CompositionPrincipes actifs
Galcanezumab (anticorps monoclonal recombinant humanisé produit sur des cellules CHO [ovaire de hamster chinois]).
Excipients
Histidine, chlorhydrate d'histidine monohydraté, chlorure de sodium, polysorbate 80, eau pour préparations injectables.
Quantité totale de sodium: 1 ml (resp. 1 stylo prérempli) contient 3.5 mg de sodium.
Indications/Possibilités d’emploiTraitement prophylactique de la migraine chez l'adulte, pour autant qu'il soit indiqué.
Posologie/Mode d’emploiL'indication pour le traitement doit être posée par un médecin expérimenté dans le domaine du traitement de la migraine, qui doit ensuite accompagner le traitement.
Afin d'assurer la traçabilité des médicaments biologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie usuelle
La dose recommandée est de 120 mg en injection sous-cutanée une fois par mois, avec au début du traitement une dose initiale unique de 240 mg (2 injections).
Les patients doivent être informés de ratrapper une dose oubliée dès que possible et de reprendre ensuite l'administration mensuelle à partir de la date de la dernière injection.
En cas d'absence de réponse au traitement ou au-moins une fois par année, il faut procéder à une réévaluation de la nécessité de poursuivre le traitement.
Les données relatives à la sécurité et à l'efficacité au-delà de 12 mois sont limitées.
Emgality est injecté par voie sous-cutanée dans l'abdomen ou la cuisse par le patient lui-même, ou dans l'arrière du bras ou dans la fesse par un professionnel de santé ou un soignant formé à l'utilisation du stylo prérempli.
Si plusieurs injections sont administrées dans une même région du corps, on évitera de faire les injections au même endroit. Emgality ne doit pas être injecté à des endroits où la peau est sensible, lésée, rougie ou durcie.
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. Emgality n'a pas été étudié chez des patients atteints d'insuffisance rénale sévère (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique
Emgality n'a pas été étudié chez des patients atteints d'insuffisance hépatique. Aucun ajustement de la dose n'est recommandé chez les patients atteints d'insuffisance hépatique car le galcanezumab, une immunoglobuline G humaine, n'est pas métabolisé par les enzymes du cytochrome P450 et sa clairance ne se fait pas de manière significative par le foie (voir «Pharmacocinétique»).
Patients âgés
Les informations chez les patients âgés de 65 ans et plus sont limitées. Il n'est pas recommandé d'ajuster la dose chez les personnes âgées car on ne dispose pas de données suffisantes pour déterminer si ces patients réagissent différemment de patients plus jeunes.
Enfants et adolescents
La sécurité et l'efficacité d'Emgality chez l'enfant et l'adolescent n'ont pas été étudiées. Emgality ne doit donc pas être utilisé dans cette classe d'âge.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients, selon la composition.
Mises en garde et précautionsHypersensibilité sévère
Des réactions d'hypersensibilité sévère, y compris des cas d'anaphylaxie, d'angioedème et d'urticaire, ont été rapportées. En cas de réaction d'hypersensibilité sévère, l'administration d'Emgality doit être immédiatement arrêtée et un traitement approprié doit être instauré. Des réactions d'hypersensibilité sévères peuvent survenir plusieurs jours après l'administration et persister.
Risque cardiovasculaire
Les patients souffrant de certaines maladies cardiovasculaires graves ont été exclus des études cliniques (voir «Efficacité clinique»), c'est pourquoi aucune donnée de sécurité n'est disponible pour cette population
Population pédiatrique (moins de 18 ans)
La sécurité et l'efficacité d'Emgality chez l'enfant et l'adolescent n'ont pas été étudiées. Emgality ne doit donc pas être utilisé dans cette classe d'âge.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par 1 ml (resp. par stylo prérempli), c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsAucune étude d'interaction n'a été réalisée.
Aucune interaction pharmacocinétique n'est attendue en raison des caractéristiques du galcanezumab.
Grossesse, allaitementGrossesse
On dispose de données insuffisantes sur l'utilisation d'Emgality chez la femme enceinte. Des études de toxicité sur le développement menées chez des lapines et des rates gestantes n'ont pas révélé de signes d'atteinte foetale. On sait que l'immunoglobuline humaine (IgG) traverse la barrière placentaire; par conséquent, le galcanezumab peut être transmis de la mère au foetus en développement. Emgality ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
On ignore si Emgality est excrété dans le lait maternel. Comme de nombreux médicaments, notamment des anticorps, sont excrétés dans le lait maternel, un risque pour le nouveau-né/l'enfant en bas âge ne peut être exclu. Il faut par conséquent décider soit d'arrêter l'allaitement, soit de renoncer au traitement par Emgality, en tenant compte du bénéfice du traitement avec Emgality pour la mère et du bénéfice de l'allaitement pour le nourrisson.
Fertilité
On ne dispose pas de données sur l'effet d'Emgality sur la fertilité humaine. Les études réalisées chez l'animal n'ont pas mis en évidence d'effets délétères sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été réalisée à ce sujet. Emgality peut avoir une influence sur l'aptitude à conduire des véhicules et à utiliser des machines. Des vertiges peuvent survenir suite à l'administration de galcanezumab (voir «Effets indésirables»).
Effets indésirablesLes effets indésirables les plus fréquemment rapportés ont été des douleurs au site d'injection et des réactions au site d'injection avec 240 mg. La majorité des événements liés au site d'injection (érythème, prurit, ecchymose, gonflement) ont été d'intensité légère à modérée et n'ont pas entraîné l'arrêt du traitement avec Emgality. La majorité des réactions au site d'injection ont été rapportées le premier jour de traitement et ont généralement disparu en quelques jours. Chez la plupart des patients ayant rapporté une douleur au site d'injection, l'événement est survenu dans l'heure suivant l'injection et a disparu le même jour.
Des cas sévères d'urticaire ont été rarement rapportés dans les études cliniques avec le galcanezumab (voir «Mises en garde et précautions»).
Au total, 2586 patients ont reçu Emgality pour la prophylaxie des migraines dans des études cliniques, ce qui correspond à une exposition de 1250.6 patients-années. Au total, 1647 patients ont reçu ≥6 doses mensuelles (120 ou 240 mg) de galcanezumab; 279 patients ont reçu 12 doses mensuelles (120 ou 240 mg).
Trois études de phase 3 contrôlées avec placebo sur la prophylaxie de la migraine ont été évaluées de façon combinée afin d'examiner la sécurité d'Emgality par rapport au placebo jusqu'à 6 mois après le début du traitement. Dans la phase en double aveugle, 1435 patients au total ont reçu le galcanezumab, ce qui correspond à une exposition de 536.3 patients-années.
Liste des effets indésirables rapportés dans les études cliniques et après la mise sur le marché
Les fréquences des effets indésirables sont indiquées comme suit: «très fréquents» (≥1/10), «fréquents» (<1/10 à ≥1/100), «occasionnels» (<1/100 à ≥1/1000), «rares» <1/1000 à ≥1/10'000), «très rares» (<1/10'000).
Affections du système immunitaire
Rare: anaphylaxie, angioedème.
Affections de l'oreille et du labyrinthe
Fréquent: vertige (0.7 % avec 120 mg, 1.2 % avec 240 mg)
Affections gastro-intestinales
Fréquent: constipation (1.0 % avec 120 mg, 1.5 % avec 240 mg).
Affections de la peau et du tissu sous-cutané
Fréquent: prurit (0.7 % avec 120 mg, 1.2 % avec 240 mg), éruption cutanée.
Occasionnel: urticaire (0.3 % avec 120 mg, 0.1 % avec 240 mg).
Troubles généraux et anomalies au site d'administration
Très fréquents: douleurs au site d'injection (10.1 % avec 120 mg, 11.6 % avec 240 mg), réactions au site d'injection (9.9% avec 120 mg, 14.5 % avec 240 mg).
Description d'effets indésirables spécifiques et informations complémentaires
Immunogénicité
Dans les études cliniques, l'incidence du développement d'anticorps contre le principe actif pendant la phase de traitement en double aveugle était de 4.8 % (33/688) parmi les patients sous traitement par galcanezumab 120 mg une fois par mois et tous, sauf un, avaient une activité neutralisante in vitro. Après 12 mois de traitement, 12.5 % (16/128) des patients traités par le galcanezumab 120 mg ont développé des anticorps contre le principe actif. Les titres étaient pour la plupart faibles et ont été testés positifs pour une activité neutralisante in vitro
La présence d'anticorps contre le principe actif n'a pas eu d'incidence sur la pharmacocinétique, l'efficacité ou la sécurité du galcanezumab. Les données disponibles ne sont toutefois pas suffisantes pour que l'on puisse tirer des conclusions fiables en la matière.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageChez l'homme, des doses allant jusqu'à 600 mg ont été administrées par voie sous-cutanée sans toxicité dose-limitante. En cas de surdosage, il est recommandé de surveiller le patient afin de détecter les signes ou symptômes d'effets indésirables et d'instaurer immédiatement un traitement symptomatique approprié.
Propriétés/EffetsCode ATC
N02CD02
Mécanisme d'action
Le galcanezumab est un anticorps monoclonal IgG4 recombinant humanisé. Il se lie au peptide relié au gène de la calcitonine (calcitonin gene-related peptide, CGRP) et empêche ainsi son activité biologique, sans bloquer le récepteur du CGRP. Des concentrations sanguines élevées de CGRP ont été associées à la migraine. En outre, chez certaines personnes qui ont des antécédents de migraine, des perfusions de CGRP peuvent déclencher des crises de type migraineux.
Pharmacodynamique
Le galcanezumab se lie au CGRP avec une forte affinité (KD = 31 pM) et avec une spécificité élevée (> 10'000 fois comparé aux peptides apparentés tels que l'adrénomédulline, l'amyline, la calcitonine et l'intermédine).
Efficacité clinique
L'efficacité et la sécurité d'Emgality ont été démontrées dans trois études de phase III, randomisées, contrôlées versus placebo, en double aveugle chez des patients adultes (N = 2886). Les 2 études sur la migraine épisodique (EVOLVE-1 et EVOLVE-2) ont inclus des patients qui répondaient aux critères de diagnostic de migraine avec ou sans aura de la classification internationale des céphalées (International Classification of Headache Disorders, ICHD), avec 4 à 14 jours de migraine par mois. L'étude sur la migraine chronique (REGAIN) a inclus des patients qui répondaient aux critères ICHD de migraine chronique avec ≥15 jours de céphalées par mois, dont au moins 8 jours avec les caractéristiques d'une migraine. Les patients ayant présenté un événement cardiovasculaire aigu récent (y compris infarctus du myocarde, angor instable, pontage coronarien (CABG), accident vasculaire cérébral, thrombose veineuse profonde) et/ou ceux considérés comme présentant un risque cardiovasculaire grave ont été exclus des études cliniques évaluant le galcanezumab. Les patients âgés de plus de 65 ans ont également été exclus. Les études EVOLVE-1 et EVOLVE-2 comportaient une période de traitement de 6 mois, en double aveugle, contrôlée versus placebo; l'étude REGAIN comportait une période de traitement de 3 mois, en double aveugle, contrôlée versus placebo, suivie d'une période d'extension ouverte de 9 mois. Les patients ont reçu le placebo, Emgality 120 mg/mois (avec une dose de charge initiale de 240 mg le premier mois) ou Emgality 240 mg/mois et ont été autorisés à prendre des médicaments pour le traitement aigu de la migraine. Au début de l'étude, le nombre moyen de jours de migraine par mois était de 9.13 par mois dans les études EVOLVE-1 et EVOLVE-2 (migraine épisodique) et de 19.41 par mois dans l'étude REGAIN (migraine chronique), et il était similaire au sein de chaque étude dans tous les groupes de traitement. Dans l'étude REGAIN, environ 15 % des patients ont continué un traitement concomitant par topiramate ou propranolol (autorisé conformément au protocole de prophylaxie de la migraine), et 64 % présentaient au début de l'étude une consommation excessive de médicaments contre les céphalées aiguës. Environ un tiers des patients dans les études étaient en échec d'au-moins un traitement prophylactique antérieur pour des raisons d'efficacité et environ 16 % des patients étaient en échec d'au-moins deux traitements prophylactiques antérieurs pour des raisons d'efficacité.
Dans les 3 études, le critère principal d'efficacité était la variation moyenne du nombre de jours de migraine par mois (migraine headache days, MHD) par rapport aux valeurs initiales. Les deux groupes de traitement, Emgality 120 mg et 240 mg, ont montré des améliorations statistiquement significatives et cliniquement pertinentes par rapport aux valeurs initiales comparativement au groupe placebo (voir Tableau).
Tableau: Efficacité et critères fonctionnels dans les études de phase 3
|
Résultats
|
EVOLVE-1 – Migraine épisodique
|
EVOLVE-2 - Migraine épisodique
|
REGAIN – Migraine chronique
| |
Emgality
|
Placebo
N=425
|
Emgality
|
Placebo
N=450
|
Emgality
|
Placebo
N=538
| |
120 mg N=210
|
240 mg N=208
|
120 mg N=226
|
240 mg N=220
|
120 mg N=273
|
240 mg N=274
| |
Résultats d'efficacité (évaluées sur les mois 1 à 6 pour EVOLVE-1 et 2 et sur les mois 1 à 3 pour REGAIN)
| |
Réduction moyenne du nombre de jours par mois avec migraine par rapport à la valeur initiale
|
-4.73a
|
-4.57a
|
-2.81
|
-4.29a
|
-4.18a
|
-2.28
|
-4.83a
|
-4.62a
|
-2.74
| |
Pourcentage moyen de patients qui ont atteint un taux de réponse ≥50 %
|
62.3a
|
60.9a
|
38.6
|
59.3a
|
56.5a
|
36.0
|
27.6a
|
27.5a
|
15.4
| |
Réduction moyenne du nombre de jours par mois avec migraine au cours desquels une médication aiguë a été utilisée.
Variation par rapport à la valeur initiale
|
-3.96a
|
-3.76a
|
-2.15
|
-3.67a
|
-3.63a
|
-1.85
|
-4.74b
|
-4.25a
|
-2.23
| |
Résultats rapportés par les patients (évalués sur les mois 4 à 6 pour EVOLVE-1 et 2 et au mois 3 pour REGAIN)
| |
Influence fonctionnelle de la migraine sur les activités de tous les jours.
Amélioration moyenne par rapport à la valeur initiale
(MSQ Role Function- Restrictive)c
|
32.43a
|
32.09a
|
24.69
|
28.47a
|
27.04a
|
19.65
|
21.81b
|
23.05a
|
16.76
|
a p < 0.001 (statistiquement significatif versus placebo après ajustement pour comparaisons multiples)
b p < 0.001 (non statistiquement significatif versus placebo après ajustement pour comparaisons multiples)
c N = 189 pour Emgality 120 mg, N = 184 pour Emgality 240 mg et N = 377 pour le placebo dans EVOLVE-1
N = 213 pour Emgality 120 mg, N = 210 pour Emgality 240 mg et N = 396 pour le placebo dans EVOLVE-2
N = 252 pour Emgality 120 mg, N = 253 pour Emgality 240 mg et N = 494 pour le placebo dans REGAIN
Comparativement aux patients sous placebo, les patients sous Emgality 120 mg ou 240 mg dans les études EVOLVE-1 et -2 (migraine épisodique) ont présenté une diminution du nombre de jours de migraine par mois significativement plus importante, par rapport aux valeurs initiales, au 1er mois et tous les mois suivants jusqu'au 6ème mois et de même dans l'étude REGAIN (migraine chronique) au 1er mois et tous les mois suivants jusqu'au 3ème mois (voir Illustration 1). En outre, les patients sous Emgality (dose initiale de 240 mg) présentaient au mois 1 significativement moins de MHD par semaine, comparativement au placebo à la semaine 1 et toutes les semaines suivantes.
Illustration 1: Réduction des jours de migraine par mois au fil du temps
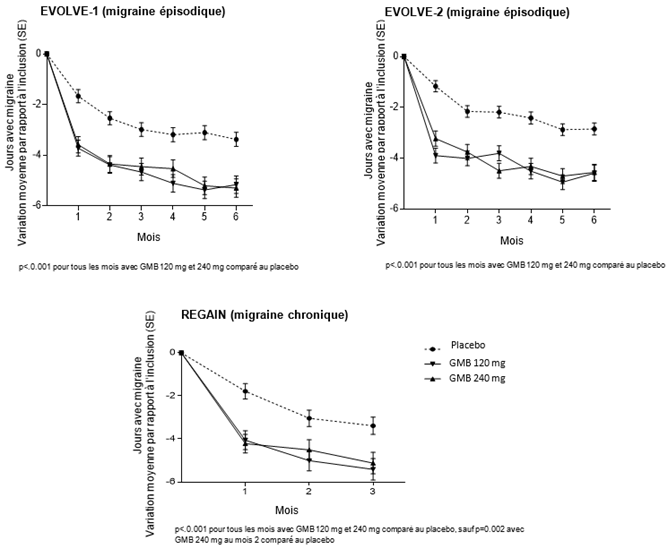
Les patients sous Emgality 120 mg et 240 mg ont présenté dans les trois études des taux de réponse plus élevés que les patients sous placebo. Le taux de réponse est le pourcentage moyen de patients atteignant un seuil défini en termes de réduction du nombre de jours de migraine par mois (≥50 %, ≥75 % et 100 %) pendant toute la durée de la période de traitement en double aveugle (voir Tableau ci-dessus).
Le nombre moyen de jours de migraine par mois lors desquels une médication aiguë a été utilisée était respectivement de 7.38 et 7.54 par mois au début de l'étude dans les études EVOLVE-1 et EVOLVE-2 (migraine épisodique), et de 15.16 par mois dans l'étude REGAIN (migraine chronique), et il était similaire dans tous les groupes de traitement au sein de chaque étude. Les deux groupes de traitement, Emgality 120 mg et 240 mg, ont présenté des réductions plus importantes, par rapport au groupe sous placebo, du nombre de jours de migraine par mois au cours desquels une médication aiguë a été utilisée (voir Tableau ci-dessus).
L'impact fonctionnel de la migraine a été évalué à l'aide du domaine Role Function-Restrictive du Migraine-Specific Quality of Life Questionnaire (MSQ) Version 2.1, qui mesure l'effet de la migraine sur le travail ou les activités de tous les jours, les rapports avec la famille et les amis, les loisirs, la productivité, la concentration, l'énergie et la fatigue. La valeur va de 0 à 100, les valeurs plus élevées indiquant une meilleure fonction.
Dans les trois études, les patients sous Emgality 120 mg et 240 mg ont présenté des améliorations plus importantes de la fonction comparativement aux patients sous placebo (voir Tableau ci-dessus). Au début de l'étude, le MSQ Role Function-Restrictive Score moyen était respectivement de 51.52 et de 51.72 dans les études EVOLVE-1 et 2 (migraine épisodique) et de 38.74 dans l'étude REGAIN (migraine chronique), et il était similaire dans tous les groupes de traitement au sein de chaque étude.
Dans les données consolidées des études EVOLVE-1 et EVOLVE-2, chez les patients en échec d'un ou plusieurs traitements prophylactiques pour des raisons d'efficacité, la différence entre les traitements sur la réduction du nombre moyen de jours de migraine par mois observé était de -2.69 jours (p < 0.001) entre galcanezumab 120 mg et placebo et de -2.78 jours (p < 0.001) entre galcanezumab 240 mg et placebo. Chez les patients en échec de deux ou plusieurs traitements prophylactiques, la différence entre les traitements a été de -2.64 jours (p <0.001) entre 120 mg et placebo et de -3.04 jours (p <0.001) entre 240 mg et placebo. Chez les patients en échec d'un ou plusieurs traitements prophylactiques pour des raisons d'efficacité, la différence de traitement sur la réduction du nombre moyen de jours de migraine par mois observé était de -3.54 jours (p < 0.001) entre galcanezumab 120 mg et placebo et de -1.37 jours (p < 0.05) entre galcanezumab 240 mg et placebo. Chez les patients en échec de deux ou plusieurs traitements prophylactiques, la différence de traitement était de -4.48 jours (p < 0.001) entre 120 mg et placebo et de -1.86 jours (p < 0.01) entre 240 mg et le placebo.
Dans l'étude REGAIN, 64 % des patients étaient en situation de consommation excessive de médicaments contre les céphalées aiguës à l'inclusion. Chez ces patients, la différence de traitement sur la réduction du MHD était de -2.53 jours (p < 0.001) entre galcanezumab 120 mg et placebo et de -2.26 jours (p < 0.001) entre galcanezumab 240 mg et placebo. Dans les données consolidées des études EVOLVE-1 et EVOLVE-2, près de 19 % des patients étaient en situation de consommation excessive de médicaments contre les céphalées aiguës au début de l'étude. Chez ces patients, la différence de traitement sur la réduction du MHD était de -3.55 jours (p < 0.001) entre galcanezumab 120 mg et placebo et de -3.07 jours (p < 0.001) entre galcanezumab 240 mg et placebo.
Etude clinique de phase III dans une population, dans laquelle 2 à 4 traitements prophylactiques de la migraine précédents ont montré une efficacité insuffisante ou n'ont pas été tolérés
L'étude clinique CONQUER a été menée chez des patients atteints de migraine chronique ou épisodique, chez lesquels, durant les 10 dernières années, 2 à 4 traitements prophylactiques de la migraine précédents ont montré une efficacité insuffisante ou n'ont pas été tolérés. Cette étude confirme les principales conclusions des précédentes études d'efficacité dans la migraine, c'est-à-dire que le traitement par galcanezumab a conduit à une réduction moyenne du nombre de jours de migraine par mois dans tout le groupe (4.1 jours par rapport à 1.0 jour dans le groupe placebo, p < 0.0001). Une réduction moyenne du nombre de jours de migraine par mois a également été observée dans la sous-population atteinte de migraine épisodique (2.9 jours dans le groupe galcanézumab par rapport à 0.3 jour dans le groupe placebo; p < 0.0001) et dans la sous-population atteinte de migraine chronique (5.9 jours dans le groupe galcanezumab par rapport à 2.2 jours dans le groupe placebo; p < 0.0001).
PharmacocinétiqueAbsorption
Sur la base d'une analyse pharmacocinétique de population, la concentration sérique maximale (Cmax) de galcanezumab après administration d'une dose de charge de 240 mg est d'environ 30 μg/ml (coefficient de variation [CV] de 27 %). La concentration sérique de galcanezumab à l'état d'équilibre a été atteinte après la première dose. Après une dose mensuelle de 120 mg, une Cmax (Cmax, ss) d'environ 28 μg/ml (CV de 35 %) a été atteinte à l'état d'équilibre, avec un temps pour atteindre la Cmax, ss d'environ 5 jours et une aire sous la courbe à l'état d'équilibre (AUCtau,ss) de 15'900 μg×h/ml.
L'emplacement du site d'injection (abdomen, cuisse, fesses et bras) n'a pas eu d'effet significatif sur l'absorption du galcanezumab.
Distribution
Sur la base d'une analyse pharmacocinétique de population, le volume de distribution apparent du galcanezumab était de 7.3 l.
Métabolisme
Le galcanezumab est un anticorps monoclonal IgG4 humanisé, on s'attend donc à une décomposition similaire à celle des IgG endogènes, en petits peptides et en acides aminés via des voies cataboliques.
Élimination
Sur la base d'une analyse pharmacocinétique de population, la clairance apparente du galcanezumab était d'environ 0.008 l/h et la demi-vie du galcanezumab était de 27 jours.
Linéarité/non-linéarité
L'exposition au galcanezumab augmente proportionnellement à la dose.
Sur la base d'une analyse pharmacocinétique de population incluant des doses comprises entre 5 et 300 mg, le taux d'absorption, la clairance apparente et le volume de distribution apparent étaient indépendants de la dose.
Cinétique pour certains groupes de patients
Age, sexe, poids, race, origine ethnique
Sur la base d'une analyse pharmacocinétique de population, aucun ajustement posologique n'est nécessaire en fonction de l'âge, du sexe, du poids, de la race ou de l'origine ethnique car ces facteurs n'ont montré aucun effet cliniquement significatif sur la clairance apparente ou le volume apparent de distribution du galcanezumab.
Troubles de la fonction rénale ou hépatique
Aucune étude de pharmacologie clinique spécifique destinée à évaluer les effets de l'insuffisance rénale et de l'insuffisance hépatique sur les paramètres pharmacocinétiques du galcanezumab n'a été menée.
L'élimination rénale des anticorps monoclonaux IgG est faible. De même, les anticorps monoclonaux IgG sont principalement éliminés par catabolisme intracellulaire et une insuffisance hépatique ne devrait pas influencer la clairance du galcanezumab. Sur la base d'une étude pharmacocinétique de population, la concentration de bilirubine (2 à 46 µmol/l) ou la clairance de la créatinine (24 à 308 ml/min) n'a pas montré d'effet significatif sur la clairance apparente du galcanezumab. Aucun patient atteint d'insuffisance rénale sévère n'a été inclus dans les études.
Données précliniquesToxicité en cas d'administration répétée
Les données non cliniques n'ont pas montré de risque particulier pour l'être humain, sur la base d'études de toxicité en administration répétée menées chez des rats et des singes Cynomolgus ainsi que d'évaluations de pharmacologie de la sécurité menées chez des singes Cynomolgus à des expositions 10 à 80 fois supérieures à l'exposition chez les patients traités avec une dose de 240 mg.
Mutagénicité / Carcinogénicité
Aucune étude non clinique n'a été réalisée pour évaluer le potentiel carcinogène ou mutagène du galcanezumab. Il n'existe aucune preuve suggérant qu'un traitement chronique par galcanezumab soit susceptible d'augmenter le risque de carcinogénicité dur la base des données des études de pharmacologie et de toxicologie chronique menées avec le galcanezumab, ainsi que d'après une évaluation de la littérature sur le CGRP.
Toxicité sur la reproduction
Aucun effet sur les paramètres de fertilité tels que les organes reproducteurs, les cycles menstruels, les analyses de sperme, l'accouplement et la fertilité n'a été observé chez les rats ayant reçu par voie sous-cutanée une dose de 250 mg/kg de galcanezumab (exposition correspondant à environ 4 à 20 fois l'exposition chez l'être humain avec 240 mg).
Dans des études sur le développement embryo-foetal menées chez le rat et le lapin, à des expositions allant jusqu'à 30 fois l'exposition chez l'être humain avec 240 mg, aucune malformation ou toxicité embryo-foetale n'a été observée. Dans une étude sur le développement prénatal et postnatal menée chez le rat, il n'y a eu aucun effet sur la survie, la croissance, la maturation sexuelle, le comportement ou la reproduction dans la descendance qui avait été exposée au galcanezumab in utero et pendant la lactation à des doses allant jusqu'à 18 fois l'exposition chez l'être humain avec 240 mg.
Dans une étude de toxicologie juvénile chez des rats auxquels le galcanezumab a été administré deux fois par semaine du 21e au 90e jour après la naissance, de légères modifications des paramètres osseux sont apparues à des expositions allant jusqu'à environ 50 fois l'exposition chez l'être humain avec 240 mg. Le risque pour l'être humain est considéré comme faible.
Remarques particulièresIncompatibilités
Aucune étude de compatibilité n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver au réfrigérateur (2-8°C). Ne pas congeler.
Conserver dans l'emballage d'origine, afin de protéger le contenu de la lumière.
Lorsqu'il n'est pas possible de le conserver au réfrigérateur, Emgality peut être conservé pendant une période maximale de 7 jours à des températures ne dépassant pas 30°C. Après avoir conservé le médicament en dehors du réfrigérateur, ne plus le remettre au réfrigérateur. Il faut jeter le stylo prérempli après 7 jours s'il est conservé non réfrigéré.
Remarques concernant la manipulation
Ne pas agiter.
Avant d'administrer Emgality, le laisser pendant au moins 30 minutes à température ambiante (15-25°C).
Emgality est une solution limpide et incolore à légèrement jaunâtre.
Contrôler le liquide avant l'emploi. Ne pas injecter Emgality si la solution est trouble ou colorée, contient des particules visibles ou a été congelée.
Le stylo prérempli est à usage unique.
Le manuel d'utilisation du stylo prérempli doit être lu attentivement et scrupuleusement respecté.
Numéro d’autorisation67026 (Swissmedic)
PrésentationEmgality solution pour injection en stylo prérempli à 120 mg: 1 et 2 stylos préremplis [B]
Titulaire de l’autorisationEli Lilly (Suisse) SA, 1214 Vernier/GE.
Mise à jour de l’informationFévrier 2024
|