymis est contre-indiqué chez les patients qui présentent une hypersensibilité au letermovir ou à l'un des excipients.
Pimozide
L'administration concomitante de Prevymis et de pimozide peut entraîner la hausse des taux de pimozide en raison de l'inhibition du cytochrome P450 (CYP3A) par le letermovir; cette hausse peut provoquer un allongement du QT et des torsades de pointes (voir sections «Mises en garde et précautions» et «Interactions»).
Alcaloïdes de l'ergot de seigle
L'administration concomitante de Prevymis et d'alcaloïdes de l'ergot de seigle peut entraîner la hausse des taux d'alcaloïdes de l'ergot de seigle (ergotamine et dihydroergotamine) en raison de l'inhibition du CYP3A par letermovir; cette hausse peut provoquer un ergotisme (voir sections «Mises en garde et précautions» et «Interactions»).
Ciclosporine avec pitavastatine ou simvastatine
L'administration concomitante de Prevymis associé à la ciclosporine peut entraîner une hausse significative du taux de pitavastatine ou de simvastatine, ce qui provoquer une myopathie ou une rhabdomyolyse (voir la section «Mises en garde et précautions», Risque d'effets indésirables ou de diminution de l'effet thérapeutique du fait des interactions médicamenteuses, ainsi que la section «Interactions» de Prevymis avec d'autres médicaments).
Mises en garde et précautionsRisque d'effets indésirables ou de diminution de l'effet thérapeutique du fait des interactions médicamenteuses
L'utilisation concomitante de Prevymis et de certains médicaments peut donner lieu à des interactions médicamenteuses connues ou potentiellement significatives. Les conséquences suivantes sont possibles:
·des effets indésirables cliniquement significatifs liés à une exposition accrue aux traitements concomitants ou à Prevymis;
·une diminution significative des concentrations plasmatiques du traitement concomitant; cette diminution peut atténuer l'effet thérapeutique du traitement concomitant.
Le Tableau 1 résume les mesures de prévention ou de contrôle de ces interactions médicamenteuses connues ou potentiellement significatives, en incluant des recommandations posologiques (voir sections «Contre-indications» et «Interactions»).
Prevymis doit être utilisé avec prudence en association avec des médicaments qui sont des substrats du CYP3A à marge thérapeutique étroite (p.ex. alfentanil, fentanyl et quinidine), car son administration concomitante peut entraîner des élévations des concentrations plasmatiques des substrats du CYP3A. Une surveillance étroite et/ou un ajustement posologique des substrats du CYP3A co-administrés sont recommandés. Veuillez consulter l'information professionnelles des substrats du CYP3A concernés. (Voir Tableau 1 et la section «Interactions»).
Intolérance au lactose
Les comprimés contiennent du lactose monohydraté. Les patients présentant une intolérance héréditaire rare au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose et du galactose ne doivent pas prendre ce médicament.
Sodium
Prevymis 240 mg solution à diluer pour perfusion contient 22,91 mg (soit 1,00 mmol) de sodium par dose. Cet élément doit être pris en compte chez les patients qui suivent un régime pauvre en sel.
Prevymis 480 mg solution à diluer pour perfusion contient 45,82 mg (soit 1,99 mmol) de sodium par dose. Cet élément doit être pris en compte chez les patients qui suivent un régime pauvre en sel.
InteractionsDes études d'interactions médicamenteuses ont été réalisées chez des patients sains avec Prevymis et des médicaments souvent administrés conjointement ou des médicaments souvent utilisés comme médicaments-tests dans le cadre d'études d'interactions pharmacocinétiques (voir Tableau 1 et Tableau 2).
Effets d'autres médicaments sur Prevymis
Les données in vitro ont montré que le letermovir est un substrat des OATP1B1/3, de la P-gp et des UGT1A1 et UGT1A3. Les inhibiteurs des transporteurs OATP1B1/3 peuvent augmenter les taux plasmatiques de letermovir.
Inhibiteurs des OATP1B1 ou 3
Si Prevymis est administré conjointement à la ciclosporine (un puissant inhibiteur des OATP1B1/3), la dose recommandée de Prevymis est de 240 mg une fois par jour (voir section «Posologie/Mode d'emploi». D'autres exemples d'inhibiteurs des OATP1B1 sont le gemfibrozil, l'érythromycine, la clarithromycine, ainsi que plusieurs inhibiteurs de la protéase du VIH (p.ex. atazanavir, lopinavir, ritonavir, siméprévir).
Inhibiteurs de la P-gp
Il est peu probable que l'inhibition de la P-gp provoque des variations cliniquement pertinentes des concentrations plasmatiques de letermovir.
Inhibiteurs des UGT1A1 ou 3
Il est peu probable que l'inhibition de l'UGT exerce un effet cliniquement pertinent sur les concentrations plasmatiques de letermovir.
Inducteurs et inhibiteurs du CYP
Certes les enzymes CYP3A, CYP2D6 et CYP2J2 ont été identifiés comme pouvant médier le métabolisme du letermovir in vitro; cependant, les donnée in vivo chez l'être humain indiquent que la voie d'élimination par métabolisme oxydatif est négligeable.
Effet du letermovir sur d'autres médicaments
Substrats du CYP
Le letermovir exerce une induction et une inhibition dépendante du temps du CYP3A in vitro. L'administration concomitante de Prevymis et du midazolam a augmenté l'exposition au midazolam, ce qui semble indiquer que l'effet net du letermovir sur le CYP3A est une inhibition modérée (voir Tableau 2). Ces résultats indiquent que l'administration concomitante de Prevymis et de substrats du CYP3A peut entraîner une hausse des taux plasmatiques des substrats du CYP3A administrés simultanément (voir sections «Contre-indications», «Mises en garde et précautions», «Interactions», et Tableau 1).
In vitro, le letermovir est un inhibiteur réversible du CYP2C8. La modélisation pharmacocinétique basée sur les données physiologiques prévoit une hausse des taux plasmatiques des substrats du CYP2C8 lorsqu'ils sont administrés conjointement à Prevymis (voir Tableau 1 et Tableau 2).
L'administration concomitante de Prevymis et du voriconazole a entraîné une diminution de l'exposition au voriconazole; cet effet est très probablement dû à l'induction des voies d'élimination du voriconazole via CYP2C9 et CYP2C19. L'administration concomitante de Prevymis et des substrats de CYP2C9 et CYP2C19 peut diminuer les taux plasmatiques de ces derniers (voir Tableau 1). D'autres exemples de substrats du CYP2C9 ou 2C19 sont la warfarine, la phénytoïne, le diazépam, le lansoprazole, l'oméprazole, l'ésoméprazole, le pantoprazole et le tolbutamide.
In vitro, le letermovir est un inducteur du CYP2B6; la pertinence clinique de cette observation n'est pas connue.
Substrats de transporteurs
Le letermovir s'est avéré agir in vitro en inhibiteur des transporteur d'efflux P-gp, BCRP (breast cancer resistance protein), BSEP (bile salt export pump), MRP2 (multidrug-resistance-associated protein 2), OAT3 et des transporteurs de captation hépatique OATP1B1/3.
L'administration concomitante de Prevymis et des substrats des transporteurs OATP1B1/3 (comme l'atorvastatine, un substrat connu du CYP3A, d'OATP1B1/3 et, potentiellement, de BCRP) peut provoquer une augmentation cliniquement pertinente des taux plasmatiques des substrats des OATP1B1/3 (voir Tableau 1). L'amplitude des interaction médiées par les OATP1B1/3 peut être plus importante quand Prevymis est co-administré avec la ciclosporine.
Dans les études cliniques, l'administration orale conjointe de Prevymis et de digoxine (un substrat de la P-gp) ou d'aciclovir (un substrat d'OAT3) n'a pas entraîné de variation cliniquement significative des taux plasmatiques de digoxine ou d'aciclovir (voir Tableau 2).
On ne connaît pas la pertinence clinique de l'effet du letermovir sur les substrats de BCRP, BSEP et MRP2, cet effet n'ayant pas été évalué dans le cadre d'études cliniques.
Si la posologie d'un médicament co-administré est ajustée à cause d'un traitement par Prevymis, cette posologie doit être réajustée au terme du traitement par Prevymis.
Si Prevymis est co-administré avec la ciclosporine, l'effet conjugué sur les substrats du CYP3A peut s'avérer similaire à celui d'un inhibiteur puissant du CYP3A. Veuillez consulter l'information professionnelle pour la posologie du substrat du CYP3A en présence d'un inhibiteur puissant du CYP3A.
Si Prevymis est co-administré avec la ciclosporine, l'effet conjugué sur les principes actifs qui sont simultanément des substrats du CYP3A et d'OATP1B1/3 peut s'avérer différent de celui obtenu en cas d'administration de Prevymis sans ciclosporine. Veuillez consulter l'information professionnelle des médicaments co-administrés et celle de la ciclosporine.
Le Tableau 1 présente une liste des interactions médicamenteuses avérées ou potentiellement significatives sur le plan clinique. Les interactions médicamenteuses indiquées se basent sur des études menées sur Prevymis ou sont déduites des interactions médicamenteuses prévisibles en cas de co-administration de Prevymis (voir section «Mises en garde et précautions»).
Tableau 1: Interactions avec d'autres médicaments et recommandations posologiques: un ajustement posologique peut être conseillé du fait de résultats d'études d'interactions médicamenteuses ou d'interactions prévisibles*
|
Classe et/ou voie d'élimination du médicament administré conjointement: nom du principe actif
|
Effet sur la concentration†
Moyennes (intervalle de confiance à 90%) de l'AUC et Cmax (mécanisme d'action probable)
|
Recommandations concernant l'administration conjointe de Prevymis
| |
Anti-arythmiques
| |
Amiodarone§
|
↑ Amiodarone
|
L'administration conjointe de Prevymis et d'amiodarone augmente les concentrations plasmatiques de l'amiodarone. Une surveillance étroite des effets indésirables liés à l'amiodarone est recommandée pendant la période de coadministration. Surveillez fréquemment les concentrations d'amiodarone.
| |
Antidiabétiques
| |
Glyburide§
|
↑ Glyburide
|
Prevymis peut augmenter la concentration plasmatique du glyburide. Il est recommandé de surveiller fréquemment les concentrations de glucose#.
| |
Antifongiques
| |
Fluconazole
(Dose unique de 400 mg p.o./ letermovir dose unique de 480 mg p.o.)
|
↔ Letermovir
AUC 1.11 (1.01, 1.23)
Cmax 1.06 (0.93, 1.21)
↔ Fluconazole
AUC 1.03 (0.99, 1.08)
Cmax 0.95 (0.92, 0.99)
|
Pas d'ajustement posologique requis.
| |
Posaconazole‡
(dose unique de 300 mg)/ letermovir (480 mg par jour)
|
↔ Posaconazole
AUC 0.98 (0.82, 1.17)
Cmax 1.11 (0.95, 1.29)
|
Pas d'ajustement posologique requis.
| |
Voriconazole‡
(200 mg 2× par jour/ letermovir (480 mg par jour)
|
↓ Voriconazole
AUC 0.56 (0.51, 0.62)
Cmax 0.61 (0.53, 0.71)
(Induction de CYP2C9/19)
|
Si l'administration concomitante s'avère nécessaire, il est recommandé de procéder à une surveillance étroite d'une éventuelle perte d'efficacité et d'effectuer un suivi thérapeutique pharmacologique du voriconazole.#
| |
Antiviraux
| |
Aciclovir‡
(dose unique de 400 mg)/ letermovir (480 mg par jour)
|
↔ Aciclovir
AUC 1.02 (0.87, 1.2)
Cmax 0.82 (0.71, 0.93)
|
Pas d'ajustement posologique requis.
| |
Inhibiteurs de la HMG-CoA réductase
| |
Pitavastatine§, simvastatine§
|
↑Pitavastatine
↑Simvastatine
(Inhibition de CYP3A et/ou OATP1B1/3, et inhibition potentielle de la BCRP intestinale)
|
Il est recommandé de suspendre le traitement par inhibiteurs de la HMG-CoA réductase pendant le traitement par Prevymis#.
Lorsque Prevymis est administré conjointement à la ciclosporine, l'utilisation de pitavastatine ou de simvastatine est contre-indiquée (voir section «Contre-indications»).
| |
Atorvastatine‡
(dose unique de 20 mg)/ letermovir (480 mg par jour)
|
↑ Atorvastatine
AUC 3.29 (2.84, 3.82)
Cmax 2.17 (1.76, 2.67)
(Inhibition de CYP3A et OATP1B1/3)
|
Il est recommandé de suspendre le traitement par inhibiteurs de la HMG-CoA réductase pendant le traitement par Prevymis.
| |
Autres inhibiteurs de la HMG-CoA réductase§
Exemples: fluvastatine,
lovastatine,
pravastatine,
rosuvastatine
|
↑ Concentrations d'inhibiteurs de la HMG-CoA réductase
(Inhibition de CYP3A et/ou OATP1B1/3, et inhibition potentielle de la BCRP intestinale)
| |
Immunosuppresseurs
| |
Ciclosporine
(dose unique de 50 mg)/ letermovir (240 mg par jour)
|
↑ Ciclosporine
AUC 1.66 (1.51, 1.82)
Cmax 1.08 (0.97, 1.19)
(Inhibition de CYP3A)
|
Si Prevymis est administré conjointement à la ciclosporine, la posologie de Prevymis doit être réduite à 240 mg une fois par jour (voir sections «Posologie/Mode d'emploi» et «Pharmacocinétique»).
Pendant et à l'arrêt de l'utilisation de Prevymis, les concentrations de ciclosporine dans le sang total doivent être fréquemment surveillées et la dose de ciclosporine doit être ajustée en conséquence.#
| |
Ciclosporine
(dose unique de 200 mg)/ letermovir (240 mg par jour)
|
↑ Letermovir
AUC 2.11 (1.97, 2.26)
Cmax 1.48 (1.33, 1.65)
(Inhibition de OATP1B1/3)
| |
Mycophénolate mofétil
(dose unique de 1 g)/ letermovir (480 mg par jour)
|
↔ Acide mycophénolique
AUC 1.08 (0.97, 1.20)
Cmax 0.96 (0.82, 1.12)
↔ Letermovir
AUC 1.18 (1.04, 1.32)
Cmax 1.11 (0.92, 1.34)
|
Le mycophénolate mofétil ne requiert pas d'ajustement posologique.
| |
Sirolimus‡
(dose unique de 2 mg)/ letermovir (480 mg par jour)
|
↑ Sirolimus
AUC 3.40 (3.01, 3.85)
Cmax 2.76 (2.48, 3.06)
(Inhibition de CYP3A)
|
Pendant et à l'arrêt de l'utilisation de Prevymis, les concentrations de sirolimus dans le sang total doivent être fréquemment surveillées et la dose de sirolimus doit être ajustée en conséquence.#
Lorsque Prevymis est co-administré avec la ciclosporine, se référer à l'information professionnelle du sirolimus pour connaître les recommandations posologiques particulières en cas d'utilisation du sirolimus avec la ciclosporine.#
| |
Tacrolimus
(dose unique de 5 mg)/ letermovir (480 mg par jour)
Tacrolimus
(dose unique de 5 mg)/ letermovir (80 mg deux fois par jour)
|
↑ Tacrolimus
AUC 2.42 (2.04, 2.88)
Cmax 1.57 (1.32, 1.86)
(Inhibition de CYP3A)
↔ Letermovir
AUC 1.02 (0.97, 1.07)
Cmax 0.92 (0.84, 1.00)
|
Pendant et à l'arrêt de l'utilisation de Prevymis, les concentrations de tacrolimus dans le sang total doivent être fréquemment surveillées et la dose de tacrolimus doit être ajustée en conséquence.
Lorsque Prevymis est co-administré avec la ciclosporine, se référer à l'information professionnelle du tacrolimus pour connaître les recommandations posologiques particulières en cas d'utilisation du tacrolimus avec la ciclosporine.#
| |
Contraceptifs oraux
| |
Ethinylestradiol (EE) (0,03 mg) /lévonorgestrel (LNG)‡ (0,15 mg) dose unique/ letermovir (480 mg par jour)
|
↔ EE
AUC 1.42 (1.32, 1.52)
Cmax 0.89 (0.83, 0.96)
↔ LNG
AUC 1.36 (1.30, 1.43)
Cmax 0.95 (0.86, 1.04)
|
Prevymis peut être utilisé avec les contraceptifs hormonaux. On ne connaît l'impact clinique de la hausse probable des taux d'éthinylestradiol et de lévonorgestrel provoquée par Prevymis, lorsque ces principes actifs sont administrés de manière répétée.
| |
Inhibiteurs de la pompe à protons
| |
Oméprazole§, pantoprazole§
|
↓Oméprazole
↓Pantoprazole
|
L'administration conjointe de Prevymis et de ces inhibiteurs de la pompe à protons (IPP) peut diminuer les concentrations plasmatiques des IPP. Une surveillance clinique et un ajustement posologique peuvent être nécessaires lorsque ces IPP sont administrés conjointement à Prevymis#.
| |
Substrats du CYP2C8**
| |
Exemples: répaglinide§, rosiglitazone§
|
↑ Concentrations des substrats du CYP2C8
|
Prevymis peut augmenter les taux plasmatiques des substrats du CYP2C8.
Il est recommandé de mesurer fréquemment la glycémie en cas d'utilisation simultanée de répaglinide ou de rosiglitazone. Du fait de l'inhibition d'OATP1B par la ciclosporine, on peut s'attendre à ce que la hausse des taux plasmatiques de répaglinide soit plus élevée lorsque Prevymis est co-administré avec la ciclosporine vs lorsque Prevymis est co-administré seul. Veuillez consulter l'information professionnelle du répaglinide pour connaître les recommandations posologiques particulières.#
| |
Substrats du CYP2C9/19
| |
Exemples: phénytoïne§, warfarine§
|
↓ Concentrations des substrats du CYP2C9/19
|
Prevymis peut diminuer les taux plasmatiques des substrats du CYP2C9/19.
Surveiller fréquemment les taux de phénytoïne en cas d'utilisation conjointe de phénytoïne et de Prevymis.#
Surveiller fréquemment l'INR en cas d'utilisation conjointe de warfarine et de Prevymis.#
| |
Substrats du CYP3A††
| |
Midazolam
(dose unique de 1 mg i.v.)/ letermovir (240 mg un fois par jour p.o.)
Midazolam (dose unique de 2 mg i.v.)/ letermovir (240 mg un fois par jour p.o.)
|
↑ Midazolam
i.v.:
AUC 1.47 (1.37, 1.58)
Cmax 1.05 (0.94, 01:17)
p.o.:
AUC 2.25 (2.04, 2.49)
Cmax 1.72 (1.55, 1.92)
(Inhibition de CYP3A)
|
Prevymis peut augmenter les taux plasmatiques des substrats du CYP3A.
Lorsque le letermovir est co-administré seul avec un substrat du CYP3A, se référer à l'information professionnelle pour connaître la posologie du substrat du CYP3A en présence d'un inhibiteur modéré du CYP3A.
En cas d'administration conjointe, il est recommandé de procéder à une surveillance fréquente des patients afin de détecter toute apparition d'effets indésirables de ces médicaments. Un ajustement posologique des substrats du CYP3A peut s'avérer nécessaire# (voir section «Mises en garde et précautions»)
| |
Autres exemples: alfentanil§, fentanyl§, quinidine§
|
↑ Concentrations des substrats du CYP3A
| |
Substrats de la P-gp
| |
Digoxine‡
(dose unique de 0,5 mg)/ letermovir (240 mg deux fois par jour)
|
↔ Digoxine
AUC 0.88 (0.80, 0.96)
Cmax 0.75 (0.63, 0.89)
(induction de la P-gp)
|
Pas d'ajustement posologique requis.
| |
* Ce tableau n'est pas exhaustif.
† ↓ = diminution, ↑ = augmentation, ↔ = pas de variation cliniquement significative
‡ Étude d'interaction unidirectionnelle évaluant l'effet du letermovir sur la co-médication.
§ Ces interactions n'ont pas été étudiées.
# Se référer aux informations professionnelles correspondantes.
** Provenant d'une modélisation pharmacocinétique basée sur les données physiologiques.
†† Basé sur des études in vivo portant sur le midazolam.
|
Grossesse/AllaitementGrossesse
Il n'existe pas de données cliniques sur l'utilisation du Prevymis chez la femme enceinte. La toxicité embryofœtale a été étudiée chez le rat et le lapin dans les conditions d'exposition maternotoxiques systémiques correspondant à environ onze fois et deux fois l'AUC obtenu sous posologie recommandée chez l'être humain (RHD). Une reprotoxicité a été observée chez l'animal à des doses maternotoxiques (voir la section «Données précliniques»).
Le risque potentiel pour l'être humain n'est pas connu. Prevymis n'est pas recommandé pendant la grossesse à moins de nécessité absolue.
Allaitement
On ne sait pas si le letermovir est excrété dans le lait maternel, ni s'il influence la lactogenèse ou s'il agit sur l'enfant allaité.
Le letermovir a été détecté dans le lait après administration à des rates allaitantes, mais n'a pas eu d'effet sur la croissance et le développement des jeunes animaux allaités. En cas d'allaitement, la décision de traiter par letermovir doit prendre en compte le bénéfice de l'allaitement en regard de celui du traitement.
Effet sur l’aptitude à la conduite et l’utilisation de machinesPrevymis n'a vraisemblablement pas d'influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesExpériences tirées des études cliniques
L'évaluation de la sécurité de Prevymis provient d'une étude de phase 3 (P001) randomisée et menée en double aveugle contre placebo. Dans cette étude, 565 patients randomisés ont été traités jusqu'à 14 semaines postgreffe par Prevymis (N=373) ou par placebo (N=192). Ils ont ensuite fait l'objet d'un suivi de pharmacovigilance pendant une période allant jusqu'à 24 semaines post-greffe (voir section «Pharmacocinétique»).
Effets indésirables fréquents
Le Tableau 2 indique la fréquence des effets indésirables (indépendamment de la causalité établie par l'investigateur) apparus chez ≥10% des sujets du groupe Prevymis et dont la fréquence était ≥2% plus élevée que celle observée dans le groupe placebo. Ces effets indésirables sont classés par systèmes d'organes et fréquence. Les fréquences sont définies comme suit: très fréquents (≥1/10), fréquents (≥1/100, <1/10), occasionnels (≥1/1'000, <1/100), rares (≥1/10'000, <1/1'000), très rares (<1/10'000).
Tableau 2: Effets indésirables observés dans l'étude P001 chez ≥10% des receveurs d'une GCSH traités par Prevymis et une fréquence ≥2% plus élevée que celle observée dans le groupe placebo.
|
Fréquence
|
Effets indésirables
| |
Affections du système nerveux
| |
Très fréquents
|
Céphalées (14% vs placebo 9%)
| |
Affections respiratoires
| |
Très fréquents
|
Toux (14% vs placebo 10%)
| |
Affections gastro-intestinales
| |
Très fréquents
|
Nausées (27% vs placebo 23%), diarrhée (26% vs placebo 24%), vomissements (19% vs placebo 14%), douleurs abdominales (12% vs placebo 9%)
| |
Troubles généraux et anomalies au site d'administration
| |
Fréquents
|
Fatigue (13% vs placebo 11%), œdème périphérique (14% vs placebo 9%)
|
Le pourcentage d'arrêt du traitement en raison d'effets indésirables a été similaire dans les deux bras de l'étude (13% de patients sous Prevymis vs 12% sous placebo). Les nausées ont été l'effet indésirable le plus souvent signalé qui a provoqué le plus d'abandons de l'étude: 2% des abandons parmi les patients sous Prevymis et 1% parmi les patients sous placebo.
Événements cardiaques
La fréquence des événements cardiaques (indépendamment de la causalité établie par l'investigateur) a été plus élevée chez les sujets sous Prevymis (13%) que chez ceux sous placebo (6%). Les événements cardiaques les plus fréquents ont été la tachycardie (rapportée pour 4% des patients sous Prevymis et 2% des patients sous placebo) ainsi que la fibrillation auriculaire (rapportée pour 3% des patients sous Prevymis et 1% des patients sous placebo). Parmi les 6 sujets ayant présenté un ou plusieurs événements cardiaques, 85% de ceux sous Prevymis et 92% de ceux sous placebo ont présenté des événements évalués comme légers ou modérés.
Description de certains effets indésirables
Un cas d'hypersensibilité catégorisé comme effet indésirable non grave a été rapporté chez un patient sous Prevymis.
Anomalies des paramètres de laboratoire
Les variations des paramètres de laboratoire (hématologie, chimie, fonction hépatique et rénale, etc.) potentiellement significatives en termes clinique ont été comparables dans le groupe Prevymis et dans le groupe placebo. Le groupe Prevymis et le groupe placebo n'ont pas présenté de différences en termes d'incidence ou de temps de prise de la greffe (engraftment).
Les marqueurs biologiques de toxicité testiculaire ont fait l'objet d'une évaluation chez les patients de sexe masculin de l'étude P001 (voir la section «Données précliniques»). Les variations par rapport aux taux initiaux des taux d'hormones sexuelles masculines (inhibine B sérique, hormone lutéinisante (LH), hormone folliculo-stimulante (FSH) et testostérone) ont été comparables dans le groupe Prevymis et dans le groupe placebo.
SurdosageAucune expérience de surdosage de Prevymis n'a été rapportée chez l'être humain. Dans des études cliniques de phase 1, 86 sujets sains ont reçu des doses de Prevymis comprises entre 720 mg/jour et 1440 mg/jour pendant une période allant jusqu'à 14 jours. Le profil d'effets indésirables a été similaire à celui de la dose clinique de 480 mg/jour. Il n'existe pas d'antidote pas d'antidote spécifique en cas de surdosage de Prevymis. En cas de surdosage, il est recommandé de surveiller étroitement le patient afin de détecter toute apparition d'effets indésirables. Le cas échéant, un traitement symptomatique approprié doit être instauré.
On ignore si la dialyse permet d'éliminer Prevymis de la circulation systémique de manière significative.
Propriétés/EffetsCode ATC: J05AX18
Prevymis est un médicament antiviral contre le CMV.
Mécanisme d'action
Le letermovir est un médicament antiviral contre le CMV.
Le letermovir inhibe le complexe terminase de l'ADN du CMV qui indispensable à la réplication virale. Les analyses biochimiques et électromiscroscopiques ont montré que le letermovir affecte la formation d'unités génomiques de longueur adéquate et interfère avec la maturation des virions.
Activité antivirale
Dans un modèle d'infection en culture cellulaire, la CE50 médiane du letermovir contre un ensemble d'isolats du CMV obtenus en clinique a été de 2,1 nM (intervalle 0,7 nM à 6,1 nM; n = 74).
Résistance virale
En culture cellulaire
Les gènes UL56 et UL89 du CMV codent des sous-unités de l'ADN terminase du CMV. Des CMV mutants présentant une sensibilité réduite au letermovir ont été sélectionnés en culture cellulaire. Les mutations correspondent au gène UL56 et affectent les acides aminés situés entre les positions 231 et 369 (V231A, V231L, V236L, V236M, E237D, L241P, T244K, T244R, L257I, F261C, F261L, F261S, Y321C, C325F, C325R, C325Y, M329T, R369G, R369M, R369S). En présence de ces mutations, la CE50 est 13 à 5870 fois plus élevée que pour les virus de référence de type sauvage. Il n'y a pas de mutation de résistance connue au letermovir liée au gène UL89.
Dans les études cliniques
Dans une étude de phase 2b évaluant des doses de letermovir de 60, 120 ou 240 mg/jour vs placebo sur une période allant jusqu'à 84 jours chez 131 receveurs de GCSH, une analyse de la séquence d'ADN d'une région choisie du gène UL56 (acides aminés 231 à 369) a été réalisée sur des échantillons obtenus auprès de 12 patients traités par letermovir. Ces patients avaient été en échec prophylactique et leurs prélèvements étaient disponibles pour analyse. Un participant de l'étude (qui avait reçu 60 mg/jour) a présenté un variant génotypique (VG) résistant au letermovir (V236M).
Dans une étude de phase 3 (P001), une analyse de la séquence d'ADN de l'ensemble des régions codantes des gènes UL56 et UL89 a été réalisée sur des échantillons obtenus auprès de 22 patients traités par letermovir dans la population en full analysis set. Ces patients avaient été en échec prophylactique et leurs prélèvements étaient disponibles pour analyse. Un patient a présenté un VG résistant au letermovir (V236M).
Résistance croisée
Une résistance croisée avec un médicament d'une autre classe est peu probable. Le letermovir est pleinement actif contre les populations virales porteuses de substitutions conférant une résistance aux inhibiteurs de la polymérase de l'ADN du CMV (ganciclovir, cidofovir et foscarnet). Ces inhibiteurs de la polymérase de l'ADN sont pleinement actifs contre les populations virales porteuses de substitutions conférant une résistance au letermovir.
Pharmacogénomique
L'impact de mutations affectant le gène OATP1B1 sur la pharmacocinétique du letermovir a été étudié sur 299 participants des études cliniques. Il s'agit des variants SLCO1B1 (rs4149056, rs2306283, rs4149032) et UGT1A1 (rs4148323 et mutation des répétitions du TA dans le promoteur). Ces variants n'ont pas donné lieu à des effets cliniquement significatifs en termes d'exposition au letermovir.
Électrophysiologie cardiaque
L'effet du letermovir sur l'intervalle QTc à des doses allant jusqu'à 960 mg administrées par voie i.v. a été évalué sur 38 sujets sains dans une étude de durée de QT randomisée, en dose unique, contrôlée versus placebo et comparateur actif (moxifloxacine 400 mg par voie orale), avec essai croisé à quatre séquences. Le letermovir ne provoque pas d'allongement cliniquement significatif de l'intervalle QTc après l'administration d'une dose de 960 mg par voie i.v., ce qui correspond à des concentrations plasmatiques environ 2 fois supérieures à celles de la dose i.v. de 480 mg.
Efficacité clinique et sécurité
Adultes séropositifs au CMV receveurs [R+] d'une greffe allogénique de cellules souches hématopoïétiques
Afin d'évaluer la capacité prophylactique du letermovir dans le cadre d'une stratégie préventive contre l'infection ou la maladie à CMV, l'efficacité du letermovir a été examinée dans une étude de phase 3 (P001) multicentrique en double aveugle, contrôlé versus placebo chez des adultes séropositifs au CMV receveurs [R+] d'une GCSH allogénique. Les sujets ont été randomisés (2:1) pour recevoir soit le letermovir soit le placebo. La randomisation a été stratifiée en fonction du centre d'étude et du risque de réactivation du CMV (élevé versus faible) à l'inclusion. Le letermovir a été initié après la GCSH (jours 0 - 28 post-greffe) et poursuivi jusqu'à la semaine 14 post-greffe. Le letermovir a été administré soit par voie orale soit par voie i.v. Le suivi des patients a été centré sur le critère principal d'efficacité jusqu'à la semaine 24 post-greffe. La surveillance s'est poursuivie en continu ensuite jusqu'à la semaine 48 post-greffe.
Parmi les 565 patients traités, 373 ont reçu le letermovir (dont 99 qui ont reçu au moins une dose i.v.) et 192 ont reçu le placebo (dont 48 qui ont reçu au moins une dose i.v.). Le délai médian avant instauration du letermovir a été de 9 jours après la greffe. Au début de l'étude, 37% des patients présentaient une prise de la greffe (engraftment). L'âge médian était de 54 ans (intervalle de 18 à 78 ans). Au début de l'étude, 50% des patients recevaient un traitement myéloablatif, 52% de la ciclosporine et 42% du tacrolimus. Les motifs de greffe les plus fréquents étaient la leucémie myéloïde aiguë (38%), le syndrome myéloblastique (15%) et le lymphome (13%). Au début de l'étude, 12% des patients étaient positifs à l'ADN du CMV.
Au début de l'étude, 31% des patients étaient à haut risque de réactivation tel que défini par un ou plusieurs des critères suivants: donneur HLA (Human Leukocyte Antigen) apparenté (frère/sœur) avec au moins une incompatibilité sur l'un des trois loci suivants des gènes HLA: donneur HLA-A, B ou DR; donneur haplo-identique; donneur non apparenté avec au moins une incompatibilité sur l'un des quatre loci suivants des gènes HLA: HLA-A, B, C et DRB1; utilisation de sang de cordon ombilical comme source de cellules souches; utilisation de greffons avec cellules T déplétées ex vivo; réaction greffon contre l'hôte (GVHD) de grade 2 ou supérieur nécessitant des corticoïdes systémiques.
Efficacité
Infection à CMV cliniquement significative
Le critère principal d'efficacité de l'étude P001 a été l'apparition d'une infection à CMV cliniquement significative jusqu'à la semaine 24 post-greffe. L'infection à CMV cliniquement significative a été définie comme étant soit la survenue d'une maladie à CMV touchant un organe cible soit l'instauration d'un traitement préemptif anti-CMV (PET) en raison d'une virémie à CMV documentée (utilisation du Roche COBAS AmpliPrep/COBAS TaqMan Assay).
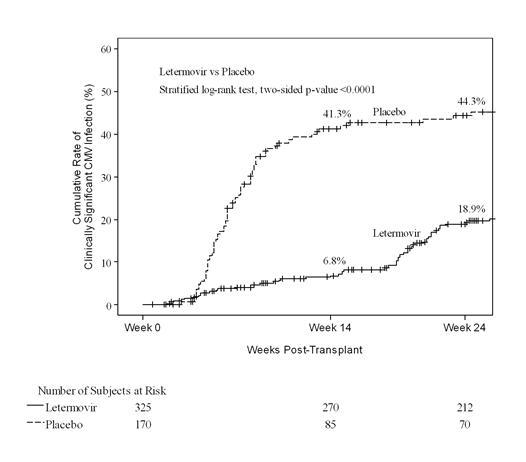
Dans l'analyse du critère principal, le letermovir a démontré une plus grande efficacité que le placebo (voir Tableau 3). La différence estimée de traitement de -23,5% était statistiquement significative (valeur de p unilatérale <0,0001).
Tableau 3: P001: Résultats d'efficacité chez les receveurs de GCSH (approche NC=F, population FAS)
|
|
Letermovir
|
Placebo
| |
|
(N=325)
|
(N=170)
| |
Paramètres
|
n (%)
|
n (%)
| |
Critère principal
(Pourcentage de patients en échec prophylactique)
|
122 (37,5)
|
103 (60,6)
| |
Raisons des échecs†
|
|
| |
Infection au CMV cliniquement significative jusqu'en semaine 24‡
|
57 (17,5)
|
71 (41,8)
| |
Instauration d'un traitement préemptif sur la base d'une virémie à CMV documentée
|
52 (16,0)
|
68 (40,0)
| |
Maladie à CMV d'un organe cible
|
5 (1,5)
|
3 (1,8)
| |
Sortie de l'étude avant la semaine 24
|
56 (17,2)
|
27 (15.9)
| |
Résultat manquant jusqu'à la fenêtre de consultation de la semaine 24
|
9 (2,8)
|
5 (2,9)
| |
Différence de traitement ajustée en fonction de la stratification (letermovir-placebo)§
|
|
| |
Différence (IC 95%)
|
-23,5 (-32,5, -14,6)
|
| |
Valeur de p
|
<0,0001
|
| |
† Les catégories d'échec sont mutuellement exclusives et se basent sur la hiérarchie des catégories dans l'ordre indiqué.
‡ Une infection à CMV cliniquement significative a été définie comme étant soit la survenue d'une maladie à CMV touchant un organe cible soit l'instauration d'un traitement préemptif en raison d'une virémie à CMV documentée et de l'état clinique du patient.
§ Les IC 95% et la valeur de p pour les différences entre traitements en pourcentage de réponse ont été calculés à l'aide de la méthode de Mantel-Haenszel ajustée en fonction des strates, en pondérant la différence en fonction de la moyenne harmonique de la taille de l'échantillon par bras pour chaque strate (risque élevé ou faible). Une valeur de p unilatérale ≤0,0249 a été utilisée pour déterminer la significativité statistique.
Remarque: FAS = population totale d'analyse (full analysis set); cette population inclut les patients randomisés qui ont reçu au moins une dose de médicament à l'étude, et exclut les patients présentant un ADN du CMV détectable en début d'étude. Approche suivie pour le traitement des valeurs manquantes: approche abandon=échec (Non-Completer = Failure; NC = F). Dans l'approche NC = F, l'échec a été défini comme l'ensemble des patients qui ont développé une infection au CMV cliniquement significative ou qui sont sortis prématurément de l'étude, ou ceux pour lesquels aucun résultat n'avait été relevé jusqu'à la fenêtre de consultation de la semaine 24 post-greffe.
N = Nombre de patients dans chaque groupe de traitement.
n (%) = Nombre (pourcentage) de patients dans chaque sous-catégorie.
|
En semaine 24 post-greffe, le taux d'infections à CMV cliniquement significatives (événements Kaplan-Meier, KM) a été de 18,9% dans le groupe letermovir vs 44,3% dans le groupe placebo (valeur de p nominale bidirectionnelle stratifiée du log-rank<0,0001) (voir Fig. 1). Chez les patients traités par le letermovir, les facteurs liés à une infection à CMV cliniquement significative entre les semaines 14 et 24 post-greffe ont été les suivants:
·risque élevé de réactivation du CMV en début d'étude,
·présence d'une GVHD et
·utilisation d'un stéroïde à un moment quelconque après randomisation.
Fig. 1: P001: Courbe de Kaplan-Meier du délai de survenue d'une infection à CMV cliniquement significative jusqu'à la semaine 24 post-greffe chez les receveurs de GCSH (population FAS)
L'efficacité a été nettement supérieure dans tous les sous-groupes du bras letermovir, y compris les suivants:
·risque faible ou élevé de réactivation du CMV,
·traitements de conditionnement, et
·traitements immunosuppresseurs concomitants.
Mortalité
Les taux d'événements de mortalité globale (analyse Kaplan-Meier) a été de 12,1% dans le groupe letermovir vs 17,2% dans le groupe placebo en semaine 24 post-greffe (valeur de p nominale bidirectionnelle stratifiée du log-rank = 0,0401) et 23,8% vs 27,6% en semaine 48 post-greffe (valeur de p nominale bidirectionnelle stratifiée du log-rank = 0,2117). Les taux d'événements de mortalité globale (analyse Kaplan-Meier) selon le sexe (hommes vs femmes) en semaine 24 post-greffe a été de 16,4% vs 6,6% dans le groupe letermovir et de 14,2% vs 25,4% dans le groupe placebo; ces taux d'événements décomposés selon le sexe sont à interpréter avec précaution; en effet, la randomisation n'a pas été stratifiée par sexe, ce qui a donné lieu en baseline à des déséquilibres entre groupes de traitement en termes de risque de mortalité spécifiquement liés au sexe.
PharmacocinétiqueIntroduction générale
La pharmacocinétique du letermovir a été caractérisée après administration par voie orale et intraveineuse chez des sujets sains et des receveurs de GCSH.
Chez les sujets sains, l'exposition au letermovir a augmenté d'une manière plus que proportionnelle à la dose après administration unique ou répétée de 240 mg et 480 mg, et ce aussi bien après administration par voie orale que par voie i.v.. Le letermovir a été rapidement absorbé, avec un délai médian jusqu'au pic de concentration plasmatique (Tmax) de 1,5 à 3 heures; ces concentrations ont ensuite diminué en deux phases. Après administration de 480 mg de letermovir une fois par jour par voie orale chez les sujets sains, les moyennes géométriques à l'état d'équilibre de l'AUC et de la Cmax ont été respectivement de 71'500 ng•h/ml et de 13'000 ng/ml. La profil de l'évolution de la concentration plasmatique du letermovir en fonction du temps a été similaire après administration p.o. et i.v.. Le letermovir a atteint l'état d'équilibre (steady state) en 9 à 10 jours, avec un ratio d'accumulation de 1,23 pour l'AUC et de 1,09 pour la Cmax.
Chez les receveurs de GCSH, l'AUC du letermovir a été estimée à partir d'analyses de pharmacocinétique de population utilisant des données de l'étude de phase 3 (voir Tableau 4). Les différences d'exposition entre les schémas thérapeutiques ne sont pas pertinentes au plan clinique; l'efficacité est restée maintenue sur l'ensemble de l'intervalle d'exposition observé dans P001.
Tableau 4: Valeurs de l'AUC (ng•h/ml) du letermovir chez les receveurs de GCSH
|
Schéma thérapeutique
|
Médiane (intervalle de prédiction à 90%)*
| |
480 mg p.o., pas de ciclosporine
|
34'400 (16'900, 73'700)
| |
480 mg i.v., pas de ciclosporine
|
100'000 (65'300, 148'000)
| |
240 mg p.o., avec ciclosporine
|
60'800 (28'700, 122'000)
| |
240 mg i.v., avec ciclosporine
|
70'300 (46'200, 106'000)
| |
* Les médianes et les intervalles de prédiction à 90% se basent sur des simulations réalisées à partir de l'analyse PK de la population de l'étude de phase 3 incluant les variabilité inter-individuelle.
|
Absorption
Sur la base des analyses de pharmacocinétique de population, la biodisponibilité absolue du letermovir chez les sujets sains a été estimée à 94% environ dans l'intervalle de doses de 240 mg à 480 mg. Chez les receveurs de GCSH, la biodisponibilité du letermovir a été estimée à 35% environ après administration orale de 480 mg de Prevymis une fois par jour sans ciclosporine. La variabilité inter-individuelle de la biodisponibilité a été estimée à environ 37%.
Effet de la ciclosporine
Chez les receveurs de GCSH, la co-administration de ciclosporine a augmenté les concentrations plasmatiques de letermovir. La biodisponibilité du letermovir a été estimée à 85% environ après administration orale de 240 mg de Prevymis une fois par jour avec ciclosporine. Si Prevymis est administré conjointement à la ciclosporine, la dose recommandée de Prevymis est de 240 mg une fois par jour (voir section «Posologie/Mode d'emploi»).
Influence des aliments
L'administration orale d'une dose unique de 480 mg de Prevymis avec un repas standard riche en graisses et en calories n'a eu aucun effet sur l'exposition totale (AUC) et a donné lieu à une augmentation de 30% environ des pics de concentration (Cmax) du letermovir par rapport à une administration à jeun. Prevymis peut être administré par voie orale avec ou sans aliments (voir section «Posologie/Mode d'emploi»).
Distribution
Sur la base des analyses de pharmacocinétique de population, le volume de distribution moyen à l'état d'équilibre (steady state) est estimé à 45,5 l après administration intraveineuse chez des receveurs de GCSH.
In vitro, le letermovir est fortement lié aux protéines plasmatiques humaines (98,7%). La répartition sang/plasma du letermovir in vitro est évaluée à 0,56 et elle est indépendante de l'intervalle de concentrations (0,1 à 10 mg/l).
Dans les études précliniques portant sur la distribution, le letermovir s'est distribué dans les organes et les tissus; les concentrations les plus élevées ont été observées dans l'appareil digestif, le canal biliaire et le foie; les concentrations observées dans le cerveau étaient faibles.
Élimination
La demi-vie terminale apparente moyenne du letermovir est de 12 heures environ après administration i.v. de 480 mg de Prevymis chez des sujets sains.
Sur la base des analyses de pharmacocinétique de population, la clairance apparente du letermovir à l'état d'équilibre est estimée à 4,84 l/h après administration intraveineuse chez des receveurs de GCSH. La variabilité inter-individuelle de la clairance a été estimée à environ 24,6%.
Métabolisme
Le composé parent inchangé constitue la plus grande partie (96,6%) des composants du médicament présents dans le plasma. Aucun métabolite majeur n'a été détecté dans le plasma.
Les principales voies d'élimination du letermovir sont l'excrétion biliaire et la glucuronidation directe. Le processus implique les transporteurs de captation hépatique OATP1B1 et 3, suivis de la glucuronidation catalysée par les UGT1A1/3.
Excrétion
Après administration orale de letermovir radiomarqué, 93,3% de la radioactivité a été retrouvée dans les fèces. La majorité du letermovir a été éliminée dans les fèces sous forme du composé parent inchangé, avec une faible proportion (6% de la dose) sous forme de métabolite acyl-glucuronide. L'excrétion urinaire du letermovir s'est avérée négligeable (<2% de la dose).
Populations particulières
Enfants et adolescents
La pharmacocinétique du letermovir n'a pas été étudiée chez les enfants et les adolescents de moins de 18 ans.
Patients âgés
Les analyses de pharmacocinétique de population n'ont révélé aucun effet de l'âge sur la pharmacocinétique du letermovir. Aucune adaptation posologique n'est requise en fonction de l'âge.
Sexe
Les analyses de pharmacocinétique de population n'ont révélé aucune différence en termes de pharmacocinétique du letermovir entre les patients de sexe masculin et féminin.
Poids
Sur la base des analyses de pharmacocinétique de population, l'AUC estimée du letermovir est plus faible de 18,7% chez les patients pesant 80-100 kg que chez les ceux pesant 67 kg. Cette différence n'est pas cliniquement significative.
Appartenance ethnique
Sur la base des analyses de pharmacocinétique de population, l'AUC estimée du letermovir est plus élevée de 33,2% chez les sujets de type asiatique que chez les ceux de type caucasien. Cette différence n'est pas cliniquement significative.
Insuffisance rénale
Chez les patients présentant une insuffisance rénale modérée (DFGe de 30 à 59 ml/min/1.73 m2) et sévère (DFGe <30 ml/min/1,73 m2), l'AUC du letermovir a été respectivement 1,9 et 1,4 fois plus élevée que chez les sujets sains. Les variations de l'exposition au letermovir en cas d'insuffisance rénale modérée ou sévère ne sont pas considérées comme cliniquement significatives. On ne dispose pas de données concernant les patients en dialyse ou présentant une DFGe <10 ml/min/1,73 m2.
Insuffisance hépatique
Chez les patients présentant une insuffisance hépatique modérée (Child-Pugh classe B [CP-B], score de 7-9) et sévère (Child-Pugh classe C [CP-C], score de 10-15), l'AUC du letermovir a été respectivement 1,6 et 3,8 fois plus élevée que chez les sujets sains. Les variations de l'exposition au letermovir chez les patients présentant une insuffisance hépatique modérée ne sont pas cliniquement significatives.
Des élévations cliniquement significatives de l'exposition au letermovir sont probables chez les patients présentant une insuffisance hépatique sévère ou une insuffisance hépatique modérée associée à une insuffisance rénale modérée à sévère.
Données précliniquesToxicité générale
Une toxicité testiculaire a été observée chez les rats à des niveaux d'exposition systémique (AUC) à ≥3 fois supérieurs aux expositions observées chez l'être humain aux doses recommandées (RHD). Cette toxicité s'est caractérisée par une dégénérescence des tubes séminifères, une oligospermie, la présence de débris cellulaires dans les épididymes et une baisse du poids des testicules et des épididymes. Le NOAEL (No Observed Adverse Effect Level) a été constaté en termes de toxicité testiculaire chez le rat à des expositions (AUC) similaires à celles obtenues chez l'être humain aux doses recommandées (RHD). Cette toxicité testiculaire paraît spécifique à l'espèce; elle n'a pas été observée chez la souris et le singe aux doses maximales testées, avec des expositions respectivement 4 fois et 2 fois supérieures à l'AUC constatée chez l'être humain aux doses recommandées (RHD). La pertinence chez l'être humain n'est pas connue.
Dans les études sur le rat et le singe, le profil de toxicité du letermovir administré par voie i.v. ou orale a été généralement similaire, hormis des vacuolisations rénales observées chez des rats ayant reçu du letermovir par voie i.v. formulé avec 1500 mg/kg/jour d'hydroxypropylbetadex, excipient de la cyclodextrine. On sait que l'hydroxypropylbetadex peut provoquer une vacuolisation du rein chez le rat après administration de doses supérieures à 50 mg/kg/jour.
Mutagénicité
Le letermovir n'a pas été génotoxique dans une batterie d'essais in vitro et in vivo, incluant des tests de mutagenèse microbienne, d'aberrations chromosomiques sur cellules CHO et dans un test in vivo de micronoyaux chez la souris.
Carcinogénicité
Aucune étude de carcinogénicité du letermovir n'a été réalisée.
Reproduction
Fertilité
Dans les études sur la fertilité et le développement précoce de l'embryon chez le rat, aucun effet du letermovir n'a été observé sur la fertilité des femelles aux doses maximales de 240 mg/kg/jour (soit 5 fois l'AUC chez l'être humain aux doses recommandées (RHD). Chez les rats mâles, l'exposition systémique ≥3 fois l'AUC chez l'être humain aux doses recommandées (RHD) a entraîné une baisse de concentration des spermatozoïdes, une baisse de motilité des spermatozoïdes et une baisse de fertilité (voir la rubrique «Toxicologie générale»).
Développement
Le letermovir a été administré p.o. à des rates gestantes aux doses de 0, 10, 50 ou 250 mg/kg/jour à leur 6e et 17e jour de gestation. Une toxicité maternelle (y compris une diminution de la prise de poids) a été observée à la dose de 250 mg/kg/jour (environ 11 fois l'AUC à la RHD); chez la progéniture, une baisse du poids du fœtus a été constatée, accompagnée des observations suivantes: retard d'ossification, fœtus légèrement œdémateux et incidence accrue de cordons ombilicaux courts et de variations et malformations des vertèbres, des côtes et du pelvis. Aucun effet sur la mère ou sur le développement n'a été observé à la dose de 50 mg/kg/jour (environ 2,5 fois l'AUC à la RHD).
Le letermovir a été administré p.o. à des lapines gestantes aux doses de 0, 25, 75 ou 225 mg/kg/jour à leur 6e et 20e jour de gestation. À la dose de 225 mg/kg/jour (environ 2 fois l'AUC à la RHD), une toxicité maternelle a été observée (notamment mortalité et avortements); chez la progéniture, une incidence accrue de malformations et de variations des vertèbres et des côtes a été observée. Aucun effet sur la mère ou sur le développement n'a été observé sous la dose de 75 mg/kg/jour (moins que l'AUC à la RHD).
Dans l'étude de développement pré et post-natal sur des rates gestantes, le letermovir a été administré par voie orale aux doses de 0, 10, 45 ou 180 mg/kg/jour (soit jusqu'à 2 fois l'AUC à la RHD) et aucune toxicité développementale n'a été observée.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Conserver hors de portée des enfants.
Ne pas conserver au-dessus de 30 °C. Conserver les comprimés dans leur emballage d'origine (blister) pour les protéger de l'humidité. Les flacons de Prevymis solution à diluer pour perfusion sont à conserver dans leur emballage d'origine et à l'abri de la lumière.
Préparation de la solution à perfuser
Prevymis solution à diluer pour perfusion est livré dans des flacons de 30 ml à dose unique qui contiennent soit 240 mg (12 ml par flacon) soit 480 mg (24 ml par flacon). Les instructions de préparation et d'administration sont identiques pour les deux doses.
Les flacons de Prevymis sont exclusivement à usage unique. Éliminer le médicament non utilisé.
Préparation
·Prevymis doit être dilué avant administration par voie intraveineuse (i.v.).
Examiner le contenu du flacon à la recherche d'une coloration et de particules avant dilution. Prevymis solution à diluer pour perfusion est une solution limpide et incolore. Ne pas utiliser le flacon si la solution présente une coloration ou contient des particules visibles.
·Ne pas secouer le flacon de Prevymis.
·Ajouter le contenu d'un flacon à dose unique de Prevymis solution à diluer pour perfusion dans une poche de perfusion de 250 ml préremplie soit de chlorure de sodium à 0,9% soit de glucose à 5% et mélanger délicatement la solution obtenue. Ne pas secouer.
·Une fois diluée, la solution de Prevymis est limpide et incolore à jaune. Les variations au sein de cette gamme de couleur n'affectent pas la qualité du produit. Lorsque le contenant et la solution le permettent, examiner visuellement la solution diluée avant administration à la recherche de particules et d'une coloration anormale. Jeter le contenu en présence d'une coloration anormale ou de particules visibles.
Conservation de la solution à perfuser
·La solution diluée ne contient pas d'agent conservateur et doit être utilisée immédiatement pour des raisons microbiologiques.
·La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 48 heures à 25 °C et pendant 48 heures entre 2 et 8 °C.
·Cette durée inclut la conservation de la solution diluée dans la poche de perfusion, ainsi que la durée de la perfusion.
Administration
·Administrer uniquement par perfusion intraveineuse. Ne pas administrer en injection rapide ou en bolus.
·Après dilution, administrer Prevymis en perfusion intraveineuse sur une durée totale de 60 minutes environ à l'aide d'un cathéter veineux périphérique ou central. La totalité du contenu de la poche de perfusion doit être administrée.
Solutions intraveineuses compatibles
Prevymis solution à diluer pour perfusion est compatible avec les solutions de chlorure de sodium à 0,9% et de glucose à 5%.
Médicaments compatibles
Une étude a porté sur la compatibilité physique de Prevymis solution à diluer pour perfusion avec d'autres médicaments injectables. La compatibilité a été objectivée par observation visuelle, turbidité et mesure particulaire. Les médicaments compatibles sont listés ci-dessous.
Prevymis ne doit pas être administré simultanément par la même ligne intraveineuse (ou cathéter) avec d'autres médicaments ou association de solvants, à l'exception de ceux listés ci-dessous.
Les médicaments compatibles† indiqués ci-dessous peuvent être administrés par injection avec Prevymis uniquement lorsqu'ils sont préparés dans du chlorure de sodium à 0,9% et injectés par le port en Y et selon les instructions approuvées dans leur information professionnelle.
Globuline anti-thymocyte, caspofungine, daptomycine, citrate de fentanyl, fluconazole, furosémide, insuline humaine, sulfate de magnésium, méthotrexate, micafungine.
† Ces préparations pour injection sont disponibles en Suisse.
Les médicaments compatibles† indiqués ci-dessous peuvent être administrés par injection avec Prevymis uniquement lorsqu'ils sont préparés dans du glucose à 5% et injectés par le port en Y et selon les instructions approuvées dans leur information professionnelle.
Amphotéricine B (complexe lipidique)*, anidulafungine, bitartrate de noradrénaline, céfazoline sodique, ceftaroline, ceftriaxone sodique, chlorure de potassium, ganciclovir sodique, sulfate de morphine, pantoprazole sodique, phosphate de potassium, télavancine, tigécycline, tacrolimus.
† Ces préparations pour injection sont disponibles en Suisse.
* L'amphotéricine B (complexe lipidique) est compatible avec Prevymis. Cependant, l'amphotéricine B (liposomale) est incompatible (voir section «Remarques particulières»).
Poches intraveineuses et matériaux des sets de perfusion compatibles
Prevymis est compatible avec les poches intraveineuses et les matériaux des sets de perfusion suivants. Les poches intraveineuses et matériaux des sets de perfusion non listés ci-dessous ne doivent pas être utilisés.
Matériaux des poches intraveineuses
Chlorure de polyvinyle (PVC), éthylène-acétate de vinyle (EVA) et polyoléfine (polypropylène et polyéthylène)
Matériaux des sets de perfusion
PVC, polyéthylène (PE), polybutadiène (PBD), caoutchouc en silicone (SR), copolymère styrènebutadiène (SBC), copolymère styrène-butadiène-styrène (SBS), polystyrène (PS)
Plastifiants
Phtalate de diéthylhexyle (DEHP), tris (2-etylhexyl)-trimellitate (TOTM), phtalate de butyle benzyle (BBP)
Cathéters
Polyuréthane radio-opaque
Médicaments incompatibles
Prevymis solution à diluer pour perfusion est physiquement incompatible avec les médicaments suivants: chlorhydrate d'amiodarone, amphotéricine B (liposomale), aztréonam, chlorhydrate de céfépime, ciprofloxacine, ciclosporine, chlorhydrate de diltiazem, filgrastim, sulfate de gentamicine, lévofloxacine, linézolide, lorazépam, chlorhydrate de midazolam, chlorhydrate de mycophénolate mofétil, ondansétron, palonosétron.
Poches intraveineuses et matériaux des sets de perfusion incompatibles
Prevymis est incompatible avec les tubulures des sets de perfusion contenant du polyuréthane.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation nationale en vigueur.
Numéro d’autorisation66652, 66653 (Swissmedic).
PrésentationPrevymis 240 mg comprimé pelliculé: 28 comprimés pelliculés sous plaquettes d'aluminium non perforées. (A)
Prevymis 480 mg comprimé pelliculé: 28 comprimés pelliculés sous plaquettes d'aluminium non perforées. (A)
Prevymis 12 ml (240 mg) solution à diluer pour perfusion: chaque boîte contient 1 flacon. (A)
Prevymis 24 ml (480 mg) solution à diluer pour perfusion: chaque boîte contient 1 flacon. (A)
Titulaire de l’autorisationMSD MERCK SHARP & DOHME SA, Lucerne.
Mise à jour de l’informationJanvier 2018.
CCDS-MK8228-MF-032017/ MK8228-CHE-2018-017039
|