Propriétés/EffetsCode ATC
N06AX27
Mécanisme d'action
L'eskétamine, l'énantiomère S de la kétamine racémique, est un antidépresseur ayant un nouveau mécanisme d'action. Il s'agit d'un antagoniste non sélectif et non compétitif du récepteur Nméthyl-Daspartate (NMDA), un récepteur ionotrope du glutamate.
On sait que les facteurs étiologiques soupçonnés de contribuer à la dépression, notamment le stress et d'autres conditions, entraînent une perturbation structurelle et fonctionnelle des synapses dans les régions du cerveau impliquées dans la régulation de l'humeur et du comportement émotionnel. Selon les informations présentes dans la littérature, l'eskétamine provoque une augmentation temporaire de la libération de glutamate en exerçant un effet antagoniste sur le récepteur NMDA, ce qui entraîne une stimulation accrue du récepteur de l'acide α-amino-3-hydroxy-5-méthyl-4-isoxazole-propionique (AMPAR) et par la suite une production accrue de signaux neurotrophiques, qui restaurent à leur tour la fonction synaptique dans ces zones du cerveau. Contrairement à d'autres traitements antidépresseurs, les récepteurs des monoamines, GABA ou opioïdes ne sont pas directement impliqués dans l'effet antidépresseur principal de l'eskétamine.
Pharmacodynamique
Voir sous Mécanisme d'action.
Efficacité clinique
L'efficacité et la sécurité de Spravato en spray nasal ont été initialement évaluées au cours de cinq études cliniques de phase 3 (TRD3001, TRD3002, TRD3003, TRD3004 et TRD3005) menées chez des patients adultes (de 18 à 86 ans) atteints de DRT qui répondaient aux critères du TDM selon le DSM-5 et qui n'ont pas répondu à au moins deux antidépresseurs oraux à une posologie et sur une durée adéquates pendant l'épisode dépressif actuel (non-répondeurs). 1833 patients adultes ont été inclus dans les études, parmi lesquels 1601 ont reçu Spravato. Par ailleurs, 202 patients ont été randomisés dans l'étude de phase 2 TRD2005 au Japon (parmi lesquels 122 patients ont reçu Spravato), 252 patients ont été randomisés dans l'étude de phase 3 TRD3006 principalement en Chine (parmi lesquels 126 patients ont reçu Spravato), et 676 patients ont été randomisés dans l'étude de phase 3 TRD3013 (parmi lesquels 334 patients ont reçu Spravato).
Dépression résistante au traitement (DRT) – études à court terme
Spravato a été évalué dans trois études de phase III de courte durée (4 semaines), randomisées, en double aveugle, multicentriques et contrôlées contre comparateur actif, menées chez des patients atteints de DRT. Les études TRANSFORM-1 (TRD3001) et TRANSFORM-2 (TRD3002) ont été réalisées chez des adultes (de 18 à < 65 ans) et l'étude TRANSFORM-3 (TRD3005) chez des adultes de ≥65 ans. Les patients des études TRD3001 et TRD3002 ont commencé, au jour 1, le traitement par Spravato à 56 mg en association avec un AD oral nouvellement initié pris quotidiennement ou un placebo en spray nasal en association avec un AD oral nouvellement initié pris quotidiennement. Les doses de Spravato ont été maintenues à 56 mg ou ajustées à 84 mg ou en correspondance au placebo en spray nasal, administrés deux fois par semaine durant une phase d'induction en double aveugle de 4 semaines. Les doses de Spravato de 56 mg et 84 mg ont été administrées selon une posologie fixe dans l'étude TRD3001 et une posologie flexible dans l'étude TRD3002. Dans l'étude TRD3005, les patients âgés de ≥65 ans ont commencé au jour 1, le traitement par Spravato à 28 mg en association avec un AD oral nouvellement initié, pris quotidiennement, un placebo en spray nasal en association avec un AD oral nouvellement initié, pris quotidiennement. Au cours de la phase d'induction de 4 semaines en double aveugle, la dose de Spravato ou du placebo en spray nasal a été ajustée à une dose plus élevée de 56 mg ou 84 mg, deux fois par semaine. Dans les études à posologie flexible (TRD3002 et TRD3005), la dose de Spravato a été ajustée à une dose plus élevée sur la base du jugement clinique, mais pouvait également être ajustée à une dose inférieure en fonction de la tolérance. Dans toutes les études, un traitement ouvert par un AD oral (IRSN: duloxétine, venlafaxine retard; ISRS: escitalopram, sertraline) a été nouvellement initié au jour 1. Le choix du traitement AD oral nouvellement initié a été fait par le médecin investigateur en fonction des antécédents de traitement individuels du patient. Dans toutes les études à court terme, le critère d'efficacité principal était la variation du score MADRS total, entre le début de l'étude (valeur de référence) et le jour 28.
Les caractéristiques démographiques et les caractéristiques liées à la maladie des patients à l'inclusion dans les études TRD3002, TRD3001 et TRD3005 sont présentées dans le tableau 3.
Tableau 3: Caractéristiques démographiques à l'inclusion des études TRD3002, TRD3001 et TRD3005 (ensembles d'analyses complets)
|
|
Étude TRD3002
(N = 223)
|
Étude TRD3001
(N = 342)
|
Étude TRD3005
(N = 137)
| |
Âge, années
| |
Médiane (fourchette)
|
47,0 (19; 64)
|
47,0 (18; 64)
|
69,0 (65; 86)
| |
Sexe, N (%)
| |
Masculin
|
85 (38,1%)
|
101 (29,5%)
|
52 (38,0%)
| |
Féminin
|
138 (61,9%)
|
241 (70,5%)
|
85 (62,0%)
| |
Appartenance ethnique, N (%)
| |
Blanc
|
208 (93,3%)
|
262 (76,6%)
|
130 (94,9%)
| |
Noir ou Afro-Américain
|
11 (4,9%)
|
19 (5,6%)
|
--
| |
Traitement AD oral antérieur sans réponse thérapeutique (c.àd. échec du traitement)
| |
Nombre de traitements AD antérieurs, N (%)
| |
2
|
136 (61,0%)
|
167 (48,8%)
|
68 (49,6%)
| |
3 ou plus
|
82 (36,8%)
|
167 (48,8%)
|
58 (42,3%)
| |
AD oral nouvellement initié le jour de la randomisation, N (%)
| |
IRSN
|
152 (68,2%)
|
196 (57,3%)
|
61 (44,5%)
| |
ISRS
|
71 (31,8%)
|
146 (42,7%)
|
76 (55,5%)
| |
Abandon de l'étude (quelle que soit la raison), n/N (%)
|
30/227 (13,2%)
|
31/346 (9,0%)
|
16/138 (11,6%)
|
Dans l'étude TRD3002 à posologie flexible, 67% des patients randomisés dans le groupe Spravato ont reçu une dose de 84 mg au jour 28. Dans l'étude TRD3002, Spravato associé à un AD oral nouvellement initié a présenté une supériorité statistique par rapport au placebo en spray nasal associé à un AD oral nouvellement initié [IRSN: duloxétine, venlafaxine retard; ISRS: escitalopram, sertraline] (tableau 4). De plus, une réduction des symptômes a déjà été observée 24 heures après l'utilisation.
Dans l'étude TRD3001, l'effet thérapeutique (défini comme la variation du score MADRS total par rapport à la valeur initiale à la fin de la période d'induction de 4 semaines) de Spravato 84 mg associé à un AD oral nouvellement initié n'a pas atteint le seuil de signification statistique par rapport au placebo en spray nasal associé à un AD oral (IRSN: duloxétine, venlafaxine retard; ISRS: escitalopram, sertraline) (tableau 4).
Dans l'étude TRD3005, 64% des patients randomisés traités par Spravato ont reçu une dose de 84 mg au jour 28, 25% une dose de 56 mg et 10% une dose de 28 mg. Dans l'étude TRD3005, l'effet thérapeutique (défini comme la variation du score MADRS total par rapport à la valeur initiale à la fin de la période d'induction de 4 semaines) de Spravato associé à un AD oral nouvellement initié n'a pas atteint le seuil de signification statistique par rapport au placebo en spray nasal associé à un AD oral [IRSN: duloxétine, venlafaxine retard; ISRS: escitalopram, sertraline] (tableau 4). Les analyses de sous-groupes indiquent une efficacité réduite dans le groupe des patients de plus de 75 ans.
Tableau 4: Résultats du critère d'efficacité principal pour la variation du score MADRS total après la semaine 4, dans les études cliniques (ANCOVA LOCF)
|
N° de l'étude
|
Groupe de traitement§
|
Nombre de patients
|
Valeur initiale moyenne (SD)
|
Variation de la LSM à la fin de la semaine 4, par rapport à la valeur initiale (SE)
|
Différence entre les LSM (IC à 95%)†
|
Valeur de p bilatérale
| |
TRD3001
|
Spravato 56 mg + AD oral
|
115
|
37,4 (4,8)
|
-18,7 (1,3)
|
-4,1
(-7,5; -0,6)#
|
N/Aδ
| |
Spravato 84 mg + AD oral
|
114
|
37,8 (5,6)
|
-17,3 (1,3)
|
-2,0
(-5,5; 1,4)#
|
0,250
| |
Placebo en spray nasal + AD oral
|
113
|
37,5 (6,2)
|
-14,8 (1,3)
|
|
| |
TRD3002
|
Spravato (56 mg ou 84 mg) + AD oral
|
114
|
37,0 (5,7)
|
-18,0 (1,3)
|
-3,5
(-6,7; -0,3)‡
|
0,034‡
| |
Placebo en spray nasal + AD oral
|
109
|
37,3 (5,7)
|
-14,5 (1,3)
|
|
| |
TRD3005 (≥65 ans)
|
Spravato (28 mg, 56 mg ou 84 mg) + AD oral
|
72
|
35,5 (5,9)
|
-10,9 (1,7)
|
-3,6
(-7,2; -0,03)#
|
0,052
| |
Placebo en spray nasal + AD oral
|
65
|
34,8 (6,4)
|
-6,9 (1,7)
|
|
| |
SD = écart type (Standard Deviation), SE = erreur type (Standard Error), LSM = moyenne des moindres carrés (Least Square Means), IC = intervalle de confiance, AD = antidépresseur
§ Eskétamine ou placebo par voie nasale; AD oral = standard (un AD nouvellement initié)
† Différence (Spravato + AD oral moins placebo en spray nasal + AD oral) entre les variations de la LSM par rapport à la valeur initiale
‡ Groupe de traitement qui était supérieur de manière statistiquement significative au traitement par placebo en spray nasal + AD oral
# Estimateur non biaisé de la médiane (c.àd. combinaison pondérée des LSM de la différence par rapport au placebo en spray nasal + AD oral) et intervalle de confiance à 95% flexible.
δ Comme 84 mg n'était pas statistiquement significatif, la valeur de p pour la comparaison Spravato 56 mg + AD oral versus placebo *AD oral n'est pas indiquée en raison de la hiérarchie des tests.
|
Évolution de la réponse au traitement au cours du temps
Dans l'étude TRD3002, un effet antidépresseur de Spravato avec une réduction des symptômes de dépression était déjà observé dans les 24 heures suivant l'administration de la première dose. Au cours des semaines qui ont suivi, une amélioration progressive a été observée et l'effet antidépresseur complet de Spravato a été atteint au jour 28. La variation moyenne du score MADRS total pour Spravato à dose flexible (56 mg ou 84 mg) plus AD oral était constamment supérieure à tous les temps de mesure (semaines 1, 2, 3 et 4) à celle de l'AD oral plus placebo administré par voie nasale. Au jour 28, 67% des patients randomisés traités par Spravato ont reçu 84 mg. Un effet de traitement uniforme a été observé dans le cadre des études TRD3001 et TRD3005.
Réponse thérapeutique et taux de rémission
La réponse thérapeutique a été définie comme une réduction du score MADRS total ≥50% par rapport à la valeur initiale (valeur de référence) pendant la phase d'induction. En se basant sur la réduction du score MADRS total par rapport à la valeur initiale, la proportion de patients des études TRD3001, TRD3002 et TRD3005 qui ont présenté une réponse au traitement par Spravato plus un AD oral était plus élevée qu'avec un AD oral plus placebo en spray nasal, pendant l'ensemble de la phase d'induction de 4 semaines en double aveugle (tableau 5).
Une rémission a été définie comme un score MADRS total ≤12. À la fin de la phase d'induction de 4 semaines en double aveugle, la proportion de patients en rémission traités par Spravato plus un AD oral était plus élevée que celle des patients traités par un AD oral plus placebo en spray nasal dans chacune des trois études (tableau 5).
Tableau 5: Taux de réponse et de rémission dans les études cliniques de 4 semaines sur la base des données LOCF
|
N° de l'étude
|
Groupe de traitement§
|
Nombre de patients (%)
| |
Taux de réponse†
|
Taux de rémission‡
| |
24 heures
|
Semaine 1
|
Semaine 2
|
Semaine 3
|
Semaine 4
|
Semaine 4
| |
TRD3001
|
Spravato 56 mg + AD oral
|
20
(19,0%)
|
21
(18,3%)
|
30
(26,1%)
|
52
(45,2%)
|
61
(53,0%)
|
40
(34,8%)
| |
Spravato 84 mg + AD oral
|
17
(16,3%)#
|
16
(14,3%)
|
26
(23,2%)
|
35
(31,0%)
|
54
(47,8%)
|
40
(35,4%)
| |
AD oral + placebo en spray nasal
|
8
(7,9%)
|
5
(4,4%)
|
15
(13,3%)
|
27
(23,9%)
|
42
(37,2%)
|
33
(29,2%)
| |
TRD3002
|
Spravato 56 mg ou 84 mg + AD oral
|
18
(16,5%)
|
15
(13,4%)
|
29
(25,9%)
|
54
(48,2%)
|
71
(63,4%)
|
54
(48,2%)
| |
AD oral + placebo en spray nasal
|
11
(10,8%)
|
13
(11,9%)
|
23
(21,1%)
|
36
(33,0%)
|
54
(49,5%)
|
33
(30,3%)
| |
TRD3005
(≥65 ans)
|
Spravato 28 mg, 56 mg ou 84 mg + AD oral
|
NA
|
4
(6,1%)
|
4
(5,6%)
|
9
(12,7%)
|
17
(23,9%)
|
11
(15,5%)
| |
AD oral + placebo en spray nasal
|
NA
|
3
(4,8%)
|
8
(12,5%)
|
10
(15,6%)
|
8
(12,5%)
|
4
(6,3%)
| |
AD = antidépresseur; NA = aucune donnée (not available)
§ Spravato ou placebo administré par voie nasale; AD oral = traitement standard (AD nouvellement initié)
† Une réponse a été définie comme une réduction du score MADRS total ≥50% par rapport à la valeur initiale
‡ Une rémission a été définie comme un score MADRS total ≤12
# La première dose était Spravato 56 mg + AD oral
|
Données à long terme
Dépression résistante au traitement (DRT) – études à long terme
Étude TRD3003 (SUSTAIN-1) - Étude sur la prévention des récidives
SUSTAIN-1 (TRD3003) était une étude à long terme randomisée, en double aveugle, multicentrique, en groupes parallèles avec contrôle actif portant sur la prévention des récidives. Au total, 705 patients ont été inclus dans l'étude, dont 437 ont été recrutés directement, 150 patients ont été inclus à partir de l'étude TRD3001 et 118 patients à partir de l'étude TRD3002. Les patients directement inclus dans l'étude ont été traités par Spravato (56 mg ou 84 mg deux fois par semaine) plus un AD oral pendant une phase d'induction ouverte (sans insu) de 4 semaines. Les patients qui ont répondu au traitement [diminution du score MADRS total de ≥50% par rapport au score initial (valeur de référence)] ont poursuivi le traitement par Spravato plus un AD oral pendant une phase d'optimisation de 12 semaines. À la fin de la phase d'induction ouverte, 52% des patients étaient en rémission (score MADRS total ≤12) et 66% des patients étaient répondeurs (amélioration de ≥50% du score MADRS total). Au total, 455 patients traités par l'eskétamine ont entamé la phase ultérieure d'optimisation; les patients en rémission stable ou présentant une réponse stable ont été randomisés pour soit poursuivre le traitement par Spravato, soit arrêter le traitement par Spravato et passer au placebo en spray nasal. Après les 16 premières semaines de traitement par Spravato plus un AD oral, 176 patients (39%) étaient en rémission stable et 121 patients (27%) présentaient une réponse stable (mais n'étaient pas en rémission stable). Une rémission stable a été définie comme un score MADRS total ≤12 pendant au moins 3 des 4 dernières semaines de la phase d'optimisation et une réponse stable a été définie comme la diminution du score MADRS total de ≥50% par rapport à la valeur initiale au cours des 2 dernières semaines de la phase d'optimisation, mais sans rémission stable.
Les valeurs initiales (valeurs de référence) des données démographiques et des caractéristiques de la maladie des patients randomisés dans la phase d'entretien en double aveugle étaient comparables dans les deux groupes; l'âge médian des patients était de 48 ans (19-64 ans), 66% étaient des femmes et 90% étaient d'origine caucasienne.
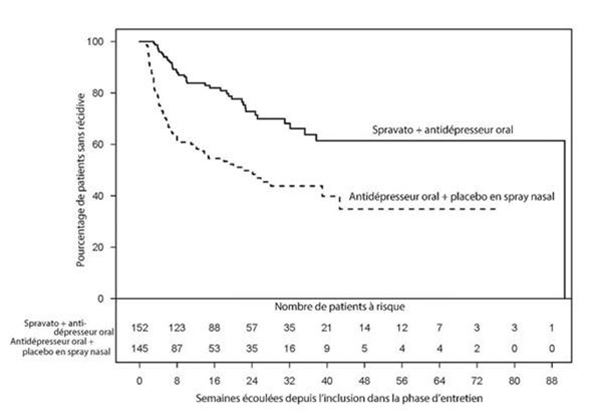
Rémission stable
Chez les patients en rémission stable qui ont poursuivi le traitement par Spravato plus un AD oral, le délai jusqu'à une récidive des symptômes de dépression a été, selon les statistiques, significativement plus long que chez les patients du groupe témoin (AD oral plus placebo en spray nasal). La récidive a été définie comme un score MADRS total ≥22 pendant deux semaines consécutives, une hospitalisation due à une aggravation de la dépression ou un autre événement cliniquement pertinent suggérant une récidive. Le délai médian avant une récidive a été de 273 jours dans le groupe témoin (AD oral plus placebo en spray nasal), alors que le délai médian pour Spravato plus AD oral n'a pas pu être estimé, car ce groupe n'a à aucun moment atteint un taux de récidive de 50% pendant l'étude.
Chez les patients en rémission stable, le hazard ratio (rapport des risques instantanés) estimé (intervalle de confiance à 95%) de Spravato en association avec un AD oral, par rapport au groupe témoin (AD oral plus placebo en spray nasal), était, sur la base des estimations pondérées, de 0,49 (intervalle de confiance à 95%: 0,29-0,84), p = 0,003. Cela signifie que les patients en rémission stable qui ont poursuivi le traitement dans le groupe sous Spravato plus AD oral ont en moyenne eu une probabilité de récidive 51% plus basse que les patients qui sont passés dans le groupe témoin (AD oral plus placebo en spray nasal).
Réponse stable
Les résultats relatifs à l'efficacité étaient également concordants chez les patients présentant une réponse stable qui ont poursuivi le traitement par Spravato plus un AD oral; le délai jusqu'à une récidive des symptômes de dépression a été, selon les statistiques, significativement plus long chez eux que chez les patients du groupe témoin (AD oral plus oral plus placebo en spray nasal). Le délai médian avant une récidive a été de 88 jours dans le groupe témoin et de 635 jours dans le groupe traité par Spravato.
Chez les patients présentant une réponse stable, le hazard ratio (rapport des risques instantanés) estimé (intervalle de confiance à 95%) de Spravato en association avec un AD oral, par rapport au groupe témoin (AD oral plus placebo en spray nasal), était, sur la base du modèle à risques proportionnels de Cox, de 0,3 (intervalle de confiance à 95%: 0,16-0,55). Cela signifie que les patients qui étaient répondeurs stables et qui ont poursuivi le traitement dans le groupe sous Spravato en association avec un AD oral ont en moyenne eu une probabilité de récidive 70% plus basse que les patients qui sont passés dans le groupe témoin (AD oral plus placebo en spray nasal).
La figure 1 montre la proportion cumulative de patients présentant une rémission stable et une réponse stable (combinés à partir de l'étude TRD3003) qui sont restés exempts de récidive.
Figure 1: Délai jusqu'à la survenue d'une récidive chez les patients présentant une réponse stable et une rémission stable dans l'étude TRD3003
Fréquence du dosage
Parmi les patients en rémission stable ou présentant une réponse stable, respectivement 23% et 55% ont reçu une dose hebdomadaire pendant la phase d'entretien et respectivement 69% et 34% ont reçu une dose toutes les deux semaines. Certains patients ont également reçu les doses aux deux fréquences, une dose hebdomadaire ou une dose toutes les deux semaines (respectivement 8% et 11%). Parmi les patients qui ont reçu Spravato après randomisation, 60% ont reçu la dose de 84 mg et 40% la dose de 56 mg.
Étude TRD3013 (ESCAPE-TRD)
L'efficacité de Spravato a été évaluée dans une étude à long terme, randomisée, en ouvert, à l'aveugle de l'évaluateur, contrôlée contre traitement actif (TRD3013), dans laquelle Spravato a été comparé à la quétiapine avec une libération prolongée du principe actif (XR) chez 676 patients adultes présentant une DRT (âgés de 18 à 74 ans) et continuant à prendre leur AD oral actuel (un IRSN ou ISRS). Les patients ont reçu un traitement par Spravato à doses flexibles (28, 56 et 84 mg) ou par Quétiapine XR, selon les recommandations posologiques figurant dans l'information professionnelle en vigueur au moment du début du traitement.
Le critère d'évaluation principal de l'efficacité était la rémission (score MADRS total ≤10) à la semaine 8 et le critère d'évaluation secondaire majeur était le maintien de l'absence de récidive jusqu'à la semaine 32, après la rémission à la semaine 8. La récidive était définie par un score MADRS total ≥22 pendant 2 semaines consécutives, un séjour à l'hôpital en raison d'une aggravation de la dépression ou un autre événement cliniquement significatif indiquant une récidive.
Les valeurs initiales démographiques et liées à la maladie des patients du groupe Spravato plus AD oral étaient similaires à celles du groupe Quétiapine XR plus AD oral. Le score MADRS total moyen (SD) au début de l'étude était de 31,4 (6,06) pour le groupe traité par Spravato plus AD oral et de 31,0 (5,83) pour le groupe traité par Quétiapine XR plus AD oral.
Spravato plus AD oral a montré une supériorité cliniquement significative et statistique comparé à Quétiapine XR plus AD oral, aussi bien en ce qui concerne la mesure d'efficacité principale (tableau 6) que la mesure d'efficacité secondaire déterminante (tableau 7).
Tableau 6: Principaux résultats d'efficacité pour l'étude TRD3013 a
|
Groupe de traitement
|
Spravato + AD oral
|
Quétiapine XR + AD oral
| |
Nombre de patients en rémission
à la semaine 8
|
91/336 (27,1%)
|
60/340 (17,6%)
| |
Écart en pourcentage (IC à 95%)
|
9,44 (3,19; 15,68)
|
–
| |
Odds ratio ajusté (IC à 95%)
|
1,74 (1,20; 2,52)
P = 0,003b
|
–
| |
IC = intervalle de confiance; AD = antidépresseur; XR = libération prolongée du principe actif.
a Un patient qui a arrêté l'intervention de l'étude avant la semaine 8 a été considéré comme un résultat négatif (c.-à-d. pas de rémission). Pour les patients pour lesquels aucun résultat MADRS n'était disponible à la semaine 8, mais qui n'ont pas arrêté l'intervention de l'étude et ne sont pas sortis de l'étude avant la semaine 8, la LOCF du score MADRS a été appliquée.
b Valeur de p pour le test de CMH (Cochran-Mantel-Haenszel), ajusté par les groupes d'âge (18–64; ≥65) et le nombre total d'arrêts du traitement.
|
Tableau 7: Résultats d'efficacité secondaires majeurs pour l'étude TRD3013a
|
Groupe de traitement
|
Spravato + AD oral
|
Quétiapine XR + AD oral
| |
Nombre de patients en rémission à la semaine 8 et sans récidive à la semaine 32
|
73/336 (21,7%)
|
48/340 (14,1%)
| |
Écart en pourcentage (IC à 95%)
|
7,61 (1,85; 13,37)
|
–
| |
Odds ratio ajusté (IC à 95%)
|
1,72 (1,15; 2,57)
P = 0,008b
|
–
| |
IC = intervalle de confiance; AD = antidépresseur; XR = libération prolongée du principe actif.
a Un patient qui a arrêté l'intervention de l'étude a été considéré comme un résultat négatif (c.-à-d. pas de rémission). Pour les patients pour lesquels aucun résultat MADRS n'était disponible à la semaine 8, mais qui n'ont pas arrêté l'intervention de l'étude et ne sont pas sortis de l'étude avant la semaine 8, la LOCF du score MADRS a été appliquée.
b Valeur de p pour le test de CMH (Cochran-Mantel-Haenszel), ajusté par les groupes d'âge (18–64; ≥65) et le nombre total d'arrêts du traitement.
|
Taux de rémission et de réponse
Le taux de rémission à la semaine 32 était de 55,0% chez les patients du groupe Spravato plus AD oral et de 37,0% dans le groupe Quétiapine XR plus AD oral, avec un odds ratio (IC à 95%) de 2,09 (1,53; 2,85). Le taux de réponse (défini comme la diminution du score MADRS total de ≥50% par rapport à la valeur initiale ou un score MADRS total ≤10) était, à la semaine 32, de 75,5% des patients dans le groupe Spravato plus AD oral et de 55,5% dans le groupe Quétiapine XR plus AD oral, avec un odds ratio (IC à 95%) de 2,48 (1,78; 3,46).
Sur la période de traitement de 32 semaines, les taux d'arrêt du traitement en raison d'événements indésirables, d'un manque d'efficacité ou en général étaient respectivement de 4,2%, 8,3% et 23,2% pour les patients du groupe Spravato plus AD oral, et respectivement de 11,5%, 15,0% et 40,3% pour les patients du groupe Quétiapine XR plus AD oral.
Étude sur la relation entre la dose et la réponse thérapeutique lors de DRT
Une étude de phase II à double randomisation, en double aveugle et contrôlée contre placebo, visant à déterminer le dosage a recruté 108 patients adultes atteints de DRT. En plus de la poursuite d'un traitement antidépresseur oral, les patients ont reçu 14 mg, 28 mg, 56 mg ou 84 mg d'eskétamine ou un placebo par voie nasale, deux fois par semaine pendant 2 semaines. Le traitement avec des doses de 28 mg, 56 mg et 84 mg de Spravato a significativement amélioré les symptômes dépressifs chez les patients atteints de DRT, ce qui s'est traduit par une modification du score MADRS total après 1 semaine. Bien que les doses de 28 mg, 56 mg et 84 mg de Spravato aient été efficaces dans le traitement de la DRT, la durée d'action de la dose de 28 mg était plus courte.
Dépression résistante au traitement – étude à court terme menée chez des patients japonais
L'efficacité de Spravato a également été évaluée dans une étude à court terme (TRD2005) randomisée, en double aveugle, contrôlée contre traitement actif (sur 4 semaines), menée chez 202 patients japonais adultes présentant une DRT. Tout en continuant leur traitement en cours par l'AD oral, les patients ont reçu un traitement d'induction de 4 semaines par Spravato en spray nasal à des doses fixes de 28 mg, 56 mg ou 84 mg, ou par placebo. Le critère d'efficacité principal était la variation du score MADRS total entre le début de l'étude (valeur de référence) et le jour 28. Au début de l'étude, les caractéristiques démographiques et liées à la maladie des patients étaient comparables entre le groupe Spravato plus AD et le groupe placebo en spray nasal plus AD.
Dans l'étude TRD2005, à la fin de la phase d'induction de 4 semaines, aucune différence statistiquement significative n'a été observée pour l'une des 3 doses de Spravato (plus AD oral) comparées au placebo (plus AD oral), quant à la variation du score MADRS total par rapport au début de l'étude.
Dépression résistante au traitement – étude à court terme menée chez des patients chinois
L'efficacité de Spravato a également été évaluée dans une étude à court terme (TRD3006) randomisée, en double aveugle, contrôlée contre traitement actif, menée (sur 4 semaines) chez 252 patients adultes (224 patients chinois, 28 patients non chinois) présentant une DRT. Les patients ont reçu un traitement d'induction de 4 semaines par Spravato à des dosages flexibles (56 mg ou 84 mg) ou par placebo, en plus d'un AD oral nouvellement initié. Le critère d'efficacité principal était la variation du score MADRS total entre le début de l'étude (valeur de référence) et le jour 28. Les caractéristiques démographiques et liées à la maladie des patients étaient comparables entre le groupe Spravato plus AD et le groupe placebo en spray nasal plus AD.
Dans l'étude TRD3006, à la fin de la phase d'induction de 4 semaines, aucune différence statistiquement significative n'a été observée entre Spravato (plus AD oral) et le placebo (plus AD oral), quant à la variation du score MADRS total par rapport au début de l'étude.
Traitement aigu de courte durée d'une urgence psychiatrique dans le cadre d'une dépression majeure
Spravato a été étudié dans deux études identiques de phase 3, randomisées, en double aveugle, multicentriques, contrôlées contre placebo et de courte durée (4 semaines), ASPIRE I (SUI3001; NCT03039192) et ASPIRE II (SUI3002; NCT03097133), menées auprès de patients adultes atteints de TDM modéré à sévère (score MADRS total > 28) et présentant des pensées suicidaires actives avec intention suicidaire. Dans ces études, les patients ont reçu, deux fois par semaine pendant 4 semaines, un traitement par 84 mg de Spravato ou par un placebo en spray nasal. Tous les patients ont reçu un traitement standard (SOC) complet qui comprenait un séjour hospitalier initial et un traitement nouvellement initié ou optimisé par un AD oral (AD en monothérapie, ou AD plus potentialisation), selon la décision du médecin investigateur. Après la première dose, une réduction unique de la dose de Spravato à 56 mg était autorisée chez les patients qui ne toléraient pas la dose de 84 mg.
Au début des études SUI3001 et SUI3002, les caractéristiques démographiques et liées à la maladie des patients étaient comparables entre le groupe traité par Spravato + SOC et le groupe sous placebo en spray nasal + SOC. L'âge médian des patients était de 40 ans (entre 18 et 64 ans), 61% étaient des femmes, 73% des Caucasiens et 6% des Noirs, et 63% des patients avaient déjà fait au moins une tentative de suicide. Avant leur recrutement dans l'étude, 92% des patients avaient été traités par un AD. Au cours de l'étude, 40% des patients ont reçu un AD en monothérapie dans le cadre du traitement standard, 54% ont reçu un AD plus potentialisation, et 6% des patients ont reçu à la fois un AD en monothérapie et un AD plus potentialisation.
Le critère d'efficacité principal était la réduction des symptômes du TDM, mesurée par la variation du score MADRS total, 24 heures après la première dose (au jour 2), par rapport à sa valeur initiale.
Dans les études SUI3001 et SUI3002, le traitement par Spravato + SOC a présenté une supériorité statistique sur le critère d'efficacité principal par comparaison au placebo en spray nasal + SOC (voir tableau 8).
Tableau 8: Principaux résultats d'efficacité concernant la variation du score MADRS total, 24 heures après la première dose par rapport à la valeur initiale (dans les études SUI3001 et SUI3002; ANCOVA*)
|
No de l'étude
|
Groupe de traitement§
|
Nombre de patients
|
Score moyen
au début de l'étude
(SD)
|
Variation de la LSM entre le début de l'étude et 24 heures après la première dose
(SE)
|
Variation de la LSM
(IC à 95%)†
Valeur de p
| |
SUI3001
|
84 mg Spravato + SOC
|
111
|
41,3 (5,87)
|
-15,9 (1,04)
|
-3,8
(-6,56; -1,09)‡
p = 0,006
| |
Placebo en spray nasal + SOC
|
112
|
41,0 (6,29)
|
-12,0 (1,02)
|
-
| |
SUI3002
|
84 mg Spravato + SOC
|
113
|
39,4 (5,21)
|
-16,0 (1,02)
|
-3,9
(-6,60; -1,11)‡
p = 0,006
| |
Placebo en spray nasal + SOC
|
113
|
39,9 (5,76)
|
-12,2 (1,05)
|
-
| |
Études combinées (SUI3001 et SUI3002)
|
84 mg Spravato + SOC
|
224
|
40,3 (5,61)
|
-16,0 (0,72)
|
-3,8
(-5,75; -1,89)‡
| |
Placebo en spray nasal + SOC
|
225
|
40,4 (6,04)
|
-12,1 (0,72)
|
-
| |
SD = écart type, SE = erreur type, LSM = moyenne des moindres carrés, IC = intervalle de confiance, SOC = traitement standard (Standard of Care).
* ANCOVA LOCF: dans l'étude SUI3001, pour 1 participant (dans le groupe de traitement placebo + SOC), on n'a disposé d'aucun score MADRS total au jour 2 (24 heures après la première dose), et son score MADRS total a été remplacé par la valeur obtenue 4 heures après la première dose («carried forward»). Dans l'étude SUI3002, pour 5 des 6 participants chez lesquels on ne disposait pas de score MADRS total au jour 2 (24 heures après la première dose), leur score MADRS total a été remplacé par la valeur obtenue 4 heures après la première dose.
§ Eskétamine ou placebo administrés par voie nasale.
† Différence (Spravato + SOC moins placebo en spray nasal + SOC) de variation de la moyenne des moindres carrés (LSM), par rapport au début de l'étude.
‡ Groupes de traitement qui ont présenté une supériorité statistiquement significative par rapport au placebo en spray nasal + SOC.
|
Les différences (IC à 95%) de variation du score MADRS total au jour 2 (24 heures après la première dose) par rapport à la valeur initiale entre le traitement par Spravato + SOC et celui par placebo + SOC ont été de -4,81 (-7,26; -2,36) dans le sous-groupe avec antécédents de tentative de suicide (N = 282) et de -2,32 (-5,54; 0,91) dans le sous-groupe sans antécédents de tentative de suicide (N = 166).
Dans une analyse posthoc des données regroupées de SUI3001 et SUI3002, Spravato plus traitement standard n'a présenté aucune supériorité par rapport au placebo plus traitement standard, au jour 2 (24 heures après la première dose), chez les 18% de patients présentant un épisode dépressif modéré (score MADRS total < 35 au début de l'étude).
Évolution au cours du temps de la réponse au traitement
Dans les deux études SUI3001 et SUI3002, une différence a été observée entre le traitement par Spravato et celui par placebo, à partir de 4 heures. Des améliorations supplémentaires ont été observées dans l'intervalle entre 4 heures et le jour 25, tant dans le groupe Spravato que dans le groupe placebo. La différence entre les groupes a généralement persisté, mais n'a pas semblé s'accentuer au cours du temps jusqu'au jour 25. La figure 3 montre l'évolution au cours du temps du score MADRS total, le critère d'efficacité principal, sur la base des données combinées des études SUI3001 et SUI3002.
Figure 3: Variation au cours du temps de la moyenne des moindres carrés (LSM) du score MADRS total, par rapport au début de l'étude, dans les études SUI3001 et SUI3002* (groupe total analysé combiné) – MMRM)
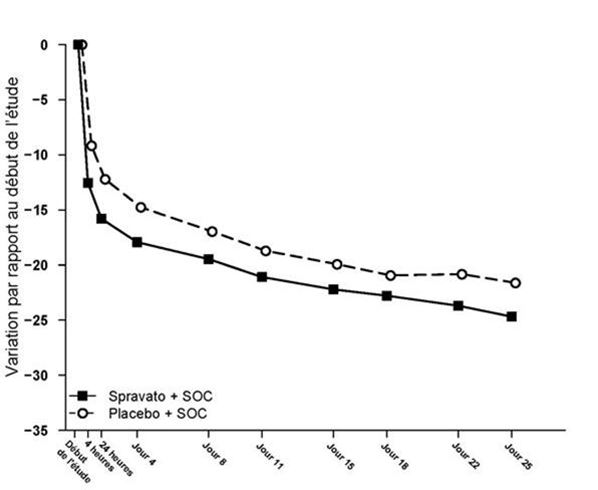
* Remarque: dans ces études, chez les patients qui ne toléraient pas la dose de 84 mg, une réduction unique de la dose de Spravato à 56 mg était autorisée après la première dose. Chez environ 16% des patients, la posologie de Spravato a été réduite de 84 mg à 56 mg, deux fois par semaine.
Taux de rémission
Dans les études de phase 3, le pourcentage de patients ayant obtenu une rémission (score MADRS total ≤12 à tout moment de l'étude) a été plus élevé dans le groupe Spravato + SOC que dans le groupe placebo + SOC, à tout moment de la phase de traitement en double aveugle (tableau 9).
Tableau 9: Patients ayant obtenu une rémission du TDM; phase de traitement en double aveugle; groupe total analysé concernant l'efficacité
|
|
SUI3001
|
SUI3002
|
Études combinées
(SUI3001 et SUI3002)
| |
|
Placebo + SOC
|
Spravato + SOC
|
Placebo + SOC
|
Spravato + SOC
|
Placebo + SOC
|
Spravato + SOC
| |
|
112
|
112
|
113
|
114
|
225
|
226
| |
Jour 1, 4 heures après la première dose
| |
Patients présentant une rémission du TDM
|
9 (8,0%)
|
12 (10,7%)
|
4 (3,5%)
|
12 (10,5%)
|
13 (5,8%)
|
24 (10,6%)
| |
Jour 2, 24 heures après la première dose
| |
Patients présentant une rémission du TDM
|
10 (8,9%)
|
21 (18,8%)
|
12 (10,6%)
|
25 (21,9%)
|
22 (9,8%)
|
46 (20,4%)
| |
Jour 25
| |
Patients présentant une rémission du TDM
|
38 (33,9%)
|
46 (41,1%)
|
31 (27,4%)
|
49 (43,0%)
|
69 (30,7%)
|
95 (42,0%)
| |
SOC = traitement standard
Remarque: la rémission est définie comme un score MADRS total ≤12. N'ont pas été considérés comme ayant obtenu une rémission les participants qui n'ont pas rempli ce critère ou dont le traitement a été arrêté pour une raison quelconque avant ce terme.
|
Effets sur la suicidalité
Le critère d'évaluation secondaire de l'efficacité était la variation sur l'échelle «Clinical Global Impression of Suicidal Severity – revised» (CGI-SSr), 24 heures après l'administration de la première dose (au jour 2).
Le CGI-SSr est une évaluation clinique à une dimension, utilisée pour classifier la sévérité actuelle des pensées et des comportements suicidaires d'un patient. Le score CGI-SSr varie de 0 à 6, les valeurs plus élevées correspondant à des pensées et des comportements suicidaires plus sévères. Dans les études SUI3001 et SUI3002, Spravato plus traitement standard n'a pas présenté de supériorité pour améliorer le CGI-SSr, par comparaison au placebo en spray nasal plus traitement standard.
L'efficacité à long terme de Spravato pour la prévention du suicide n'a pas été étudiée.
Informations complémentaires
Effet sur l'aptitude à la conduite
Les effets de Spravato sur la capacité de conduire un véhicule automobile ont été évalués dans le cadre de deux études, l'une menée chez des adultes atteints de TDM et l'autre menée chez des sujets sains. La capacité de conduite sur route a été évaluée à l'aide de l'écart type moyen de la position latérale du véhicule (SDLP, standard deviation of the lateral position), une mesure de la perturbation de la conduite.
Les effets d'une dose unique de 84 mg d'eskétamine en spray nasal, sur la conduite le jour suivant ainsi que l'effet d'une dose intranasale répétée de 84 mg de Spravato sur la conduite le jour même ont été évalués dans le cadre d'une étude en simple aveugle contrôlée contre placebo, menée auprès de 25 patients adultes atteints de TDM. Une boisson alcoolisée a servi de témoin positif pour la phase de traitement par dose unique. Le SDLP, 18 heures après l'administration d'une dose unique de 84 mg d'eskétamine en spray nasal, a été semblable à celui observé après l'administration du placebo. Pendant la phase de traitement par doses multiples, le SDLP après l'utilisation intranasale répétée de 84 mg de Spravato 6 heures après la dernière dose aux jours 11, 18 et 25 a été similaire à celui après le placebo. La limite supérieure de l'intervalle de confiance bilatéral à 95% de la différence moyenne entre une dose unique d'eskétamine et de placebo a été de 0,58 cm, soit moins que la limite de non-infériorité prédéfinie de 2,4 cm. La limite inférieure de l'intervalle de confiance à 95% de la différence moyenne entre l'éthanol et le placebo a été de 1,03 cm (p < 0,001), ce qui confirme la sensibilité de l'étude.
Les effets d'une dose unique de 84 mg d'eskétamine en spray nasal sur la conduite ont été évalués dans le cadre d'une étude randomisée, en double aveugle, croisée et contrôlée contre placebo, menée auprès de 23 sujets sains. La mirtazapine a servi de témoin positif. La capacité de conduire a été évaluée 8 heures après l'administration de l'eskétamine ou de la mirtazapine. Le SDLP après l'administration d'eskétamine en spray nasal a été semblable à celui observé après l'administration du placebo. La limite supérieure de l'intervalle de confiance bilatéral à 95% de la différence moyenne entre l'eskétamine et le placebo a été de 0,86 cm, soit moins que la limite de non-infériorité prédéfinie de 2,4 cm. La limite inférieure de l'intervalle de confiance à 95% de la différence moyenne entre la mirtazapine et le placebo a été de 1,12 cm (p = 0,001), ce qui confirme la sensibilité de l'étude. Sur les 23 sujets évalués, 21 ont passé le test avec succès. Deux sujets ont annulé le test de conduite après l'administration d'eskétamine, car ils avaient l'impression de ne pas être aptes à la conduite.
Effet sur l'intervalle QT/QTc et l'électrophysiologie cardiaque
Le traitement par Spravato n'a pas prolongé l'intervalle QTc. L'effet de Spravato (84 mg en spray nasal et 0,8 mg/kg d'eskétamine en perfusion intraveineuse pendant 40 minutes) sur l'intervalle QTc a été évalué dans le cadre d'une étude croisée randomisée, en double aveugle, contrôlée contre placebo et avec témoin positif (moxifloxacine 400 mg), en 4 phases, menée auprès de 60 sujets sains. Les concentrations maximales d'eskétamine dans le plasma après perfusion intraveineuse étaient environ 3 fois plus élevées que celles après administration de la dose de 84 mg par voie nasale. La limite supérieure de l'intervalle de confiance à 90% pour le plus grand intervalle QTc ajusté en fonction du placebo après correction en fonction des valeurs initiales (valeurs de référence), calculé grâce à la formule de correction de Fridericia (QTcF) pour les deux groupes de traitement, est restée inférieure à 10 ms, à tous les moments évalués.
|