Propriétés/EffetsCode ATC
L04AC18
Le risankizumab, inhibiteur de l'interleukine 23, est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1). Le risankizumab est produit à partir d'une lignée cellulaire de mammifère à l'aide de la technologie de l'ADN recombinant.
Mécanisme d'action
Le risankizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) qui se lie sélectivement avec une grande affinité à la sous-unité p19 de la cytokine humaine interleukine 23 (IL-23) et en inhibe l'interaction avec le complexe récepteur de l'IL-23. L'IL-23 est une cytokine présente naturellement qui participe aux réactions inflammatoires et immunitaires. L'IL-23 favorise la prolifération, le maintien et l'activation des cellules Th17, qui produisent l'IL-17A, l'IL-17F et l'IL-22 de même que d'autres cytokines pro-inflammatoires et jouent un rôle central dans la pathogenèse des maladies auto-immunes inflammatoires telles que le psoriasis. Chez les patients atteints de psoriasis en plaques, on observe une régulation positive de l'expression de l'IL-23 dans les lésions cutanées par rapport aux zones non touchées. Le risankizumab bloquant la liaison de l'IL-23 à son récepteur, il inhibe ainsi la transduction du signal cellulaire IL-23 dépendante et la libération des cytokines pro-inflammatoires.
Le risankizumab ne se lie pas à l'IL-12 humain, qui possède comme l'IL-23 une sous-unité p40.
Pharmacodynamique
Dans une étude menée chez des patients atteints de psoriasis, une diminution de l'expression des gènes associés à l'axe IL-23/IL-17 a été observée dans la peau après une dose unique de risankizumab. En outre, des réductions de l'épaisseur de l'épiderme, de l'infiltration de cellules inflammatoires et de l'expression des marqueurs du psoriasis dans les lésions psoriasiques ont été observées.
Dans une étude menée chez des patients atteints d'arthrite psoriasique et qui ont reçu le risankizumab 150 mg à la semaine 0, à la semaine 4 puis toutes les 12 semaines, le risankizumab a diminué le taux sérique d'IL-17A, d'IL-17F et d'IL-22 jusqu'à la semaine 24 par rapport à l'inclusion et par rapport au placebo.
Efficacité clinique
Psoriasis en plaques
L'efficacité et la sécurité de SKYRIZI ont été évaluées chez 2'109 patients atteints de psoriasis en plaques modéré à sévère dans le cadre de quatre études multicentriques randomisées à double insu, contrôlées par placebo et/ou par comparateur actif (ULTIMMA-1, ULTIMMA-2, IMMHANCE et IMMVENT). Dans les études ULTIMMA-1 et ULTIMMA-2, l'ustékinumab a été utilisé comme comparateur actif. L'étude IMMHANCE a évalué l'arrêt, puis la reprise du traitement par le risankizumab chez les patients ayant obtenu une réponse. Dans l'étude IMMVENT, l'adalimumab a été utilisé comme comparateur actif. À l'issue des études, les patients pouvaient être inclus dans l'étude d'extension en ouvert, LIMMITLESS. Les patients inclus étaient âgés d'au moins 18 ans et étaient atteints de psoriasis en plaques avec une surface cutanée atteinte (body surface area, BSA) ≥10%, un score d'évaluation globale statique par un médecin (static Physician Global Assessment, sPGA) ≥3, un psoriasis de grade de 0 à 4 et un score PASI (Psoriasis Area and Severity Index) ≥12. Les patients atteints de psoriasis érythrodermique, de psoriasis en gouttes ou de psoriasis pustuleux étaient exclus.
Au total, les patients avaient un score PASI initial médian de 17,8 et une surface cutanée atteinte moyenne de 20,0%. Chez 19,3% des patients, le score sPGA initial correspondait à une maladie sévère. Chez 9,8% des patients des études au total, l'anamnèse a révélé une arthrite psoriasique diagnostiquée.
Sur l'ensemble des études, 30,9% des patients n'avaient reçu auparavant aucun traitement systémique biologique ou non biologique, 38,1% avaient reçu une photothérapie, 48,3% un traitement systémique non biologique et 42,1% un traitement biologique (23,7% de tous les patients dans ces études ont reçu au moins un inhibiteur du TNF-alpha) pour leur psoriasis. Afin d'éviter tout biais dans l'évaluation de l'efficacité de SKYRIZI dans le traitement du psoriasis, l'utilisation concomitante d'un médicament antipsoriasique systémique ou topique (à l'exception des corticostéroïdes topiques sur le visage, les aisselles et/ou les organes génitaux) ou d'une photothérapie était proscrite pendant les études.
ULTIMMA-1 et ULTIMMA-2
Les études ULTIMMA-1 et ULTIMMA-2 ont inclus 997 patients (598 patients dans le groupe SKYRIZI 150 mg, 199 patients dans les groupes ustékinumab 45 mg ou 90 mg [selon le poids corporel initial] et 200 patients dans le groupe placebo). Le traitement a été administré les semaines 0 et 4, puis toutes les 12 semaines par la suite. Les résultats sont présentés dans le tableau 2 et la figure 1.
Tableau 2: Résultats obtenus chez les adultes atteints de psoriasis en plaques dans les études d'efficacité ULTIMMA 1 et ULTIMMA 2
|
|
ULTIMMA-1
|
ULTIMMA-2
| |
|
SKYRIZI
(N=304)
n (%)
|
Ustékinumab
(N=100)
n (%)
|
Placebo
(N=102)
n (%)
|
SKYRIZI
(N=294)
n (%)
|
Ustékinumab
(N=99)
n (%)
|
Placebo
(N=98)
n (%)
| |
Score sPGA «sans» ou «minime» (0 ou 1)
| |
Semaine 16
|
267 (87,8)a
|
63 (63,0)
|
8 (7,8)
|
246 (83,7)a
|
61 (61,6)
|
5 (5,1)
| |
Semaine 52
|
262 (86,2)
|
54 (54,0)
|
--
|
245 (83,3)
|
54 (54,5)
|
--
| |
Score sPGA «sans» (0)
| |
Semaine 16
|
112 (36,8)
|
14 (14,0)
|
2 (2,0)
|
150 (51,0)
|
25 (25,3)
|
3 (3,1)
| |
Semaine 52
|
175 (57,6)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
PASI 75
| |
Semaine 12
|
264 (86,8)
|
70 (70,0)
|
10 (9,8)
|
261 (88,8)
|
69 (69,7)
|
8 (8,2)
| |
Semaine 52
|
279 (91,8)
|
70 (70,0)
|
--
|
269 (91.5)
|
76 (76,8)
|
--
| |
PASI 90
| |
Semaine 16
|
229 (75,3)a
|
42 (42,0)
|
5 (4,9)
|
220 (74,8)a
|
47 (47,5)
|
2 (2,0)
| |
Semaine 52
|
249 (81,9)
|
44 (44,0)
|
--
|
237 (80,6)
|
50 (50,5)
|
--
| |
PASI 100
| |
Semaine 16
|
109 (35,9)
|
12 (12,0)
|
0 (0,0)
|
149 (50,7)
|
24 (24,2)
|
2 (2,0)
| |
Semaine 52
|
171 (56,3)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
Dans toutes les comparaisons de SKYRIZI à l'ustékinumab et au placebo, une valeur p < 0,001 a été atteinte, sauf pour le score PASI 75 la semaine 52 dans l'étude ULTIMMA-2 (p = 0,001).
a Co-critères d'évaluation principaux par rapport au placebo
|
Figure 1: Évolution du pourcentage de variation moyenne par rapport à l'inclusion du score PASI dans les études ULTIMMA-1 et ULTIMMA-2
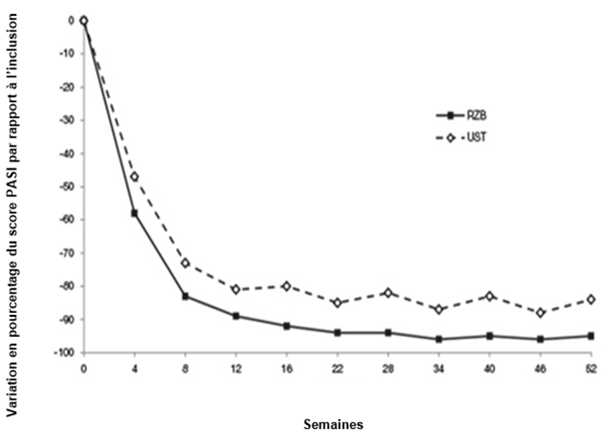
RZB = risankizumab
UST = ustékinumab
p < 0,001 à chaque point dans le temps
Une analyse en fonction de l'âge, du sexe, de l'origine ethnique, du poids corporel, du score PASI initial, de la présence concomitante d'arthrite psoriasique, des antécédents de traitement systémique non biologique, des antécédents de traitement biologique et de l'absence de réponse antérieure aux médicaments biologiques n'a montré aucune différence entre ces sous-groupes pour ce qui est de la réponse à SKYRIZI.
Chez les patients traités par SKYRIZI, une amélioration du psoriasis au niveau du cuir chevelu, des ongles, de la paume des mains et de la plante des pieds a été observée les semaines 16 et 52.
IMMHANCE
L'étude IMMHANCE a inclus 507 patients (407 patients dans le groupe SKYRIZI 150 mg et 100 patients dans le groupe placebo). Le traitement a été administré les semaines 0 et 4, puis toutes les 12 semaines par la suite.
La semaine 16, SKYRIZI s'est avéré supérieur au placebo sur les co-critères d'évaluation principaux, à savoir score sPGA «sans» ou «minime» (83,5% SKYRIZI vs. 7,0% placebo) et PASI 90 (73,2% SKYRIZI vs. 2,0% placebo). La semaine 16, un plus grand nombre de patients sous SKYRIZI n'avaient plus de lésions cutanées (score sPGA 0 [46,4% SKYRIZI vs. 1,0% placebo] ou score PASI 100 [47,2% SKYRIZI vs. 1,0% placebo]). En outre, les patients sous SKYRIZI avaient une plus grande probabilité d'obtenir un score PASI 75 que ceux sous placebo (88,7% SKYRIZI vs. 8,0% placebo).
Aucun des 31 patients de l'étude IMMHANCE atteints d'une tuberculose (TB) latente et qui n'avaient reçu aucune prophylaxie pendant l'étude n'a présenté une TB active pendant la période de suivi moyenne sous SKYRIZI de 55 semaines. Toutefois, les patients présentant une TB latente doivent recevoir un traitement antituberculeux avant l'instauration du traitement par SKYRIZI (voir «Mises en garde et précautions»).
IMMVENT
L'étude IMMVENT a inclus 605 patients (301 patients dans le groupe SKYRIZI et 304 patients dans le groupe adalimumab). Les patients randomisés dans le groupe SKYRIZI ont reçu une dose de 150 mg les semaines 0 et 4, puis toutes les 12 semaines par la suite. Les patients randomisés dans le groupe adalimumab ont reçu 80 mg la semaine 0, 40 mg la semaine 1, puis 40 mg toutes les deux semaines jusqu'à la semaine 15. A partir de la semaine 16, en fonction de leur réponse, les patients traités par adalimumab ont poursuivi le traitement ou ont changé de traitement:
·< PASI 50: changement par SKYRIZI,
·PASI 50 à < PASI 90: nouvelle randomisation pour continuer le traitement avec 40 mg d'adalimumab toutes les 2 semaines ou changement par SKYRIZI,
·PASI 90: poursuite du traitement avec 40 mg d'adalimumab toutes les 2 semaines.
Dans l'étude IMMVENT, les patients sous SKYRIZI avaient obtenu à la semaine 16 des résultats semblables à ceux rapportés dans les autres études cliniques (tableau 3 et figure 2).
Tableau 3: Résultats obtenus à la semaine 16 chez les adultes atteints de psoriasis en plaques dans l'étude d'efficacité IMMVENT
|
|
SKYRIZI
(N=301)
n (%)
|
Adalimumab
(N=304)
n (%)
| |
Score sPGA «sans» ou «minime»a
|
252 (83,7)
|
183 (60,2)
| |
PASI 75
|
273 (90,7)
|
218 (71,7)
| |
PASI 90a
|
218 (72,4)
|
144 (47,4)
| |
PASI 100
|
120 (39,9)
|
70 (23,0)
| |
Dans toutes les comparaisons, une valeur p < 0,001 a été atteinte.
a Co-critères d'évaluation principaux
|
Chez les patients sous adalimumab ayant obtenu à la semaine 16 un score PASI 50 à < PASI 90 et de nouveau randomisés, une différence entre ceux qui sont passés à SKYRIZI et ceux qui ont poursuivi le traitement par adalimumab a été observée en termes de taux de réponse PASI 90 (49,1% vs. 26,8%) dès la quatrième semaine suivant la nouvelle randomisation. Au total, 66,0% (35/53) des patients ont obtenu un score PASI 90 après 28 semaines de traitement par SKYRIZI, comparativement à 21,4% (12/56) chez ceux qui ont continué de recevoir l'adalimumab. Des autres paramètres de réponse ont été également supérieurs après le relais par SKYRIZI: 39,6% ont obtenu un score PASI 100, 39,6% un score sPGA «sans» et 73,6% un score sPGA «sans» ou «minime» après le changement par SKYRIZI, comparativement à 7,1% ayant obtenu un score PASI 100, 7,1% un score sPGA «sans» et 33,9% un score sPGA «sans» ou «minime» avec la poursuite du traitement par adalimumab.
Figure 2: Évolution du score PASI 90 après la nouvelle randomisation dans l'étude IMMVENT
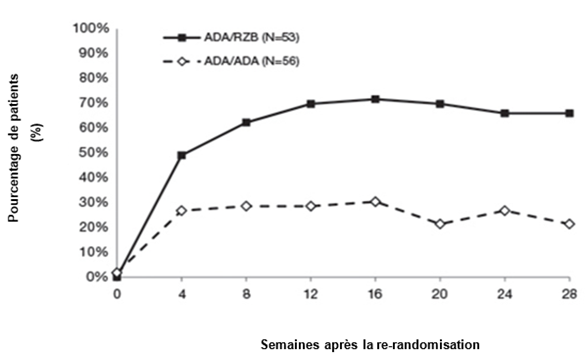
ADA/ADA: patients affectés initialement au groupe adalimumab et qui ont continué de recevoir l'adalimumab
ADA/RZB: patients affectés initialement au groupe adalimumab et qui sont passés à SKYRIZI
p < 0,05 la semaine 4 et p < 0,001 à chaque point dans le temps à partir de la semaine 8
Obtention et maintien de la réponse
Dans une analyse combinée des patients ayant reçu SKYRIZI dans le cadre des études ULTIMMA-1 et ULTIMMA-2 et qui présentaient un score PASI 100 à la semaine 16, le taux de réponse est resté à 79,8% (206/258) chez les patients qui ont poursuivi le traitement par SKYRIZI jusqu'à la semaine 52. Parmi les patients ayant obtenu un score PASI 90 à la semaine 16, 88,4% (398/450) ont maintenu leur réponse jusqu'à la semaine 52.
Après une nouvelle randomisation, les patients ayant reçu initialement SKYRIZI dans le cadre de l'étude IMMHANCE et dont le sPGA à la semaine 28 était «sans» ou «minime» ont continué d'être traités par SKYRIZI toutes les 12 semaines jusqu'à la semaine 88 incluse (N=111) ou ont arrêté le traitement (N=225).
À la semaine 104 (soit 16 semaines après la dernière administration de SKYRIZI), 81,1% (90/111) des patients ayant poursuivi le traitement par SKYRIZI ont obtenu un sPGA «sans» ou «minime», comparativement à 7,1% (16/225) des patients ayant arrêté le traitement par SKYRIZI. Chez les patients ayant arrêté le traitement par SKYRIZI, une perte du sPGA «sans» ou «minime» a été observée dès 12 semaines après une dose manquée.
À la semaine 104, 63,1% (70/111) des patients ayant poursuivi le traitement par SKYRIZI ont obtenu un score sPGA «sans», comparativement à 2,2% (5/225) des patients ayant arrêté le traitement par SKYRIZI. Une augmentation du sPGA «sans» et PASI 100 a été constatée entre la semaine 28 et la semaine 88 chez les patients ayant poursuivi le traitement par SKYRIZI. Parmi les patients ayant obtenu un sPGA «sans» ou «minime» à la semaine 28 et ayant présenté une rechute (sPGA ≥3) après l'arrêt du traitement par SKYRIZI, 83,7% (128/153) ont de nouveau obtenu un sPGA «sans» ou «minime» 16 semaines après la reprise du traitement.
Dans l'étude LIMMITLESS, chez les patients ayant terminé les études ULTIMMA-1 et ULTIMMA-2 et qui ont poursuivi le traitement par SKYRIZI, le taux de réponse PASI 90 et sPGA «sans» ou «minime» a été maintenu jusqu'à la semaine 304. Chez les patients qui sont passés de l'ustékinumab à un traitement par SKYRIZI à la semaine 52, une augmentation des réponses PASI 90 et sPGA «sans» ou «minime» a été observée de la semaine 52 à la semaine 76 et a été conservée jusqu'à la semaine 304 (figure 3 et 4).
Figure 3: Pourcentage de patients ayant obtenu une réponse PASI 90 lors d'une visite dans LIMMITLESS (Last Observation Carried Forward, LOCF)*
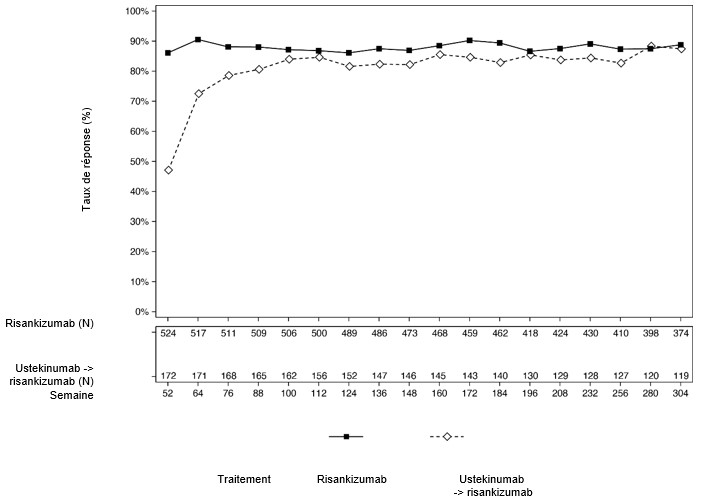
*Patients randomisés pour recevoir l'ustekinumab ou le risankizumab 150 mg dans les études ULTIMMA-1 et ULTIMMA-2 et inclus à LIMMITLESS.
Figure 4: Pourcentage de patients ayant obtenu une réponse de score sPGA «sans» ou «minime» lors d'une visite dans LIMMITLESS (Last Observation Carried Forward, LOCF)*
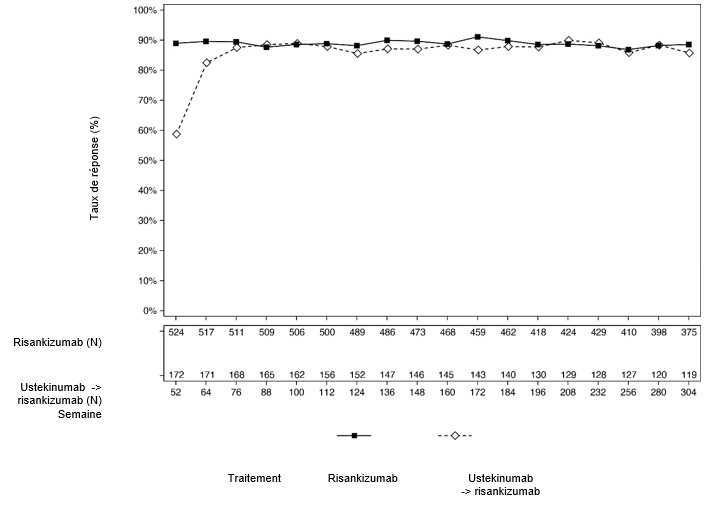
*Patients randomisés pour recevoir l'ustekinumab ou le risankizumab 150 mg dans les études ULTIMMA-1 et ULTIMMA-2 et inclus à LIMMITLESS.
Qualité de vie / Événements rapportés par les patients
Un nombre significativement supérieur de patients traités par SKYRIZI ont obtenu un score DLQI (Dermatology Life Quality Index) de 0 ou 1 [aucun effet sur la qualité de vie liée à la santé] à la semaine 16 comparativement aux patients sous placebo, adalimumab ou ustékinumab. L'amélioration s'est maintenue jusqu'à la semaine 52 dans les études ULTIMMA-1 et ULTIMMA-2 et a été maintenu chez les patients qui ont continué le traitement par SKYRIZI dans l'étude d'extension en ouvert LIMMITLESS jusqu'à la semaine 304.
Dans les études ULTIMMA-1 et ULTIMMA-2, une amélioration significativement supérieure des symptômes du psoriasis (démangeaisons, douleurs, rougeur et sensation de brûlure, mesurés au moyen du score PSS [Psoriasis Symptom Score]) a été observée comparativement au placebo à la semaine 16. Comparativement à l'ustékinumab et au placebo, une proportion significativement supérieure de patients sous SKYRIZI a obtenu un score PSS de 0 (absence de symptômes) à la semaine 16. Jusqu'à la semaine 52, 55,7% (333/598) des patients sous SKYRIZI n'ont rapporté aucune démangeaison, douleur, rougeur ou sensation de brûlure.
Arthrite psoriasique
Il a pu être démontré que chez les adultes atteints d'arthrite psoriasique active (APs), SKYRIZI améliore les signes et symptômes de la maladie, les capacités fonctionnelles ainsi que la qualité de vie liée à la santé.
La sécurité et l'efficacité de SKYRIZI ont été évaluées chez 1'407 patients de plus de 18 ans atteints d'une APs active au cours de deux études randomisés en double aveugle, contrôlées contre placebo (964 patients dans KEEPSAKE1 et 443 patients dans KEEPSAKE2).
Les participants présentaient un diagnostic d'APs depuis au moins six mois, selon les critères de classification de l'arthrite psoriasique (CASPAR), une durée médiane d'APs de 4,9 ans à l'inclusion, ≥5 articulations douloureuses à la pression et ≥5 articulations gonflées et un psoriasis en plaques actif ou un psoriasis unguéal à l'inclusion. 55,9% des patients présentaient un psoriasis en plaques actif avec un score BSA correspondant ≥3%. Des enthésites et des dactylites étaient présentes chez respectivement 63,4% et 27,9% des patients. Dans l'étude KEEPSAKE1, au cours de laquelle le psoriasis unguéal a été plus spécifiquement évalué, 67,3% des patients présentaient un psoriasis unguéal.
Dans l'étude KEEPSAKE1, tous les patients à l'étude présentaient des antécédents de réponse inadéquate ou d'intolérance à un traitement par DMARD non biologiques et étaient naïfs de traitement biologique. Dans l'étude KEEPSAKE2, 53,5% des patients à l'étude avaient présenté une réponse inadéquate ou une intolérance à un thérapie par DMARD non biologique et 46,5% des patients à l'étude avaient présenté une réponse inadéquate ou une intolérance à une thérapie biologique.
Dans les deux études, les patients étaient randomisés pour recevoir SKYRIZI 150 mg ou le placebo aux semaines 0, 4 et 16. À partir de la semaine 28, tous les patients ont reçu SKYRIZI toutes les 12 semaines. Les deux études incluent une prolongation à long terme encore en cours allant jusqu'à 204 semaines supplémentaires. 59,6% des patients aux deux études recevaient un traitement concomitant par méthotrexate (MTX), 11,6% ont reçu un traitement concomitant par DMARD non biologique autre que le MTX et 28,9% recevaient SKYRIZI en monothérapie.
Dans les deux études, le critère d'évaluation principal était la proporption de patients ayant atteint une réponse ACR20 (American College of Rheumatology) à la semaine 24.
Réponse clinique
Dans les deux études, le traitement par SKYRIZI a entraîné une amélioration significative des paramètres de l'activité de la maladie à la semaine 24 par rapport au placebo. Les principaux résultats d'efficacité figurent dans le tableau 4.
L'évaluation de la dactylite et de l'enthésite a été menée sur la base des données poolées des études KEEPSAKE1 et 2. Chez les patients ayant une dactylite préexistante, la proportion des patients ayant observé une résolution de la dactylite à la semaine 24 était plus élevée dans le groupe SKYRIZI (68,1%, p < 0,001) que dans le groupe placebo (51,0%). Chez les patients ayant une enthésite préexistante, la proportion des patients ayant observé une résolution de l'enthésite à la semaine 24 était plus élevée dans le groupe SKYRIZI (48,4%, p < 0,001) que dans le groupe placebo (34,8%).
Dans les deux études, une réponse similaire a été observée, indépendamment de l'utilisation concomitante de DMARD non biologiques, du nombre de traitements antérieurs par DMARD non biologiques, de l'âge, du sexe, de l'origine ethnique et de l'IMC. Dans l'étude KEEPSAKE2, les réponses observées étaient indépendantes du traitement biologique antérieur.
Tableau 4. Résultats d'efficacité des études KEEPSAKE1 et KEEPSAKE2
|
|
KEEPSAKE1
|
KEEPSAKE2
| |
Critère d'évaluation
|
Placebo
n = 481
n (%)
|
SKYRIZI
n = 483
n (%)
|
Placebo
n = 219
n (%)
|
SKYRIZI
n = 224
n (%)
| |
Réponse ACR20
| |
Semaine 16
|
161 (33,4)
|
272 (56,3)a
|
55 (25,3)
|
108 (48,3)a
| |
Semaine 24
|
161 (33,5)
|
277 (57,3)a
|
58 (26,5)
|
115 (51,3)a
| |
Semaine 52*
|
-
|
338/433 (78,1)
|
-
|
131/191 (68,6)
| |
Réponse ACR50
| |
Semaine 24
|
54 (11,3)
|
162 (33,4)b
|
20 (9,3)
|
59 (26,3)b
| |
Semaine 52*
|
-
|
209/435 (48,0)
|
-
|
72/192 (37,5)
| |
Réponse ACR70
| |
Semaine 24
|
23 (4,7)
|
74 (15,3)b
|
13 (5,9)
|
27 (12,0)c
| |
Semaine 52*
|
-
|
125/437 (28,6)
|
-
|
37/192 (19,3)
| |
Réponse relative à l'activité minimale de la maladie
| |
Semaine 24
|
49 (10,2)
|
121 (25,0)a
|
25 (11,4)
|
57 (25,6)a
| |
Semaine 52*
|
-
|
183/444 (41,2)
|
-
|
61/197 (31,0)
| |
*Les données sur les patients à l'étude observés sont indiquées au format n/N observés (%).
a Multiplicité contrôlée p ≤0,001 SKYRIZI par rapport au placebo
b Valeur nominale p ≤0,001 SKYRIZI par rapport au placebo
c Valeur nominale p ≤0,05 SKYRIZI par rapport au placebo
Les patients qui avaient un traitement concomitant contre l'APs avec un effet possible sur l'évaluation de l'efficacité, qui ont reçu un traitement de secours ou qui avaient des données manquantes la semaine de l'évaluation sont indiqués comme non-répondeurs dans les analyses des semaines 16 et 24. Les patients ayant des données manquantes la semaine de l'évaluation ont été exclus des analyses à la semaine 52. Toutes les données observées ont été utilisées pour les analyses de la semaine 52.
|
Dans KEEPSAKE1, une réponse ACR20 plus forte a été observée dès la semaine 4 par rapport au placebo et s'est renforcée jusqu'à la semaine 24. Le pourcentage de patients ayant obtenu une réponse ACR20 jusqu'à la semaine 24 dans l'étude KEEPSAKE1 est présenté dans la figure 5.
Figure 5. Pourcentage de patients ayant obtenu une réponse ACR20 jusqu'à la semaine 24 dans l'étude KEEPSAKE1
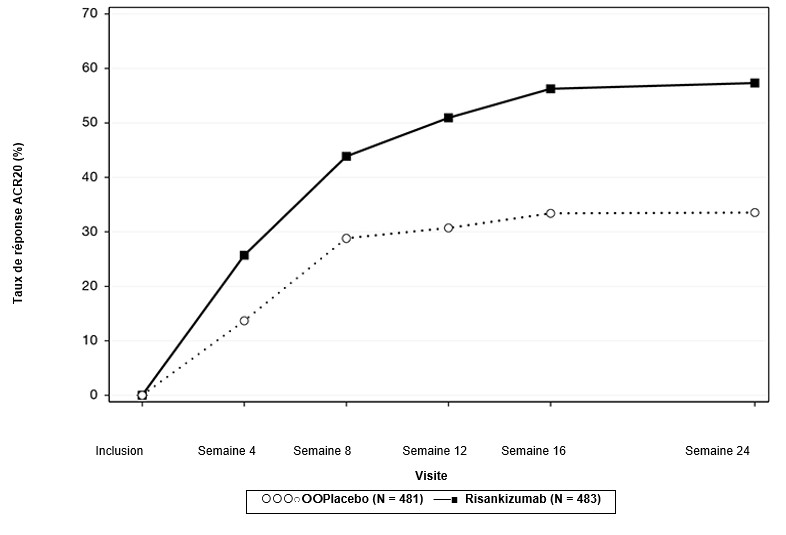
Par rapport au placebo, le traitement par SKYRIZI a produit une amélioration plus importante de toutes les composantes ACR, y compris le nombre d'articulations douloureuses/douloureuses à la pression et gonflées, l'évaluation générale de l'activité de la maladie par le médecin (PGA) et par le patient (PtGA), le Health Assessment Questionnaire-Disability Index (HAQ-DI), l'évaluation de la douleur et la protéine Créactive haute sensibilité (hsCRP).
Dans l'étude KEEPSAKE1, les patients traités par SKYRIZI ont montré une amélioration significative de la douleur par rapport à l'inclusion lors de l'évaluation de la douleur par le patient à l'aide d'une échelle visuelle analogique (EVA) à la semaine 24 (évolution moyenne: -21,0) par rapport aux patients ayant reçu le placebo (évolution moyenne: -10,2) (valeur p ≤0,001). Dans l'étude KEEPSAKE2, les patients traités par SKYRIZI ont montré une amélioration significative de la douleur par rapport à l'inclusion lors de l'évaluation de la douleur par le patient à l'aide de l'EVA à la semaine 24 (évolution moyenne: -14,7) par rapport aux patients ayant reçu le placebo (évolution moyenne: -6,5) (valeur p ≤0,001). Les améliorations ont été maintenues jusqu'à la semaine 52 dans les deux études.
Chez les patients ayant une APs active avec une atteinte psoriasique correspondant à un score BSA ≥3%, des améliorations significatives de l'atteinte cutanée ont été observées dans le groupe risankizumab par rapport au placebo.
Dans l'étude KEEPSAKE1, les patients atteints d'arthrite psoriasique et de psoriasis unguéal traités par SKYRIZI ont rapporté des améliorations statistiquement significatives du psoriasis unguéal à l'inclusion (67,3%) par rapport à la valeur initiale, mesurées par l'indice modifié de sévérité du psoriasis unguéal (mNAPSI) par rapport au placebo (-9. 76 vs. -5,57, p ≤0,001) et l'AGP du psoriasis unguéal (PGA-F) composé de 5 points (-0,8 vs. -0,4, p ≤0,001) à la semaine 24. Ces améliorations ont duré jusqu'à la semaine 52.
Réponse radiologique
Dans l'étude KEEPSAKE1, l'inhibition de la progression de lésions structurelles a été évaluée radiologiquement et exprimée en termes de variation du score total de Sharp modifié (mTSS) à la semaine 24 par rapport à l'inclusion. Le score mTSS a été modifié pour l'arthrite psoriasique en ajoutant les articulations interphalangiennes distales de la main. Par rapport au placebo, SKYRIZI a réduit numériquement la progression moyenne de lésions structurelles à la semaine 24 (le score mTSS moyen était de 0,23 dans le groupe SKYRIZI contre 0,32 dans le groupe placebo [statistiquement non significatif]). La proportion de patients à l'étude ne présentant pas de progression radiologique (définie comme un changement du mTSS ≤0 par rapport à l'inclusion) était plus élevé à la semaine 24 sous SKYRIZI (92,4%) par rapport au placebo (87,7%) (valeur p nominale = 0,016). La réponse s'est maintenue jusqu'à la semaine 52.
Capacités fonctionnelles et qualité de vie liée à la santé
Dans KEEPSAKE1 et KEEPSAKE2, les capacités fonctionnelles et l'invalidité ont été évaluées à l'aide du Health Assessment Questionnaire-Disability Index (HAQ-DI), du questionnaire d'état de santé SF-36 (V2) et du score d'évaluation fonctionnelle de la fatigue (FACIT-F, Functional Assessment of Chronic Illness Therapy-Fatigue).
Dans l'étude KEEPSAKE1, les patients traités par SKYRIZI ont présenté une amélioration statistiquement significative (-0,31) à la semaine 24 par rapport à l'inclusion des capacités fonctionnelles, mesurées à l'aide du score HAQ-DI, qu'avec le placebo (-0,11) (valeur p ≤0,001). Dans l'étude KEEPSAKE2, les patients traités par SKYRIZI ont présenté une amélioration statistiquement significative (-0,22) à la semaine 24 par rapport à l'inclusion du score HAQ-DI qu'avec le placebo (- 0,05) (valeur p ≤0,001). Dans les deux études, une proportion plus importante des patients à l'étude du groupe SKYRIZI ont atteint à la semaine 24 une réduction cliniquement significative du score HAQ-DI d'au moins 0,35 par rapport à l'inclusion que les patients du groupe placebo. Dans les deux études, l'amélioration des capacités fonctionnelles s'est maintenue jusqu'à la semaine 52.
Dans les deux études, les patients traités par SKYRIZI ont en outre démontré à la semaine 24 des améliorations significatives pour les domaines physiques du questionnaire SF-36 V2 et du score FACIT-F par rapport aux patients sous placebo. Dans les deux études, les améliorations des domaines physiques du questionnaire SF-36 V2 et du score FACIT-F se sont maintenues jusqu'à la semaine 52.
|