CompositionPrincipes actifs
Capsules molles à 20 mg: tafamidisum megluminum.
Capsules molles à 61 mg: tafamidisum.
Excipients
Capsule molle à 20 mg: bleu brillant FCF, carminum, gelatina, glycerolum, ferrum oxydatum flavum, macrogolum 400, polysorbatum 80, polyvinylis acetas phthalas, propylenglycolum, sorbitani mono-oleas, sorbitolum liquidum partim deshydricum corresp. sorbitolum (E 420, max. 44 mg), titanii dioxidum.
Capsule molle à 61 mg: butylhydroxytoluenum, gelatina, glycerolum, ferrum oxydatum rubrum, macrogolum 400, polysorbatum 20, polyvinylis acetas phthalas, povidonum (valeur K 90), propylenglycolum, sorbitolum liquidum partim deshydricum corresp. sorbitolum (E 420, max. 44 mg), titanii dioxidum.
Indications/Possibilités d’emploiVyndaqel est indiqué dans le traitement de l'amylose à transthyrétine chez les patients adultes présentant une cardiomyopathie de type sauvage ou héréditaire, afin de réduire la mortalité toutes causes confondues et les hospitalisations d'origine cardiovasculaire.
Posologie/Mode d’emploiLe traitement doit être instauré par un médecin expérimenté dans la prise en charge de patients présentant une amylose ou une cardiomyopathie.
La dose recommandée de Vyndaqel est de 61 mg de tafamidis ou de 80 mg de tafamidis méglumine (administrée sous forme de 4 capsules à 20 mg) administrée par voie orale en une prise journalière (voir «Propriétés/Effets»).
En cas d'intolérance, la dose peut être réduite à 20 mg de tafamidis méglumine à la discrétion du médecin traitant.
Tafamidis et tafamidis méglumine ne sont pas interchangeables sur la base de la spécification en milligrammes. 61 mg de tafamidis sont bioéquivalents à 80 mg de tafamidis méglumine. Pour d'autres informations concernant la bioéquivalence, voir «Propriétés/Effets» et «Pharmacocinétique».
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique légère à modérée. Vyndaqel n'a pas été étudié chez les patients atteints d'une insuffisance hépatique sévère. La prudence est donc de rigueur chez ces patients.
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale. Les données disponibles chez les patients atteints d'insuffisance rénale sévère (clairance de la créatinine inférieure ou égale à 30 ml/min) sont limitées.
Patients âgés
Aucun ajustement posologique n'est nécessaire chez les patients âgés (≥65 ans) (voir «Pharmacocinétique»).
Enfants et adolescents
Vyndaqel ne doit pas être prescrit aux enfants et aux adolescents, car son utilisation et son innocuité n'ont pas été testées et l'amylose à transthyrétine n'est pas présente dans cette population.
Prise retardée
En cas d'omission d'une prise, le patient doit la prendre dès qu'il s'en rend compte. S'il reste moins de 6 heures avant la prise suivante, la dose oubliée ne doit pas être prise et la dose suivante doit être prise à l'heure habituelle. Il ne faut pas prendre de double dose.
Mode d'administration
Voie orale.
Les capsules doivent être avalées entières, sans être croquées ni coupées. Vyndaqel peut être pris avec ou sans nourriture.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients de Vyndaqel.
Mises en garde et précautionsAucune étude n'est disponible concernant les patients ayant subi une transplantation d'organe. L'efficacité et la sécurité de Vyndaqel chez les patients après transplantation d'organe ne sont pas connues.
Ce médicament contient 44 mg de sorbitol (E 420) par capsule.
L'effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l'apport alimentaire de sorbitol (ou de fructose) doit être pris en compte.
La teneur en sorbitol dans les médicaments à usage oral peut affecter la biodisponibilité d'autres médicaments à usage oral administrés de façon concomitante.
InteractionsÉtudes in vitro
Le tafamidis est un inducteur du CYP2B6 et du CYP3A4, mais pas du CYP1A2. Le tafamidis n'inhibe pas le CYP1A2, le CYP2B6, le CYP2C8, le CYP2C9, le CYP2C19, le CYP3A4/5 ou le CYP2D6.
Des études in vitro montrent que des interactions médicamenteuses systémiques entre le tafamidis administré à des concentrations cliniquement pertinentes et des substrats de l'UDP-glucuronosyltransférase (UGT) sont peu probables. Le tafamidis pourrait inhiber l'activité intestinale de l'UGT1A1.
Le tafamidis a montré un faible potentiel d'inhibition de la Multidrug-Resistance-Protein (MDR1, également connue sous le nom de glycoprotéine-P [Pgp]) de façon systémique et dans le tractus gastro-intestinal, ainsi que d'inhibition du transporteur de cations organiques 2 (organic cation transporter 2, OCT2), des transporteurs MATE (multidrug and toxin extrusion) MATE1 et MATE2K, et des polypeptides de transport d'anions organiques 1B1 (organic anion transporting polypeptide, OATP1B1) et OATP1B3, à des concentrations cliniquement pertinentes.
Dans une étude clinique menée chez des participants sains, l'exposition à la rosuvastatine, un substrat de la BCRP (protéine de résistance au cancer du sein) a environ doublé après l'administration quotidienne répétée de 61 mg de tafamidis. Il convient de tenir compte des recommandations posologiques correspondantes pour les substrats de la BCRP sensibles (p.ex. le méthotrexate, la rosuvastatine, l'imatinib) spécifiées dans les informations professionnelles respectives.
Le tafamidis pourrait inhiber les transporteurs d'anions organiques 1 (organic anion transporter, OAT1) et OAT3 et entraîner des interactions médicamenteuses avec les substrats de ces transporteurs (p.ex. anti-inflammatoires non stéroïdiens, bumétanide [non autorisé en Suisse], furosémide, lamivudine, méthotrexate, oseltamivir, ténofovir, ganciclovir, adéfovir, cidofovir, zidovudine, zalcitabine [non autorisé en Suisse]).
Effet de Vyndaqel sur d'autres médicaments
Aucun effet significatif sur la pharmacocinétique du midazolam (un substrat du CYP3A4) ou sur la formation de son métabolite actif (1hydroxymidazolam) n'a été constaté lorsqu'une dose unique de 7.5 mg de midazolam a été administrée avant et après un traitement de 14 jours par 20 mg de tafamidis méglumine une fois par jour. L'exposition systémique totale (AUC0-∞) et la clairance totale (CL/F) du midazolam étaient équivalentes.
Effet d'autres médicaments sur Vyndaqel
Aucune étude d'interaction n'a été réalisée pour évaluer l'effet d'autres médicaments sur le tafamidis.
Autres interactions
Anomalies biologiques
Le tafamidis pourrait diminuer les concentrations sériques de thyroxine totale, sans modification de la thyroxine libre (T4) ou de l'hormone thyréostimulante (TSH). Cette observation concernant le taux de thyroxine totale résulte probablement d'une réduction de la liaison de la thyroxine à la transthyrétine (TTR) ou de la délocalisation de celle-ci du fait de la forte affinité de liaison du tafamidis au récepteur de la TTR. Aucun résultat clinique suggérant un dysfonctionnement thyroïdien n'a été observé.
Grossesse, allaitementFemmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par Vyndaqel et pendant un mois après l'arrêt du traitement, du fait de la longue demi-vie du médicament.
L'utilisation de Vyndaqel n'est pas recommandée chez les femmes en âge de procréer n'utilisant pas de contraception efficace.
Grossesse
Il n'existe pas de données suffisantes concernant l'emploi chez la femme enceinte.
Les expérimentations animales ont révélé une toxicité de reproduction (données plus précises sous la rubrique «Données précliniques»).
Le risque potentiel pour l'être humain n'est pas connu. Vyndaqel ne doit pas être administré pendant la grossesse.
Afin de surveiller les effets sur les femmes enceintes exposées au Vyndaqel, un programme visant à améliorer la surveillance des effets du tafamidis sur la grossesse a été introduit (Tafamidis Enhanced Surveillance for Pregnancy Outcomes=TESPO). Si une grossesse survient chez une femme traitée avec Vyndaqel, les professionnels de la santé doivent signaler la grossesse au titulaire de l'autorisation.
Allaitement
Les effets de Vyndaqel sur les nourrissons allaités après exposition de la mère n'ont pas été étudiés. Les études expérimentales menées chez les animaux ont montré que le tafamidis passe dans le lait maternel (voir «Données précliniques»). Il n'existe aucune donnée clinique sur la détection du tafamidis dans le lait maternel humain. Un risque pour le nouveau-né/l'enfant ne peut pas être exclu. Vyndaqel ne doit pas être administré durant l'allaitement.
Fertilité
Chez le rat, aucun effet sur la fertilité, la capacité de reproduction ou l'indice d'accouplement n'a été démontré à aucune des doses testées (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesCardiomyopathie amyloïde à transthyrétine (ATTR-CM)
Des effets indésirables chez les patients atteints de cardiomyopathie amyloïde à transthyrétine (ATTR-CM) ont été évalués à partir des études cliniques et de la surveillance après la mise sur le marché. Les données cliniques en matière de sécurité proviennent de 3 études cliniques combinées, y compris une étude contrôlée par placebo d'une durée de 30 mois (441 patients, parmi lesquels 264 ont reçu du tafamidis méglumine et 177 un placebo) (voir «Propriétés/Effets»), une étude de prolongation ouverte non contrôlée à long terme sur une période allant jusqu'à 10 ans (31 patients) et une étude de prolongation ouverte non contrôlée à long terme concernant la sécurité d'une durée de 60 mois (1'728 patients, parmi lesquels 170 avaient reçu du tafamidis méglumine dans l'étude précédente contrôlée par placebo). L'étude de prolongation à long terme regroupait des participants inclus dans l'étude contrôlée par placebo de 30 mois, qui avaient terminé un traitement en aveugle de 30 mois (cohorte A), et des participants atteints d'ATTR-CM qui n'avaient pas participé à l'étude contrôlée par placebo de 30 mois (cohorte B).
Les données cliniques combinées en matière de sécurité reflètent l'exposition de 1'853 patients atteints d'ATTR-CM à une dose quotidienne de tafamidis méglumine (20 mg ou 80 mg [administré sous forme de quatre capsules de 20 mg]) ou de tafamidis (61 mg) sur une durée moyenne de 31.1 mois (entre 1 jour et 111 mois). La population comprenait des patients adultes ayant reçu un diagnostic d'ATTR-CM et dont la majorité (environ 92%) présentait une insuffisance cardiaque au début de l'étude et avait été répartie dans les classes NYHA (New York Heart Association) II ou III.
Dans l'étude contrôlée par placebo d'une durée de 30 mois, les événements indésirables suivants sont survenus plus fréquemment chez les patients traités par une dose quotidienne de 80 mg ou 20 mg de tafamidis méglumine que chez les patients traités par placebo:
·Flatulence (8 patients [4.5%] sous 80 mg de tafamidis méglumine et 2 patients [2.3%] sous 20 mg de tafamidis méglumine par rapport à 3 patients [1.7%] sous placebo) et
·Élévation des tests hépatiques (6 patients [3.4%] sous 80 mg de tafamidis méglumine et 2 patients [2.3%] sous 20 mg de tafamidis méglumine par rapport à 2 patients [1.1%] sous placebo).
·Problèmes d'équilibre (15 patients [8.5%] sous 80 mg de tafamidis méglumine et 2 patients [2.3%] sous 20 mg de tafamidis méglumine par rapport à 2 patients [1.1%] sous placebo).
·Ulcère cutané (7 patients [4.0%] sous 80 mg de tafamidis méglumine et 3 patients [3.4%] sous 20 mg de tafamidis méglumine par rapport à 1 patient [0.6%] sous placebo).
·Cataracte (9 patients [5.1%] sous 80 mg de tafamidis méglumine et 3 patients [3.4%] sous 20 mg de tafamidis méglumine par rapport à 2 patients [1.1%] sous placebo).
·Kératose actinique (3 patients [1.7%] sous 80 mg de tafamidis méglumine et 3 patients [3.4%] sous 20 mg de tafamidis méglumine). Cet effet indésirable n'a pas été observé dans le groupe traité par placebo.
Aucun lien de causalité entre la prise de tafamidis et la survenue de ces événements indésirables n'a été démontré.
Les effets indésirables les plus fréquents dans les données cliniques combinées concernant la sécurité étaient la diarrhée (7.7%), le prurit (3.4%) et le rash cutané (2.2%).
Les effets indésirables sont rangés ci-dessous par classe de système d'organes de la classification MedDRA et par fréquence selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (≥1/100 à <1/10), «occasionnels» (≥1/1'000 à <1/100), «rares» (≥1/10'000 à <1/1'000), «très rares» (<1/10'000), «Fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Affections gastro-intestinales
Fréquents: diarrhée*.
Affections de la peau et du tissu sous-cutané
Fréquents: prurit*, rash cutané*.
* Effets indésirables identifiés après la mise sur le marché; catégorie de fréquence basée sur l'incidence dans les études cliniques.
Polyneuropathie amyloïde à transthyrétine (ATTR-PN) (Indication non autorisée en Suisse)
Effets indésirables lors du traitement de l'ATTR-PN par 20 mg de tafamidis méglumine
Les effets indésirables énoncés ci-dessous pour Vyndaqel ont été observés dans une étude de phase III de 18 mois (Fx-005), pertinente pour obtenir une autorisation de mise sur le marché, multicentrique, randomisée, en double aveugle et contrôlée par placebo, menée chez des patients adultes (n=128) atteints de polyneuropathie amyloïde à transthyrétine (ATTR-PN) symptomatique de stade 1, qui ont été traités par une dose quotidienne de 20 mg de tafamidis méglumine (n=65) ou par placebo (n=63). Vyndaqel n'est pas autorisé en Suisse pour cette indication.
Infections et infestations
Très fréquents: infections des voies urinaires (23%), infections vaginales (12%).
Affections gastro-intestinales
Très fréquents: diarrhée (26%), douleurs dans la région abdominale supérieure (12%).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes données disponibles concernant le surdosage sont limitées. Deux patients diagnostiqués avec une ATTR-CM et inclus dans des études cliniques ont accidentellement ingéré une dose unique de 160 mg de tafamidis méglumine sans qu'aucun événement indésirable associé ne survienne. La dose maximale de tafamidis méglumine administrée à des volontaires sains lors d'une étude clinique a été de 480 mg en une prise. À cette dose, un orgelet bénin a été rapporté en tant qu'événement indésirable lié au traitement.
Propriétés/EffetsCode ATC
N07XX08
Mécanisme d'action
Le tafamidis est un stabilisateur sélectif de la TTR. Le tafamidis se lie de façon non coopérative aux deux sites de fixation de la thyroxine présents sur la forme native tétramérique de la TTR, empêchant sa dissociation en monomères, c'est-à-dire l'étape limitant la vitesse dans l'amyloïdogénèse. L'inhibition de la dissociation du tétramère de TTR constitue la base de l'utilisation de tafamidis pour réduire la mortalité toutes causes confondues et les hospitalisations d'origine cardiovasculaire des patients atteints d'ATTR-CM.
Pharmacodynamique
La stabilité du tétramère de TTR dans des conditions de dénaturation a pu être évaluée au moyen d'un test de stabilisation de TTR utilisé comme marqueur pharmacodynamique.
Le tafamidis a stabilisé à la fois le tétramère TTR de type sauvage et les tétramères de 14 variants de TTR, comme l'ont montré les essais cliniques après une prise unique quotidienne. Le tafamidis a également stabilisé le tétramère TTR de 25 variants supplémentaires testés ex-vivo, démontrant ainsi la stabilisation de la TTR de 40 génotypes TTR amyloïdogéniques.
Électrophysiologie cardiaque
À environ 2.2 fois la concentration plasmatique maximale (Cmax) à l'état d'équilibre à la dose recommandée, le tafamidis n'allonge pas l'intervalle QTc dans une mesure cliniquement pertinente.
Efficacité clinique
ATTR-CM
L'efficacité a été démontrée dans une étude multicentrique, internationale, en double aveugle, contrôlée par placebo et randomisée sur 3 groupes de 441 patients atteints d'ATTR-CM de type sauvage ou héréditaire.
Les patients ont été randomisés pour recevoir soit du tafamidis méglumine 20 mg (n=88) ou 80 mg [administré sous forme de quatre capsules de 20 mg] (n=176), soit le placebo correspondant (n=177) en plus des traitements habituels (p.ex. diurétiques) une fois par jour sur une durée de 30 mois. L'attribution des traitements a été stratifiée en fonction des variants du génotype TTR ou de l'absence de tels variants et en fonction de la gravité de la maladie à l'inclusion (classe NYHA). Le Tableau 1 présente les données démographiques et les caractéristiques à l'inclusion des patients.
Tableau 1: Données démographiques et caractéristiques à l'inclusion des patients
|
Caractéristique
|
Tafamidis méglumine combiné, doses de 20 mg et 80 mg
n=264
|
Placebo
n=177
| |
Âge - années
| |
Moyenne (écart type)
|
74.5 (7.2)
|
74,1 (6,7)
| |
Valeur médiane (minimum, maximum)
|
75 (46, 88)
|
74 (51, 89)
| |
Sexe — nombre (%)
| |
Hommes
|
241 (91.3)
|
157 (88.7)
| |
Femmes
|
23 (8.7)
|
20 (11.3)
| |
Génotype TTR — nombre (%)
| |
ATTRm
|
63 (23.9)
|
43 (24.3)
| |
ATTRwt
|
201 (76.1)
|
134 (75.7)
| |
Classe de la NYHA — nombre (%)
|
|
| |
Classe I de la NYHA
|
24 (9.1)
|
13 (7.3)
| |
Classe II de la NYHA
|
162 (61.4)
|
101 (57.1)
| |
Classe III de la NYHA
|
78 (29.5)
|
63 (35.6)
|
Abréviations: ATTRm = amylose héréditaire à transthyrétine, ATTRwt = amylose à transthyrétine de type sauvage.
L'analyse principale a utilisé une combinaison hiérarchique appliquant la méthode de Finkelstein-Schoenfeld (F-S) à la mortalité toutes causes confondues et à la fréquence des hospitalisations d'origine cardiovasculaire, définie comme le nombre de fois où un sujet est hospitalisé pour la prise en charge d'une morbidité cardiovasculaire. La méthode a été utilisée pour comparer chaque patient avec tous les autres patients d'une même strate, par paires. Pour ce faire, la hiérarchie suivante a été observée: mortalité totale (mortalité toutes causes confondues), suivie de l'hospitalisation d'origine cardiovasculaire s'il n'y a pas de possibilité de différenciation basée sur la mortalité.
L'analyse a montré une réduction significative (p=0.0006) de la mortalité toutes causes confondues et de la fréquence des hospitalisations d'origine cardiovasculaire dans les groupes ayant reçu 20 mg et 80 mg de tafamidis méglumine comparé au placebo (voir Tableau 2).
Tableau 2: Analyse principale de la mortalité toutes causes confondues et de la fréquence des hospitalisations d'origine cardiovasculaire selon la méthode de Finkelstein-Schoenfeld (F-S)
|
Analyse principale
|
Tafamidis méglumine combiné, doses de 20 mg et 80 mg
n=264
|
Placebo
n=177
| |
Nombre de patients vivants* (%) au mois 30
|
186 (70.5)
|
101 (57.1)
| |
Nombre moyen d'hospitalisations d'origine cardiovasculaire pendant 30 mois (par patient et par an) parmi les patients vivants au mois 30†
|
0.297
|
0.455
| |
Valeur de p d'après la méthode de F-S
|
0.0006
|
* La transplantation cardiaque et le recours à un dispositif d'assistance cardiaque mécanique sont considérés comme des indicateurs d'une phase terminale imminente. Ainsi, ces patients sont considérés comme l'équivalent d'un décès dans l'analyse et ne sont pas comptés dans la catégorie «Nombre de patients vivants au mois 30» même s'ils sont encore en vie lors de l'examen de suivi effectué après 30 mois.† Moyenne descriptive chez les patients ayant survécu après 30 mois.
L'analyse des différentes composantes de l'analyse principale (mortalité toutes causes confondues et hospitalisations d'origine cardiovasculaire) a également montré des réductions significatives pour le tafamidis méglumine comparé au placebo.
Le rapport de risque selon le modèle à risques proportionnels de Cox était de 0.698 (IC à 95% 0.508, 0.958) pour la mortalité toutes causes confondues pour les deux doses de tafamidis méglumine combinées. Cela représente une réduction de 30.2% (p=0.0259) du risque de mortalité par rapport au groupe placebo. La Figure 1 présente la courbe de Kaplan-Meyer indiquant le délai avant mortalité toutes causes confondues.
Figure 1: Mortalité toutes causes confondues*
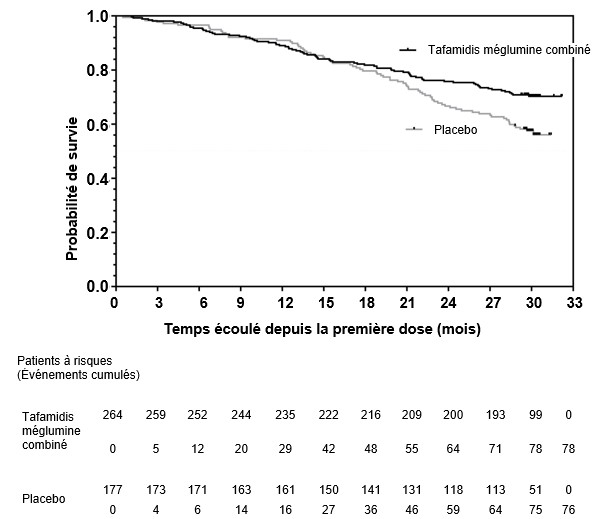
* Classification des transplantations cardiaques et des dispositifs d'assistance cardiaque comme des décès. Rapport de risque d'après le modèle à risques proportionnels de Cox avec traitement, génotype TTR (héréditaire et type sauvage) et classification NYHA à l'inclusion (classes I et II de la NYHA combinées et classe III de la NYHA) comme facteurs.
Le nombre d'hospitalisations d'origine cardiovasculaire a été significativement moindre avec le tafamidis méglumine comparativement au placebo. La réduction du risque est de 32.4% (voir Tableau 3).
Tableau 3: Fréquence des hospitalisations d'origine cardiovasculaire
|
|
Tafamidis méglumine combiné, doses de 20 mg et 80 mg
n=264
|
Placebo
n=177
| |
Nombre total (%) de patients hospitalisés pour des troubles d'origine cardiovasculaire
|
138 (52.3)
|
107 (60.5)
| |
Nombre annuel d'hospitalisations d'origine cardiovasculaire*
|
0.4750
|
0.7025
| |
Différence de traitement entre les données combinées sur le tafamidis méglumine et le placebo (rapport de risque relatif)*
|
0.6761
| |
Valeur de p*
|
<0.0001
|
Abréviations: NYHA = New York Heart Association
* Analyse basée sur un modèle de régression de Poisson avec traitement, génotype TTR (héréditaire et type sauvage), classification NYHA à l'inclusion (classes I et II de la NYHA combinées et classe III de la NYHA), rapport entre traitement et génotype TTR ainsi que rapport entre traitement et classification NYHA à l'inclusion comme facteurs.
Les résultats de la méthode F-S, représentés par le taux de succès pour le critère d'efficacité combiné et ses composantes (mortalité toutes causes confondues et fréquence des hospitalisations d'origine cardiovasculaire), ont été en faveur du tafamidis méglumine dans les sous-groupes suivants (type sauvage, type héréditaire et classes I et II de la NYHA) par rapport au placebo, sauf pour la fréquence des hospitalisations d'origine cardiovasculaire chez les patients de classe III de la NYHA (voir Figure 2).
Figure 2: Résultats de la méthode F-S et composantes par sous-groupe et par dose
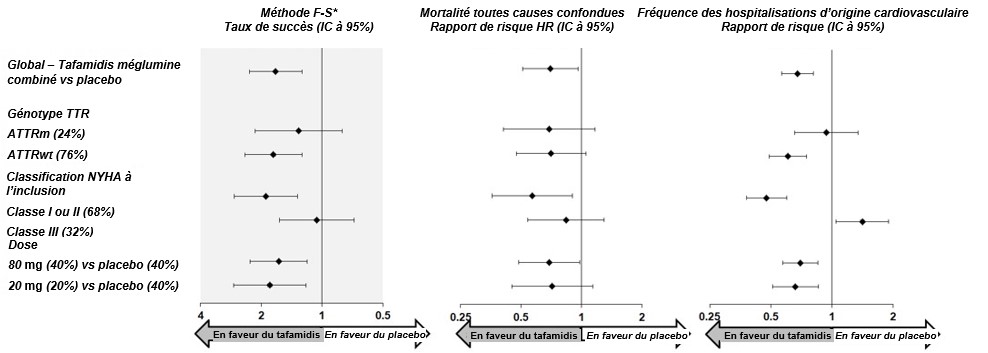
Abréviations: ATTRm = Variant héréditaire de l'amylose à transthyrétine, ATTRwt = amylose à transthyrétine de type sauvage, F-S = Finkelstein-Schoenfeld, IC = intervalle de confiance.
*Résultats de la méthode F-S présentés en utilisant le taux de succès (basé sur la mortalité toutes causes confondues et la fréquence des hospitalisations d'origine cardiovasculaire).
Classification des transplantations cardiaques et des dispositifs d'assistance cardiaque dans les décès.
En appliquant la méthode F-S à chaque groupe posologique, le tafamidis méglumine a réduit le critère combiné de la mortalité toutes causes confondues et de la fréquence des hospitalisations d'origine cardiovasculaire aussi bien pour la dose de 20 mg que pour la dose de 80 mg comparativement au placebo (p=0.0048 et p=0.0030).
L'effet thérapeutique du tafamidis méglumine, accompagné de critères d'évaluation secondaires concernant la capacité fonctionnelle et l'état de santé, a été évalué au moyen du Test de marche de six minutes (6 Minute Walk Test, 6MWT) et du score global du questionnaire général sur la cardiomyopathie de Kansas City (Kansas City Cardiomyopathy Questionnaire-Overall Summary, KCCQ-OS).
Un effet thérapeutique significatif en faveur du tafamidis méglumine a été observé pour la première fois au mois 6 et s'est maintenu jusqu'au mois 30, aussi bien pour la distance du 6MWT que pour le score KCCQ-OS. Les biomarqueurs associés à l'insuffisance cardiaque (NT-proBNP et Troponine I) ont privilégié le tafamidis méglumine par rapport au placebo.
PharmacocinétiqueLe profil pharmacocinétique du tafamidis a été étudié dans des études de phase I chez des volontaires sains et des patients atteints d'ATTR-PN et d'ATTR-CM.
Absorption
Après une prise journalière de tafamidis méglumine ou de tafamidis par voie orale, le pic de concentration maximale (Cmax) est atteint au cours d'une durée médiane (tmax) de 4 heures après une prise à jeun. L'administration concomitante d'un repas riche en matières grasses et en calories a affecté la vitesse d'absorption, mais pas la quantité absorbée. Ces résultats confirment la possibilité de prendre du tafamidis méglumine ou du tafamidis avec ou sans nourriture.
L'exposition à l'état d'équilibre (Cmax et AUC) de 61 mg de tafamidis équivaut à 80 mg de tafamidis méglumine (administré sous forme de 4 capsules de 20 mg), que les patients présentant une ATTR-CM ont reçu au cours de l'étude randomisée en double aveugle contrôlée par placebo (voir Tableau 4, voir «Propriétés/Effets»).
Tableau 4: Pharmacocinétique comparative d'une capsule de 61 mg de tafamidis et de tafamidis méglumine administré sous forme de quatre capsules de 20 mg
|
Paramètres
(Unités)
|
Comparaison
(Test vs référence)
|
Moyenne géométrique adaptée
|
Test versus référence
| |
Test
|
Référence
|
Rapport (%)a
(Test/référence)
|
IC à 90%a
pour rapport
| |
AUCtau
(µg.h/ml)
|
Tafamidis capsule à 61 mg (test)
versus
Tafamidis méglumine
quatre capsules à 20 mg (référence)
|
170.0
|
166.2
|
102.28
|
(97.99, 106.76)
| |
Cmax
(µg/ml)
|
8.553
|
9.087
|
94.12
|
(89,09, 99,42)
|
Abréviations: IC = intervalle de confiance; AUCtau = aire sous la courbe du temps 0 jusqu'au temps tau, intervalle de dosage avec tau = 24 heures dans le cas d'une dose journalière; Cmax = concentration sérique maximale.
a Indication des ratios et IC à 90% exprimés en pourcentage.
Distribution
Le tafamidis est fortement lié aux protéines plasmatiques (>99%). Le volume apparent de distribution à l'état d'équilibre est de 16 litres pour le tafamidis méglumine et de 18.5 litres pour le tafamidis.
L'étendue de la liaison du tafamidis aux protéines plasmatiques a été évaluée à l'aide de plasma animal et humain. L'affinité du tafamidis pour la TTR est 1'000 fois supérieure à celle pour l'albumine. Le tafamidis se lie donc de préférence à la TTR malgré la concentration plasmatique significativement plus élevée de l'albumine (600 μM) par rapport à la TTR (3.6 μM).
Métabolisme
Il n'existe aucune évidence d'une excrétion biliaire du tafamidis chez l'homme. Les données précliniques suggèrent cependant que le tafamidis est métabolisé par glucuronidation et excrété dans la bile. Cette voie de métabolisme et de transformation est plausible chez l'homme, car environ 59% de la dose totale administrée est détectée en grande partie sous forme de substance active inchangée dans les selles et environ 22% sous forme de métabolite glucuronide dans l'urine.
Élimination
Sur la base des résultats de pharmacocinétique de population, la clairance orale apparente du tafamidis méglumine est de 0.228 l/h (celle du tafamidis est de 0.263 l/h). La demi-vie moyenne parmi la population a été d'environ 49 heures. Le degré d'accumulation du médicament à l'état d'équilibre est environ 2.5 fois plus élevé après l'administration de doses journalières répétées qu'après l'administration d'une dose unique.
La demi-vie moyenne et la clairance orale étaient similaires après l'administration unique et l'administration répétée de 20 mg de tafamidis méglumine, indiquant l'absence d'induction ou d'inhibition du métabolisme du tafamidis.
Les résultats après l'administration d'une solution buvable de 15 mg à 60 mg de tafamidis méglumine une fois par jour pendant 14 jours ont montré que l'état d'équilibre était atteint au jour 14.
Linéarité/non-linéarité
L'exposition au tafamidis méglumine administré une fois par jour a augmenté avec l'augmentation de la dose jusqu'à 480 mg en dose unique et jusqu'à 80 mg par jour en doses multiples. En général, l'augmentation était proportionnelle ou légèrement moins que proportionnelle à la dose.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Les données de pharmacocinétique montrent une diminution de l'exposition systémique (environ 40%) et une augmentation de la clairance totale (0.52 l/h versus 0.31 l/h) de Vyndaqel (tafamidis méglumine) chez les patients présentant une insuffisance hépatique modérée (score de Child-Pugh 7 à 9 inclus) comparativement aux sujets sains. Étant donné que les patients présentant une insuffisance hépatique modérée ont des taux plus faibles de TTR que les sujets sains, une exposition au tafamidis en fonction des niveaux de TTR serait suffisante pour stabiliser le tétramère de TTR chez ces patients. Chez les patients présentant une insuffisance hépatique légère, l'exposition au tafamidis a été similaire à celle des sujets sains.
L'exposition au tafamidis chez les patients présentant une insuffisance hépatique sévère n'est pas connue.
Troubles de la fonction rénale
Le tafamidis n'a pas été spécifiquement évalué chez les patients atteints d'insuffisance rénale. Le tafamidis est principalement métabolisé par glucuronidation et probablement excrété par voie hépatobiliaire. Les répercussions de la clairance de la créatinine sur la pharmacocinétique du tafamidis ont été évaluées dans une analyse pharmacocinétique de population chez des patients dont la clairance de la créatinine était >18 ml/min. Les estimations pharmacocinétiques n'ont indiqué aucune différence en termes de clairance orale apparente du tafamidis chez les patients dont la clairance de la créatinine était <80 ml/min et ceux dont la clairance de la créatinine était ≥80 ml/min. Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale. Les données disponibles chez les patients atteints d'insuffisance rénale sévère (clairance de la créatinine ≤30 ml/min) sont limitées.
Patients âgés
Sur la base des résultats de la pharmacocinétique de population, les patients ≥65 ans ont une clairance orale apparente à l'état d'équilibre en moyenne inférieure de 15% à celle des sujets de moins de 65 ans. Cependant, cette différence de clairance entraîne une augmentation <20% de la Cmax et de l'AUC moyennes par rapport aux sujets plus jeunes et n'est pas cliniquement significative.
Données précliniquesSur la base des études conventionnelles concernant la pharmacologie de la sécurité, la toxicité en cas d'administration répétée chez le rat et le chien, la fertilité et le développement embryonnaire précoce, la génotoxicité et le potentiel cancérigène, aucun risque particulier pour l'homme n'a été décelé dans les données précliniques.
Lors d'études de toxicité et de cancérogénicité consistant à administrer des doses répétées chez la souris et le rat, le foie et/ou les reins sont apparus comme les organes cibles de la toxicité. Des effets sur le foie ont été observés en cas d'expositions correspondant environ à ≥0.7 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et de 80 mg de tafamidis méglumine (voir également «Carcinogénicité»).
Carcinogénicité
Il n'y a eu aucune preuve d'une augmentation de l'incidence des néoplasies dans une étude de carcinogénicité de deux ans chez les rats avec des expositions jusqu'à 18 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et 80 mg de tafamidis méglumine. Des lésions non néoplasiques du foie (y compris une hypertrophie centrolobulaires et une nécrose) ont été observées à des expositions correspondant à ≥3.4 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et 80 mg de tafamidis méglumine.
Aucune preuve d'augmentation de l'incidence de néoplasie n'a été établie chez la souris transgénique (Tg)rasH2 après une administration quotidienne répétée pendant 26 semaines avec des expositions multipliant jusqu'à 9.6 fois et 9.9 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et 80 mg de tafamidis méglumine. Dans l'étude en question, des lésions non néoplasiques significatives ont été identifiées dans les reins (néphrose) et le foie (hypertrophie centrolobulaire et nécrose unicellulaire) des souris (Tg)rasH2 à des doses équivalentes à ≥2.8 fois et ≥2.9 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et 80 mg de tafamidis méglumine.
Toxicité sur la reproduction
Fertilité
Chez le rat, aucun effet sur la fertilité, la capacité de reproduction ou l'indice d'accouplement n'a été démontré à aucune des doses de tafamidis testées. Des rats ont reçu une dose journalière de tafamidis avant l'accouplement (pendant au moins 15 jours chez les femelles et 28 jours chez les mâles), et pendant la période d'accouplement jusqu'à la veille de la fin de la période d'accouplement (mâles) et jusqu'à la nidation (7e jour de la gestation chez les femelles). La dose était de (5, 15 et 30 mg/kg/jour). Étant donné l'absence d'effet sur la reproduction à la dose la plus élevée étudiée, les doses sans effet observé DSEO (no observed effect level, NOEL), paternelle et maternelle, pour la toxicité du tafamidis sur la reproduction sont supérieures à 30 mg/kg/jour (dose équivalente de tafamidis chez l'homme supérieure à 4.8 mg/kg/jour), soit 5.5 fois et 6.9 fois plus élevées que les doses cliniques de 61 mg de tafamidis et de 80 mg de tafamidis méglumine.
Toxicité sur le développement
Dans une étude de toxicité sur le développement embryofœtal chez le lapin, l'administration orale de tafamidis (0.5, 2 ou 8 mg/kg/jour) du 7e au 19e jour de la gestation a entraîné une légère augmentation des variations squelettiques à ≥2 mg/kg/jour (environ ≥2.1 fois et ≥2.2 fois l'AUC à l'état d'équilibre chez l'homme à des doses cliniques de 61 mg de tafamidis et de 80 mg de tafamidis méglumine). Des malformations du squelette, des diminutions de la survie embryofœtale, et des diminutions du poids fœtal ont été observées à 8 mg/kg/jour (environ 9.1 fois et 9.3 fois l'AUC à l'état d'équilibre chez l'homme à des doses cliniques de 61 mg de tafamidis et de 80 mg de tafamidis méglumine). Dans une étude de toxicité sur le développement embryofœtal chez le rat, l'administration orale de tafamidis (15, 30 ou 45 mg/kg/jour) du 7e au 17e jour de la gestation a entraîné une réduction du poids fœtal sans effets sur la morphologie du fœtus à ≥30 mg/kg/jour (environ ≥9.5 fois et ≥9.7 fois l'AUC chez l'homme à des doses cliniques de 61 mg de tafamidis et de 80 mg de tafamidis méglumine).
Dans l'étude du développement pré et postnatal chez le rat, les mères portantes ont reçu des doses orales de tafamidis de 5, 15 et 30 mg/kg/jour dès le 7e jour de gestation jusqu'au 20e jour de lactation. Une diminution de la survie et du poids des ratons a été observée à 15 mg/kg/jour et à 30 mg/kg/jour. Une diminution du poids des ratons mâles a été associée à un retard de la maturation sexuelle (séparation préputiale) à 15 mg/kg/jour. Une altération des performances dans le test du labyrinthe aquatique concernant l'apprentissage et la mémoire a été observée à 15 mg/kg/jour. La dose sans effet nocif observable DSENO (no observable adverse effect level, NOAEL) pour la viabilité et la croissance de progéniture de la génération F1 après administration d'une dose de tafamidis aux mères pendant la gestation et l'allaitement était de 5 mg/kg (dose équivalente chez l'homme = 0.8 mg/kg), soit environ 0.92 fois la dose clinique de 61 mg de tafamidis et environ 1.2 fois la dose clinique de 80 mg de tafamidis méglumine. Il a été démontré que le tafamidis passe dans le lait des rates en lactation.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 25 °C.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Tout médicament non utilisé doit être éliminé conformément à la réglementation en vigueur.
Numéro d’autorisation67083, 67518 (Swissmedic).
PrésentationCapsules molles à 20 mg: 30. [B]
Capsules molles à 61 mg: 30. [B]
Titulaire de l’autorisationPfizer AG, Zürich.
Mise à jour de l’informationOctobre 2023.
LLD V014
|