Propriétés/EffetsCode ATC
L04AB04
AMGEVITA est un biosimilaire.
Mécanisme d'action
L'adalimumab est un anticorps monoclonal humain fabriqué dans des cellules CHO par technologie de l'ADN recombinant. Il a été mis au point avec des chaînes lourdes et légères humaines par la méthode «phage display». On obtient ainsi un anticorps doté de régions variables des chaînes lourdes et légères sans séquence peptidique animale, ce qui permet une spécificité pour le facteur de nécrose tumorale (TNF) humain, et doté de régions constantes humaines IgG1 (chaîne lourde) et kappa (chaîne légère). L'adalimumab se lie avec une affinité et une spécificité élevées au facteur de nécrose tumorale soluble (TNF-α), mais pas à la lymphotoxine (TNF-β). Il comporte 1330 acides aminés et son poids moléculaire est de 148 kilodaltons environ.
L'adalimumab se lie spécifiquement au TNF et neutralise la fonction biologique du TNF par inhibition de son interaction avec les récepteurs du TNF p55 et p75 à la surface des cellules. Le TNF est une cytokine naturelle importante pour les réponses inflammatoires et immunitaires normales. Chez les patients atteints de polyarthrite rhumatoïde, de rhumatisme psoriasique ou de spondylarthrite ankylosante (maladie de Bechterew), on observe des concentrations élevées de TNF dans le liquide synovial, qui jouent un rôle important aussi bien dans le cadre de l'inflammation pathologique que dans le cadre de la destruction de l'articulation, signes caractéristiques de la polyarthrite rhumatoïde.
L'adalimumab module aussi des réactions biologiques induites ou régulées par le TNF, entre autres les modifications des concentrations en molécules d'adhésion responsables de la migration des leucocytes (ELAM-1, VCAM-1 et ICAM-1, avec une CI50 de 1-2× 10-10 M).
Pharmacodynamique
Après traitement par l'adalimumab, en comparaison avec les valeurs initiales, on a observé chez les patients atteints de polyarthrite rhumatoïde une régression rapide des valeurs des paramètres de la phase aiguë de l'inflammation (protéine C réactive [CRP]), vitesse de sédimentation des érythrocytes (VSE) et cytokines sériques (IL-6). La concentration sérique des métalloprotéinases matricielles (MMP-1 et MMP-3), qui entraînent le remodelage tissulaire responsable de la destruction du cartilage, a également diminué après administration d'adalimumab. On constate souvent, chez les patients atteints de polyarthrite rhumatoïde, de rhumatisme psoriasique ou de spondylarthrite ankylosante (maladie de Bechterew), une anémie légère à modérée, une diminution du nombre de lymphocytes et une augmentation des nombres de neutrophiles et de thrombocytes. Chez les patients traités par l'adalimumab, on observe généralement une amélioration de ces signes hématologiques d'une inflammation chronique.
Une régression rapide des taux de CRP après le traitement par l'adalimumab a été observée aussi chez les patients atteints de maladie de Crohn, de colite ulcéreuse, de la forme polyarticulaire de l'arthrite juvénile idiopathique et de l'hidradénite suppurée. Chez les patients atteints de maladie de Crohn, une diminution (statistiquement non significative) des cellules exprimant des marqueurs d'inflammation dans le côlon, y compris une diminution significative de l'expression du TNF-α, a été observée.
Efficacité clinique
Polyarthrite rhumatoïde
L'adalimumab a été étudié chez plus de 3000 patients dans le cadre de toutes les études cliniques sur la polyarthrite rhumatoïde. Certains patients ont été traités sur une période de plus de 60 mois. L'efficacité et la tolérance de l'adalimumab dans le traitement de la polyarthrite rhumatoïde ont été étudiées dans cinq études randomisées, en double aveugle et bien contrôlées.
L'étude 1 a évalué 271 patients souffrant d'une polyarthrite rhumatoïde active modérée à sévère, ayant ≥18 ans ou plus, en échec d'un traitement par au moins un, mais pas plus de quatre antirhumatismaux de fond, pour lesquels on avait observé une efficacité insuffisante du méthotrexate à une dose de 12,5 à 25 mg (10 mg en cas d'intolérance au méthotrexate) par semaine, et pour lesquels la dose de méthotrexate était restée constante pendant l'étude, entre 10 et 25 mg par semaine. Les patients avaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. La polyarthrite rhumatoïde avait été diagnostiquée en appliquant les critères de l'American College of Rheumatology (ACR). Pendant 24 semaines, les patients ont reçu, toutes les deux semaines, des doses de 20, 40 ou 80 mg d'adalimumab ou un placebo.
L'étude 2 a évalué 544 patients présentant une polyarthrite rhumatoïde active modérée à sévère, ayant ≥18 ans ou plus, en échec d'un traitement comportant au moins un antirhumatismal de fond. Les patients présentaient ≥10 articulations enflées et ≥12 articulations sensibles à la pression et avaient également été diagnostiqués selon les critères de l'ACR. Pendant 26 semaines, les patients ont reçu par injection sous-cutanée 20 ou 40 mg d'adalimumab toutes les deux semaines, en alternance avec un placebo la semaine suivante, ou un placebo chaque semaine. Le placebo a été administré chaque semaine au même moment. Les patients n'ont pas reçu de traitement concomitant par DMARDs.
L'étude 3 a évalué 619 patients souffrant d'une polyarthrite rhumatoïde active modérée à sévère, ayant 18 ans ou plus, chez lesquels le méthotrexate à la dose de 12,5 à 25 mg (10 mg en cas d'intolérance au méthotrexate) par semaine, avait eu une efficacité insuffisante et pour lesquels la dose de méthotrexate était restée constante pendant l'étude, entre 12,5 et 25 mg par semaine. À la différence de l'étude 1, l'inclusion des patients de l'étude 3 ne supposait pas obligatoirement l'échec d'un traitement par antirhumatismaux de fond (méthotrexate exclus). Les patients avaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. La polyarthrite rhumatoïde avait été diagnostiquée en appliquant les critères de l'ACR. Cette étude comprenait trois groupes. Le groupe 1 a reçu une injection de placebo chaque semaine pendant 52 semaines. Le deuxième groupe a reçu 20 mg d'adalimumab chaque semaine pendant 52 semaines. Le troisième groupe a reçu 40 mg d'adalimumab toutes les deux semaines et une injection de placebo la semaine suivante. Enfin, 457 patients ont été inclus dans une phase d'extension en ouvert de 5 ans au maximum pendant laquelle ils ont reçu 40 mg d'adalimumab toutes les deux semaines.
L'étude 4 a évalué 636 patients souffrant de polyarthrite rhumatoïde active modérée à sévère, ayant 18 ans ou plus. Ces patients remplissaient les critères diagnostiques de la polyarthrite rhumatoïde de l'ACR depuis au moins trois mois et présentaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. Ces patients soit n'avaient jamais été traités par antirhumatismaux de fond soit pouvaient poursuivre leur traitement rhumatologique en cours, à condition qu'il ait été stable depuis au moins 28 jours. Les patients ont été randomisés dans le groupe de traitement 40 mg d'adalimumab ou placebo toutes les deux semaines, pendant 24 semaines.
Dans l'étude 5 concernant la polyarthrite rhumatoïde précoce, 525 patients adultes (≥18 ans) atteints d'une polyarthrite rhumatoïde active modérée à sévère précoce (durée de la maladie inférieure à 3 ans), et n'ayant pas été traités par le méthotrexate, ont été évalués. Dans cette étude, l'efficacité de l'adalimumab associé au méthotrexate a été comparée au méthotrexate en monothérapie en ce qui concerne la réduction des signes et des symptômes et du taux de progression des lésions articulaires lors de la polyarthrite rhumatoïde. Les patients ont été randomisés dans le groupe de traitement 40 mg d'adalimumab toutes les deux semaines associé au méthotrexate ou dans le groupe traité par méthotrexate toutes les deux semaines en monothérapie. La durée du traitement a été de 104 semaines.
Les résultats de ces cinq études sont exprimés sous forme du pourcentage de patients présentant une amélioration de leur polyarthrite rhumatoïde selon les critères de réponse de l'ACR. Le critère d'évaluation principal des études 1, 2 et 3 et le critère secondaire de l'étude 4 étaient le pourcentage de patients ayant atteint un taux de réponse ACR20 à la semaine 24 ou 26. Le critère d'évaluation principal de l'étude 5 portant sur la polyarthrite rhumatoïde précoce était le pourcentage de patients atteignant une réponse ACR50 à la semaine 52. Les études 3 et 5 ont défini comme critère d'évaluation principal supplémentaire le retard de progression de la maladie (constaté par radiographie) à la semaine 52. Dans l'étude 3, les changements de la qualité de vie ont été également étudiés comme critère d'évaluation principal.
Taux de réponse ACR
Le pourcentage de patients traités par l'adalimumab ayant atteint des taux de réponse ACR20, ACR50 et ACR70 a été similaire dans les études 1, 2, 3 et 4. Les résultats pour la dose de 40 mg d'adalimumab administrée toutes les deux semaines sont résumés dans le tableau 4.
Tableau 4: Taux de réponse ACR au cours des études contrôlées par placebo (en pourcentage des patients)
|
Taux de réponse
|
Étude 1a*
|
Étude 2a*
|
Étude 3a*
|
Étude 4
| |
|
Placebo/
MTXc
n=60
|
Adalimumabb/
MTXc
n=63
|
Placebo
n=110
|
Adalimumabb
n=113
|
Placebo/
MTXc
n=200
|
Adalimumabb/
MTXc
n=207
|
Traitement
standard/
Placebo
n=318
|
Traitement
standard/
Adalimumab
n=318
| |
ACR20
| |
6 mois
|
13,3%
|
65,1%
|
19,1%
|
46,0%
|
29,5%
|
63,3%
|
34,9%
|
53,0%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
24,0%
|
58,9%
|
NA
|
NA
| |
ACR50
| |
6 mois
|
6,7%
|
52,4%
|
8,2%
|
22,1%
|
9,5%
|
39,1%
|
11,1%
|
29,2%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
9,5%
|
41,5%
|
NA
|
NA
| |
ACR70
| |
6 mois
|
3,3%
|
23,8%
|
1,8%
|
12,4%
|
2,5%
|
20,8%
|
3,2%
|
14,9%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
4,5%
|
23,2%
|
NA
|
NA
|
a Etude 1 au bout de 24 semaines, étude 2 au bout de 26 semaines et étude 3 au bout de 24 et 52 semaines.
b 40 mg d'adalimumab administrés toutes les deux semaines
c MTX=méthotrexate
* p<0,01 adalimumab contre placebo
NA=sans objet
Les patients ayant reçu, au cours de l'étude 2, 40 mg d'adalimumab par semaine, ont atteint au bout de 6 mois des taux de réponse ACR20, ACR50 et ACR70 statistiquement significatifs à hauteur de 53,4%, 35,0% et 18,4%.
Au cours des études 1-4, une amélioration de toutes les composantes individuelles des critères de réponse de l'ACR (nombre d'articulations sensibles à la pression et enflées, évaluation de l'activité de la maladie et des douleurs par le médecin et le patient, évaluation au moyen de l'index de handicap du HAQ et concentrations de CRP [mg/dl]) a été observée par comparaison avec le placebo au bout de 24 et 26 semaines. Au cours de l'étude 3, ces améliorations se sont aussi maintenues pendant 52 semaines. De plus, les taux de réponse ACR se sont maintenus jusqu'à la semaine 104 chez la majorité des patients ayant participé à la phase d'extension en ouvert. Les résultats à deux ans de l'étude montrent que chez 24% des patients traités par l'adalimumab, un effet clinique, défini comme un taux de réponse ACR70 maintenu pendant 6 mois, a pu être obtenu. Un effet clinique durable jusqu'à 5 ans a pu être démontré pendant les phases non contrôlées de l'étude 3. Le taux de réponse ACR observé à la semaine 52 a pu être maintenu lorsque l'adalimumab a été administré sans interruption pendant 5 ans, avec un taux de réponse ACR20 de 75,5% dans le sous-groupe de 220 patients évalués après 5 ans. Le taux de réponse ACR70 après 5 ans était de 34,7%. Chez 25,7% des patients, la dose de méthotrexate administrée concomitamment a pu être réduite sans diminution de l'effet clinique; le même phénomène a été observé pour les corticostéroïdes chez 29,9% de ces mêmes patients.
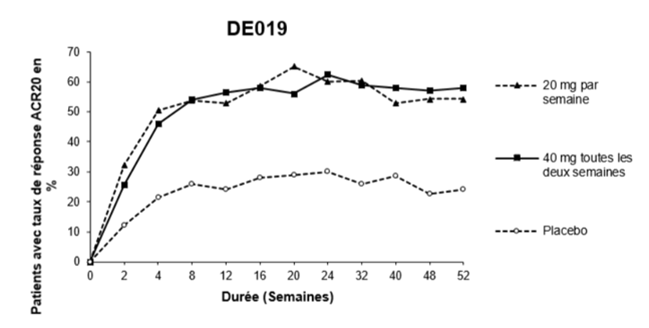
La figure 1 ci-dessous illustre la persistance du taux de réponse ACR20 obtenu avec l'adalimumab au cours de l'étude 3. Dans le cadre de cette étude, 84,7% des patients ayant atteint un taux de réponse ACR20 à la semaine 24 l'ont maintenu jusqu'à la semaine 52.
Figure 1: Taux de réponse ACR20 sur 52 semaines pendant l'étude 3
Au cours de l'étude 4, les taux de réponse ACR20 des patients traités par l'adalimumab plus le traitement standard ont été meilleurs de manière statistiquement significative que ceux des patients prenant un placebo plus le traitement standard (p<0,001).
Dans les quatre études, les patients traités par l'adalimumab ont atteint les taux de réponse ACR20, ACR50 et ACR70 plus rapidement et plus souvent que les patients recevant le placebo. Au cours de l'étude 1, une différence statistiquement significative a été observée pour les taux de réponse ACR20 à la semaine 1 (premier examen dans le cadre de l'étude) entre les patients traités par l'adalimumab (26,0%) et les patients recevant un placebo (5,0%). Des différences statistiquement significatives des taux de réponse ACR20 ont aussi été observées au cours des études 2, 3 et 4 à la semaine 2 (premier examen dans le cadre de l'étude) entre les patients traités par l'adalimumab (36,4%, 29,1% et 33,7%) et les patients recevant le placebo (7,3%, 13,0% et 8,6%). On a obtenu un tableau similaire au cours des quatre études pour le temps écoulé jusqu'à ce que les premiers taux de réponse ACR50 et ACR70 soient atteints.
Pour certains patients ne prenant pas concomitamment du méthotrexate, une augmentation de la fréquence d'administration de l'adalimumab à 40 mg par semaine pourrait apporter un bénéfice supplémentaire. Cette observation a été confirmée lors d'une étude en ouvert au long cours pendant laquelle la fréquence d'administration a été augmentée pour les patients ne répondant que partiellement au traitement, de 40 mg toutes les deux semaines à 40 mg par semaine.
Dans l'étude 5, la thérapie associant l'adalimumab et le méthotrexate chez des patients atteints de polyarthrite rhumatoïde précoce et n'ayant pas été traités par méthotrexate a entraîné un taux de réponse rapide et significativement plus élevé à la semaine 52 qu'avec le méthotrexate en monothérapie, avec un taux de réponse maintenu jusqu'à la semaine 104 (voir tableau 5).
Tableau 5: Taux de réponse dans l'étude 5 (en pourcentage du nombre de patients)
|
Taux de réponse*
|
MTX
n=257
|
Adalimumab/MTX
n=268
| |
ACR20
| |
Semaine 52
|
62,6%
|
72,8%
| |
Semaine 104
|
56,0%
|
69,4%
| |
ACR50
| |
Semaine 52
|
45,9%
|
61,6%
| |
Semaine 104
|
42,8%
|
59,0%
| |
ACR70
| |
Semaine 52
|
27,2%
|
45,5%
| |
Semaine 104
|
28,4%
|
46,6%
|
* p<0,05, adalimumab/méthotrexate comparé au méthotrexate pour ACR20
* p<0,001, adalimumab/méthotrexate comparé au méthotrexate pour ACR50 et 70
Tous les critères de réponse ACR ont montré une amélioration à la semaine 52 sous traitement par l'adalimumab/méthotrexate, qui s'est maintenue jusqu'à la semaine 104. Au cours de l'étude sur deux ans, 48,5% des patients ayant été traités par l'association adalimumab/méthotrexate ont atteint une réponse clinique majeure (ACR70 pendant six mois). En comparaison, 27,2% des patients traités par méthotrexate en monothérapie (p<0,001) ont atteint ces résultats.
Tableau 6: Taux de réponse DAS28 dans l'étude 5 sur la polyarthrite rhumatoïde précoce
|
Taux de réponse DAS28
|
MTX
n=257
|
Adalimumab/MTX
n=268
| |
Différence moyenne par rapport au début de l'étude
| |
Valeur initiale (valeur moyenne)
|
6,3
|
6,3
| |
Semaine 52 (valeur moyenne ± écart type)
|
-2,8 ± 1,4
|
-3,6 ± 1,3*
| |
Semaine 104 (valeur moyenne ± écart type)
|
-3,1 ± 1,4
|
-3,8 ± 1,3*
| |
Rémission (DAS28<2,6)
| |
Semaine 52 (pourcentage du nombre de patients)
|
20,6%
|
42,9%*
|
* p<0,001, adalimumab/méthotrexate comparé au méthotrexate
Réponse radiologique
Au cours de l'étude 3, dans laquelle les patients traités par l'adalimumab étaient en moyenne atteints de polyarthrite rhumatoïde depuis environ 11 ans, les lésions articulaires structurelles ont été évaluées par radiographie et exprimées en fonction du changement du score global de Sharp (SST) modifié et de ses composantes, le score d'érosion et le joint space narrowing score = JSN (score évaluant l'amincissement de l'espace articulaire). Une différence statistiquement significative a été observée au bout de 6 mois et s'est maintenue jusqu'à 12 mois pour le changement du score global de Sharp modifié ainsi que pour le score d'érosion. Au bout de 52 semaines, les patients traités par adalimumab/méthotrexate présentaient moins de modifications radiologiques que les patients traités par le méthotrexate seul. Cet effet de ralentissement de la progression des lésions structurelles a pu être maintenu pendant 5 ans.
Parmi les patients initialement traités par 40 mg d'adalimumab toutes les deux semaines, 55% ont subi un examen radiologique après 5 ans. Le ralentissement de la progression des lésions structurelles a pu être maintenu, et, chez 50% de ces patients, la progression des lésions structurelles a pu être complètement arrêtée, ce qui s'est manifesté par une modification du score TSS de zéro ou moins. Les patients ayant été traités par le méthotrexate pendant la phase en double aveugle de l'étude ont présenté une progression minimale des lésions structurelles lorsqu'ils ont été traités par l'adalimumab pendant la phase en ouvert de l'étude.
Tableau 7: Modification radiologique à 12 mois dans l'étude 3 avec traitement de fond par le méthotrexate
|
|
Placebo
n=200
|
Adalimumaba
n=207
|
Différence entre l'adalimumaba et le placebo
|
Valeur p
| |
Changement du score global de Sharp modifié (moyenne)
|
2,7
|
0,1
|
-2,6
|
=0,001b
| |
Modification des érosions (moyenne)
|
1,6
|
0,0
|
-1,6
|
=0,001
| |
Pas de nouvelles érosions (% de patients)
|
46,2
|
62,9
|
16,7
|
=0,001
| |
Modification du score JSN (moyenne)
|
1,0
|
0,1
|
-0,9
|
=0,002
|
a 40 mg, administrés toutes les deux semaines
b sur la base des valeurs moyennes, mesurées par le SST
Dans l'étude 5 portant sur la polyarthrite rhumatoïde précoce, les patients traités par l'adalimumab avaient une durée de la maladie moyenne inférieure à 9 mois et n'avaient auparavant pas été traités par le méthotrexate. Les lésions articulaires structurelles ont été évaluées par radiographie et exprimées en fonction du changement du score global de Sharp modifié. Les résultats après 52 semaines sont décrits dans le tableau 8. Un changement statistiquement significatif du score global de Sharp modifié et du score d'érosion a été constaté à la semaine 52 et a été maintenu jusqu'à la semaine 104.
Tableau 8: Différences radiologiques moyennes à la semaine 52 dans l'étude 5
|
|
MTX
n=257
IC 95%
|
Adalimumab/MTX
n=268
IC 95%
|
Valeur p*
| |
Score global de Sharp
|
5,7 (4,2-7,3)
|
1,3 (0,5-2,1)
|
<0,001
| |
Score d'érosion
|
3,7 (2,7-4,7)
|
0,8 (0,4-1,2)
|
<0,001
| |
JSN Score
|
2,0 (1,2-2,8)
|
0,5 (0-0,1)
|
<0,001
|
* Comparaison entre adalimumab/méthotrexate et méthotrexate au moyen du test U de Mann-Whitney
Le pourcentage de patients sans progression radiologique de la maladie (accroissement par rapport aux valeurs initiales du score global de Sharp modifié ≤0,5) était significativement plus élevé sous adalimumab/méthotrexate en association comparé au méthotrexate en monothérapie à la semaine 52 (respectivement 63,8% et 37,4%, p<0,001) et à la semaine 104 (respectivement 61,2% et 33,5%, p<0,001).
Dans le cadre de la phase d'extension en ouvert de l'étude 3, 77% des patients à l'origine traités par l'adalimumab ont été évalués par radiographie au bout de deux ans. L'inhibition de la progression des lésions structurelles a été maintenue. 54% des patients étudiés ne présentaient pas de progression des lésions structurelles, ce qui s'est reflété par une modification du SST de 0 ou inférieure.
Qualité de vie et capacités fonctionnelles physiques
Pour l'évaluation de la qualité de vie en rapport avec l'état de santé, critère prescrit de l'étude 3 après 52 semaines, l'index de handicap contenu dans le Health Assessment Questionnaire (HAQ = questionnaire d'évaluation de l'état de santé) a été utilisé au cours des quatre études adéquates et bien contrôlées. Au cours des quatre études, des améliorations statistiquement significatives et plus fortes de l'index de handicap du HAQ ont été observées pour l'ensemble des doses/traitements par l'adalimumab par comparaison avec le placebo entre les valeurs initiales et les valeurs au bout de 6 mois. Au cours de l'étude 3, l'amélioration moyenne (IC) du HAQ entre avant le traitement et à la semaine 52 a été de -0,60 (-0,65, -0,55) pour les patients traités par adalimumab/méthotrexate et de -0,25 (-0,33, -0,17) chez les patients traités par placebo/méthotrexate (p<0,001). Chez 82% des patients traités par adalimumab/méthotrexate, et pour lesquels une amélioration de 0,5 ou plus au questionnaire HAQ a été obtenue à la semaine 52, cette amélioration a été maintenue jusqu'au mois 60 de la phase d'extension en ouvert.
Dans l'étude 5, l'étude contrôlée portant sur la polyarthrite rhumatoïde précoce par comparaison avec le méthotrexate, l'amélioration de l'index de handicap du HAQ et de la composante physique du SF 36 dans la semaine 52 était plus importante (p<0,001) sous l'association adalimumab/méthotrexate que sous le méthotrexate en monothérapie et a été maintenue jusqu'à la semaine 104.
Par ailleurs, la qualité de vie globale en rapport avec l'état de santé a été évaluée pour les quatre études adéquates et bien contrôlées à l'aide du Short Form Health Survey (SF 36 = analyse courte de l'état de santé). Au cours de ces quatre études, des améliorations statistiquement significatives plus fortes du score résumé pour les composantes physiques du questionnaire SF 36 ont été observées pour l'ensemble des doses/fréquences d'injection de l'adalimumab par comparaison avec le placebo entre les valeurs initiales et le mois 6, et ont été maintenues jusqu'à la semaine 52 au cours de l'étude 3. Les scores résumés des composantes mentales du questionnaire SF 36 au cours des études 2 et 4 ont été statistiquement significativement plus élevés au mois 6 pour l'adalimumab par comparaison avec le placebo. On a observé une amélioration statistiquement significative plus forte des scores de douleur et de vitalité du questionnaire SF 36 au cours des quatre études pour la dose de 40 mg d'adalimumab toutes les deux semaines par comparaison avec le placebo, entre les valeurs initiales et le mois 6. Ces résultats ont été renforcés par les scores atteints dans le cadre du Functional Assessment of Chronic Illness Therapy (FACIT = évaluation fonctionnelle du traitement de maladies chroniques), selon lesquels, pour les trois études analysées, on a eu une diminution statistiquement significative de la fatigue au mois 6, qui a été maintenue jusqu'à la semaine 52 au cours de l'étude 3. Le score SF 36 a été calculé jusqu'à la semaine 156 (3 ans) et l'amélioration a été maintenue pendant cette période pour les patients restés dans l'étude.
Arthrite juvénile idiopathique polyarticulaire (AJIp)
La sécurité et l'efficacité de l'adalimumab ont été évaluées dans une étude multicentrique randomisée, en double aveugle, par groupes parallèles, auprès de 171 enfants (de 4 à 17 ans) atteints d'AJI polyarticulaire. L'analyse a été effectuée après avoir stratifié les patients en deux groupes: ceux qui recevaient du méthotrexate (MTX) et ceux qui n'en recevaient pas. Les patients ont reçu des doses stables d'AINS et/ou de prednisone (≤0,2 mg/kg/jour ou au maximum 10 mg par jour). Dans la phase initiale en ouvert («open-label lead-in», OL LI), d'une durée de 16 semaines, tous les patients ont reçu toutes les deux semaines une dose d'adalimumab de 24 mg/m2 (sans dépasser une dose maximale de 40 mg). La répartition des patients est présentée dans le tableau 9.
Tableau 9: Répartition des patients par âge et par dose d'adalimumab administrée pendant la phase OL LI
|
Groupe d'âge
|
Nombre de patients au début de l'étude
n (%)
|
Dose minimale, moyenne et maximale
| |
4 à 7 ans
|
31 (18,1)
|
10, 20 et 25 mg
| |
8 à 12 ans
|
71 (41,5)
|
20, 25 et 40 mg
| |
13 à 17 ans
|
69 (40,4)
|
25, 40 et 40 mg
|
Les patients ayant atteint une réponse ACR30 pédiatrique à 16 semaines ont été randomisés pour la phase d'étude en double aveugle (double blind, DB), dans laquelle ils ont reçu toutes les 2 semaines l'adalimumab (24 mg/m2 jusqu'à une dose unique maximale de 40 mg) ou un placebo. Ce traitement a été poursuivi pour une durée maximale de 32 semaines ou jusqu'à la survenue d'une nouvelle poussée de la maladie. Les critères définissant une nouvelle poussée de la maladie ont englobé une aggravation de ≥30% par rapport au début de l'étude pour ≥3 des 6 critères ACR pédiatriques, une activité de la maladie dans ≥2 articulations et pas plus d'un critère sur 6 présentant une amélioration de >30%. Au bout de 32 semaines ou en présence d'une nouvelle poussée de la maladie, les patients étaient éligibles pour une participation à la phase d'extension en ouvert («open-label extension», OLE).
Au bout de 16 semaines (à l'issue de la phase OL LI), 94,1% (80 patients sur 85) du groupe sous adalimumab et MTX et 74,4% (64 patients sur 86) du groupe sous adalimumab en monothérapie avaient atteint une réponse ACR30. Les résultats de la période en double aveugle sont présentés dans le tableau 10.
Tableau 10: Réponse ACR30 pédiatrique dans l'étude sur l'AJI
Résultats d'efficacité
|
En double aveugle,
32 semaines
|
Adalimumab/MTX
(n=38)
|
Placebo/MTX
(n=37)
|
Adalimumab
(n=30)
|
Placebo
(n=28)
| |
Nouvelle poussée de la maladie après 32 semainesa (n/N)
|
36,8% (14/38)
|
64,9% (24/37)b
|
43,3% (13/30)
|
71,4% (20/28)c
| |
Temps moyen écoulé jusqu'à une nouvelle poussée de la maladie
|
>32 semaines
|
20 semaines
|
>32 semaines
|
14 semaines
|
a La réponse ACR30/50/70 pédiatrique à 48 semaines était significativement supérieure à celle des patients ayant reçu le placebo
b p=0,015
c p=0,031
Parmi les patients ayant atteint une réponse à 16 semaines (n=144), la réponse ACR30/50/70/90 pédiatrique s'est maintenue jusqu'à six ans dans la phase OLE chez ceux qui ont reçu de l'adalimumab pendant toute la durée de l'étude. 19 patients au total (11 qui avaient 4 à 12 ans au début de l'étude et 8 qui avaient 13 à 17 ans au début de l'étude) ont été traités pendant 6 ans ou plus.
Il est apparu que la réponse globale au traitement associant l'adalimumab et le MTX était supérieure et que moins de patients ayant reçu ce traitement ont développé des anticorps en comparaison avec le groupe sous adalimumab seul. Compte tenu de ces résultats, il est recommandé d'utiliser l'adalimumab en association avec le MTX. L'utilisation de l'adalimumab en monothérapie n'est recommandée que chez les patients pour lesquels le MTX est inapproprié (voir «Posologie/Mode d'emploi»).
Arthrite psoriasique
L'efficacité de l'adalimumab a été étudiée chez 413 patients. Dans l'étude principale, 313 patients adultes atteints d'arthrite psoriasique modérée à sévère, n'ayant pas répondu suffisamment au traitement par anti-inflammatoires non stéroïdiens ont été traités. 158 (50,5%) des patients traités prenaient du méthotrexate au moment de la randomisation. L'adalimumab a été administré à la dose de 40 mg toutes les 2 semaines sur une période de 24 semaines. Après la fin des études, 383 patients ont été inclus dans une phase d'extension en ouvert pendant laquelle l'adalimumab a été administré toutes les deux semaines. 382 des patients inclus ont été traités par l'adalimumab au moins au début de cette étude d'extension. Concernant les patients évaluables à 48 et 144 semaines, voir plus bas.
Taux de réponse ACR et PASI
Le tableau 11 montre que l'adalimumab était supérieur au placebo dans tous les paramètres de progression de la maladie (p<0,001). Chez les patients atteints d'arthrite psoriasique traités par l'adalimumab, une efficacité clinique a été observée dès le premier contrôle (2 semaines); cette efficacité était significative après 12 semaines et s'est maintenue pendant les 24 semaines de traitement.
Les patients chez lesquels au moins 3% de la surface corporelle étaient atteints de psoriasis ont été évalués d'après le Psoriatic Area and Severity Index (PASI). Les lésions cutanées dues au psoriasis se sont améliorées chez ces patients par rapport au placebo, selon le PASI.
Le taux de réponse était comparable lors du traitement avec ou sans méthotrexate.
Les taux de réponse ACR se sont maintenus pendant la phase d'extension en ouvert jusqu'à 136 semaines.
Tableau 11: Taux de réponse ACR et PASI dans une étude contrôlée contre placebo chez des patients atteints d'arthrite psoriasique (en pourcentage du nombre de patients)
|
Taux de réponse*
|
Placebo
|
Adalimumab
| |
|
N=162
|
N=151
| |
ACR20
| |
Semaine 12
|
14%
|
58%
| |
Semaine 24
|
15%
|
57%
| |
ACR50
| |
Semaine 12
|
4%
|
36%
| |
Semaine 24
|
6%
|
39%
| |
ACR70
| |
Semaine 12
|
1%
|
20%
| |
Semaine 24
|
1%
|
23%
| |
|
N=69
|
N=69
| |
PASI 50
| |
Semaine 12
|
15%
|
72%
| |
Semaine 24
|
12%
|
75%
| |
PASI 75
| |
Semaine 12
|
4%
|
49%
| |
Semaine 24
|
1%
|
59%
|
* p<0,001 pour toutes les comparaisons entre l'adalimumab et le placebo
Au cours des études portant sur le rhumatisme psoriasique, les modifications radiologiques ont été évaluées. Des radiographies des mains, des poignets et des pieds ont été réalisées au début de l'étude (Baseline) et à la semaine 24 de la période en double aveugle pendant laquelle les patients ont reçu soit l'adalimumab, soit un placebo, ainsi qu'à la semaine 48, pendant laquelle tous les patients recevaient l'adalimumab. Un score de Sharp global modifié (mTSS) incluant les articulations interphalangiennes distales (différent du score total de Sharp pour la polyarthrite rhumatoïde) a été utilisé.
Comparativement au placebo, l'adalimumab a réduit la vitesse de progression des lésions articulaires périphériques. Une modification du score de Sharp global modifié de 0,8 ± 2,5 (moyenne ± écart type) par rapport à la valeur initiale a été observée dans le groupe placebo (à la semaine 24), contre 0,0 ± 1,9 dans le groupe adalimumab (à la semaine 48, n=133); (p<0,001).
84% des patients qui ont été traités par l'adalimumab et qui n'ont présenté aucune progression radiologique entre l'inclusion et la semaine 48 (n=102) n'ont pas non plus présenté de progression radiologique jusqu'à la semaine 144.
L'évaluation de l'index de handicap du HAQ et du questionnaire Short Form Health Survey (SF 36) a montré, dans le groupe de patients traités par l'adalimumab, une amélioration statistiquement significative des capacités fonctionnelles physiques à la semaine 24 comparativement aux patients traités par le placebo.
L'amélioration des capacités fonctionnelles physiques s'est maintenue au cours de la phase d'extension en ouvert de l'étude, jusqu'à la semaine 136.
Maladie de Crohn
La sécurité et l'efficacité d'une dose multiple d'adalimumab ont été étudiées chez plus de 1500 patients atteints d'une forme active modérée à sévère de la maladie de Crohn (indice d'activité de la maladie de Crohn [CDAI = Crohn's Disease Activity Index] ≥220 et ≤450) dans le cadre d'études randomisées, contrôlées contre placebo et en double aveugle. L'administration concomitante d'aminosalicylates, de corticostéroïdes ou d'immunomodulateurs à doses constantes était permise, et l'un de ces médicaments au moins a été administré à 80% des patients.
L'induction d'une rémission clinique (définie par un CDAI <150) a été évaluée dans le cadre de deux études (CLASSIC I et GAIN). 299 patients n'ayant pas reçu de traitement par antagonistes du TNF au préalable, répartis par randomisation dans quatre groupes de traitement, ont participé à l'étude CLASSIC I; un placebo a été administré au groupe placebo à la semaine 0 et 2, le groupe 160/80 a reçu 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, le groupe 80/40 a été traité par 80 mg à la semaine 0 et 40 mg à la semaine 2, et le groupe 40/20 a été traité par 40 mg à la semaine 0 et 20 mg à la semaine 2. Dans l'étude GAIN, 325 patients qui ne répondaient plus à l'infliximab ou ne le toléraient pas, ont été randomisés pour recevoir soit 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, soit un placebo à la semaine 0 et 2.
Le maintien de la rémission clinique a été évalué dans le cadre de l'étude CHARM. Celle-ci portait sur 854 patients qui ont reçu en ouvert 80 mg d'adalimumab à la semaine 0 et 40 mg d'adalimumab à la semaine 2. À la semaine 4, les patients ont été randomisés et ont reçu soit 40 mg d'adalimumab toutes les deux semaines, soit 40 mg d'adalimumab toutes les semaines, soit un placebo. L'étude a duré au total 56 semaines. Les patients présentant une réponse clinique (diminution du CDAI ≥70) à la semaine 4 ont été stratifiés et analysés (séparément de ceux qui ne présentaient aucune réponse clinique à la semaine 4). Une réduction progressive des corticostéroïdes était permise après la semaine 8.
Résultats cliniques
En comparaison avec le placebo, un pourcentage statistiquement significativement plus élevé de patients traités par 160/80 mg d'adalimumab dans le cadre des études CLASSIC I et GAIN a affiché une rémission clinique à la semaine 4, indépendamment du fait que les patients aient suivi au préalable un traitement par des antagonistes du TNF ou aient déjà été exposés à un traitement par l'infliximab (voir tableau 12).
Tableau 12: Induction d'une rémission et d'une réponse cliniques (en pourcentage de patients)
|
|
CLASSIC I: Patients n'ayant pas été traités par l'infliximab
|
GAIN: Patients ayant été traités par l'infliximab
| |
Placebo
N=74
|
Adalimumab
160/80 mg
N=76
|
Placebo
N=166
|
Adalimumab
160/80 mg
N=159
| |
Semaine 4
| |
Rémission clinique
|
12%
|
36%*
|
7%
|
21%*
| |
Réponse clinique
(CR-100)
|
24%
|
50%**
|
25%
|
38%**
| |
Réponse clinique
(CR-70)
|
34%
|
58%**
|
34%
|
52%**
|
Toutes les valeurs p sont des comparaisons appariées des pourcentages pour l'adalimumab vs placebo.
* p<0,001
** p<0,01
58% (499/854) des patients participant à l'étude CHARM ont montré une réponse clinique à la semaine 4 et ont été évalués dans le cadre de l'analyse primaire. 48% des patients qui présentaient une réponse clinique à la semaine 4 avaient déjà reçu au préalable une autre thérapie anti-TNF. Aux semaines 26 et 56, un pourcentage statistiquement significativement plus élevé des groupes recevant une thérapie d'entretien par l'adalimumab et ayant affiché une réponse clinique dans la semaine 4 a atteint une rémission clinique, par comparaison avec le groupe bénéficiant d'une thérapie d'entretien par placebo. Par ailleurs, toujours par comparaison avec le groupe ayant reçu une thérapie d'entretien par placebo, on a observé que dans les groupes ayant été traités par une thérapie d'entretien par l'adalimumab et ayant bénéficié au début du traitement simultanément de corticostéroïdes, le pourcentage de patients avec une rémission clinique et en mesure d'interrompre l'administration de corticostéroïdes pendant au moins 90 jours aux semaines 26 et 56 était statistiquement significativement plus élevé (voir tableau 13).
Une analyse post hoc a montré une diminution statistiquement significative des hospitalisations et opérations intra-abdominales liées à la maladie sous adalimumab par rapport au placebo pendant la phase en double aveugle.
Tableau 13: Maintien de la rémission et de la réponse cliniques (en pourcentage de patients)
|
|
Placebo
|
40 mg d'adalimumab
toutes les deux semaines
|
40 mg d'adalimumab
toutes les semaines
| |
Semaine 26
|
N=170
|
N=172
|
N=157
| |
Rémission clinique
|
17%
|
40%*
|
47%*
| |
Réponse clinique (CR-100)
|
27%
|
52%*
|
52%*
| |
Réponse clinique (CR-70)
|
28%
|
54%*
|
56%*
| |
Patients présentant une rémission sans stéroïdes ≥90 joursa
|
3% (2/66)
|
19% (11/58)**
|
15% (11/74)**
| |
Semaine 56
|
N=170
|
N=172
|
N=157
| |
Rémission clinique
|
12%
|
36%*
|
41%*
| |
Réponse clinique (CR-100)
|
17%
|
41%*
|
48%*
| |
Réponse clinique (CR-70)
|
18%
|
43%*
|
49%*
| |
Patients présentant une rémission sans stéroïdes ≥90 joursa
|
5% (3/66)
|
29% (17/58)*
|
20% (15/74)**
|
* p<0,001 pour adalimumab vs placebo (comparaisons appariées des pourcentages)
** p<0,002 pour adalimumab vs placebo (comparaisons appariées des pourcentages)
a Parmi ceux ayant reçu initialement des corticostéroïdes
Les résultats de rémission clinique présentés dans le tableau 13 sont restés relativement constants, en dépit d'une exposition préalable aux antagonistes du TNF.
Parmi les patients qui ont affiché une réponse à la semaine 4 et atteint une rémission dans le courant de l'étude, les patients issus des groupes de thérapie d'entretien par l'adalimumab ont pu maintenir cette rémission significativement plus longtemps que les patients issus du groupe de thérapie d'entretien par placebo (voir figure 2).
Figure 2: Jours de rémission clinique chez les patients qui ont atteint une rémission clinique durant l'étude CHARM (Intent-to-Treat-Population)
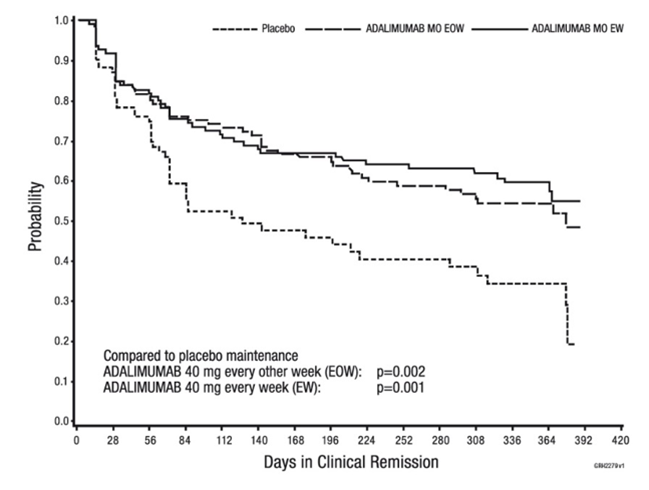
Parmi les patients qui n'affichaient aucune réponse à la semaine 4, 43% des patients des groupes de thérapie d'entretien par l'adalimumab jusqu'à la semaine 12, contre un taux de 30% chez les patients du groupe de thérapie d'entretien par placebo. Ces résultats permettent de conclure que la poursuite d'une thérapie d'entretien jusqu'à la semaine 12 peut présenter un avantage pour certains patients qui n'avaient affiché aucune réponse à la semaine 4. La poursuite de la thérapie au-delà des 12 semaines n'a conduit à aucune autre augmentation significative des réponses (voir «Posologie/Mode d'emploi»).
Dans l'étude CLASSIC I, 117/276 patients et dans les études GAIN et CHARM, 272/777 patients ont poursuivi le traitement par l'adalimumab pendant au moins 3 ans dans une étude ouverte de suivi. Respectivement 88 et 189 patients étaient toujours en rémission clinique après 3 ans. Le taux de réponse clinique (CR-100) s'est maintenu chez respectivement 102 et 233 patients.
Dans l'étude M05-769 (EXTEND) endoscopique, randomisée et contrôlée contre placebo, 135 patients ont été examinés avec comme critère principal la guérison de la muqueuse (définie comme la disparition des ulcérations de la muqueuse). Après une phase d'induction de l'adalimumab de 4 semaines, les patients ont été randomisés. À la semaine 12, 27,4% des patients traités par l'adalimumab présentaient une guérison de la muqueuse contre 13,1% de ceux sous placebo (p=0,056); à la semaine 52, 24,2% des patients sous adalimumab présentaient une guérison de la muqueuse contre 0% de ceux sous placebo (p<0,001).
Résultats de la perspective des patients/Patient-Reported Outcomes
Le résultat total issu du «questionnaire portant sur les maladies inflammatoires de l'intestin» (Inflammatory Bowel Disease Questionnaire = IBDQ), questionnaire spécialement conçu pour la maladie, à la semaine 4, a indiqué une amélioration statistiquement significative, en comparaison au placebo, chez les patients qui avaient été randomisés pour bénéficier d'un traitement par l'adalimumab 160/80 mg dans le cadre des études CLASSIC I et GAIN. En comparaison avec le groupe placebo, les groupes traités par l'adalimumab dans le cadre de l'étude CHARM ont affiché une amélioration statistiquement significative du score total IBDQ par rapport aux valeurs initiales observées dans les semaines 26 et 56.
Qualité de vie et capacités fonctionnelles physiques
La qualité de vie en rapport avec l'état de santé et les capacités fonctionnelles physiques ont été évaluées dans l'étude sur l'arthrite psoriasique à l'aide du Health Assessment Questionnaire (HAQ). Les patients traités par l'adalimumab ont montré, par rapport aux patients traités par placebo, des améliorations statistiquement significatives et plus fortes de l'index de handicap du HAQ entre les valeurs initiales et la semaine 24.
Les résultats du Short Form Health Survey (SF 36) renforcent ces résultats avec un Physical Component Summary (PCS) Score ainsi que des Pain and Vitality Domain Scores statistiquement significatifs.
Maladie de Crohn chez l'enfant et l'adolescent
La sécurité et l'efficacité ont été évaluées dans une étude randomisée, effectuée en double aveugle, chez 192 enfants et adolescents de 6 à 17 ans inclus, qui étaient atteints d'une maladie de Crohn (MC) modérée à sévère, définie par un score PCDAI (indice d'activité de la maladie de Crohn) >30.
Cette étude a inclus des patients chez lesquels un traitement conventionnel de la MC (y compris glucocorticoïde et/ou immunosuppresseur) avait échoué; elle a aussi inclus des patients ayant présenté une perte de la réponse clinique ou une intolérance sous infliximab.
Tous les patients ont reçu en ouvert un traitement d'induction dont la dose était adaptée au poids corporel déterminé au début de l'étude: 160 mg la semaine 0 et 80 mg la semaine 2 chez les patients pesant ≥40 kg; 80 mg et 40 mg chez les patients pesant <40 kg.
À 4 semaines, les patients ont été randomisés dans un rapport 1:1 pour recevoir sur la base de leur poids corporel à cette date un traitement normalement dosé ou faiblement dosé. La dose standard était de 20 mg toutes les 2 semaines chez les patients pesant <40 kg et de 40 mg chez les patients pesant ≥40 kg. La dose réduite était de 10 mg toutes les 2 semaines chez les patients pesant <40 kg et de 20 mg chez les patients pesant ≥40 kg.
Résultats d'efficacité
Le critère primaire de l'étude était l'obtention d'une rémission clinique à 26 semaines, définie comme un score PCDAI ≤10.
À 26 semaines, les taux de rémission clinique et de réponse clinique (définie comme une réduction d'au moins 15 points au score PCDAI versus score initial) étaient de 38,7% et de 59,1% respectivement avec la dose standard (n=93), par rapport à 28,4% et 48,4% respectivement avec la dose réduite (n=95). Les différences entre les deux groupes pour les taux de rémission et les taux de réponse clinique à 26 semaines n'étaient pas statistiquement significatives (p=0,075 et p=0,073).
À 52 semaines, les taux de rémission clinique et de réponse clinique étaient de 33,3% et 41,9% respectivement avec la dose standard, par rapport à 23,2% et 28,4% avec la dose réduite. La différence concernant la réponse clinique à 52 semaines était statistiquement significative (p=0,038).
Parmi les patients traités par la dose standard, l'administration de glucocorticoïdes a été arrêtée chez 84,8% à 26 semaines et chez 69,7% à 52 semaines (n=33). À 52 semaines, 30,0% des patients avaient arrêté les immunosuppresseurs (arrêt selon l'appréciation par le médecin investigateur, à 26 semaines ou plus tard, si le patient remplissait le critère d'une réponse clinique) (n=60). La rémission des fistules (définie comme la fermeture de toutes les fistules drainantes présentes au début de l'étude et objectivée lors d'au moins deux visites consécutives au cours de l'étude) était de 46,7% à 26 semaines et de 40,0% à 52 semaines chez les patients traités par la dose standard (n=15).
Parmi les patients traités par la dose réduite, l'administration de glucocorticoïdes a été arrêtée chez 65,8% à 26 semaines et chez 60,5% à 52 semaines. L'arrêt des immunosuppresseurs était de 29,8% à 52 semaines (n=57). La rémission des fistules était de 38,1% à 26 semaines et de 23,8% à 52 semaines chez les patients traités par la dose réduite (n=21).
Pour l'indice de masse corporelle et la taille, des augmentations (améliorations) statistiquement significatives versus valeurs initiales ont été observées à 26 et à 52 semaines dans les deux groupes de traitement.
Des améliorations statistiquement et cliniquement significatives versus valeurs initiales ont également été observées dans les deux groupes de traitement pour les paramètres de la qualité de vie (y compris IMPACT III).
Colite ulcéreuse
La sécurité et l'efficacité ont été examinées chez des patients adultes atteints de colite ulcéreuse active modérée à sévère (score Mayo de 6 à 12, avec sous-score endoscopique de 2 à 3) dans deux études randomisées, en double aveugle, avec contrôle contre placebo. Les patients pouvaient prendre en même temps une médication permanente sous forme d'aminosalicylates, de glucocorticoïdes et/ou d'immunomodulateurs.
L'induction d'une rémission clinique (définie comme un score Mayo ≤2 sans sous-score >1) a été examinée chez 390 patients naïfs d'inhibiteurs du TNF. Ces patients ont été traités aux semaines 0 et 2 par un placebo, par 160 mg et 80 mg d'adalimumab ou par 80 mg et 40 mg d'adalimumab, puis aux semaines 4 et 6 par un placebo ou par 40 mg d'adalimumab. Tous les patients sont ensuite passés à un traitement d'entretien par 40 mg d'adalimumab toutes les 2 semaines.
À la semaine 8, on a pu constater une rémission clinique chez 18% des patients traités avec une dose d'induction de 160 mg/80 mg d'adalimumab, vs 9% des patients ayant reçu le placebo (p=0,031). Aucune supériorité statistiquement significative de l'adalimumab n'a été observée avec la dose d'induction de 80 mg/40 mg (10%, p=0,833).
L'efficacité au cours de la phase d'induction et de la phase d'entretien (52 semaines au total) a été évaluée chez 248 patients traités par 160 mg/80 mg/40 mg toutes les 2 semaines versus 246 patients sous placebo. Une rémission à 8 et à 52 semaines a été constatée chez 16,5% (p=0,019) et 17,3% (p=0,004) des patients sous adalimumab vs 9,3% et 8,5% des patients sous placebo. Les taux de réponse persistante, de rémission et de guérison de la muqueuse sont résumés dans le tableau 14.
Tableau 14: Taux de réponse persistante, de rémission et de guérison de la muqueuse dans l'étude UC II, pourcentage de patients (intervalle de confiance à 95%)
|
|
Placebo et intervalle de confiance à 95%
|
40 mg d'adalimumab toutes les deux semaines et intervalle de confiance à 95%
| |
Semaines 8 et 52
| |
Réponse persistante
|
12% (IC: 8,1 à 16,3)
|
24%** (IC: 18,5 à 29,1)
| |
Rémission persistante
|
4% (IC: 1,6 à 6,5)
|
8%* (IC: 5,0 à 11,9)
| |
Guérison durable de la muqueuse
|
11% (IC: 6,7 à 14,4)
|
19%* (IC: 13,7 à 23,4)
|
a Intervalle de confiance pour la proportion, sur la base d'une approximation de la distribution binomiale par loi normale
La rémission clinique signifie un score Mayo ≤2 sans aucun sous-score >1
* p <0,05 pour adalimumab versus placebo
** p <0,001 pour adalimumab versus placebo
La guérison de la muqueuse signifie un sous-score endoscopique de 0 ou de 1
Une réponse signifie un score Mayo réduit de ≥3 points et inférieur de ≥30% au score initial ainsi qu'un sous-score d'hémorragies rectales de 0 ou de 1 ou réduit de ≥1 point versus valeur initiale.
Sur les 125 patients ayant atteint une réponse à 8 semaines, 59 (47%) étaient encore répondeurs à 52 semaines, 36 (29%) étaient en rémission, 51 (41%) présentaient une guérison de la muqueuse et 18 (20% des 90 patients répondeurs à 8 semaines et initialement sous corticostéroïdes) étaient en rémission sans utilisation de corticostéroïdes depuis ≥90 jours.
Une réduction statistiquement significative des taux d'hospitalisations de toutes causes et des taux d'hospitalisations imputables à la colite ulcéreuse a été observée dans l'analyse incluant les deux études UC.
Le traitement anti-TNF par l'infliximab avait échoué auparavant chez presque 40% des patients de l'étude UC II. L'efficacité de l'adalimumab était plus faible chez ces patients que chez les patients naïfs de traitements par anti-TNF. Dans ce sous-groupe, une rémission à 52 semaines a été atteinte dans 10% des patients sous adalimumab vs 3% sous placebo (p=0,039).
Les patients des études UC I et II ont pu continuer à être traités dans le cadre de l'étude à long terme en ouvert (UC III). 3 ans après le traitement par l'adalimumab, 75% (301/402) étaient encore en rémission clinique d'après le score Mayo partiel.
Une amélioration de la qualité de vie, évaluée à l'aide du score IBDQ total (Inflammatory Bowel Disease Questionnaire) spécifique de la maladie, était atteinte à 52 semaines vs placebo (p=0,007).
Spondylarthrite ankylosante (maladie de Bechterew)
L'efficacité de 40 mg d'adalimumab toutes les deux semaines en injection sous-cutanée a été étudiée dans deux études randomisées, en double aveugle, contrôlées contre placebo pendant 24 semaines chez 393 patients atteints de spondylarthrite ankylosante active (score d'activité de la maladie [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)]) >4 (valeurs initiales moyennes de 6,3 aussi bien dans le groupe adalimumab que dans le groupe placebo) n'ayant pas répondu suffisamment aux traitements conventionnels. 79 patients (20,1%) ont reçu simultanément un agent antirhumatismal de fond et 37 patients (9,4%) des glucocorticoïdes. La période en aveugle fut suivie d'une phase d'extension en ouvert pendant laquelle les patients ont reçu 40 mg d'adalimumab par injection sous-cutanée toutes les deux semaines pendant une durée maximale de 28 semaines supplémentaires.
Dans l'étude de plus grande ampleur avec 315 patients, les résultats ont indiqué des améliorations statistiquement significatives des signes et des symptômes de la spondylarthrite ankylosante chez les patients ayant reçu de l'adalimumab, par rapport aux patients sous placebo. Une réponse significative a été constatée pour la première fois après 2 semaines de traitement et s'est maintenue jusqu'à la semaine 24.
Les taux de réponse selon l'Assessment in Ankylosing Spondylitis (ASAS) 20/50/70 ont été atteints à la semaine 12 chez 58%, 38% et 23% des patients sous adalimumab contre 21%, 10% et 5% des patients sous placebo (p<0,001 adalimumab versus placebo). Une réponse relativement comparable a été constatée à la semaine 24.
Comme constaté avec le BASDAI, le traitement par l'adalimumab a entraîné une amélioration des signes et symptômes. Chez 45% des patients traités par l'adalimumab, une réduction d'au moins 50% des valeurs initiales du BASDAI a été atteinte à la semaine 12, comparé aux 16% des patients traités par le placebo (p<0,01). Des résultats comparables ont été constatés à la semaine 24.
De plus, la diminution moyenne des taux initiaux de protéine C réactive (CRP) à la semaine 12 était plus importante avec le traitement par l'adalimumab (-1,3 mg/dl) qu'avec le placebo (-0,1 mg/dl), (p<0,001).
Des résultats comparables (non tous statistiquement significatifs) ont été constatés dans une étude plus réduite, randomisée, en double aveugle, contrôlée contre placebo, chez 82 patients adultes atteints de spondylarthrite ankylosante active.
Dans les études portant sur la spondylarthrite ankylosante, les résultats rapportés par les patients ont été évalués à l'aide du Generic Health Status Questionnaire Short Form-36 (SF 36) et du Disease Specific Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL). Des améliorations significatives plus importantes de l'ASQoL et de la composante physique du SF 36 ont été observées à la semaine 12 chez les patients traités par l'adalimumab par rapport aux patients du groupe placebo, et se sont maintenues jusqu'à la semaine 24.
Psoriasis
L'efficacité et la sécurité de l'adalimumab ont été examinées au cours d'études randomisées, en double aveugle, contrôlées, menées chez plus de 1600 patients atteints de psoriasis en plaques chronique modéré à sévère, ayant 18 ans ou plus et candidats à un traitement systémique ou une photothérapie.
Au cours de l'étude 1, 1212 patients atteints de psoriasis en plaques chronique sur ≥10% de leur surface corporelle (Body Surface Area, BSA) et présentant un score PASI (Psoriatic Area and Severity Index) ≥12, ont été évalués sur trois périodes de traitement. Pendant la période A, les patients ont reçu par voie sous-cutanée un placebo ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1. Les patients ayant obtenu au bout de 16 semaines de traitement au minimum une réponse PASI 75 (définie par une amélioration d'au moins 75% du score PASI par rapport à la valeur initiale) ont poursuivi leur traitement en ouvert pendant la période B supplémentaire. Ils ont reçu 40 mg d'adalimumab toutes les deux semaines. Après 17 semaines de traitement en ouvert, les patients ayant conservé au minimum une réponse PASI 75 à la semaine 33 et ayant reçu un traitement actif pendant la période A, sont passés à la période de traitement C. Ils ont reçu durant 19 semaines supplémentaires 40 mg d'adalimumab ou un placebo toutes les 2 semaines. Le score PASI initial moyen était de 18,9 pour tous les groupes de traitement. Le score PGA (Physician's Global Assessment) initial pour tous les groupes allait de «modéré» (52,6%) à «sévère» (41,3%) et «très sévère» (6,1%).
L'étude 2 a comparé l'efficacité et la sécurité de l'adalimumab versus méthotrexate et placebo chez 271 patients atteints de psoriasis en plaques chronique avec un BSA de 10% et un score PASI ≥10. Pendant 16 semaines, les patients ont reçu un placebo, du méthotrexate (7,5-20 mg) ou une dose initiale de 80 mg d'adalimumab par voie sous-cutanée à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1. Le score PASI initial moyen était de 19,7 pour tous les groupes de traitement. Le score PGA initial allait pour tous les groupes de «léger» (0,4%) à «modéré» (47,8%), «sévère» (45,6%) et «très sévère» (6,3%).
1469 patients provenant des études de phase II et III ont été inclus dans une étude d'extension ouverte à 3 périodes, comportant une période de poursuite du traitement (104-252 semaines), une période d'interruption du traitement (jusqu'à une récidive ou au maximum de 52 semaines), puis une période de reprise du traitement (16 semaines).
Dans l'étude 3, 148 patients atteints de psoriasis en plaques chronique avec un BSA ≥5% ont été évalués pendant au moins 1 an. Les patients ont reçu un placebo ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1, ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab chaque semaine à partir de la semaine 1.
Résultats cliniques
Le critère d'évaluation principal des études 1, 2 et 3 était le pourcentage de patients ayant atteint à la semaine 16 (études 1 et 2) ou à la semaine 12 (étude 3) une réduction de leur score PASI d'au moins 75% par rapport à la valeur initiale (PASI 75). Les études 1–3 menées sur le psoriasis ont également porté entre autres sur le score PGA et d'autres valeurs PASI.
Étude 1: en plus du critère d'évaluation principal ci-dessus, l'étude 1 avait comme second critère d'évaluation principal la perte d'une réponse appropriée à la semaine 33 et pendant ou avant la semaine 52. La perte d'une réponse appropriée était définie comme une réponse < PASI 50 par rapport à la valeur initiale avec un minimum d'augmentation de 6 points du score PASI par rapport à la semaine 33.
Des données contrôlées portant sur un traitement par l'adalimumab versus placebo sont disponibles pour une durée de traitement de 52 semaines. Dans une étude comparative contrôlée contre placebo, menée auprès des patients ayant obtenu une disparition prolongée des troubles jusqu'à la semaine 33 sous adalimumab, 95,1% des patients ayant poursuivi le traitement par l'adalimumab sont restés sans récidive jusqu'à la semaine 52, contre 71,6% des patients sous placebo (c.-à-d. après l'arrêt de l'administration de l'adalimumab à la semaine 33).
Parmi les patients présentant une diminution de réponse appropriée après la re-randomisation dans le groupe placebo et ayant ensuite été inclus dans l'étude d'extension en ouvert, 38% (25/66) et 55% (36/66) ont retrouvé une réponse PASI 75 après respectivement 12 et 24 semaines de thérapie active.
Cette réponse tardive après récidive est probablement liée à une évolution sévère du psoriasis dans ce sous-groupe de patients.
Dans les études 1 et 2 sur le psoriasis, plus de patients traités par l'adalimumab que de patients sous placebo ont atteint à la semaine 16 une réduction d'au moins 75% par rapport au score PASI initial. D'autres paramètres cliniques significatifs incluant le score PASI 100 (p.ex. disparition complète des signes cutanés du psoriasis) et le score PGA «clair ou minime» ont été également améliorés par rapport au placebo.
Étude 2: Dans l'étude 2 sur le psoriasis, les patients recevant l'adalimumab ont présenté de meilleurs résultats pour les scores PASI 75, PASI 100 et PGA «clair ou minime» que les patients sous méthotrexate.
Tableau 15: Étude 1 sur le psoriasis - Efficacité à la semaine 16 (% de patients)
|
|
Placebo
N=398
|
40 mg d'adalimumab 1 sem./2
N=814
| |
≥PASI 75
|
6,5
|
70,9a
| |
PASI 100
|
0,8
|
20,0a
| |
PGA: clair/minime
|
4,3
|
62,2a
|
a p<0,001, adalimumab vs placebo
Tableau 16: Étude 2 sur le psoriasis - Efficacité à la semaine 16 (% de patients)
|
|
Placebo
N=53
|
MTX
N=110
|
40 mg d'adalimumab 1 sem./2
N=108
| |
≥PASI 75
|
18,9
|
35,5
|
79,6a, b
| |
PASI 100
|
1,9
|
7,3
|
16,7a, b
| |
PGA: clair/minime
|
11,3
|
30,0
|
73,1 a, b
|
a p<0,001, adalimumab vs placebo
b p<0,001 adalimumab vs méthotrexate
Étude de prolongation: au total, 233 patients qui avaient obtenu une réponse PASI-75 à la semaine 16 et à la semaine 33 et avaient reçu dans l'étude 1 sur le psoriasis un traitement continu par l'adalimumab pendant 52 semaines ont poursuivi le traitement par l'adalimumab dans l'étude de prolongation ouverte. La réponse PASI-75 ou la réponse PGA définie comme un score PGA «clair» ou «minime» ont été respectivement de 74,7% et 59,0% chez ces patients après 108 semaines supplémentaires de traitement en ouvert (au total 160 semaines). Dans une analyse de Non Responder Imputation (NRI), dans laquelle tous les patients qui avaient interrompu leur participation à l'étude en raison d'effets indésirables ou d'absence d'efficacité ou chez lesquels la dose avait été augmentée, ont été considérés comme non-répondeurs, la réponse PASI 75 et la réponse PGA définie comme un score PGA «clair» ou «minime» ont été respectivement de 69,6% et 55,7% chez ces patients après 108 semaines supplémentaires de poursuite du traitement en ouvert (au total 160 semaines).
Dans l'étude de prolongation, 347 patients ayant présenté une réponse durable ont participé à une évaluation de l'interruption du traitement et de la reprise du traitement. Pendant la période d'interruption du traitement, les symptômes psoriasiques sont réapparus chez 54,2% (188/347) des patients en moyenne en l'espace d'environ 5 mois (diminution du score PGA à «modéré» ou plus grave). Aucun de ces patients n'a connu de phénomène de rebond pendant la période d'interruption. Au total, 76,5% (218/285) des patients ayant participé à la période ultérieure de reprise du traitement ont présenté une réponse PGA définie comme un score PGA «clair» ou «minime» 16 semaines après la reprise du traitement, qu'ils aient eu une récidive pendant l'arrêt (69,1% [123/178] ou non 88,8% [95/107]). Le profil de sécurité observé pendant la période de reprise du traitement a été similaire à celui observé avant l'interruption du traitement.
Étude 3: Les résultats de l'étude 3 sur le psoriasis confirment l'efficacité démontrée dans les études 1 et 2. Les patients de l'étude 1 ayant présenté une réponse PASI 75 et ayant été re-randomisés à la semaine 33 dans le groupe adalimumab, ont présenté avant ou à la semaine 52 une diminution de la réponse appropriée inférieure à celle présentée par les patients re-randomisés dans le groupe placebo (4,9% vs 28,4%, p<0,001).
Chez les patients d'une étude d'extension en ouvert qui avaient subi un échec thérapeutique secondaire – c'est-à-dire qui ne présentaient plus de réponse PASI 50 versus valeur initiale après avoir atteint auparavant une réponse au moins partielle – et chez lesquels la dose de 40 mg toutes les deux semaines a été augmentée à 40 mg par semaine, une réponse PASI 75 a été observée à 12 et à 24 semaines chez 28,3% (63/223) et 39,5% (88/223) respectivement.
Qualité de vie
Les résultats rapportés par les patients (Patient Reported Outcomes, PRO) ont été évalués à l'aide de divers paramètres. La qualité de vie a été déterminée au cours des études 1 et 2 à l'aide de l'indice spécifique de la maladie DLQI (Dermatology Life Quality Index). Dans l'étude 1, les patients sous adalimumab ont présenté aux semaines 4 et 16 des améliorations du score total DLQI, du degré de la maladie, des douleurs et du prurit en comparaison avec les patients sous placebo. Les résultats DLQI ont été maintenus jusqu'à la semaine 52.
Dans l'étude 2, les patients sous adalimumab ont présenté à la semaine 16 des améliorations du score total DLQI, du degré de la maladie et du prurit en comparaison avec les patients sous placebo ou sous méthotrexate, ainsi que des améliorations cliniquement significatives des douleurs comparativement aux patients sous placebo.
La qualité de vie générale en rapport avec l'état de santé a été définie à l'aide du questionnaire Short Form Health Survey (SF 36) dans l'étude 1. Les patients sous adalimumab ont présenté une amélioration significativement plus importante des scores résumés physique (Physical Component Summary, PCS) et mental (Mental Component Summary, MCS) du SF 36.
Psoriasis chez l'enfant et l'adolescent
L'efficacité d'adalimumab a été évaluée dans une étude randomisée, en double aveugle, contrôlée, auprès de 114 patients pédiatriques à partir d'un âge de 4 ans qui étaient atteints de formes sévères de psoriasis en plaques chronique et chez lesquels un traitement topique et une héliothérapie ou photothérapie s'étaient avérés insuffisants (les formes sévères de psoriasis en plaques chroniques sont définies comme suit: PGA ≥4 ou atteinte de >20 % de la surface corporelle (BSA, body surface area) ou atteinte de >10 % de BSA avec présence de lésions très épaisses ou PASI ≥20 ou PASI ≥10 avec atteinte cliniquement significative du visage, de la zone génitale ou des mains/pieds).
Les patients ont reçu adalimumab 0,8 mg/kg (sans dépasser 40 mg) toutes les deux semaines, 0,4 mg/kg (sans dépasser 20 mg) toutes les deux semaines ou du méthotrexate par voie orale à raison de 0,1 à 0,4 mg/kg (sans dépasser 25 mg) une fois par semaine. Aucune comparaison n'a été faite avec le MTX à haute dose administré par voie sous-cutanée.
La distribution des âges des patients est présentée dans le tableau 17.
Tableau 17: Distribution des patients par âges (patients assignés par randomisation au traitement par adalimumab 0,8 mg/kg toutes les deux semaines)
|
Groupes d'âge (ans)
|
Nombre de patients au début de l'étude, n (%)
| |
>6 à 9
|
7 (18,4)
| |
>9 à 12
|
8 (21,1)
| |
>12 à 15
|
13 (34,2)
| |
>15
|
10 (26,3)
|
17 (45 %) des patients avaient reçu auparavant un traitement systémique (étanercept et/ou agents non biologiques tels qu'acitrétine, ciclosporine, méthotrexate, etc). Les patients non-répondeurs aux traitements antérieurs par le méthotrexate étaient exclus de cette étude.
Les critères d'évaluation primaires correspondaient à un PASI 75 et à un score PGA «sans»/«minime». À 16 semaines, davantage de patients assignés par randomisation au groupe traité par adalimumab 0,8 mg/kg présentaient des preuves supérieures d'efficacité par rapport aux patients assignés au groupe traité par le MTX.
Tableau 18: Résultats d'efficacité à 16 semaines contre le psoriasis en plaques
|
|
MTXa
N=37
|
Adalimumab 0,8 mg/kg toutes les deux semaines
N=38
| |
PASI 75b
|
12 (32,4%)
|
22 (57,9%)
| |
PGA: sans/minimiec
|
15 (40,5%)
|
23 (60,5%)
| |
a
MTX=méthotrexate
b p=0,027, Adalimumab 0,8 mg/kg versus MTX
c p=0,083, Adalimumab 0,8 mg/kg versus MTX
|
On ne dispose pas de données comparatives versus méthotrexate au-delà de 16 semaines.
Chez les patients ayant atteint un PASI 75 et un score PGA «sans» ou «minime», le traitement a été interrompu pour une durée allant jusqu'à 36 semaines. Ces patients ont été observés quant à la survenue d'une récidive (perte de la réponse d'après le PGA). Le traitement des patients a ensuite été repris pour 16 semaines de plus avec 0,8 mg d'adalimumab par kg toutes les deux semaines. Les taux de réponse observés après la reprise du traitement étaient: 78,9 % (15/19) des patients avec une réponse PASI 75 et 52,6 % (10/19) des patients avec un score PGA «sans» ou «minime».
Dans la phase d'extension en ouvert de l'étude, une réponse PASI 75 et un score PGA «sans» ou «minime» se sont maintenus jusqu'à 52 semaines de plus. Il n'y a eu aucun élément nouveau concernant la sécurité.
Hidradénite suppurée
L'efficacité et la sécurité d'adalimumab ont été évaluées dans des études randomisées effectuées en double aveugle avec contrôle versus placebo et dans une étude d'extension en ouvert auprès de 727 patients adultes atteints d'hidradénite suppurée modérée à sévère. Les patients présentaient une contre-indication, une réponse insuffisante ou une intolérance à une antibiothérapie systémique et leur maladie était de stade II ou III selon la classification de Hurley, avec au moins 3 abcès ou nodules enflammés.
Deux études randomisées de phase III (HS-I et -II) effectuées en double aveugle avec contrôle versus placebo, totalisant 633 patients adultes, comprenaient chacune une phase initiale de 12 semaines de traitement en double aveugle (période A) suivie d'une période de traitement de 24 semaines en double aveugle (période B). Dans la période A, les patients ont reçu adalimumab (160 mg la semaine 0, puis 80 mg la semaine 2 et 40 mg par semaine au cours des semaines 4 à 11), ou un placebo. Au bout de 12 semaines, les patients ayant reçu adalimumab pendant la période A ont été à nouveau randomisés pour la période B, durant laquelle ils ont reçu, jusqu'à la semaine 35, adalimumab à 40 mg par semaine, adalimumab à 40 mg toutes les 2 semaines ou un placebo. Les patients ayant reçu un placebo pendant la période A ont été randomisés pour la période B, durant laquelle ils ont reçu adalimumab à 40 mg par semaine (HS-I) ou un placebo (HS-II).
Un traitement concomitant par un antibiotique oral était autorisé dans l'étude HS-II.
Les patients des deux études HS ont pu participer à une étude d'extension en ouvert avec administration hebdomadaire d'adalimumab à 40 mg. Chez toutes les populations recevant de l'adalimumab, l'exposition moyenne était de 762 jours. Dans les 3 études, les patients effectuaient quotidiennement des nettoyages avec une solution topique antiseptique.
Efficacité clinique
La réduction des lésions inflammatoires et la prévention de l'aggravation des abcès et des fistules drainantes ont été évaluées à l'aide du score HiSCR (Hidradenitis Suppurativa Clinical Response; réduction d'au moins 50% du nombre de tous les abcès et nodules inflammatoires sans augmentation du nombre d'abcès ou de fistules drainantes versus valeurs initiales).
À 12 semaines, on a pu constater dans les deux études (HS-I et -II) qu'un pourcentage significativement plus élevé de patients traités par adalimumab que de patients sous placebo avaient atteint une réponse HiSCR. Dans l'étude HS-II, un pourcentage significativement plus élevé de patients sous adalimumab ont présenté une réduction significative des douleurs cutanées liées à l'HS (voir le tableau 19). De plus, le risque de nouvelle poussée de la maladie au cours du traitement initial de 12 semaines était significativement réduit chez les patients traités par adalimumab.
Tableau 19: Études HS-I et -II, efficacité à 12 semaines
|
|
Étude HS-I
|
Étude HS-II
| |
Placebo
|
Adalimumab 40 mg par semaine
|
Placebo
|
Adalimumab 40 mg par semaine
| |
Efficacité clinique du traitement de l'hidradénite suppurée (HiSCR) a
|
N=154
40 (26,0%)
|
N=153
64 (41,8%)*
|
N=163
45 (27,6%)
|
N=163
96 (58,9%)***
|
* p<0,05, *** P<0,001, adalimumab vs placebo
a Sur l'ensemble des patients randomisés.
Chez les patients traités par adalimumab une fois par semaine, le taux de réponse HiSCR total s'est maintenu jusqu'à la semaine 96.
Uvéite
La sécurité et l'efficacité d'adalimumab ont été évaluées dans le cadre de deux études randomisées, en double aveugle, avec contrôle versus placebo (études UV 1 [M10-877] et UV 2 [M10-880]) sur une durée maximale de 80 semaines chez des patients adultes atteints d'uvéite non infectieuse de localisation intermédiaire/postérieure/panuvéite (ou «uvéite non infectieuse de la partie postérieure de l'œil»). Les patients atteints uniquement d'uvéite antérieure étaient exclus. Les patients ont reçu soit un placebo soit adalimumab (dose initiale de 80 mg, puis 40 mg toutes les deux semaines à partir de la semaine suivante).
Une co-médication par un immunosuppresseur conventionnel (ciclosporine A, méthotrexate, mycophénolate mofétil, azathioprine, tacrolimus) à dose stable était autorisée. Dans les deux études, le critère d'efficacité primaire était «le temps jusqu'à un échec thérapeutique». L'échec thérapeutique était défini en tant que critère composite incluant les éléments suivants: 1) lésions vasculaires chorio-rétiniennes inflammatoires et/ou rétiniennes inflammatoires, 2) grade des cellules de la chambre antérieure, 3) degré d'opacification du corps vitré (vitreous haze) et 4) la meilleure acuité visuelle après correction (BCVA).
L'étude UV 1 a inclus 217 patients souffrant d'uvéite active malgré une corticothérapie (prednisone orale à raison de 10 à 60 mg par jour). Tous les patients recevaient lors de leur inclusion à l'étude une dose standardisée de prednisone de 60 mg par jour, avec schéma obligatoire de réduction de la dose jusqu'à l'arrêt complet de la corticothérapie à la semaine 15.
L'étude UV 2 a inclus 226 patients présentant une uvéite inactive qui exigeait une corticothérapie (prednisone orale à raison de 10 à 35 mg par jour) au début de l'étude. Le traitement par le médicament à l'étude a été commencé parallèlement à la poursuite de la corticothérapie en cours, après quoi la dose du corticostéroïde a été réduite progressivement jusqu'à l'arrêt complet de la corticothérapie à la semaine 19.
Réponse clinique
Les résultats des deux études montrent de façon statistiquement significative que le temps écoulé jusqu'au premier échec thérapeutique était plus long chez les patients traités par l'adalimumab que chez les patients sous placebo (voir le tableau 20). De même, les deux études ont mis en évidence une efficacité rapide et persistante d'adalimumab versus placebo pour retarder le premier échec thérapeutique (voir la figure 3).
Tableau 20: Temps écoulé jusqu'au premier échec thérapeutique dans les études UV 1 et 2
|
Traitement évalué
|
N
|
Échec
N (%)
|
Délai médian (en mois)
jusqu'à l'échec
|
HRa
|
IC à 95%
pour HRa
|
Valeur pb
| |
Temps écoulé jusqu'à un échec thérapeutique pendant ou après la semaine 6 dans l'étude UV 1
| |
Analyse principale (ITT)
| |
Placebo
|
107
|
84 (78,5)
|
3,0
|
--
|
--
|
--
| |
Adalimumab
|
110
|
60 (54,5)
|
5,6
|
0,50
|
0,36; 0,70
|
<0,001
| |
Temps écoulé jusqu'à un échec thérapeutique pendant ou après la semaine 2 dans l'étude UV 2
| |
Analyse principale (ITT)
| |
Placebo
|
111
|
61 (55,0)
|
8,3
|
--
|
--
|
--
| |
Adalimumab
|
115
|
45 (39,1)
|
n.e.c
|
0,57
|
0,39; 0,84
|
0,004
|
Remarque: Un échec thérapeutique pendant ou après la semaine 6 dans l'étude UV 1 ou un échec thérapeutique pendant ou après la semaine 2 dans l'étude UV 2 n'a pas été pris en compte en tant qu'événement. Les arrêts prématurés dus à d'autres raisons qu'un échec thérapeutique ont été censurés lors de leur survenue.
a HR pour l'adalimumab vs placebo à partir de la régression en modèle à risques proportionnels, avec le traitement en tant que facteur.
b Valeur p bilatérale à partir du test du log-rank.
c n.e. = non estimable. Moins de la moitié des patients entrant en ligne de compte ont eu un événement au cours de la durée d'étude de 80 semaines.
Figure 3: Courbes de Kaplan-Meier pour le temps écoulé jusqu'au premier échec thérapeutique pendant ou après la semaine 6 dans l'étude UV 1 ou pendant ou après la semaine 2 dans l'étude UV 2
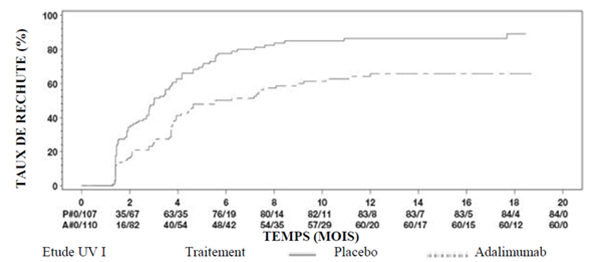
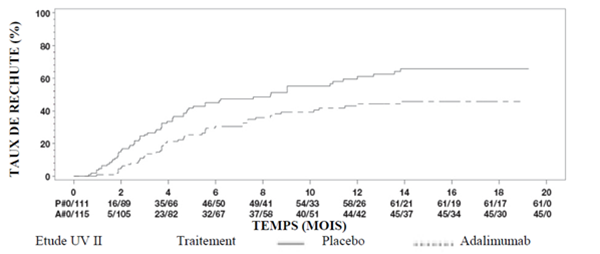
Une analyse de sensibilité du critère primaire, comptabilisant comme échecs thérapeutiques les patients avec arrêt prématuré du traitement et les patients avec une co-administration interdite de corticostéroïdes, a révélé un effet puissant du traitement en faveur d'adalimumab (UV I: HR = 0,66; IC à 95%: 0,48 à 0,88, p = 0,005; UV II: HR = 0,59; IC à 95%: 0,42 à 0,81; p = 0,001).
Dans les deux études, tous les éléments du critère primaire ont contribué de façon cumulative à la différence totale entre le groupe sous adalimumab et le groupe sous placebo.
Dans l'étude UV 1, des différences statistiquement significatives ont été constatées en faveur de l'adalimumab pour la variation du nombre de cellules dans la chambre antérieure, le grade d'opacification du corps vitré et l'acuité visuelle (logMAR BCVA) (variation moyenne par rapport à la meilleure valeur atteinte avant la semaine 6, analyse LOCF; valeurs p: 0,011; <0,001 et 0,003).
Les résultats de l'étude UV II vont en tendance dans le même sens sans être statistiquement significatifs.
Dans l'étude UV I, l'évolution moyenne concernant le grade des cellules de la chambre antérieure, le degré d'opacification du corps vitré et l'acuité visuelle (logMAR BCVA) était caractérisée par des améliorations similaires dans les deux bras de l'étude pendant les 4 à 6 premières semaines de traitement avec réduction progressive de la corticothérapie concomitante. À la suite de cette période, l'augmentation du degré d'inflammation et la perte d'acuité visuelle étaient plus faibles sous adalimumab que sous placebo. Dans l'étude UV II, l'augmentation du degré d'inflammation et la perte d'acuité visuelle étaient également plus faibles que sous placebo.
Sur un total de 417 participants inclus à l'étude d'extension à long terme non contrôlée des études UV I et UV II, 46 participants ont été exclus de l'analyse primaire de l'efficacité, pour laquelle ils avaient été jugés inappropriés (p.ex. développement de complications secondaires à une rétinopathie diabétique, opération d'une cataracte ou vitrectomie). Sur les 371 patients restants, 276 patients analysables ont poursuivi le traitement en ouvert par l'adalimumab jusqu'à la semaine 78. D'après les données enregistrées, 222 (80,4%) des patients recevant en même temps des corticostéroïdes à raison de ≤7,5 mg par jour ont atteint un contrôle de la maladie (aucune lésion inflammatoire active, grade des cellules de la chambre antérieure ≤0,5+, grade d'opacification du corps vitré ≤0,5+), tandis que 184 (66,7%) des patients ont atteint un contrôle de la maladie sans utilisation de corticostéroïdes. À 78 semaines, BCVA était améliorée ou inchangée chez 88,4% des yeux (réduction de <5 lettres). Parmi les patients ayant quitté l'étude avant la semaine 78, cette décision était due à des effets indésirables chez 11% et à une réponse insuffisante à l'adalimumab chez 5%.
Qualité de vie
Dans l'étude UV 1, le traitement par l'adalimumab a permis de préserver la fonction dépendante de la capacité visuelle et la qualité de vie en fonction de la santé (évaluée à l'aide du NEI VFQ-25).
Étude sur la polyarthrite rhumatoïde (PR) comparant AMGEVITA et le médicament de référence
L'efficacité et la sécurité d'AMGEVITA ont été évaluées comparativement au médicament de référence au cours de l'étude comparative randomisée, en double aveugle, contrôlée contre traitement actif (étude 1 RA-ABP), menée auprès de 526 patients ≥18 ans atteints de polyarthrite rhumatoïde active modérée à sévère et présentant une réponse insuffisante au méthotrexate. Les patients ont reçu 40 mg d'AMGEVITA ou la même quantité du médicament de référence par voie sous-cutanée toutes les deux semaines pendant jusqu'à 22 semaines.
Le critère d'évaluation principal, la réponse ACR20, s'est situé dans la plage d'équivalence prédéfinie et a montré l'équivalence clinique entre AMGEVITA et le médicament de référence.
Le profil de sécurité général d'AMGEVITA ressemble à celui du médicament de référence.
Étude sur le psoriasis en plaques (Ps) comparant AMGEVITA et le médicament de référence
L'efficacité et la sécurité d'AMGEVITA ont été évaluées comparativement au médicament de référence au cours d'une étude randomisée, en double aveugle, contrôlée contre traitement actif (étude 1 Ps-ABP) menée auprès de 350 patients ≥18 ans atteints de psoriasis en plaques modéré à sévère chez lesquels une thérapie systémique ou une photothérapie était envisagée. Les patients ont reçu soit AMGEVITA, soit le médicament de référence, à la dose initiale ou de charge de 80 mg par voie sous-cutanée à la semaine 1/au jour 1, puis 40 mg par voie sous-cutanée toutes les deux semaines, en commençant une semaine après l'administration de la dose initiale. L'amélioration du score PASI (en pourcentage) par rapport à la valeur initiale à la semaine 16 s'est située dans la plage d'équivalence prédéfinie et a donc démontré l'équivalence clinique entre AMGEVITA et le médicament de référence.
|