CompositionPrincipes actifs
Romosozumab (produit à partir de cellules CHO [ovariennes de hamster chinois] génétiquement modifiées).
Excipients
Acétate de calcium, hydroxyde de sodium (pour ajustement du pH), saccharose, polysorbate 20, eau pour préparations injectables q.s. ad solutionem pro 1,17 ml.
Indications/Possibilités d’emploiLe romosozumab est utilisé dans le traitement de l'ostéoporose sévère chez les femmes post-ménopausées présentant un risque élevé de fracture (voir Propriétés/Effets).
Une fois le traitement par romosozumab terminé, il est recommandé de passer à un traitement anti-résorption afin de prolonger le bénéfice obtenu par le romosozumab.
Posologie/Mode d’emploiLe traitement doit être initié et contrôlé par des médecins spécialisés dans la prise en charge de l'ostéoporose.
Pour garantir la traçabilité des médicaments biotechnologiques, il est recommandé de documenter le nom commercial et le numéro de lot pour chaque traitement.
Posologie usuelle
La dose de romosozumab recommandée est de 210 mg (administrée sous forme de deux injections sous-cutanées de 105 mg chacune). L'administration doit se faire une fois par mois pendant une durée de 12 mois.
Les patientes doivent recevoir une supplémentation adéquate en calcium et en vitamine D (voir Mises en garde et précautions).
Une carte d'alerte du patient doit être délivrée aux patientes traitées par EVENITY.
Pour d'autres indications cliniques, voir Propriétés/Effets.
Instructions posologiques particulières
Patientes présentant des troubles de la fonction hépatique
Aucune étude clinique n'a été menée pour évaluer l'effet des troubles de la fonction hépatique (voir Pharmacocinétique).
Patientes présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patientes présentant des troubles de la fonction rénale (voir Pharmacocinétique).
Les données cliniques relatives à l'utilisation du romosozumab chez des patientes atteintes d'insuffisance rénale sévère (DFGe<30 ml/min/1,73m2) ou sous dialyse sont très limitées. L'utilisation de romosozumab chez ces patientes requiert une évaluation bénéfice/risque particulièrement attentive (voir Mises en garde et précautions) et le taux de calcium doit être surveillé (voir Mises en garde et précautions).
Patientes âgées (âgées de ≥65 ans)
Aucun ajustement de la dose n'est nécessaire chez les patientes âgées (voir Pharmacocinétique). Globalement, aucune différence n'a été observée entre les patientes jeunes et plus âgées en termes de sécurité et d'efficacité.
Enfants et adolescents
La sécurité et l'efficacité du romosozumab n'ont pas encore été établies chez les patientes pédiatriques (<18 ans). Aucune donnée n'est disponible.
Prise retardée
En cas d'oubli d'une dose de romosozumab, l'administrer dès que possible. L'administration de romosozumab peut ensuite être reprogrammée une fois par mois à partir de la date d'administration de la dernière dose.
Mode d'administration
Voie sous-cutanée
Pour administrer une dose de 210 mg, 2 injections de romosozumab doivent être effectuées par voie sous-cutanée dans l'abdomen, la cuisse ou le haut du bras. La deuxième injection ne doit pas être appliquée exactement au même endroit que la première.
L'administration doit être effectuée par une personne formée aux techniques d'injection.
Les instructions de manipulation et d'élimination sont disponibles dans la rubrique Remarques particulières.
Contre-indications·Hypocalcémie non corrigée (voir Mises en garde et précautions).
·Hypersensibilité au principe actif ou à l'un des excipients selon la Composition (voir Mises en garde et précautions).
·Antécédents d'infarctus du myocarde ou d'accident vasculaire cérébral (voir Mises en garde et précautions).
Mises en garde et précautionsHypocalcémie
Une hypocalcémie transitoire a été observée chez les patientes traitées par romosozumab. Toute éventuelle hypocalcémie doit être corrigée avant de débuter le traitement par romosozumab. Les patientes doivent être surveillées pour détecter les signes et symptômes d'une hypocalcémie. Les patientes doivent recevoir une supplémentation adéquate en calcium et vitamine D (voir Contre-indications et Effets indésirables).
Les patientes atteintes d'insuffisance rénale sévère (débit de filtration glomérulaire estimé [DFGe] compris entre 15 et 29 ml/min/1,73 m2) et les patientes sous dialyse présentent un risque accru de développer une hypocalcémie. Les taux de calcium doivent être surveillés chez ces patients.
Hypersensibilité
Dans des études cliniques, des réactions d'hypersensibilité cliniquement significatives, comme l'angio-œdème, l'érythème polymorphe et l'urticaire, sont survenues dans le groupe recevant le romosozumab. En cas de réactions anaphylactiques ou d'autres réactions allergiques cliniquement significatives, un traitement approprié doit être instauré et l'utilisation de romosozumab doit être interrompue (voir Contre-indications et Effets indésirables).
Infarctus du myocarde et accident vasculaire cérébral
Dans des études randomisées et contrôlées regroupées, une hausse des événements cardiovasculaires sévères (infarctus du myocarde et accident vasculaire cérébral) a été observée chez les patientes traitées par romosozumab par rapport au groupe témoin (voir Effets indésirables).
EVENITY est contre-indiqué chez les patientes ayant des antécédents d'infarctus du myocarde ou d'accident vasculaire cérébral (voir Contre-indications).
L'évaluation permettant de déterminer si EVENITY doit être utilisé chez une patiente donnée doit tenir compte de son risque de fracture au cours de l'année suivante et de son risque cardiovasculaire en fonction de ses facteurs de risque (par ex. maladie cardiovasculaire avérée, hypertonie, hyperlipidémie, diabète, tabagisme, insuffisance rénale sévère, âge). EVENITY ne doit être utilisé que si le bénéfice est supérieur aux risques.
Les patientes qui développent des symptômes indiquant un infarctus du myocarde ou un accident vasculaire cérébral pendant le traitement par EVENITY doivent immédiatement consulter un médecin et le traitement doit être interrompu en fonction de l'analyse bénéfice/risque individuelle.
Ostéonécrose de la mâchoire
Des ostéonécroses de la mâchoire (ONM) susceptibles de survenir spontanément, mais qui se développent le plus souvent après une extraction dentaire et/ou une infection locale accompagnées d'un retard de cicatrisation, ont été rapportées dans de rares cas chez des patientes sous romosozumab. Les patientes chez qui l'on soupçonne une ONM ou qui développent une ONM sous romosozumab doivent être traitées par un dentiste ou un chirurgien maxillo-facial. L'arrêt du traitement par romosozumab doit être envisagé selon le rapport bénéfice/risque individuel.
Fractures atypiques du fémur
Des fractures atypiques du col du fémur peu traumatiques, qui peuvent survenir spontanément, ont été rapportées dans de rares cas par des femmes sous romosozumab. Pour toutes les patientes qui présentent de nouvelles ou inhabituelles douleurs au niveau de la cuisse, de la hanche ou de l'aine, une fracture atypique doit être soupçonnée et évaluée pour exclure une fracture incomplète du fémur. Les patientes présentant une fracture atypique du fémur doivent également être examinées pour détecter d'éventuels signes et symptômes de fracture dans le membre controlatéral. L'arrêt du traitement par romosozumab doit être envisagé en fonction de l'évaluation du rapport bénéfice/risque individuel.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsAucune étude n'a été menée sur les interactions médicamenteuses avec le romosozumab.
Grossesse, allaitementGrossesse
EVENITY n'est pas indiqué chez la femme en âge de procréer ou enceinte. Aucune étude n'ayant été menée sur le romosozumab chez la femme enceinte, on ne sait pas si son administration pendant la grossesse peut nuire au fœtus.
Dans des études sur la reproduction menées chez le rat, les effets liés au romosozumab se sont limités à une légère augmentation de l'incidence des rétrécissements des appendices ventraux au niveau de la 6ème vertèbre cervicale. Ce résultat a été évalué comme un retard de développement d'un appendice squelettique qui ne survient pas chez l'homme. Les études réalisées sur des animaux ne permettent pas toujours de prédire les effets sur l'humain.
Dans toutes les études, les anomalies squelettiques (y compris la syndactylie et la polydactylie) ont touché 1 portée sur 75. Les données suggèrent que ces observations n'étaient pas liées au romosozumab. Aucun effet indésirable n'a été constaté sur la croissance post-natale et le développement. La syndactylie est très fréquente en cas de sclérostéose, mais ne survient pas chez les patients présentant une mutation des gènes hétérozygotes. Le risque de malformation des mains et des pieds chez le fœtus humain est faible suite à l'administration de romosozumab, car les doigts et les orteils humains se forment au premier trimestre de grossesse, lorsque le passage placentaire des immunoglobulines est limité.
Allaitement
EVENITY n'est pas indiqué chez la femme allaitante. On ne sait pas si le romosozumab passe dans le lait maternel humain.
Fertilité
Il n'existe pas de données sur les effets du romosozumab sur la fertilité chez l'humain. Les études effectuées chez l'animal avec des rats mâles et femelles n'ont montré aucun effet sur le critère d'évaluation de la fertilité à des doses allant jusqu'à 300 mg/kg (100 fois la dose clinique) (voir Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesLes effets indésirables décrits ci-après reposent sur les données regroupées du traitement de 12 mois par EVENITY de patientes atteintes d'ostéoporose ayant participé à des études cliniques contrôlées de phases II et III.
Les effets indésirables les plus fréquents (≥1/10) parmi les données d'innocuité regroupées étaient la rhinopharyngite et l'arthralgie.
Liste des effets indésirables
Les effets indésirables sont classés par classe de systèmes d'organes MedDRA et par fréquence en utilisant la convention suivante:
«très fréquent» (≥1/10)
«fréquent» (≥1/100, <1/10),
«occasionnel» (≥1/1000, <1/100)
«rare» (≥1/10000, <1/1000)
«très rare» (<1/10000)
Infections et infestations
Très fréquent: Rhinopharyngite (13,6%)
Affections du système immunitaire
Fréquent: Hypersensibilitéa, éruption cutanée, dermatite
Occasionnel: Urticaire
Rare: Angio-œdème, érythème polymorphe
Troubles du métabolisme et de la nutrition
Occasionnel: Hypocalcémieb
Affections du système nerveux
Fréquent: Céphalées
Occasionnel: Accident vasculaire cérébrald
Affections respiratoires, thoraciques et médiastinales
Fréquent: Toux
Affections oculaires
Occasionel: Cataracte
Affections cardiaques
Occasionnel: Infarctus du myocarded
Affections musculosquelettiques et du tissu conjonctif
Très fréquent: Arthralgie (12,4%)
Fréquent: Cervicalgie, spasmes musculaires
Troubles généraux et anomalies au site d'administration
Fréquent: Œdème périphérique, réactions au site d'injectionc
a Voir Contre-indications et Mises en garde et précautions
b Définie comme un taux de calcium sérique corrigé par l'albumine inférieur au seuil de référence inférieur. Voir Contre-indications et Mises en garde et précautions
c Les réactions les plus fréquentes au site d'injection étaient la douleur et l'érythème.
d Voir Infarctus du myocarde et accident vasculaire cérébral ci-dessous.
Les effets indésirables du romosozumab décrits ci-dessus sont survenus avec une incidence ≥2% dans tout le groupe ayant reçu du romosozumab et, au sein de la population contrôlée contre placebo pendant 12 mois, ils ont été plus fréquents dans le groupe recevant le romosozumab que dans le groupe recevant le placebo. En outre, après examen de toutes les données cliniques, les événements suivants sont considérés comme des effets indésirables: réactions au site d'injection, hypersensibilité, hypocalcémie.
Description de certains effets indésirables
Immunogénicité
Comme pour toutes les protéines thérapeutiques, une immunogénicité est possible. L'immunogénicité du romosozumab a été évaluée à l'aide d'un essai immunologique de dépistage permettant de détecter les anticorps anti-romosozumab de liaison. Chez les patientes qui ont réagi positivement à l'essai immunologique de dépistage, un test de liaison compétitive a été mené pour détecter les anticorps neutralisants.
Chez les femmes post-ménopausées à qui l'on a administré du romosozumab mensuellement, l'incidence des anticorps anti-romosozumab était de 18,6% (1162 sur 6244) pour les anticorps de liaison et de 0,9% (58 sur 6244) pour les anticorps neutralisants. La présence d'anticorps anti-romosozumab n'a eu aucune influence sur l'efficacité et la sécurité du romosozumab.
Infarctus du myocarde et accident vasculaire cérébral
Dans l'étude contrôlée par principe actif sur le romosozumab dans le traitement de l'ostéoporose chez les femmes post-ménopausées, 16 femmes (0,8%) du groupe recevant le romosozumab ont eu un infarctus du myocarde contre 5 femmes (0,2%) dans le groupe recevant l'alendronate au cours de la phase de traitement par romosozumab de 12 mois en double aveugle. En outre, 13 femmes (0,6%) du groupe recevant du romosozumab ont eu un accident vasculaire cérébral contre 7 femmes (0,3%) dans le groupe recevant de l'alendronate.
Dans l'étude contrôlée par placebo sur le romosozumab dans le traitement de l'ostéoporose chez les femmes post-ménopausées (y compris des femmes atteintes d'ostéoporose sévère et moins sévère), il n'y a pas eu de différence entre les infarctus du myocarde et les accidents vasculaires cérébraux confirmés au cours de la phase de traitement par romosozumab de 12 mois en double aveugle; 9 femmes (0,3%) du groupe recevant du romosozumab ont eu un infarctus du myocarde contre 8 femmes (0,2%) du groupe recevant le placebo. En outre, 8 femmes (0,2%) du groupe recevant le romosozumab ont eu un accident vasculaire cérébral, contre 10 femmes (0,3%) dans le groupe placebo.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'y a pas eu d'expérience de surdosage dans les études cliniques.
Propriétés/EffetsCode ATC
M05BX06
Mécanisme d'action/Pharmacodynamique
Le romosozumab est un anticorps monoclonal humanisé (IgG2) qui se lie à la sclérostine pour l'inhiber. Le romosozumab a une double action sur les os: Il augmente l'ossification et diminue la résorption osseuse. Le romosozumab augmente la masse osseuse trabéculaire et corticale, tout en améliorant la structure et la solidité des os.
Le rosomozumab entraîne une augmentation de l'ossification et une diminution de la résorption osseuse. Chez la femme post-ménopausée atteinte d'ostéoporose, le romosozumab a augmenté le marqueur d'ossification, à savoir le propeptide N-Terminal du procollagène de type 1 (P1NP), en début de traitement, l'augmentation maximale d'environ 145% par rapport au placebo ayant été atteinte 2 semaines après le début du traitement; puis, les valeurs sont revenues au niveau du placebo au bout de 9 mois et ont chuté à 15% environ au-dessous du niveau du placebo après 12 mois. Le romosozumab a diminué le marqueur de résorption osseuse, à savoir le télopeptide C-Terminal du collagène de type 1 (CTX), la réduction maximale d'environ 55% par rapport au placebo étant intervenue 2 semaines après le début du traitement. Les taux de CTX sont restés sous le niveau du placebo et étaient environ 25% inférieurs à ceux du placebo au bout de 12 mois.
Après l'arrêt du romosozumab chez la femme post-ménopausée atteinte d'ostéoporose, les taux de P1NP sont revenus aux valeurs de départ en l'espace de 12 mois; en 3 mois, le taux de CTX est passé au-dessus des valeurs de départ, puis est revenu aux taux de départ au bout de 12 mois, ce qui signifie que l'effet est réversible. Le renouvellement du traitement par romosozumab (chez un nombre limité de patientes) après une pause thérapeutique de 12 mois a entraîné une hausse du taux de P1NP et une baisse de CTX dues au romosozumab dans des proportions similaires à celles observées pendant le premier traitement.
Chez les femmes qui avaient antérieurement pris de l'alendronate par voie orale, le romosozumab a également entraîné une augmentation de l'ossification et une baisse de la résorption osseuse.
Efficacité clinique
Chez la femme post-ménopausée atteinte d'ostéoporose, EVENITY réduit le risque de fractures vertébrales et cliniques d'une part et non vertébrales d'autre part. EVENITY augmente la masse osseuse chez la femme post-ménopausée atteinte d'ostéoporose.
Traitement de l'ostéoporose chez la femme post-ménopausée
L'efficacité et la sécurité d'EVENITY ont été contrôlées dans deux études pivots, l'une contrôlée par alendronate (ARCH) et l'autre contrôlée par placebo (FRAME).
Etude 20110142 (ARCH)
L'efficacité et la sécurité d'EVENITY dans le traitement de l'ostéoporose chez la femme post-ménopausée ont été démontrées dans une étude de supériorité multicentrique, internationale, randomisée, en double aveugle, contrôlée par alendronate sur 4093 femmes post-ménopausées ayant entre 55 et 90 ans (âge moyen de 74,3 ans).
Les femmes incluses avaient soit un score DMO-T ≤ −2,50 au niveau de la hanche totale ou du col du fémur et au moins 1 fracture vertébrale modérément sévère ou sévère, soit un score DMO-T ≤ -2,00 au niveau de la hanche totale ou du col du fémur et au moins 2 fractures vertébrales modérément sévères ou sévères, soit elles avaient subi une fracture du fémur proximal 3 à 24 mois avant la randomisation.
Au début de l'étude, les scores DMO-T moyens étaient respectivement de -2,6, -2,80 et -2,90 au niveau du rachis lombaire, de la hanche totale et du col du fémur; 96,1% des femmes présentaient une fracture vertébrale au début de l'étude et 99% avaient déjà subi une fracture ostéoporotique. Les femmes ont été randomisées (1:1) pour recevoir un traitement de 12 mois en double aveugle par injections sous-cutanées mensuelles d'EVENITY ou par alendronate administré par voie orale une fois par semaine. Suite à la phase d'étude de 12 mois en double aveugle, les femmes ont reçu de l'alendronate dans les deux groupes, sans lever le caractère aveugle du traitement initial. L'analyse principale a été effectuée après que toutes les femmes ont achevé la visite de l'étude du 24è mois; des fractures cliniques ont été confirmées chez au moins 330 femmes et sont survenues après une période de suivi médiane de 33 mois d'étude. Les femmes ont reçu une supplémentation quotidienne en calcium et vitamine D.
Les principaux critères d'évaluation de l'efficacité étaient l'incidence des nouvelles fractures vertébrales jusqu'au 24è mois et l'incidence des fractures cliniques (fractures non vertébrales ou cliniques et vertébrales) au moment de l'analyse principale.
Effet sur les nouvelles fractures vertébrales et cliniques
Comme indiqué dans le tableau 1, EVENITY entraîne une baisse significative de l'incidence des nouvelles fractures vertébrales jusqu'au mois 24 et de l'incidence des fractures cliniques jusqu'au moment de l'analyse principale par rapport au traitement par alendronate seul. Le risque de subir les deux types de fractures était déjà réduit au bout de 12 mois.
Tableau 1. Effet d'EVENITY sur l'incidence et le risque de nouvelles fractures vertébrales et cliniques chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Taux de femmes présentant une fracture
|
Réduction du risque absolu
(%) (IC à 95%)
|
Réduction du risque relatif
(%) (IC à 95%)
|
Valeur P nominale
|
Valeur P
ajustéea
| |
Alendronate/
Alendronate
(%)
|
Romosozumab/
Alendronate
(%)
| |
Nouvelles fractures vertébrales
| |
Jusqu'au mois 12 b
|
85/1703
(5,0)
|
55/1696
(3,2)
|
1,84
(0,51; 3,17)
|
36
(11; 54)
|
0,008
|
NAc
| |
Jusqu'au mois 24b
|
147/1834 (8,0)
|
74/1825
(4,1)
|
4,03
(2,50; 5,57)
|
50
(34; 62)
|
< 0,001
|
< 0,001
| |
Fractures cliniquesd
| |
Analyse principale
Suivi médian d'environ 33 mois
|
266/2047 (13,0)
|
198/2046
(9,7)
|
NAe
|
27
(12; 39)
|
< 0,001
|
< 0,001
| |
Jusqu'au mois 12
|
110/2047 (5,4)
|
79/2046
(3,9)
|
1,8
(0,5; 3,1)
|
28
(4; 46)
|
0,027
|
NAc
| |
Jusqu'au mois 24
|
197/2047 (9,6)
|
146/2046
(7,1)
|
2,7
(0,8; 4,5)
|
26
(9; 41)
|
0,005
|
NAc
|
a Les valeurs P ajustées reposent sur la procédure d'Höchberg et doivent être comparées avec un niveau de signification de 0,05.
b La réduction du risque absolu et la réduction du risque relatif reposent sur la méthode de Mantel-Haenszel ajustée pour Altersstrata, le score DMO-T au niveau de la hanche totale au début de l'étude (≤2,5, > -2,5) et la présence de fractures vertébrales sévères au début de l'étude. La comparaison des traitements repose sur un modèle de régression logistique ajusté pour Altersstrata, le score DMO-T au niveau de la hanche totale au début de l'étude et la présence de fractures vertébrales sévères au début de l'étude.
NAc: Le critère d'évaluation ne faisant pas partie du test séquentiel, l'ajustement de la valeur P n'est pas applicable.
d Toutes les fractures symptomatiques, y compris les fractures non vertébrales et vertébrales douloureuses, font partie des fractures cliniques. La comparaison entre les traitements repose sur un modèle à risque proportionnel (régression de Cox) ajusté pour Altersstrata, le score DMO-T au niveau de la hanche totale au début de l'étude et la présence de fractures vertébrales sévères au début de l'étude.
e NA: Non disponible, car l'exposition au moment de l'analyse principale était différente parmi les participantes de l'étude.
Effet sur d'autres types/groupes de fractures
Tableau 2. Effet d'EVENITY sur l'incidence et le risque d'autres types/groupes de fractures jusqu'à l'analyse principale (suivi médian de 33 mois environ) chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Taux de femmes présentant une fracture
|
Réduction du risque relatif
(%) (IC à 95%)
|
Valeur P nominalea
|
Valeur P
ajustée
| |
Alendronate/
Alendronate
(%)
|
Romosozumab/
Alendronate
(%)
| |
Non vertébrale
|
217/2047 (10,6)
|
178/2046 (8,7)
|
19 (1; 34)
|
0,019d
|
0,040c
| |
Hanche
|
66/2047 (3,2)
|
41/2046 (2,0)
|
38 (8; 58)
|
0,015
|
NAe
| |
Ostéoporotique majeure f
|
209/2047 (10,2)
|
146/2046 (7,1)
|
32 (16; 45)
|
< 0,001
|
NAe
| |
Non vertébrale majeureg
|
196/2047 (9,6)
|
146/2046 (7,1)
|
27 (10; 41)
|
0,004
|
NAe
|
a Les valeurs P nominales reposent sur un modèle à risque proportionnel (régression de Cox) ajusté pour Altersstrata, le score DMO-T au niveau de la hanche totale au début de l'étude et la présence de fractures vertébrales sévères au début de l'étude.
b Les valeurs P ajustées reposent sur une association des méthodes suivantes: Procédure d'Höchberg, procédure de test avec hypothèses posées a priori et procédure de test séquentielle par groupes; elles doivent être comparées avec un niveau de signification de 0.05.
c Bilatérale
d Unilatérale
NAe: Le critère d'évaluation ne faisant pas partie du test séquentiel, l'ajustement de la valeur P n'est pas applicable.
f Fractures de la hanche, de l'avant-bras ou du haut du bras sans lien avec une fracture pathologique, quelle que soit la sévérité du traumatisme, et fractures cliniques vertébrales
g Fractures du bassin, du fémur distal, du tibia proximal, des côtes, du haut du bras proximal, de l'avant-bras et fractures de la hanche
En outre, EVENITY a réduit l'incidence des fractures non vertébrales majeures (major nonvertebral) après 12 mois seulement par rapport à l'alendronate.
Effet sur la densité minérale osseuse (DMO)
Chez les femmes post-ménopausées atteintes d'ostéoporose, EVENITY a entraîné une hausse significative de la DMO au niveau du rachis lombaire, de la hanche totale et du col du fémur au bout de 12 mois seulement par rapport à l'alendronate. Suite à la phase en double aveugle, la DMO a augmenté chez les patientes qui étaient passées d'EVENITY à l'alendronate par rapport aux patientes qui avaient continué à être traitées par alendronate jusqu'au mois 24 (voir tableau 3 et illustration 2).
Au bout de 12 mois de traitement, le romosozumab a augmenté la DMO au niveau du rachis lombaire par rapport aux valeurs de départ chez 98% des femmes post-ménopausées. 91% des femmes traitées par romosozumab ont obtenu une hausse d'au moins 5% de la DMO au niveau du rachis lombaire et 68% ont obtenu une hausse d'au moins 10% par rapport aux valeurs de départ en l'espace de 12 mois.
Les valeurs de départ d'âge et de DMO, ainsi que la région géographique n'ont eu aucune influence sur la hausse constante observée de la densité minérale osseuse (DMO) au niveau du rachis lombaire et de la hanche totale.
Tableau 3. Variation moyenne en pourcentage de la DMO entre le début de l'étude et les mois 12 et 24 chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Alendronate vs. Alendronate
Moyenne (IC à 95%)
N = 2047a
|
Romosozumab vs. Alendronate
Moyenne (IC à 95%)
N = 2046a
|
Différence entre les traitements pour l'Alendronate vs. Alendronate
| |
Après 12 mois
| |
Rachis lombaire
|
5,0 (4,73; 5,21)
|
13,7 (13,36; 13,99)
|
8,7b (8,31; 9,09)
| |
Hanche totale
|
2,8 (2,67; 3,02)
|
6,2 (5,94; 6,39)
|
3,3b (3,03; 3,60)
| |
Col du fémur
|
1,7 (1,46; 1,98)
|
4,9 (4,65; 5,23)
|
3,2b (2,90; 3,54)
| |
Après 24 mois
| |
Rachis lombaire
|
7,2 (6,90; 7,53)
|
15,3 (14,89; 15,69)
|
8,1b (7,58; 8,57)
| |
Hanche totale
|
3,5 (3,23; 3,68)
|
7,2 (6,95; 7,48)
|
3,8b (3,42; 4,10)
| |
Col du fémur
|
2,3 (1,96; 2,57)
|
6,0 (5,69; 6,37)
|
3,8b (3,40; 4,14)
|
a Nombre de femmes randomisées
b Valeur P <0,001 basée sur un modèle ANCOVA
Illustration 1. Modification en pourcentage de la DMO au niveau du rachis lombaire, de la hanche totale et du col du fémur à partir du début de l'étude, pendant 24 mois, chez des femmes post-ménopausées atteintes d'ostéoporose
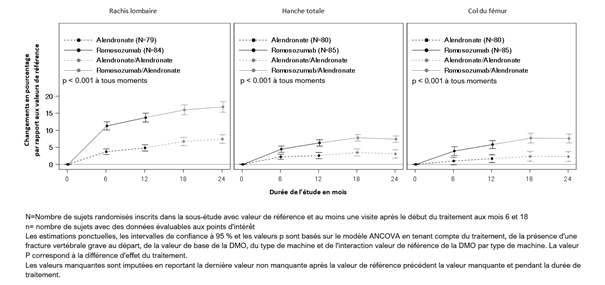
La différence significative obtenue au cours des 12 premiers mois en termes de densité minérale osseuse a perduré après le passage à l'aldendronate ou sa poursuite jusqu'au mois 36. Au mois 6, des différences entre les traitements ont été observées au niveau du rachis lombaire, de la hanche totale et du col du fémur.
Etude 20070337 (FRAME)
L'efficacité et la sécurité du romosozumab dans le traitement de l'ostéoporose chez la femme post-ménopausée ont été démontrées dans une étude à groupes parallèles multicentrique, internationale, randomisée, en double aveugle et contrôlée par placebo sur 7180 femmes post-ménopausées ayant entre 55 et 90 ans (âge moyen de 70,9 ans). 40,8% des femmes qui ont participé présentaient une ostéoporose sévère avec une fracture antérieure au départ.
Les femmes qui avaient un score T de densité minérale osseuse (DMO) ≤ -2,50 à > -3,5 au niveau de la hanche totale ou du col du fémur ont été incluses. Au début de l'étude, les scores T de DMO moyens étaient respectivement de -2,2, -2,47 et -2,75 au niveau du rachis lombaire, de la hanche totale et du col du fémur et 18,3% des femmes présentaient une fracture vertébrale au début de l'étude. Les femmes ont été randomisées pour recevoir un traitement de 12 mois en aveugle, soit par injections sous-cutanées de romosozumab, soit par placebo une fois par mois. Après la phase d'étude de 12 mois en double aveugle, les femmes sont passées à une étude ouverte, où 60 mg de dénosumab ont été administrés par voie sous-cutanée tous les 6 mois pendant une durée de 12 mois, tandis que le caractère aveugle du traitement de départ était maintenu. La dernière phase a été étendue à une étude ouverte de 12 mois supplémentaires sur le dénosumab. Les femmes ont reçu une supplémentation quotidienne en calcium et vitamine D.
Les co-critères d'évaluation principaux de l'efficacité étaient l'incidence des nouvelles fractures vertébrales jusqu'aux mois 12 et 24.
Effet sur les nouvelles fractures vertébrales, cliniques et non vertébrales
Le romosozumab a diminué significativement l'incidence des nouvelles fractures vertébrales jusqu'au mois 12 (p< 0,001), comme indiqué dans le tableau 4. En outre, chez les femmes qui avaient reçu du romosozumab la première année, le risque de fracture est resté réduit pendant toute la deuxième année lorsqu'elles sont passées du romosozumab au dénosumab par rapport à celles qui sont passées du placebo au dénosumab (mois 24; p < 0,001).
Le romosozumab a également réduit significativement l'incidence des fractures cliniques jusqu'au mois 12 (voir tableau 4). L'effet du romosozumab sur l'incidence et le risque de nouvelles fractures vertébrales, cliniques et non vertébrales jusqu'aux mois 12 et 24 est représenté dans le tableau 4.
Tableau 4. Effet du romosozumab sur l'incidence et le risque de nouvelles fractures vertébrales et cliniques et de nouvelles fractures non vertébrales jusqu'aux mois 12 et 24 chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Taux de femmes présentant une fracture
|
Réduction du risque absolu
(%) (IC à 95%)
|
Réduction du risque relatif
(%) (IC à 95%)
|
Valeur P nominale
|
Valeur P
ajustéea
| |
Placebo
(%)
|
Romosozumab
(%)
| |
Jusqu'au mois 12
| |
Nouvelle vertébraleb
|
59/3322 (1,8)
|
16/3321 (0,5)
|
1,30 (0,79; 1,80)
|
73 (53; 84)
|
< 0,001
|
< 0,001
| |
Cliniquec
|
90/3591 (2,5)
|
58/3589 (1,6)
|
1,2 (0,4; 1,9)
|
36 (11;54)
|
0,008
|
0,008
| |
Non vertébrale
|
75/3591 (2,1)
|
56/3589 (1,6)
|
0,8 (0,1; 1,4)
|
25 (−5; 47)
|
0,096
|
0,096
| |
|
Placebo vs. Dénosumab (%)
|
Romosozumab vs. Dénosumab (%)
|
| |
Jusqu'au mois 24
| |
Nouvelle vertébraleb
|
84/3327 (2,5)
|
21/3325 (0,6)
|
1,89 (1,30; 2,49)
|
75 (60; 84)
|
< 0,001
|
< 0,001
| |
Cliniquec,d
|
147/3591 (4,1)
|
99/3589 (2,8)
|
1,4 (0,5; 2,4)
|
33 (13; 48)
|
0,002
|
0,096
| |
Non vertébrale
|
129/3591 (3,6)
|
96/3589 (2,7)
|
1,0 (0,2; 1,9)
|
25 (3; 43)
|
0,029
|
0,057
|
a Les valeurs P ajustées reposent sur une procédure de test séquentielle et doivent être comparées avec un niveau de signification de 0,05.
b La réduction du risque absolu et du risque relatif repose sur la méthode de Mantel-Haenszel ajustée pour les facteur de stratification d'âge et de prévalence des fractures vertébrales. Les comparaisons entre les traitements reposent sur un modèle de régression logistique ajusté pour les facteurs de stratification.
c Toutes les fractures symptomatiques, y compris les fractures non vertébrales et vertébrales douloureuses, font partie des fractures cliniques. Les comparaisons entre les traitements reposent sur un modèle à risque proportionnel (régression de Cox) ajusté pour les facteurs de stratification d'âge et de prévalence des fractures vertébrales.
d Non significatif, car la signification statistique n'a pas été atteinte pour un critère d'évaluation qui se trouvait antérieurement dans la séquence de test; valeur-P nominale: 0,002.
Pour tous les types de fractures, la réduction du risque a été allongée jusqu'au mois 36.
Effet sur d'autres types/groupes de fractures
L'effet du romosozumab sur les autres types/groupes de fractures jusqu'au mois 24 est indiqué dans le tableau 5.
Tableau 5. Effet du romosozumab sur l'incidence et le risque d'autres types/groupes de fractures jusqu'aux mois 12 et 24 chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Taux de femmes présentant une fracture
|
Réduction du risque absolu
(%)
(IC à 95%)
|
Réduction du risque relatif
(%) (IC à 95%)
|
Valeur P nominalea
|
Valeur P
ajustée
| |
Placebo
(%)
|
Romosozumab (%)
| |
Jusqu'au mois 12
| |
Non vertébrale majeured
|
55/3591 (1,5)
|
37/3589 (1,0)
|
0,6
(0,1; 1,2)
|
33
(−2; 56)
|
0,060
|
0,096
| |
Vertébrale, nouvelle ou s'étant aggravée
|
59/3322 (1,8)
|
17/3321 (0,5)
|
1,3
(0,76; 1,77)
|
71
(51; 83)
|
< 0,001
|
0,096
| |
Hanche
|
13/3591 (0,4)
|
7/3589 (0,2)
|
0,3
(0,0; 0,6)
|
46
(-35; 78)
|
0,18
|
0,18
| |
Ostéoporotique majeuree
|
63/3591 (1,8)
|
38/3589 (1,1)
|
0,9
(0,3; 1,5)
|
40
(10; 60)
|
0,012
|
NAc
| |
Vertébrale, multiple, nouvelle/s'étant aggravée
|
9/3322 (0,3)
|
1/3321 (< 0,1)
|
0,24
(0,05; 0,43)
|
89
(13; 99)
|
0,011
|
NAc
| |
|
Placebo vs. Dénosumab
(%)
|
Romosozumab vs. Dénosumab
(%)
|
|
| |
Jusqu'au mois 24
| |
Non vertébrale majeured
|
101/3591 (2,8)
|
67/3589 (1,9)
|
1,1
(0,3; 1,8)
|
33
(9; 51)
|
0,009
|
0,096
| |
Vertébrale, nouvelle ou s'étant aggravée
|
84/3327 (2,5)
|
22/3325 (0,7)
|
1,86
(1,27; 2,46)
|
74
(58; 84)
|
< 0,001
|
0,096
| |
Hanche
|
22/3591 (0,6)
|
11/3589 (0,3)
|
0,4
(0,0; 0,7)
|
50
(-4; 76)
|
0,059
|
0,12
| |
Ostéoporotique majeuree
|
110/3591 (3,1)
|
68/3589 (1,9)
|
1,2
(0,5; 2,0)
|
38
(16; 54)
|
0,002
|
NAc
| |
Vertébrale, multiple, nouvelle/s'étant aggravée
|
17/3327 (0,5)
|
1/3325 (< 0,1)
|
0,48 (0,23; 0,73)
|
94
(56; 99)
|
< 0,001
|
NAc
|
a Les valeurs P nominales reposent sur un modèle de régression logistique (nouvelles fractures vertébrales ou fractures vertébrales s'aggravant et fractures vertébrales multiples nouvelles/s'aggravant) ou sur un modèle à risque proportionnel (régression de Cox) (fractures non vertébrales, non vertébrales majeures [major nonvertebral], de la hanche et ostéoporotiques majeures [major osteoporotic]) ajusté pour les facteurs de stratification d'âge et de prévalence des fractures vertébrales.
b Les valeurs P ajustées reposent sur une procédure de test séquentielle et doivent être comparées avec un niveau de signification de 0,05.
c NA: Le critère d'évaluation ne faisant pas partie du test séquentiel, l'ajustement de la valeur P n'est pas applicable.
d Bassin, fémur distal, tibia proximal, côtes, haut du bras proximal, avant-bras et hanche
e Fractures vertébrales cliniques et fractures de la hanche, de l'avant-bras et du haut du bras
Effet sur la densité minérale osseuse (DMO)
Chez les femmes post-ménopausées atteintes d'ostéoporose, le romosozumab a entraîné une hausse significative de la densité minérale osseuse au niveau du rachis lombaire, de la hanche totale et du col du fémur au bout de 6 et 12 mois par rapport au placebo (tableau 6). Au bout de 12 mois de traitement, le romosozumab a augmenté la DMO au niveau du rachis lombaire par rapport aux valeurs de départ chez 99% des femmes post-ménopausées. 92% des femmes traitées par romosozumab ont obtenu une hausse d'au moins 5% de la DMO au niveau du rachis lombaire et 68% ont obtenu une hausse d'au moins 10% par rapport aux valeurs de départ en l'espace de 12 mois. Cet effet a perduré après le passage à un autre traitement de l'ostéoporose: Suite au passage du romosozumab au dénosumab, la DMO a continué à augmenter jusqu'au mois 24. Chez les patientes qui sont passées du placebo au dénosumab, la DMO a également augmenté sous dénosumab. Chez les femmes qui ont reçu du romosozumab puis du dénosumab, la hausse de la DMO au niveau du rachis lombaire, de la hanche totale et du col du fémur était plus importante au bout de 24 mois que chez celles qui ont reçu un placebo puis du dénosumab (tableau 6). La modification en pourcentage de la DMO au niveau du rachis lombaire, de la hanche totale et du col du fémur à partir du début de l'étude et pendant une durée de 24 mois est représentée dans l'illustration 4.
Les valeurs de départ d'âge et de DMO, ainsi que la région géographique n'ont eu aucune influence sur la hausse constante observée de la densité minérale osseuse (DMO) au niveau du rachis lombaire et de la hanche totale.
Tableau 6. Variation moyenne en pourcentage de la DMO entre le début de l'étude et les mois 12 et 24 chez des femmes post-ménopausées atteintes d'ostéoporose
|
|
Placebo
Moyenne (IC à 95%)
N = 3591a
|
Placebo
Moyenne (IC à 95%)
N = 3589a
|
Différence entre les traitements vs. Placebo
Moyenne (IC à 95%)
| |
Après 12 mois
| |
Rachis lombaire
|
0,4 (0,2; 0,5)
|
13,1 (12,8; 13,3)
|
12,7b (12,4; 12,9)
| |
Hanche totale
|
0,3 (0,1; 0,4)
|
6,0 (5,9; 6,2)
|
5,8b (5,6; 6,0)
| |
Col du fémur
|
0,3 (0,1; 0,5)
|
5,5 (5,2; 5,7)
|
5,2b (4,9; 5,4)
| |
|
Placebo vs. Dénosumab
Moyenne (IC à 95%)
N = 3591 a
|
Romosozumab vs. Dénosumab
Moyenne (IC à 95%)
N = 3589 a
|
Différence entre les traitements pour le placebo vs. Dénosumab
| |
Après 24 mois
| |
Rachis lombaire
|
5,5 (5,3; 5,7)
|
16,6 (16,3; 16,8)
|
11,1b (10,8; 11,4)
| |
Hanche totale
|
3,2 (3,1; 3,3)
|
8,5 (8,3; 8,7)
|
5,3b (5,1; 5,5)
| |
Col du fémur
|
2,3 (2,1; 2,6)
|
7,3 (7,0; 7,5)
|
4,9b (4,7; 5,2)
|
a Nombre de femmes randomisées
b Valeur P <0,001 basée sur un modèle ANCOVA
Illustration 2. Modification en pourcentage de la DMO au niveau du rachis lombaire, de la hanche totale et du col du fémur à partir du début de l'étude, pendant la durée de 24 mois, chez des femmes post-ménopausées atteintes d'ostéoporose
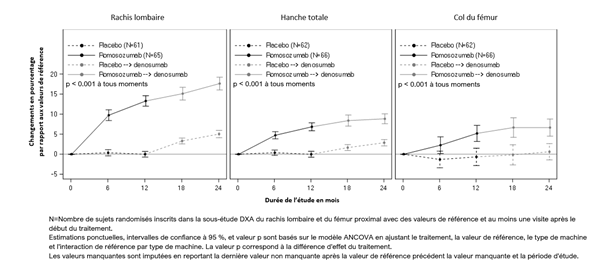
La différence significative obtenue au cours des 12 premiers mois en termes de densité minérale osseuse a perduré jusqu'au mois 36 après le passage au dénosumab.
Histologie osseuse et histomorphométrie
Au total, 139 femmes post-ménopausées atteintes d'ostéoporose se sont prêtées à 154 biopsies osseuses transiliaques au bout de 2, 12 et/ou 24 mois. Parmi les biopsies prélevées, 154 (100,0%) convenaient pour une analyse histologique qualitative et 138 (89,6%) pour une analyse histomorphométrique complète. Les analyses histologiques qualitatives des échantillons des femmes traitées par romosozumab ont démontré une architecture et une qualité osseuses normales à tous les points temporels. Il n'y a pas eu de signes d'os fibreux, de défauts de minéralisation ni de fibroses médullaires.
L'effet du romosozumab par rapport au placebo a été analysé par histomorphométrie sur des biopsies prélevées au bout de 2 et 12 mois. Chez les femmes traitées par romosozumab, les indices histomorphométriques ont montré une hausse de l'ossification et une baisse de la résorption osseuse au bout de 2 mois. Au bout de 12 mois, les indices indiquaient à la fois une diminution de l'ossification et de la résorption sous romosozumab, tandis que les volumes osseux et la densité trabéculaire augmentaient.
Femmes en transition d'une thérapie par bisphosphonates
Etude 20080289 (STRUCTURE)
Pour évaluer la sécurité et l'efficacité du romosozumab chez la femme post-ménopausée atteinte d'ostéoporose en transition d'un traitement par bisphosphonates, une étude multicentrique, randomisée et ouverte a été menée sur 436 femmes post-ménopausées ayant entre 56 et 90 ans (âge moyen de 71,5 ans). Dans cette étude, la sécurité et les évolutions de la DMO ont été évaluées par absorptiométrie biphotonique à rayons X (DXA) au bout de 12 mois de traitement par romosozumab par rapport à un traitement de 12 mois par tériparatide. L'étude a également porté sur la densité osseuse de la hanche pendant 12 mois, selon la méthode des éléments finis (FEM), en utilisant l'imagerie qualitative par tomodensitométrie.
Pour être incluses, les femmes devaient remplir les critères suivants au début de l'étude: score DMO-T ≤ -2,50 au niveau du rachis lombaire, de la hanche totale ou du col du fémur et anamnèse de fractures non vertébrales après l'âge de 50 ans ou de fractures vertébrales à un moment donné. Au début de l'étude, les scores DMO-T moyens étaient respectivement de -2,85, -2,24 et -2,46 au niveau du rachis lombaire, de la hanche totale et du col du fémur.
Au bout de 12 mois, la DMO atteinte sous romosozumab était 9,8% plus élevée que la valeur de départ au niveau du rachis lombaire (IC à 95%: 9,0; 10,5), 2,9% au niveau de la hanche totale (IC à 95%: 2,5; 3,4) et 3,2% au niveau du col du fémur (IC à 95%: 2,6; 3,8). Par rapport au tériparatide, les différences entre les traitements en termes de DMO au bout de 12 mois atteignaient 4,4% (IC à 95%: 3,4; 5,4) au niveau du rachis lombaire, 3,4% (IC à 95%: 2,8; 4,0) au niveau de la hanche totale et 3,4% au niveau du col du fémur (IC à 95%: 2,6; 4,2; valeur p < 0,0001 pour toutes les comparaisons). Des différences entre les traitements ont été observées au bout de 6 mois seulement.
Au bout de 12 mois, la densité osseuse estimée sous rosomozumab était 2.5% plus élevée que la valeur de départ au niveau de la hanche totale (IC à 95%: 1,7; 3,2). Par rapport au tériparatide, la différence entre les traitements en termes de densité osseuse estimée au niveau de la hanche totale au bout de 12 mois atteignait 3.2% (IC à 95%: 2,1; 4,3; valeur p < 0,0001). Les effets indésirables observés dans cette étude correspondaient généralement à ceux constatés chez les femmes qui n'avaient pas pris antérieurement de traitement par bisphosphonates (voir Effets indésirables).
PharmacocinétiqueAbsorption
L'administration d'une dose unique de 210 mg de romosozumab à des volontaires en bonne santé (N = 90, plage de 21 à 65 ans) a entraîné une concentration sérique maximale (Cmax) moyenne (écart type [ET]) de 22,2 (5,8) µg/ml et une surface moyenne sous la courbe (ASC) de concentration par rapport au temps de 389 (127) µg x jour/ml. La durée médiane avant d'atteindre la concentration maximale de romosozumab (tmax) était de 5 jours (plage de 2 à 7 jours). Suite à l'administration par voie sous-cutanée d'une dose de 210 mg, la biodisponibilité était de 81%.
Métabolisme
Le romosozumab est un anticorps monoclonal humanisé (IgG2) ayant une affinité et une spécificité élevées pour la sclérostine et qui est donc éliminé par une voie d'élimination saturable rapide (clairance non linéaire à médiation cellulaire, régulée par la dégradation du complexe romosozumab/sclérostine) et par une voie d'élimination non spécifique lente médiée par le système réticulo-endothélial.
Élimination
Après avoir atteint la concentration Cmax, les taux sériques ont diminué avec une demi-vie efficace moyenne de 12,8 jours. Après une administration mensuelle, l'état d'équilibre a généralement été atteint en 3 mois avec un cumul minimal (moins du double). La présence d'anticorps anti-romosozumab de liaison a diminué l'exposition du romosozumab jusqu'à 25% et jusqu'à 63% pour les anticorps neutralisants, ce qui est considéré comme cliniquement non significatif (voir Propriétés/Effets).
Linéarité/non-linéarité
Suite à une administration par voie sous-cutanée, le romosozumab présente une pharmacocinétique non linéaire en fonction de la dose suite à sa liaison à la sclérostine.
Cinétique pour certains groupes de patients
Aucun ajustement de la dose n'est nécessaire pour les patients présentant des caractéristiques particulières. Sur la base d'une analyse de population pharmacocinétique, l'âge, le sexe, l'origine ethnique («personnes d'origine asiatique / non asiatique») et le stade de la maladie (faible masse osseuse ou ostéoporose) n'ont pas eu d'influence cliniquement pertinente sur la pharmacocinétique du romosozumab (modification de l'exposition à l'état stable < 20%). L'exposition du romosozumab a diminué proportionnellement à la prise de poids. Cette baisse d'exposition du romosozumab n'a toutefois eu qu'un effet minime sur la hausse de la densité minérale osseuse (DMO) au niveau du rachis lombaire, en tenant compte des analyses d'effet par rapport à l'exposition, (évolution < 15%) et a été jugée cliniquement non pertinente.
Troubles de la fonction hépatique
Aucune étude clinique n'a été menée pour évaluer l'effet d'une insuffisance hépatique.
Troubles de la fonction rénale
Suite à une administration d'une dose de 210 mg de romosozumab dans une étude clinique menée auprès de 16 patients et patientes atteints d'insuffisance rénale sévère (clairance de la créatinine < 30%) ou de patientes dialysées atteintes d'une insuffisance rénale au stade terminal (IRT), la concentration Cmax moyenne des patients et patientes atteints d'une insuffisance rénale sévère était de -31% supérieure et l'ASC moyenne était 43% supérieure à celle des personnes en bonne santé. L'exposition moyenne au romosozumab était comparable chez les patientes dialysées atteintes d'une IRT par rapport aux sujets sains.
L'analyse pharmacocinétique de population a permis de conclure à une exposition au romosozumab croissante proportionnelle au degré de sévérité de l'insuffisance rénale. D'après un modèle exposition-réponse des variations de la densité minérale osseuse (DMO) et la comparaison aux expositions obtenues aux doses cliniques tolérées, cette hausse n'a toutefois pas eu de pertinence clinique, de sorte que ces patientes n'ont pas besoin d'un ajustement de la dose.
Patientes âgées (Âge ≥65)
L'âge (entre 20 et 89 ans) n'a pas eu d'effet sur la pharmacocinétique du romosozumab.
Enfants et adolescents
Le profil pharmacocinétique n'a pas été étudié chez les patientes pédiatriques.
Données précliniquesLes données non cliniques provenant d'études conventionnelles de pharmacovigilance, toxicité à doses répétées et carcinogénicité et de toxicité pour la reproduction, la fertilité et le développement n'ont pas révélé de risque particulier pour l'humain.
Chez des rats et des singes auxquels on avait administré 26 injections par voie sous-cutanées une fois par semaine, à des expositions systémiques respectivement 37 et 90 fois supérieures à celle appliquée à l'homme après l'administration d'une dose mensuelle par voie sous-cutanée de 210 mg de romosozumab (d'après la comparaison des ASC), aucun effet indésirable n'a été constaté.
Chez les rats adolescents qui ont reçu un substitut d'anticorps anti-rongeur dirigé contre la sclérostine à des doses pharmacologiquement efficaces, une hausse temporaire du taux de croissance longitudinale a été observée, ce qui correspond à une hausse prédictive de la longueur des os < 1%. Chez les rats adolescents qui ont reçu du romosozumab pendant 6 mois, aucun effet n'a été observé sur la longueur du fémur à des expositions jusqu'à 19 fois supérieures à l'exposition systémique observée chez l'humain après l'administration d'une dose sous-cutanée mensuelle de 210 mg de romosozumab (en se basant sur la comparaison des ASC).
Dans des études de sécurité sur les os menées sur des rates et des singes ayant subi une ovariectomie, le traitement de 12 mois a amplifié l'ossification et diminué la résorption osseuse dans le cadre d'une administration hebdomadaire de romosozumab. La hausse de la masse osseuse et l'amélioration de la géométrie osseuse au niveau des os corticaux et de la micro-architecture osseuse trabéculaire qui s'en sont suivies ont été associées à une plus grande résistance osseuse à des expositions 0,5 à 21 fois supérieures à l'exposition systémique de l'humain après l'administration d'une dose mensuelle par voie sous-cutanée de 210 mg de romososumab (en se basant sur la comparaison des ASC). La qualité du tissu osseux était normale, voire meilleure, et aucun signe de défaut de minéralisation, d'accumulation d'ostéoïde ou d'os fibreux n'a été détecté.
Carcinogénicité
Dans une étude de carcinogénicité, des doses allant jusqu'à 50 mg/kg/semaine ont été administrées par injections sous-cutanées à des rats et des rates Sprague-Dawley ayant entre 8 semaines et 98 semaines. Ces doses ont entraîné des expositions systémiques jusqu'à 19 fois supérieures à l'exposition systémique observée chez l'homme après l'administration d'une dose mensuelle par voie sous-cutanée de 210 mg de romosozumab (en se basant sur la comparaison des ASC). Le romosozumab a entraîné une hausse dose-dépendante de la masse osseuse, accompagnée d'un épaississement osseux macroscopique à toutes les doses. Le romosozumab n'a eu aucun effet sur la mortalité ou la tumorogénicité chez les rats et les rates.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C). Ne pas congeler.
Le stylo prérempli peut être conservé jusqu'à 25 °C dans son emballage d'origine pendant une durée allant jusqu'à 30 jours. S'il n'est pas utilisé dans les 30 jours, il doit être éliminé, même s'il a été conservé au réfrigérateur.
Conserver le récipient dans son carton pour le protéger de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Vérifier visuellement si la solution comporte des particules ou présente une coloration avant l'administration. Si la solution est teintée ou trouble ou si elle contient des particules, ne pas utiliser le romosozumab.
Laisser le romosozumab revenir à température ambiante pendant 30 minutes avant l'injection par voie sous-cutanée. Ainsi, l'injection est moins douloureuse. Ne pas le réchauffer par un autre moyen.
Ne pas agiter.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale en vigueur.
Numéro d’autorisation67033 (Swissmedic)
PrésentationEvenity, solution injectable en stylo prérempli: 2 [B]
Titulaire de l’autorisationUCB Pharma SA, 1630 Bulle
Mise à jour de l’informationJuillet 2024
|