CompositionPrincipe actif
Fibrinogène humain.
Fabriqué à partir de plasma de donneurs humains.
Excipients
Poudre:
Chlorhydrate de L-arginine.
Glycine.
Chlorure de sodium.
Citrate de sodium dihydraté.
Correspond à 132 mg de sodium par flacon.
Solvant:
Eau pour solutions injectables 50 ml.
Indications/Possibilités d’emploiTraitement et prophylaxie péri-opératoire d'épisodes hémorragiques chez les patients présentant une hypo- ou afibrinogénémie congénitale avec une tendance hémorragique.
Comme thérapie complémentaire dans le traitement d'une hémorragie sévère incontrôlée lors d'un déficit acquis en fibrinogène.
Posologie/Mode d’emploiLe traitement devra être instauré sous la surveillance d'un médecin expérimenté dans le traitement des troubles de la coagulation.
Posologie
La dose et la durée de la thérapie de substitution dépendent de la sévérité de la maladie, de la localisation et de l'ampleur de l'hémorragie ainsi que de l'état clinique du patient.
Le dosage individuel est calculé en se basant sur le taux de fibrinogène (fonctionnel). La posologie et la fréquence d'administration doivent être déterminées individuellement pour chaque patient en fonction du dosage régulier du taux de fibrinogène plasmatique et de la surveillance continue de l'état clinique du patient, ainsi que de la surveillance des autres thérapies simultanées de substitution utilisées.
En cas d'intervention chirurgicale majeure, un suivi précis de la thérapie de substitution par les tests de coagulation est essentiel.
a) Prophylaxie chez les patients présentant une hypo- ou afibrinogénémie congénitale avec une tendance hémorragique connue
Pour prévenir des hémorragies excessives au cours d'interventions chirurgicales, le traitement prophylactique est recommandé pour augmenter le taux de fibrinogène jusqu'à 1 g/l et le maintenir à ce niveau jusqu'à stabilisation de l'hémostase, puis un taux au-dessus de 0,5 g/l est à maintenir jusqu'à guérison complète des plaies.
En cas d'intervention chirurgicale ou de traitement d'un épisode hémorragique, la formule de calcul pour la dose initiale est la suivante:

Le calcul avec l'IVR donné (0,018 g/l par mg/kg de poids corporel) s'applique aux patients âgés entre 12 et 65 ans.
La posologie ultérieure (dose et fréquence des injections) devra être adaptée à l'état clinique et aux résultats des tests de laboratoire du patient.
La demi-vie biologique du fibrinogène est de 3-4 jours. Ainsi, en l'absence de consommation, un traitement répété avec le fibrinogène humain n'est habituellement pas nécessaire. A cause de l'accumulation survenant en cas d'administration répétée lors de prophylaxie, la dose et la fréquence doivent être déterminées en fonction des objectifs thérapeutiques du médecin pour un patient donné.
Patients âgés
Les études cliniques menées avec Fibryga n'ont pas inclus de patients âgés de 65 ans et plus, ne permettant ainsi pas de fournir des données concluantes si ces patients répondent différemment au traitement que des patients plus jeunes.
Enfants et adolescents
En cas d'intervention chirurgicale ou de traitement d'un épisode hémorragique, la dose pour les adolescents devrait être calculée selon la formule pour adultes décrite ci-dessus, tandis que la dose pour enfants doit être calculée comme suit pour les enfants ≥1 à < 12 ans:

La dose suivante doit être déterminée en fonction de l'état clinique du patient et des résultats de laboratoire.
b) Traitement des hémorragies
Hémorragies chez des patients présentant une hypo- ou afibrinogénémie congénitale
Les épisodes hémorragiques devraient être traités selon les formules pour adultes/adolescents ou enfants ci-dessus afin d'augmenter le taux cible recommandé de fibrinogène plasmatique de 1 g/l. Ce taux doit être maintenu jusqu'à ce que l'hémostase soit sous contrôle.
Hémorragies chez des patients présentant une hypofibrinogénémie acquise
Adultes
D'une manière générale, 1-2 g sont administrés initialement avec des perfusions ultérieures selon besoin. En cas d'hémorragies graves, par ex. en cas d'interventions chirurgicales majeures, des quantités plus importantes (4-8 g) de fibrinogène peuvent être nécessaires.
Enfants et adolescents
La posologie devrait être déterminée en fonction du poids corporel et de la situation clinique et s'élève normalement à 20-30 mg/kg.
Mode d'administration
Perfusion ou injection intraveineuse.
Fibryga doit être administré lentement par voie intraveineuse. La vitesse maximum recommandée est de 5 ml par minute chez les patients présentant une hypo- ou afibrinogénémie congénitale et de 10 ml par minute chez les patients présentant un déficit acquis en fibrinogène.
Pour des instructions concernant la reconstitution du médicament avant administration, voir la rubrique «Remarques particulières».
Contre-indicationsFibryga ne doit pas être utilisé en cas d'hypersensibilité au principe actif ou à l'un des excipients conformément à la composition.
Mises en garde et précautionsThromboembolie
Il existe un risque de thrombose, chez les patients traités pour un déficit congénital ou acquis, avec du fibrinogène humain tout particulièrement à forte dose ou à doses répétées. Les patients traités par du fibrinogène humain devront être surveillés étroitement pour détecter les signes ou symptômes de thrombose.
Le bénéfice potentiel d'un traitement par du fibrinogène à base de plasma humain doit être soigneusement évalué par rapport au risque thromboembolique chez les patients avec antécédents de maladie coronarienne ou infarctus du myocarde, les patients avec une maladie hépatique, les patients en période péri- ou post-opératoire, chez les nouveau-nés ou chez les patients à risque d'événements thromboemboliques ou de coagulation intravasculaire disséminée. La prudence et une surveillance étroite sont impératives.
Réactions allergiques ou anaphylactiques
L'injection/perfusion doit être interrompue immédiatement en cas de survenue de réaction allergique ou anaphylactique. En cas de choc anaphylactique, appliquer la procédure médicale standard.
Teneur en sodium
Fibryga contient jusqu'à 132 mg de sodium par flacon, ce qui équivaut à 6,6% de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte. Cela est à prendre en compte chez les patients suivant un régime pauvre en sodium.
Sécurité vis-à-vis des virus
Les mesures standard de prévention des infections liées à l'utilisation de médicaments fabriqués à partir de sang ou de plasma humain comprennent la sélection des donneurs, le dépistage des dons individuels et des pools de plasma pour la présence de marqueurs spécifiques d'infection et l'inclusion d'étapes d'inactivation/élimination efficace des virus dans le procédé de fabrication. Malgré cela, lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés, la possibilité de transmission d'agents infectieux ne peut être totalement exclue. Cela concerne également des virus inconnus ou émergents et d'autres agents pathogènes.
Les mesures prises sont jugées efficaces contre les virus enveloppés, tels que le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC), et contre le virus non-enveloppé de l'hépatite A (VHA). Les mesures prises peuvent être d'efficacité limitée vis-à-vis des virus non-enveloppés tels que le parvovirus B19. L'infection par le parvovirus-B19 peut avoir des conséquences sévères chez la femme enceinte (infection du fœtus) et chez les personnes présentant une immunodéficience ou une érythropoïèse augmentée (anémie hémolytique, p. ex.).
Chaque fois que Fibryga est administré à un patient, il est recommandé d'enregistrer le nom et le numéro de lot du produit afin de conserver une traçabilité entre le patient et le lot du produit.
Une vaccination appropriée (hépatites A et B) des patients recevant régulièrement/de façon répétée des produits dérivés du plasma humain est recommandée.
Immunogénicité
Des réactions d'anticorps ont été observées dans le cas de thérapies de substitution avec des facteurs de coagulation dans d'autres déficits congénitaux. Aucune donnée n'est disponible à ce jour avec les concentrés de fibrinogène.
InteractionsAucune interaction entre les produits à base de fibrinogène humain et d'autres médicaments n'est connue.
Grossesse, allaitementGrossesse
Aucune expérimentation animale n'a été réalisée avec Fibryga. Dans la mesure où le principe actif est d'origine humaine, il est catabolisé de la même manière que les propres protéines des patients. Ces composants physiologiques du sang humain ne devraient pas induire d'effets néfastes sur la reproduction ou sur le fœtus.
L'innocuité de fibrinogène humain chez les femmes enceintes n'a pas été évaluée au cours d'études cliniques contrôlées.
Néanmoins l'expérience clinique dans le traitement des complications obstétricales avec des produits à base de fibrinogène suggère qu'aucun effet néfaste n'est à attendre soit sur l'évolution de la grossesse, soit sur le développement du fœtus ou du nouveau-né.
Allaitement
On ignore si Fibryga est excrété dans le lait humain. L'innocuité du fibrinogène humain chez les femmes qui allaitent n'a pas été évaluée par des essais cliniques contrôlés jusqu'à présent.
Fertilité
Aucune donnée relative à la fertilité n'est disponible.
Effet sur l’aptitude à la conduite et l’utilisation de machinesFibryga n'a aucune influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Il n'existe pas de données solides sur la fréquence des effets indésirables liés à ce médicament constatés au cours d'études cliniques.
Les effets indésirables suivants ont été décrits au cours d'études cliniques: pyrexie, exanthème médicamenteux, phlébite et thrombose.
Les effets indésirables suivants ont été rapportés pour Fibryga et d'autres concentrés de fibrinogène:
|
Classe de système d'organe selon MedDRA
|
Effets indésirables
|
Fréquence
| |
Affections du système immunitaire
|
Réactions allergiques ou anaphylactiques
Réactions cutanées
|
Cas isolés
| |
Affections vasculaires
|
Episodes thromboemboliques (dont infarctus du myocarde et embolie pulmonaire)
Thrombophlébite
|
Cas isolés
| |
Troubles généraux et anomalies au site d'administration
|
Elévation de la température corporelle (Pyrexie)
|
Cas isolés
|
Pour la sécurité vis-à-vis des agents transmissibles, voir rubrique «Mises en garde et précautions».
Enfants et adolescents
26 patients âgés de 1 à moins de 18 ans ont été inclus dans l'analyse de sécurité pour le déficit congénital en fibrinogène: 12 adolescents âgés de 12 à moins de 18 ans, 8 enfants âgés de 6 à moins de 12 ans et 6 enfants âgés de 1 à moins de 6 ans.
Le profil de sécurité général est identique chez les adultes, les adolescents et les enfants.
On ne dispose pas de données sur l'utilisation de Fibryga chez les patients pédiatriques présentant un déficit acquis en fibrinogène.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté.
Une surveillance régulière du taux de fibrinogène plasmatique est indiquée pour éviter tout surdosage (voir rubrique «Posologie/Mode d'emploi»).
En cas de surdosage, le risque de complications thromboemboliques est accru.
Propriétés/EffetsCode ATC
B02BB01
Classe pharmacothérapeutique: antihémorragique, fibrinogène humain
Mécanisme d'action
L'administration de fibrinogène humain augmente le taux de fibrinogène plasmatique et peut corriger temporairement le défaut de coagulation chez les patients porteurs d'un déficit en fibrinogène.
Pharmacodynamique
Le fibrinogène humain (facteur I de coagulation) se transforme en fibrine en présence de thrombine, du facteur XIII activé (FXIIIa) et d'ions calcium, et forme un réseau tridimensionnel de fibrine stable et élastique.
Efficacité clinique
Une étude pharmacocinétique ouverte, prospective, randomisée et contrôlée à deux groupes croisés de phase II, d'une dose unique chez 22 patients présentant un déficit congénital en fibrinogène (afibrinogénémie) (voir rubrique «Pharmacocinétique») a également évalué la fermeté maximale du caillot (Maximum Clot Firmness, MCF) comme marqueur substitutif de l'efficacité hémostatique (FORMA-01). La valeur MCF a été déterminée par thromboélastométrie (ROTEM). Pour chaque patient, la valeur MCF a été déterminée avant (référence) et une heure après l'administration d'une dose unique de Fibryga. Les valeurs MCF étaient sensiblement plus élevées après administration de Fibryga par rapport à la mesure initiale (voir tableau ci-dessous).
Tableau 1: Fermeté maximale du caillot (MCF, en mm) (population en ITT) n = 22
|
Point temporel
|
Valeur moyenne ± ÉT
|
Médiane (écart)
| |
Avant la perfusion
|
0 ± 0
|
0 (0-0)
| |
1 heure après la perfusion
|
9,7 ± 3,0
|
10,0 (4,0-16,0)
| |
Variation moyenne (analyse primaire)*
|
9,7 ± 3,0
|
10,0 (4,0-16,0)
|
MCF = fermeté maximale du caillot (Maximum Clot Firmness); ITT = Intention-to-treat.
* p <0,0001 (95%intervalle de confiance 8,37; 10,99)
Une étude ouverte, multicentrique, prospective non contrôlée de phase III (FORMA-02) a été menée chez 25 patients présentant un déficit congénital en fibrinogène (afibrinogénémie et hypofibrinogénémie) âgés de 12 à 54 ans (6 adolescents, 19 adultes). Elle a inclus le traitement de 89 épisodes hémorragiques et 12 interventions chirurgicales. Il y a eu un changement significatif de la valeur MCF mesurée par ROTEM et du taux plasmatique de fibrinogène par rapport aux valeurs initiales. La dose médiane de Fibryga par perfusion pour le traitement des épisodes hémorragiques était de 57,5 mg/kg et la médiane de la dose totale était de 59,4 mg/kg. La dose totale médiane de Fibryga par opération était de 85,8 mg/kg. L'efficacité hémostatique générale a été jugée un succès (évaluation de l'efficacité bonne ou excellente) pour 98,9 % des épisodes hémorragiques traités et 100 % des opérations par un comité d'attribution indépendant utilisant un système d'évaluation objectif.
Une autre étude ouverte, multicentrique, prospective, non contrôlée de phase 3 (FORMA-04) a été menée chez 14 enfants présentant un déficit congénital en fibrinogène (afibrinogénémie et hypofibrinogénémie), âgés de 1 à 10 ans (6 enfants de moins de 6 ans et 8 enfants de 6 à moins de 12 ans). Elle a inclus le traitement de 10 épisodes hémorragiques et 3 interventions chirurgicales ainsi que l'étude de la pharmacocinétique après une dose unique. Il y a eu un changement significatif de la valeur MCF mesurée par ROTEM et du taux plasmatique de fibrinogène par rapport aux valeurs initiales. La dose médiane de Fibryga par perfusion pour le traitement des épisodes hémorragiques était de 70,2 mg/kg et la médiane de la dose totale était de 73,9 mg/kg. La dose totale médiane de Fibryga par opération était de 108 mg/kg. L'efficacité hémostatique globale a été jugée un succès (évaluation de l'efficacité bonne ou excellente) pour 100 % des épisodes hémorragiques traités et des opérations par un comité d'attribution indépendant utilisant un système d'évaluation objectif.
L'étude FORMA-05 est une étude prospective, randomisée, contrôlée qui étudie l'efficacité hémostatique et la sécurité de Fibryga par rapport à un cryoprécipité chez des patients présentant un déficit acquis en fibrinogène pendant une chirurgie cytoréductive pour la maladie tumorale abdominale pseudomyxome péritonéal. 23 patients ont été inclus dans l'analyse intérimaire per protocole (PP), dont 10 ont été traités avec Fibryga et 13 par cryoprécipité. L'administration de fibrinogène durant l'opération consistait en 4 g de Fibryga ou 2 pools de respectivement 5 unités de cryoprécipité, répétés au besoin. Durant l'opération (7,7 ± 1,4 heures) 6,4 ± 2,8 g de Fibryga (85 ± 36 mg/kg de poids corporel), respectivement 3,9 ± 1,7 pools de 5 unités de cryoprécipité ont été administrés. Dans les deux groupes, une seule unité de concentré érythrocytaire a été administrée en moyenne pendant l'opération et 0 unité de concentré érythrocytaire durant les 24 heures suivant l'opération, sans autre transfusion de plasma frais congelé ou de concentrés plaquettaires (voir tableau 2). Tous les traitements à base de fibrinogène, analysés pour les 23 opérations, ont été classifiés comme des succès par un comité d'attribution indépendant utilisant un système d'évaluation objectif (évaluation de l'efficacité bonne ou excellente).
Tableau 2: Transfusion de concentrés érythrocytaires* [unités] pendant l'opération et durant les premières 24 heures suivant l'opération (analyse PP)
|
Période
|
Groupe Fibryga (n=10)
Médiane (écart)
|
Groupe cryoprécipité (n=13)
Médiane ( écart)
| |
Pendant l'opération
|
1 (0-2)
|
1 (0-5)
| |
Premières 24 heures après l'opération
|
0 (0-2)
|
0 (0-1)
|
PP = per protocole.
* aucune transfusion de plasma frais congelé (PFC) et aucun concentré plaquettaire administré.
Sécurité et efficacité en pédiatrie
Pour le traitement du déficit congénital en fibrinogène, Fibryga a été administré à 20 patients âgés de 1 à moins de 18 ans (6 adolescents de 12 à moins de 18 ans, 8 enfants de 6 à moins de 12 ans et 6 enfants de 1 à moins de 6 ans) dans deux études cliniques (FORMA-02 et FORMA-04). L'efficacité hémostatique a été jugée un succès pour tous les épisodes hémorragiques traités (10 épisodes hémorragiques chez les adolescents, 5 chez les enfants âgés de 6 à moins de 12 ans et 5 chez les enfants âgés de 1 à moins de 6 ans) par un comité d'attribution indépendant. La prophylaxie a également été jugée un succès pour les 4 opérations effectuées chez ces patients (1 chez les adolescents et 3 chez les enfants de 1 à moins de 6 ans).
PharmacocinétiqueLe fibrinogène humain est un constituant normal du plasma humain et se comporte comme le fibrinogène endogène. La demi-vie biologique du fibrinogène est de 3-4 jours dans le plasma. Fibryga est administré par voie intraveineuse et est immédiatement disponible à une concentration plasmatique correspondant à la dose administrée.
Une étude croisée ouverte, prospective, randomisée contrôlée de phase II à deux bras menée chez 22 patients présentant un déficit congénital en fibrinogène (afibrinogénémie) et âgés de 12 à 53 ans (6 adolescents, 16 adultes), a comparé chez les mêmes patients les propriétés pharmacodynamiques d'une dose unique de Fibryga à celles d'un autre concentré de fibrinogène commercialisé (FORMA-01). Chaque patient a reçu une dose intraveineuse unique de 70 mg/kg de Fibryga et du produit comparatif. Pour déterminer l'activité du fibrinogène des échantillons de sang ont été prélevés avant la perfusion et jusqu'à 14 jours après la perfusion. Les paramètres pharmacocinétiques de Fibryga dans l'analyse per protocole (PP) (n=21) sont résumés dans le tableau ci-dessous.
Tableau 3: Paramètres pharmacocinétiques (n = 21) de l'activité du fibrinogène (population PP*)
|
Paramètre
|
Valeur moyenne ± ÉT
|
Écart
| |
Demi-vie (h)
|
75,9 ± 23,8
|
40,0-157,0
| |
Cmax (mg/dl)
|
139,0 ± 36,9
|
83,0-216,0
| |
AUCnorm pour dose de 70 mg/kg (mg*h/ml)
|
113,7 ± 31,5
|
59,7-175,5
| |
Clairance (ml/h/kg)
|
0,67 ± 0,2
|
0,4-1,2
| |
Durée moyenne de persistance (h)
|
106,3 ± 30,9
|
58,7-205,5
| |
Volume de répartition à la phase de plateau (steady state) (ml/kg)
|
70,2 ± 29,9
|
36,9-149,1
|
* Un patient a été exclu de la population PP, ayant reçu <90% de la dose prévue de Fibryga et du produit comparatif.
Cmax = concentration plasmatique maximale; AUCnorm = aire normalisée sous la courbe pour la dose administrée; ÉT = écart-type
Par ailleurs, la récupération incrémentielle in vivo (In Vivo Recovery [IVR]) a été déterminée à partir des taux obtenus jusqu'à 4 heures après la perfusion. L'IVR incrémentielle médiane était une augmentation de 1,8 mg/dl (écart: 1,08-2,62 mg/dl) par mg/kg. L'IVR médiane indique qu'une dose de 70 mg/kg augmente la concentration plasmatique du fibrinogène du patient d'environ 125 mg/dl.
Cinétique pour certains groupes de patients
Aucune différence statistiquement significative de l'activité du fibrinogène n'a été observée entre les participants masculins et féminins à l'étude. Dans l'analyse PP, une petite différence a été constatée dans la demi-vie chez les patients de moins de 18 ans (n = 5), soit 72,8 ± 16.5 heures comparées à 76,9 ± 26,1 heures dans le groupe adulte (n = 16).
Absorption
Non pertinent.
Distribution
Volume de répartition à la phase de plateau (steady state) (ml/kg): cf. Tableau 3. Le volume de répartition s'élevait à 70,2 ± 29,9 ml/kg.
Métabolisme
Non connu.
Elimination
Clairance (ml/h/kg): Cf. Tableau 3. La clairance était presque identique dans les 2 groupes d'âge et s'élevait à 0,68 ± 0,18 ml/h/kg respectivement 0,66 ± 0,21 ml/h/kg.
Enfants et adolescents
Les propriétés pharmacocinétiques de Fibryga ont été étudiées plus en détail dans l'étude FORMA-04 menée chez 13 enfants âgés de 1 à 10 ans présentant un déficit congénital en fibrinogène (afibrinogénémie). Chaque patient a reçu une dose intraveineuse unique de 70 mg/kg de Fibryga. Les paramètres pharmacocinétiques de Fibryga sont résumés dans le tableau ci-dessous. L'IVR incrémentielle médiane était une augmentation de 1,4 mg/dl (écart: 1,3-2,1 mg/dl) par mg/kg.
Tableau 4: Paramètres pharmacocinétiques (n = 13) de l'activité du fibrinogène
|
Paramètre
|
Valeur moyenne ± ÉT
|
Écart
| |
Demi-vie (h)*
|
63,3 ± 12,0
|
45,6-91,6
| |
Cmax (mg/dl)
|
107,2 ± 16,8
|
93,0-154,0
| |
AUCnorm pour dose de 70 mg/kg (mg*h/ml)*
|
92,0 ± 20,0
|
69,7-134,2
| |
Clairance (ml/h/kg)*
|
0,8 ± 0,2
|
0,5-1,0
| |
Durée moyenne de persistance (h)*
|
88,0 ± 16,8
|
63,6-126,7
| |
Volume de répartition à la phase de plateau (steady state) (ml/kg)*
|
67,6 ± 7,1
|
52,8-76,8
|
*Calculé pour 10 patients sur 13 en raison du nombre insuffisant de valeurs mesurables chez 3 patients.
IVR = récupération in vivo (In Vivo Recovery); Cmax = concentration plasmatique maximale; AUCnorm = aire sous la courbe normalisée pour la dose administrée; ET = écart-type
Données précliniquesPharmacologie de sécurité
En se basant sur les études de pharmacologie de sécurité, les données pré-cliniques ne révèlent aucun danger particulier pour l'homme.
Toxicité à long terme (ou toxicité en cas d'administration répétée)
Des études de toxicité avec une administration répétée n'ont pas été réalisées. Des études de toxicité avec une administration répétée chez l'animal ne sont pas réalisables en raison de l'interférence prévue avec induction d'anticorps contre la protéine humaine.
Mutagénicité
Des études de mutagénicité n'ont pas été réalisées. Étant donné qu'il s'agit d'une protéine humaine extraite du plasma, utilisée en prophylaxie péri-opératoire et en thérapie de substitution, aucun effet mutagénique n'est escompté.
Carcinogénicité
Des études de cancérogénicité n'ont pas été réalisées. Étant donné qu'il s'agit d'une protéine humaine extraite du plasma, utilisée en prophylaxie péri-opératoire et en thérapie de substitution, aucun effet cancérogène n'est escompté.
Toxicité sur la reproduction
Des études de toxicité sur la reproduction n'ont pas été réalisées. Pour l'indication clinique et l'emploi prévus du Fibryga, les effets toxicologiques sur la reproduction sont à exclure.
Autres données (toxicité locale, phototoxicité, immunotoxicité)
La tolérance locale intraveineuse, intra-artérielle et para-veineuse a été testée sur les lapins. L'examen macroscopique des sites d'application après administration intra-artérielle a indiqué une légère formation d'une croûte et rougeur. L'examen histo-pathologique n'a donné aucun signe de lésions morphologiques. Fibryga a montré une tolérance excellente pour une administration intraveineuse, qui est la voie d'administration prévue pour l'utilisation clinique.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, Fibryga ne doit pas être mélangé avec d'autres médicaments.
Stabilité
Fibryga ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
La stabilité chimique et physique de la solution reconstituée a été démontrée pendant 24 heures à température ambiante (max. 25 °C). D'un point de vue microbiologique, l'utilisation immédiate après reconstitution est recommandée. Si la solution reconstituée n'est pas utilisée immédiatement, les délais et conditions de conservation sont de la responsabilité de l'utilisateur. La solution reconstituée ne doit pas être congelée ou conservée au réfrigérateur. Les flacons partiellement utilisés doivent être éliminés.
Remarques concernant le stockage
Tenir hors de portée des enfants.
Ne pas conserver au-dessus de 25 °C. Ne pas congeler. Conserver le flacon dans son carton pour le protéger de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique «Stabilité après ouverture».
Remarques concernant la manipulation
La solution reconstituée doit être presque incolore et légèrement opalescente. Ne pas utiliser les solutions troubles ou présentant des dépôts.
Fibryga est à usage unique. Aucun des composants ne doit être réutilisé.
Pour des raisons de sécurité microbiologique, la solution doit être administrée immédiatement après reconstitution. La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 24 heures à température ambiante (25 °C max.). Après reconstitution, ne pas congeler ou mettre la solution de Fibryga au réfrigérateur.
Reconstitution
1.Veiller à ce que le flacon de poudre (Fibryga) et le flacon de solvant soient à température ambiante. Cette température doit être maintenue pendant la reconstitution. Si un bain-marie est utilisé pour le réchauffement, il faut veiller à éviter que l'eau n'entre en contact avec les bouchons en caoutchouc ou les opercules amovibles des récipients. La température du bain-marie ne doit pas dépasser 37 °C.
2.Retirer les opercules amovibles du flacon de poudre (Fibryga) et du flacon de solvant afin d'exposer la partie centrale du bouchon pour la perfusion. Nettoyer les bouchons en caoutchouc avec un tampon alcoolisé et laisser sécher les bouchons.
3.Ouvrir l'emballage du dispositif de transfert (nextaro) en détachant la pellicule du couvercle (fig. 1). Laisser le dispositif de transfert dans la coque d'emballage transparente afin de maintenir sa stérilité. Ne pas toucher le perforateur.
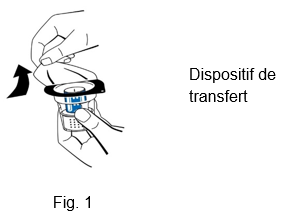
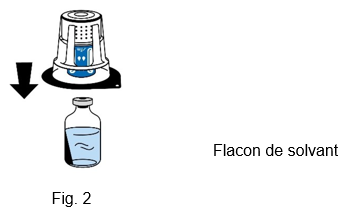
4.Placer le flacon de solvant sur une surface plane et propre et le tenir fermement. Sans retirer la coque d'emballage, placer la partie bleue du dispositif de transfert sur le haut du flacon de solvant. Appuyer fermement, tout droit, jusqu'à ce que le dispositif s'enclenche (fig. 2). Ne pas effectuer de rotation pendant la fixation.
Remarque:
Le dispositif de transfert doit d'abord être fixé sur le flacon de solvant, et ensuite sur le flacon de poudre lyophilisée. Dans le cas contraire, il y a une perte du vide et le solvant ne peut pas être transféré.
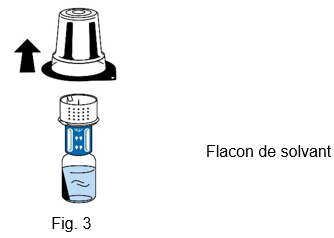
5.Tenir le flacon de solvant et retirer délicatement la coque d'emballage du dispositif de transfert (nextaro) en la tirant tout droit vers le haut. Veiller à ce que le dispositif de transfert reste fermement connecté au flacon de solvant (fig. 3).
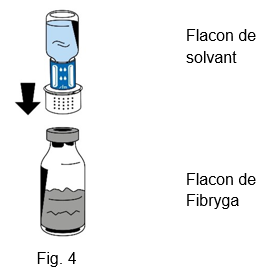
6.Placer le flacon de poudre (Fibryga) sur une surface plane et propre et le tenir fermement. Prendre le flacon de solvant avec le dispositif de transfert fixé dessus et le retourner. Placer la partie blanche du connecteur du dispositif de transfert sur le haut du flacon de poudre (Fibryga) et appuyer fermement jusqu'à ce qu'il s'enclenche (fig. 4). Ne pas tourner pendant la fixation. Le solvant s'écoule automatiquement dans le flacon de poudre (Fibryga).
7.En laissant le flacon de solvant fixé, faire pivoter avec précaution le flacon de Fibryga jusqu'à ce que la poudre soit entièrement dissoute. Ne pas secouer le flacon afin d'éviter la formation de mousse. La poudre devrait être complètement dissoute au bout de 5 minutes environ. La dissolution de la poudre ne doit pas prendre plus de 20 minutes. Si la poudre n'est pas dissoute au bout de 20 minutes, le produit doit être éliminé.
8.Dans les rares cas où pendant le transfert du solvant des amas flottants de poudre non reconstituée continueraient à flotter, ou en cas de prolongation inattendue du temps nécessaire à la reconstitution, il est possible d'accélérer le processus de dissolution au moyen d'une agitation horizontale plus vigoureuse du flacon.
9.Une fois la reconstitution terminée, dévisser le dispositif de transfert (partie bleue) en tournant dans le sens contraire des aiguilles d'une montre de façon à séparer les deux parties (fig. 5). Ne pas toucher le connecteur Luer lock sur la partie blanche du dispositif de transfert.
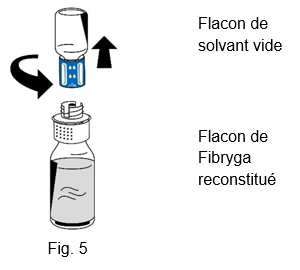
10.Éliminer le flacon de solvant vide avec la partie bleue du dispositif de transfert.
Administration
1.Raccorder délicatement la seringue au connecteur Luer lock de la partie blanche du dispositif de transfert (fig. 6).
2.Retourner le flacon de Fibryga et aspirer la solution dans la seringue (fig. 7).
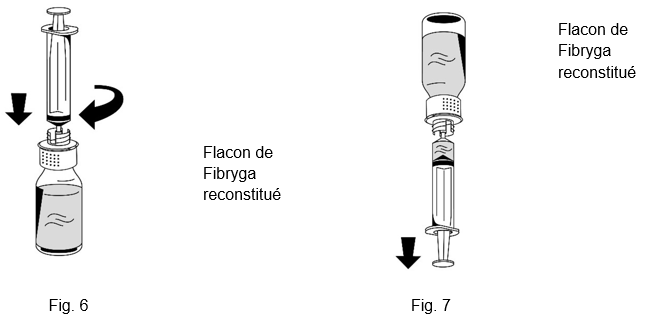
3.Une fois la solution transférée, tenir fermement le cylindre de la seringue (en laissant le piston de la seringue dirigé vers le bas) et détacher la seringue du dispositif de transfert (fig. 8).
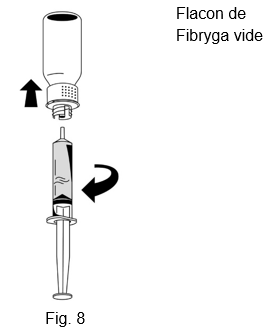
4.Éliminer la partie blanche du dispositif de transfert avec le flacon de Fibryga vide.
Un ensemble standard pour perfusion est recommandé pour l'administration intraveineuse de la solution reconstituée à température ambiante.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation nationale en vigueur.
Numéro d’autorisation66836 (Swissmedic).
Présentation1 emballage de Fibryga (B) avec:
1 flacon pour injection avec poudre
1 flacon pour injection avec solvant (eau pour préparations injectables)
1 dispositif de transfert nextaro
Le flacon de poudre est en verre incolore de type II, scellé avec bouchon en caoutchouc de bromobutyle et un opercule en aluminium.
Le flacon de solvant est en verre incolore de type II, scellé avec bouchon en caoutchouc de halobutyle et un opercule en aluminium.
Titulaire de l’autorisationOctapharma AG, Seidenstrasse 2, CH-8853 Lachen.
Mise à jour de l’informationNovembre 2023.
|