CompositionPrincipe actif: adalimumab (produit par des cellules ovariennes de hamster chinois génétiquement modifiées).
Excipients: acide acétique glacial, saccharose, polysorbate 80 (produit à partir de maïs génétiquement modifié), hydroxyde de sodium (pour ajuster le pH), eau pour préparations injectables q.s. ad solutionem pro 0,8 ml.
Forme galénique et quantité de principe actif par unitéAMGEVITA 40 mg solution injectable en seringue préremplie
Chaque seringue unidose préremplie contient 40 mg d'adalimumab dans 0,8 ml de solution (50 mg/ml).
AMGEVITA 40 mg solution injectable en stylo prérempli (SureClick)
Chaque stylo unidose prérempli contient 40 mg d'adalimumab dans 0,8 ml de solution (50 mg/ml).
Indications/Possibilités d’emploiPolyarthrite rhumatoïde
AMGEVITA est indiqué pour la réduction des signes et symptômes et pour le ralentissement de la progression des lésions structurelles et pour l'amélioration des capacités fonctionnelles du corps chez les patients adultes souffrant de polyarthrite rhumatoïde active modérée à sévère n'ayant répondu qu'insuffisamment au traitement par agents antirhumatismaux de fond (DMARD).
AMGEVITA peut être utilisé en monothérapie ou en association avec le méthotrexate ou d'autres agents antirhumatismaux de fond; l'association d'AMGEVITA avec la ciclosporine, l'azathioprine et d'autres traitements anti-TNFα n'a pas été étudiée.
Chez les patients chez lesquels une polyarthrite rhumatoïde modérée à sévère a été diagnostiquée depuis peu (<3 ans) et qui n'ont pas été traités par le méthotrexate auparavant, l'efficacité d'AMGEVITA en association avec le méthotrexate a été démontrée.
Arthrite juvénile idiopathique polyarticulaire
AMGEVITA est indiqué, en association avec le méthotrexate, pour le traitement de l'arthrite juvénile idiopathique polyarticulaire active chez les adolescents à partir de 13 ans présentant une surface corporelle minimale de 1,7 m2 qui n'ont pas atteint une réponse suffisante à un ou à plusieurs antirhumatismaux modificateurs de la maladie (DMARD), y compris au méthotrexate, ou sont intolérants à un tel traitement. AMGEVITA peut être utilisé en monothérapie lors d'une intolérance au méthotrexate ou lorsqu'un traitement par le méthotrexate n'est plus possible. L'adalimumab n'a pas été étudié chez les enfants de moins de 4 ans.
Arthrite psoriasique
AMGEVITA est indiqué pour la réduction des signes et symptômes de l'arthrite psoriasique chez les patients répondant insuffisamment au traitement par agents antirhumatismaux de fond. AMGEVITA ralentit la vitesse de progression des lésions structurelles et améliore les capacités fonctionnelles physiques des patients atteints de la forme polyarticulaire symétrique de la maladie. AMGEVITA peut être utilisé en monothérapie ou en association à des agents antirhumatismaux de fond.
Spondylarthrite ankylosante (maladie de Bechterew)
AMGEVITA est indiqué pour le traitement des patients adultes atteints de spondylarthrite ankylosante active n'ayant répondu qu'insuffisamment aux traitements conventionnels.
Maladie de Crohn
AMGEVITA est indiqué pour le traitement de patients adultes atteints de la maladie de Crohn présentant une activité pathologique moyenne à forte et n'ayant répondu qu'insuffisamment aux traitements conventionnels, ainsi que pour le traitement de patients adultes ne répondant plus à l'infliximab ou ne le supportant pas.
Colite ulcéreuse
AMGEVITA est indiqué pour le traitement de la colite ulcéreuse active modérée à sévère chez les patients adultes n'ayant pas atteint une réponse suffisante sous un traitement conventionnel, y compris par des glucocorticoïdes et/ou la 6mercaptopurine (6-MP) ou l'azathioprine (AZA), ou présentant une intolérance ou une contre-indication à un tel traitement.
Psoriasis
AMGEVITA est indiqué en monothérapie pour le traitement de patients adultes atteints de psoriasis en plaques chronique modéré à sévère et candidats à un traitement systémique ou une puvathérapie.
Posologie/Mode d’emploiIl est recommandé que le traitement par AMGEVITA soit mené et surveillé par des spécialistes expérimentés dans le domaine du diagnostic et du traitement de la polyarthrite rhumatoïde (PR), de la forme polyarticulaire de l'arthrite juvénile idiopathique (AJIp), de la spondylarthrite ankylosante, de l'arthrite psoriasique ou du psoriasis. Après une formation adaptée aux méthodes d'injection par voie sous-cutanée, les patients peuvent s'injecter eux-mêmes AMGEVITA lorsque le médecin estime que cela est opportun et lorsque le suivi médical nécessaire est assuré.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Adultes (18-64 ans)
Polyarthrite rhumatoïde
La dose recommandée d'AMGEVITA pour les patients adultes atteints de polyarthrite rhumatoïde est une injection sous-cutanée de 40 mg d'adalimumab administrée une fois toutes les deux semaines. Le méthotrexate, les glucocorticoïdes, les salicylés, les anti-inflammatoires non stéroïdiens, les antalgiques et d'autres agents antirhumatismaux de fond (à l'exception de la ciclosporine, de l'azathioprine et d'autres traitements anti-TNF-α) peuvent continuer à être administrés pendant le traitement par AMGEVITA.
Chez certains patients, lors d'une monothérapie par AMGEVITA, on observe une diminution de l'effet du médicament. Dans ces cas, une augmentation de la fréquence d'administration à 40 mg d'adalimumab par semaine peut être avantageuse.
Arthrite psoriasique
La dose recommandée d'AMGEVITA pour les patients atteints d'arthrite psoriasique est une injection sous-cutanée de 40 mg d'adalimumab administrée une fois toutes les deux semaines.
Les glucocorticoïdes, les salicylés, les anti-inflammatoires non stéroïdiens, les antalgiques et d'autres agents antirhumatismaux de fond peuvent continuer à être administrés pendant un traitement par AMGEVITA.
Spondylarthrite ankylosante (maladie de Bechterew)
La dose recommandée d'AMGEVITA pour les patients atteints de spondylarthrite ankylosante est une injection sous-cutanée de 40 mg d'adalimumab administrée une fois toutes les deux semaines.
Les données disponibles indiquent qu'une réponse clinique est généralement obtenue dans une période de 12 semaines de traitement. La poursuite du traitement chez les patients qui n'ont pas obtenu de réponse pendant cette période de temps doit être étudiée à nouveau avec soin.
Des anti-inflammatoires non stéroïdiens peuvent continuer à être administrés pendant un traitement par AMGEVITA.
Maladie de Crohn
Le schéma posologique recommandé pour le traitement par AMGEVITA de patients adultes atteints de la maladie de Crohn consiste à injecter par voie sous-cutanée 160 mg à la semaine 0 (la dose peut être administrée sous forme de quatre injections le même jour ou de deux injections par jour pendant deux jours consécutifs), 80 mg à la semaine 2, ensuite 40 mg une semaine sur deux.
Des aminosalicylates, des corticostéroïdes ou des immunomodulateurs (p.ex., 6mercaptopurine et azathioprine) peuvent continuer à être administrés pendant le traitement par AMGEVITA.
Colite ulcéreuse
La dose d'induction d'AMGEVITA recommandée chez l'adulte atteint de colite ulcéreuse modérée à sévère est de 160 mg à la semaine 0 (la dose peut être administrée sous forme de quatre injections le même jour ou de deux injections par jour pendant deux jours consécutifs) et de 80 mg à la semaine 2. Après le traitement d'induction, la dose recommandée est de 40 mg en injection sous-cutanée toutes les deux semaines.
Au cours du traitement d'entretien, l'administration de glucocorticoïdes peut être arrêtée par réductions progressives.
Les patients subissant une perte d'efficacité après avoir atteint une réponse primaire peuvent profiter d'une augmentation de la fréquence d'administration à 40 mg d'AMGEVITA une fois par semaine.
La poursuite du traitement doit être réévaluée régulièrement. Les données disponibles suggèrent qu'une réponse clinique est généralement atteinte en l'espace de 2 à 8 semaines de traitement. Le traitement par AMGEVITA ne doit pas être poursuivi chez les patients n'ayant pas atteint de réponse au bout de cette période.
On ne dispose pas de données contrôlées au-delà d'une période de 52 semaines.
Psoriasis
La dose recommandée pour les patients adultes atteints de psoriasis est une dose initiale de 80 mg d'adalimumab administrée par injection sous-cutanée, suivie d'une injection sous-cutanée de 40 mg d'adalimumab administrée toutes les deux semaines (en commençant 1 semaine après la dose initiale).
Le traitement devrait être arrêté après 16 semaines chez les patients n'ayant pas répondu au traitement pendant cette période.
Chez les patients ayant répondu initialement au traitement et ayant arrêté le traitement après 33 semaines, une récidive a été observée à la semaine 52 seulement chez un peu plus d'un quart des patients; c'est pourquoi une interruption du traitement devrait être envisagée après 33 semaines.
Il convient de procéder périodiquement à une évaluation du rapport bénéfice/risque du traitement.
Patients âgés (plus de 65 ans)
Il n'est pas nécessaire d'ajuster la dose.
Enfants et adolescents
AMGEVITA est disponible exclusivement en seringue préremplie de 40 mg et en stylo prérempli de 40 mg. Il n'est pas possible de traiter par AMGEVITA les enfants et les adolescents nécessitant une dose inférieure à la dose complète de 40 mg. Si une autre dose s'avère nécessaire, il convient d'utiliser d'autres médicaments à base d'adalimumab permettant une telle option.
Arthrite juvénile idiopathique polyarticulaire (à partir de l'âge de 13 ans)
Chez les patients âgés de 13 à 17 ans qui souffrent d'arthrite juvénile idiopathique polyarticulaire et présentant une surface corporelle minimale de 1,7 m2, la dose recommandée d'AMGEVITA est de 40 mg toutes les 2 semaines. Ceci correspond à l'association d'un poids et d'une taille du patient indiquée dans le tableau 1.
Tableau 1: Posologie d'AMGEVITA en milligrammes (mg) dans le traitement de l'arthrite juvénile idiopathique polyarticulaire en fonction de la taille et du poids du patient
|
Taille (cm)
|
Poids corporel total (kg)
| |
45
|
50
|
55
|
60
|
65
|
70
| |
140
|
-
|
-
|
-
|
-
|
-
|
40*
| |
150
|
-
|
-
|
-
|
-
|
40*
|
40*
| |
160
|
-
|
-
|
40*
|
40*
|
40*
|
40*
| |
170
|
-
|
40*
|
40*
|
40*
|
40*
|
40*
| |
180
|
40*
|
40*
|
40*
|
40*
|
40*
|
40*
|
* La dose unitaire maximale est de 40 mg (0,8 ml).
Les données disponibles suggèrent qu'une réponse clinique est généralement atteinte en l'espace de 12 semaines de traitement. L'utilité d'une poursuite du traitement doit être soigneusement réévaluée chez les patients n'ayant pas atteint une réponse après cette période.
L'adalimumab n'a pas été étudié chez les enfants de moins de 4 ans atteints d'arthrite juvénile idiopathique polyarticulaire. Les données disponibles sur le traitement par l'adalimumab chez les patients pédiatriques pesant moins de 15 kilogrammes sont limitées.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients cités dans la composition.
Tuberculose active ou autres infections graves telles que septicémie et infections opportunistes (voir «Mises en garde et précautions»).
Insuffisance cardiaque modérée à sévère (classe III-IV selon la classification de la NYHA).
Mises en garde et précautionsInfections
Comme avec les autres antagonistes du TNF, les patients doivent être étroitement surveillés avant, pendant et après un traitement par AMGEVITA afin de détecter toute survenue d'infections (tuberculose incluse).
Il est recommandé de ne pas instaurer de traitement par AMGEVITA à des patients présentant des infections actives (y compris infections chroniques ou locales) tant que ces infections ne sont pas maîtrisées.
Les patients présentant une nouvelle infection alors qu'ils sont déjà sous traitement par AMGEVITA doivent être surveillés étroitement et un bilan diagnostique complet doit être pratiqué.
Lorsqu'une nouvelle infection grave apparaît chez un patient, il est recommandé d'interrompre l'administration d'AMGEVITA jusqu'à ce que cette infection soit maîtrisée. Le médecin doit envisager avec prudence l'administration d'AMGEVITA pour des patients présentant des antécédents d'infections récidivantes ou des troubles sous-jacents les prédisposant aux infections.
Des infections graves dues à des infections bactériennes, mycobactériennes, fongiques invasives (histoplasmose disséminée ou extrapulmonaire, blastomycose, aspergillose, coccidioïdomycose), virales, parasitaires ou à d'autres infections opportunistes, ont été rapportées chez des patients ayant reçu un traitement anti-TNF-α.
Septicémie, infections à Candida, listériose, légionellose et infections à Pneumocystis ont été également rapportées chez des patients ayant reçu un traitement anti-TNF-α, adalimumab inclus. Des cas d'infections nécessitant une hospitalisation et ayant une issue fatale ont été rapportés. De nombreuses infections graves sont survenues chez des patients suivant parallèlement un traitement immunosuppresseur ayant pu les rendre, en plus de leur maladie sous-jacente, particulièrement susceptibles aux infections.
Tuberculose
Des cas de tuberculose, y compris d'apparition nouvelle et de réactivation de la tuberculose ont été rapportés chez des patients ayant reçu de l'adalimumab. Les rapports comprenaient des cas de tuberculose pulmonaire et extrapulmonaire (p.ex. disséminée).
Avant d'instaurer un traitement par AMGEVITA, il convient de rechercher chez tous les patients la présence éventuelle d'une tuberculose active ou inactive (latente). Il est recommandé d'inclure à cette recherche une anamnèse médicale précise permettant de détecter tout contact antérieur éventuel avec des personnes avec une tuberculose active ainsi que tout traitement immunosuppresseur antérieur ou en cours. La réalisation de tests de dépistage adaptés (p.ex. radiographie du thorax et test à la tuberculine) conformes aux recommandations locales est recommandée. Le traitement de la tuberculose active doit être instauré avant le début du traitement par AMGEVITA. Lorsqu'un test tuberculinique cutané est réalisé pour une tuberculose latente, une induration de 5 mm ou plus doit être considérée comme positive, même lorsqu'une vaccination par le bacille de Calmette et Guérin (BCG) a été antérieurement pratiquée.
On doit particulièrement veiller à la possibilité d'une tuberculose non détectée chez les patients provenant de pays où la prévalence de la tuberculose est élevée, ou ayant voyagé dans ces pays, de même que chez les patients en contact étroit avec des personnes atteintes de tuberculose active.
En cas de diagnostic d'une tuberculose latente, un traitement antituberculeux prophylactique doit être instauré conformément aux recommandations locales avant de commencer un traitement par AMGEVITA. L'instauration d'une prophylaxie de la tuberculose avant un traitement par AMGEVITA doit également être envisagée pour les patients présentant plusieurs facteurs significatifs de risque de tuberculose malgré un test de tuberculose négatif, et chez les patients atteints de tuberculose latente ou active lors d'antécédents médicaux, chez qui l'administration d'un traitement adéquat ne peut être confirmée. La décision d'instaurer un traitement antituberculeux chez ces patients devra dépendre autant du risque de tuberculose latente que du risque lié au traitement antituberculeux. Si nécessaire, il conviendra de consulter un médecin expérimenté en matière de traitement de la tuberculose.
L'administration d'un traitement antituberculeux en cas de tuberculose latente réduit le risque de réactivation de la tuberculose chez les patients sous traitement par AMGEVITA. Malgré une prophylaxie de la tuberculose, des cas de réactivation de la tuberculose sont apparus chez des patients traités par l'adalimumab. Des patients chez lesquels le test de dépistage de la tuberculose latente était négatif ont également développé une tuberculose active et quelques patients ayant été précédemment traités avec succès contre une tuberculose active, ont à nouveau développé une tuberculose pendant un traitement par antagonistes du TNF.
Il convient de surveiller les signes et symptômes d'une tuberculose active chez les patients recevant AMGEVITA, en particulier parce que le test de dépistage de la tuberculose latente peut éventuellement donner des résultats faussement négatifs. En particulier chez les patients gravement malades ou immunosupprimés, le test à la tuberculine peut donner des résultats faussement négatifs.
Il convient de recommander aux patients de consulter leur médecin en cas de survenue de signes/symptômes suggérant une tuberculose (p.ex. toux persistante, consomption/perte de poids, fièvre faible, apathie), pendant ou après le traitement par AMGEVITA.
Autres infections opportunistes
Des infections opportunistes, y compris des infections fongiques invasives, ont été observées sous adalimumab. Ces infections n'ont pas toujours été détectées chez les patients recevant des antagonistes du TNF, ce qui a retardé l'instauration d'un traitement approprié, avec parfois une issue fatale.
Les patients recevant un traitement anti-TNF-α sont plus susceptibles de contracter des infections fongiques graves, telles que l'histoplasmose, la coccidioïdomycose, la blastomycose, l'aspergillose, les infections à Candida et autres infections opportunistes. Chez les patients qui présentent des signes et symptômes tels que fièvre ou malaise, perte de poids, sueurs, toux, dyspnée et/ou infiltrats pulmonaires ou une autre maladie systémique grave avec ou sans choc concomitant, la consultation d'un médecin doit avoir lieu très rapidement.
Il convient de surveiller, chez les patients vivant ou ayant voyagé dans des régions à risque de mycoses endémiques, les signes et symptômes d'une infection fongique systémique potentielle, en cas d'infections fongiques invasives. Ces patients sont à risque de développer une histoplasmose ou une autre maladie fongique invasive, c'est pourquoi un traitement antimycosique empirique devrait être envisagé jusqu'à ce que le ou les agents pathogènes soient identifiés.
Des tests de dépistage des anticorps et antigènes de l'histoplasmose peuvent donner des résultats négatifs chez certains patients présentant une infection active.
Si possible, chez ces patients, la décision d'un traitement empirique par un antimycosique doit être prise en consultation avec un médecin expérimenté dans le diagnostic et le traitement des maladies fongiques invasives, en prenant en considération les risques d'une infection fongique grave ainsi que les risques d'une thérapie antimycosique.
Chez les patients développant une infection fongique grave, il est recommandé d'interrompre le traitement anti-TNF-α jusqu'à ce que l'infection soit maîtrisée.
Réactivation de l'hépatite B
Une relation a été établie entre l'utilisation d'antagonistes du TNF, adalimumab inclus, et la réactivation du virus de l'hépatite B (VHB) chez des patients porteurs chroniques de ce virus. Dans quelques cas, la réactivation du VHB sous traitement par antagonistes du TNF a eu des conséquences fatales. La plupart de ces patients prenaient concomitamment d'autres médicaments immuno-suppresseurs pouvant également contribuer à une réactivation du VHB. Il convient, pour les patients présentant un risque d'infection par le VHB, de rechercher des signes de cette infection avant de commencer le traitement par des antagonistes du TNF. Il convient de prescrire avec prudence les antagonistes du TNF aux patients identifiés comme étant porteurs du VHB. Il convient de surveiller de très près, chez les patients porteurs du VHB et ayant besoin d'un traitement par antagonistes du TNF, les signes et les symptômes d'une infection active par le VHB pendant le traitement et pendant quelques mois après la fin du traitement. Aucune donnée suffisante n'est disponible concernant la sécurité et l'efficacité chez les patients porteurs du VHB traités concomitamment par un antiviral et des antagonistes du TNF pour empêcher la réactivation de l'hépatite B. En cas de réactivation du VHB, le traitement par AMGEVITA doit être stoppé et un traitement antiviral efficace doit être instauré.
Événements neurologiques
Les anti-TNF, dont l'adalimumab, ont été mis en rapport avec de rares cas d'apparition ou d'aggravation de symptômes cliniques ou de preuves radiologiques de maladies démyélinisantes du SNC, y compris de sclérose en plaques (p.ex. paresthésie et troubles de la fonction oculaire, voir «Effets indésirables»), névrite optique et maladies démyélinisantes périphériques (y compris syndrome de Guillain-Barré). Le médecin prescripteur doit soigneusement évaluer l'utilité d'un traitement par AMGEVITA chez les patients présentant des troubles démyélinisants du système nerveux central ou périphérique, préexistants ou survenus récemment. L'arrêt du traitement par AMGEVITA doit être envisagé si de tels problèmes se manifestent.
Il existe un rapport connu entre l'uvéite intermédiaire et les maladies démyélinisantes du système nerveux central, y compris la sclérose en plaques. Les patients atteints d'affections rhumatismales et présentant également une uvéite intermédiaire non infectieuse doivent être examinés quant à la présence d'une maladie démyélinisante du système nerveux avant de commencer le traitement par AMGEVITA.
Réactions allergiques
Des réactions allergiques en rapport avec l'adalimumab (p.ex. éruption cutanée d'origine allergique, réaction anaphylactoïde, exanthème médicamenteux fixe, réaction médicamenteuse non spécifique, urticaire, œdème angioneurotique) n'ont été observées qu'occasionnellement au cours des essais cliniques. Après la mise sur le marché, des cas de réactions allergiques graves, y compris d'anaphylaxie, ont été spontanément rapportés à la suite de l'administration d'adalimumab. En cas de réaction anaphylactique ou d'autre réaction allergique grave, il est recommandé d'arrêter immédiatement l'administration d'AMGEVITA et d'instaurer un traitement approprié.
Caoutchouc naturel sec
Le capuchon protecteur de l'aiguille du stylo prérempli est en caoutchouc naturel sec (un dérivé du latex), lequel peut provoquer des réactions allergiques.
Tumeurs malignes
Pendant les phases contrôlées des études cliniques portant sur les antagonistes du TNF, un nombre plus important de tumeurs malignes, lymphomes inclus, a été observé chez les patients recevant des antagonistes du TNF que dans les groupes de contrôle. Toutefois, la taille des groupes de contrôle et la durée limitée des phases contrôlées des études ne permettent pas de tirer des conclusions décisives. De plus, chez les patients atteints d'une maladie inflammatoire fortement active, le risque de lymphome augmente, ce qui rend plus difficile une bonne évaluation des risques.
Des tumeurs malignes, dont certaines d'issue fatale, ont été signalées chez des enfants et adolescents ayant été traités par des inhibiteurs du TNF. Environ la moitié de ces cas étaient des lymphomes, tant hodgkiniens que non hodgkiniens. Les autres cas comprenaient diverses autres tumeurs malignes, dont certaines, rares, qui s'observent d'ordinaire en relation avec l'immunosuppression. Les tumeurs malignes se sont manifestées après en médiane 30 mois de traitement. La plupart des patients recevaient simultanément des immunosuppresseurs. Ces cas figurent dans des rapports post-marketing provenant de différentes sources, y compris des registres et rapports post-marketing spontanés.
Il existe de très rares rapports post-marketing de lymphome T hépatosplénique (en anglais: hepatosplenic T-cell lymphoma HSTCL) chez des patients traités par l'adalimumab. Il s'agit d'un type de lymphome agressif rare qui a souvent une issue fatale. Les patients en question étaient dans certains cas de jeunes adultes qui avaient été traités antérieurement par l'infliximab en association avec de l'azathioprine ou de la 6-mercaptopurine en raison d'affections intestinales inflammatoires. Le risque potentiel d'une administration concomitante d'azathioprine ou de 6mercaptopurine avec AMGEVITA doit être soupesé avec soin. Le rapport causal entre l'adalimumab et le lymphome T hépatosplénique n'est pas clarifié, mais ne peut pas être exclu.
En l'état actuel des connaissances, on ne peut pas exclure un risque possible d'apparition de lymphomes ou d'autres tumeurs malignes chez les patients traités par un antagoniste du TNF. Tous les patients, notamment ceux ayant des antécédents de traitement immunosuppresseur intense ou ceux atteints de psoriasis et ayant reçu auparavant une puvathérapie, doivent être examinés à la recherche d'un cancer cutané non-mélanome avant et pendant le traitement par AMGEVITA.
Des cas de leucémie aiguë et chronique ont été rapportés en relation avec l'utilisation, après leur mise sur le marché, d'inhibiteurs du TNF lors de la polyarthrite rhumatoïde et d'autres indications. Les patients atteints de polyarthrite rhumatoïde présentent probablement un risque accru (jusqu'à 2 fois plus élevé) de développer une leucémie par rapport à la population générale, même sans traitement par des inhibiteurs du TNF.
Les données actuellement disponibles ne permettent pas de déterminer si le traitement par l'adalimumab influence le risque de développer des dysplasies ou un cancer du côlon. Tous les patients atteints de colite ulcéreuse qui présentent un risque accru de dysplasies ou de cancer du côlon (p.ex. patients souffrant depuis longtemps de colite ulcéreuse ou présentant une cholangite sclérosante primitive) ou qui ont des antécédents de dysplasies ou de cancer du côlon doivent être soumis régulièrement à des examens à la recherche de dysplasies éventuelles avant le traitement et pendant toute l'évolution de la maladie. Ces examens doivent inclure des coloscopies et des biopsies conformément aux recommandations nationales.
Les lymphomes intraoculaires peuvent ressembler à une uvéite et doivent être exclus avant le traitement par AMGEVITA chez les patients atteints d'affections rhumatismales et présentant également une uvéite intermédiaire non infectieuse.
Immunosuppression
Dans le cadre d'une étude pendant laquelle 64 patients atteints de polyarthrite rhumatoïde ont été traités par l'adalimumab, aucun signe d'affaiblissement de la réaction d'hypersensibilité tardive, de diminution de la concentration d'immunoglobulines ni de modification du nombre de lymphocytes T effecteurs, de lymphocytes B, de lymphocytes NK, de monocytes/macrophages ni de neutrophiles n'a été observé.
Vaccinations
Au cours d'une étude randomisée, en double aveugle et contrôlée contre placebo, les réponses anticorps à des vaccins contre les pneumocoques et le virus Influenza (H1N1, H3N2, B) administrés simultanément ont été évaluées chez 226 patients adultes atteints de polyarthrite rhumatoïde et traités par l'adalimumab. 86% des patients du groupe adalimumab ont présenté des titres d'anticorps protecteurs contre au moins 3 des 5 antigènes pneumococciques, comparés à 82% dans le groupe placebo. Au total, 37% des patients traités par l'adalimumab et 40% des patients sous placebo ont présenté un titre au moins deux fois plus élevé d'anticorps dirigés contre au moins 3 des 5 antigènes pneumococciques. Dans la même étude, 98% des patients du groupe adalimumab et 95% des patients du groupe placebo ont présenté un titre d'anticorps protecteurs contre au moins 2 des 3 antigènes du virus de l'influenza. Au total, 52% des patients traités par l'adalimumab et 63% des patients sous placebo ont présenté un titre au moins quatre fois plus élevé d'anticorps dirigés contre au moins 2 des 3 antigènes du virus de l'influenza.
Chez les patients pédiatriques, il est recommandé de procéder si possible à la mise à jour de toutes les immunisations conformément aux directives applicables avant de commencer le traitement par AMGEVITA.
Les patients sous AMGEVITA peuvent être vaccinés, à condition que les vaccins utilisés ne soient pas des vaccins vivants. L'administration concomitante de vaccins vivants et d'AMGEVITA n'est pas recommandée, étant donné qu'on ne dispose pas de données correspondantes. Concernant la transmission secondaire d'une infection lors de l'administration simultanée de vaccins vivants et d'adalimumab, aucune donnée n'est disponible.
L'administration de vaccins vivants à des nouveau-nés ayant été exposés à l'adalimumab in utero n'est pas recommandée pendant les 5 mois après la dernière administration d'AMGEVITA durant la grossesse.
Insuffisance cardiaque
Une aggravation de l'insuffisance cardiaque et une augmentation de la mortalité due à l'insuffisance cardiaque ont été observées dans des études cliniques sur d'autres anti-TNF. Des cas d'aggravation d'une insuffisance cardiaque décompensée ont également été rapportés parmi les patients traités par l'adalimumab. AMGEVITA doit être utilisé avec prudence chez les patients présentant une insuffisance cardiaque légère (classes NYHA I et II). AMGEVITA est contre-indiqué chez les patients présentant une insuffisance cardiaque modérée à sévère (classes NYHA III et IV). Si une apparition ou une aggravation des symptômes d'une insuffisance cardiaque se produit chez un patient, le traitement par AMGEVITA doit être arrêté.
Administration concomitante d'un inhibiteur d'antirhumatismaux biologiques de fond (DMARD) ou d'autres antagonistes du TNF
Des infections sévères ont été observées dans les études cliniques lors de l'administration concomitante d'anakinra et d'étanercept, un autre agent thérapeutique anti-TNFα. L'association était sans bénéfice comparée à l'utilisation d'étanercept pris seul.
La nature des effets indésirables observés avec l'association d'étanercept et d'anakinra laisse supposer l'apparition d'effets semblables lors de l'utilisation concomitante d'anakinra et d'autres agents thérapeutiques anti-TNFα.
Par conséquent, l'association d'AMGEVITA et d'anakinra n'est pas conseillée.
L'administration concomitante d'AMGEVITA avec d'autres DMARDS biologiques (p.ex. anakinra ou abatacept) ou d'autres antagonistes du TNF n'est pas recommandée à cause du risque éventuel d'infections et d'autres interactions pharmacologiques éventuelles.
Événements hématologiques
De rares cas de pancytopénie, y compris d'anémie aplasique, ont été observés sous un traitement par des antagonistes du TNF. Des effets indésirables concernant le système hématologique, y compris une cytopénie cliniquement significative (p.ex. thrombocytopénie, leucopénie), ont été observés lors d'un traitement par l'adalimumab. Il doit être indiqué à tous les patients de consulter sans attendre un médecin dans le cas où ils développent des signes ou des symptômes indiquant une dyscrasie sanguine (p.ex. fièvre persistante, hématomes, saignements, pâleur) lors d'un traitement par AMGEVITA. L'arrêt du traitement par AMGEVITA doit être envisagé chez des patients atteints d'anomalies hématologiques confirmées.
Autoanticorps
Le traitement par AMGEVITA peut entraîner la formation d'auto-anticorps. Les effets d'un traitement au long cours par AMGEVITA sur l'apparition de maladies auto-immunes ne sont pas connus. Si un patient sous traitement par AMGEVITA développe des symptômes lupiques, il convient d'arrêter le traitement par AMGEVITA (voir «Effets indésirables», «Auto-anticorps»).
Anticorps contre adalimumab
(Voir «Effets indésirables»).
Utilisation en gériatrie
La fréquence des infections graves était plus élevée chez les patients de plus de 65 ans traités par l'adalimumab que chez les patients de moins de 65 ans. Parmi les patients traités par l'adalimumab dans les études cliniques, 9,6% avaient 65 ans ou plus et environ 2,0% avaient 75 ans ou plus. Étant donné que l'incidence des infections est en général plus élevée chez les patients âgés, la prudence est de mise lors du traitement de ces patients.
InteractionsL'effet de l'adalimumab a été étudié chez des patients atteints de polyarthrite rhumatoïde prenant également du méthotrexate. Les données obtenues n'indiquent pas qu'un ajustement de la dose d'adalimumab ou de méthotrexate est nécessaire (voir «Pharmacocinétique»).
L'adalimumab a été étudié en monothérapie ainsi qu'en association avec le méthotrexate chez des patients atteints de polyarthrite rhumatoïde, de la forme polyarticulaire de l'arthrite juvénile idiopathique et de rhumatisme psoriasique. La formation d'anticorps a été moins importante en cas d'utilisation concomitante d'adalimumab et de méthotrexate que lors de la monothérapie. L'utilisation d'adalimumab sans méthotrexate a entraîné une production d'anticorps plus élevée, une clairance plus élevée et une diminution de l'efficacité de l'adalimumab.
Les interactions entre l'adalimumab et d'autres médicaments que le méthotrexate n'ont pas été étudiées dans le cadre d'études de pharmacocinétique. Aucune interaction n'a été observée dans le cadre des essais cliniques relatifs au traitement de la polyarthrite rhumatoïde lors de l'administration de l'adalimumab avec d'autres agents antirhumatismaux de fond fréquemment utilisés (sulfasalazine, hydroxychloroquine, léflunomide et or parentéral), avec des glucocorticoïdes, des salicylés, des anti-inflammatoires non stéroïdiens ou des antalgiques. L'association de l'adalimumab avec la ciclosporine, l'azathioprine et d'autres traitements anti-TNFα n'a pas été étudiée.
Grossesse/AllaitementLes données cliniques concernant l'exposition de la femme enceinte à l'adalimumab sont limitées.
Une cohorte prospective du registre sur l'exposition pendant la grossesse a inclus 257 femmes atteintes de polyarthrite rhumatoïde ou de maladie de Crohn qui avaient été traitées par l'adalimumab au moins pendant le premier trimestre de la grossesse ainsi que 120 femmes atteintes de polyarthrite rhumatoïde ou de maladie de Crohn qui n'avaient pas reçu d'adalimumab.
Le taux de malformations congénitales graves (critère d'évaluation principal) sur la totalité des grossesses, à l'exception des cas perdus de vue au cours du suivi, était de 10,1% (25/247) chez les femmes traitées par l'adalimumab et de 8,1% (9/111) chez les femmes non traitées. Les données limitées du registre sur l'exposition pendant la grossesse n'indiquent pas un modèle des malformations congénitales graves. Des différences entre les groupes d'exposition pourraient avoir eu une influence sur l'incidence des malformations congénitales. Aucune différence concernant les critères secondaires – avortement spontané, malformations congénitales minimes, naissance prématurée, taille corporelle à la naissance et infections sévères ou opportunistes – n'est clairement ressortie entre les femmes traitées par l'adalimumab et les femmes non traitées. Aucun cas d'enfant mort-né ou de maladie maligne n'a été rapporté. L'évaluation des données peut être influencée par les limitations méthodologiques du registre, p.ex. par la petite taille de l'échantillon et par la conception sans randomisation.
En raison de son effet inhibiteur sur le TNFα, l'utilisation de l'adalimumab durant la grossesse pourrait avoir des effets sur les réactions immunitaires normales du nouveau-né.
L'adalimumab ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue.
Les femmes en âge de procréer doivent envisager l'utilisation de méthodes de contraception appropriées pendant le traitement par l'adalimumab et les cinq mois suivant la fin de ce traitement.
L'administration de vaccins vivants à des nouveau-nés ayant été exposés à l'adalimumab in utero n'est pas recommandée pendant les 5 mois qui suivent la dernière administration d'AMGEVITA pendant la grossesse.
Utilisation au cours de l'allaitement
Des informations limitées provenant de trois cas présentés dans la littérature publiée indiquent que l'adalimumab passe en très faible concentration dans le lait maternel et y est présent à des concentrations comprises entre 0,1% et 1% de la concentration sérique maternelle. Les avantages de l'allaitement pour le développement et la santé doivent être pris en considération autant que la nécessité clinique du traitement par l'adalimumab pour la mère et tous les effets secondaires potentiels de l'adalimumab ou de la maladie maternelle chez l'enfant allaité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesÉtudes cliniques
L'adalimumab a été étudié chez 9238 patients au cours d'études contrôlées et en ouvert pendant une durée allant jusqu'à 60 mois ou plus. Les données présentées sont fondées sur les études pivots contrôlées et incluent 5963 patients traités par l'adalimumab et 3689 patients ayant reçu un placebo ou un comparateur actif pendant les phases contrôlées des études.
La part des patients ayant interrompu le traitement en raison d'effets indésirables au cours de la phase contrôlée en double aveugle des études pivots a été de 6,0% dans le groupe adalimumab et de 5,5% dans le groupe contrôle.
Les effets indésirables (tant cliniques que touchant des paramètres biologiques) rapportés avec l'adalimumab pendant les études cliniques et dans des rapports spontanés après la commercialisation chez les patients adultes et pédiatriques sont classés ci-dessous par système d'organes et par fréquence (très fréquents ≥1/10, fréquents ≥1/100, <1/10, occasionnels ≥1/1000, <1/100, rares ≥1/10'000, <1/1000 et cas isolés (la fréquence ne peut pas être estimée sur la base des données disponibles)). Les informations concernant spécifiquement les enfants sont présentées à la suite de la liste générale. Au sein de chaque groupe de fréquence, les effets secondaires sont classés par degré de gravité décroissant. En se basant sur le regroupement de termes médicaux apparentés, le calcul des fréquences englobe tous les événements qui sont au moins en relation causale possible avec le traitement par l'adalimumab. La fréquence la plus élevée des effets indésirables observés dans chaque indication au cours des essais cliniques est indiquée. Un * placé après un système d'organes signale que d'autres informations figurent dans les rubriques «Contre-indications», «Mises en garde et précautions» ainsi que «Effets indésirables». Un ** signale que l'effet indésirable survient chez les patients traités par un antagoniste du TNF (y compris adalimumab). Les pourcentages sont indiqués pour les effets indésirables très fréquents.
Effets indésirables rapportés au cours des études cliniques et rapportés spontanément
Infections et infestations*
Fréquents: infections de l'appareil respiratoire (y compris infections des voies respiratoires inférieures et supérieures, pneumonie, sinusite, pharyngite, nasopharyngite et pneumopathie virale à Herpes), infections buccales (y compris herpès simplex et herpès labial), infections de la peau et des tissus mous (y compris paronychie, impétigo, fasciite nécrosante, panniculite et zona), infections des voies urinaires (y compris pyélonéphrite), infections systémiques (y compris septicémie et candidose).
Occasionnels: infections de l'oreille, infections intestinales (y compris hépatite, gastroentérite virale), infections articulaires, infections de l'appareil génital (y compris mycoses vulvo-vaginales), mycoses, infections bactériennes, abcès.
Rares: infections abdominales (y compris diverticulite), infections des yeux (y compris herpès simplex), infections opportunistes et tuberculose (y compris histoplasmose et infections par le complexe Mycobacterium avium), méningite virale, infestations parasitaires sévères.
Tumeurs bénignes, malignes et non précisées (y compris kystes et polypes)*
Occasionnels: néoplasie bénigne.
Rares: lymphome, mélanome malin, cancer cutané, sauf mélanome (y compris carcinome squameux), néoplasies solides d'organes (y compris cancer du sein, des ovaires et des testicules), leucémie.
Cas isolés: carcinome à cellules de Merkel (tumeur neuro-endocrine cutanée), lymphome hépatosplénique à lymphocytes T.
Affections hématologiques et du système lymphatique*
Fréquents: leucopénie (y compris neutropénie et agranulocytose).
Occasionnels: anémie, thrombocytopénie, lymphadénopathie, leucocytose.
Rares: purpura thrombocytopénique idiopathique, pancytopénie.
Affections du système immunitaire*
Occasionnels: hypersensibilité, réactions allergiques sérieuses, y compris anaphylaxie et angiœdème.
Rares: sarcoïdose, allergies (y compris saisonnières).
Affections endocriniennes
Rares: hypothyroïdie, goitre.
Troubles du métabolisme et de la nutrition
Occasionnels: taux de cholésterol élevé, hypokaliémie, anorexie, appétit accru, hyperglycémie.
Rares: natrémie anormale, hypercalcémie, hypocalcémie, hyperuricémie.
Affections psychiatriques
Occasionnels: variations d'humeur (y compris dépression), anxiété (y compris nervosité et agitation), insomnie.
Affections du système nerveux*
Fréquents: céphalées, paresthésies, obnubilation.
Occasionnels: troubles de la conscience, tremblements, événements cérébrovasculaires.
Rares: maladies démyélinisantes (p.ex. sclérose en plaques, névrite optique, syndrome de Guillain-Barré), troubles des réflexes, syndrome lumbago-sciatique, syncope, troubles de l'équilibre, troubles de l'attention, paralysie faciale.
Affections oculaires
Occasionnels: conjonctivite, yeux gonflés, troubles visuels, glaucome.
Rares: blépharite, iritis, panophtalmie, iridocyclite.
Affections de l'oreille et du labyrinthe
Occasionnels: troubles de l'oreille (y compris douleur et tuméfaction), vertiges, acouphènes.
Rares: surdité.
Affections cardiaques*
Occasionnels: tachycardie, palpitations, infarctus du myocarde.
Rares: arythmie, fibrillation auriculaire, insuffisance coronaire, bruits cardiaques, arrêt cardiaque, insuffisance cardiaque décompensée, épanchement péricardique.
Affections vasculaires
Occasionnels: hypertension, flush.
Rares: sténose aortique, artériopathie oblitérante, vasculite, hématomes, lymphœdèmes, thrombophlébite, anévrisme aortique.
Affections respiratoires, thoraciques et médiastinales*
Fréquents: toux.
Occasionnels: asthme, dyspnée, dysphonie, rhinorrhée, râles crépitants, épistaxis.
Rares: œdème pharyngé, pneumonie interstitielle, pleurésie, pneumonie, obstruction des voies respiratoires supérieures, ulcères du nez, insuffisance respiratoire, irritation de la gorge, embolie pulmonaire, épanchement pleural.
Affections gastro-intestinales
Fréquents: diarrhée et troubles de la motilité, douleurs abdominales, douleurs oropharyngées, nausées.
Occasionnels: dyspepsie, saignements gastro-intestinaux, reflux gastro-œsophagien, syndrome de Gougerot-Sjögren, ulcération buccale, maladie inflammatoire intestinale.
Rares: gastrite, obstruction intestinale, pancréatite, diverticules, dysphagie, douleurs dentaires, saignements de gencives, prurit oral, chéilite, décolorations de la muqueuse buccale, œdème facial, perforation intestinale.
Affections hépatobiliaires*
Fréquents: élévation des enzymes hépatiques.
Occasionnels: hépatotoxicité (y compris nécrose hépatique, stéatose hépatique).
Rares: cholélithiase, bilirubine élévée dans le sang, réactivation d'une hépatite B, défaillance hépatique.
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée (y compris érythème squameux), prurit, dermatite.
Occasionnels: chute des cheveux, acné, eczéma, psoriasis (y compris psoriasis palmo-plantaire pustuleux), hématomes (y compris purpura et ecchymoses), hyperhidrose, sudation nocturne, troubles de la pigmentation cutanée, urticaire.
Rares: dermatite acnéiforme, anomalies capillaires et unguéales, indurations de la peau, irritation cutanée, vascularite cutanée, érythème polymorphe, angiœdème, réaction cutanée lichénoïde**.
Cas isolés: syndrome de Stevens-Johnson, détérioration des symptômes d'une dermatomyosite.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: arthrite, douleurs musculosquelettiques.
Occasionnels: crampes musculaires (y compris élévation de la créatine phosphokinase sérique).
Rares: syndrome lupus-like, lupus érythémateux systémique, rhabdomyolyse, tendinite, myosite, sensation de lourdeur.
Affections du rein et des voies urinaires
Occasionnels: hématurie, troubles de la vessie et de l'urètre.
Rares: douleurs rénales, nycturie, protéinurie, insuffisance rénale.
Affections des organes de reproduction et du sein
Occasionnels: troubles vulvo-vaginaux, troubles du cycle menstruel.
Rares: kystes et sensibilité mammaires, dysfonction érectile, troubles utérins.
Troubles généraux et anomalies au site d'administration*
Très fréquents: réactions au site d'injection (y compris douleur, tuméfaction, rougeur ou prurit) dans 13% (contrôle 7%).
Fréquents: fatigue (y compris asthénie et malaise).
Occasionnels: douleurs thoraciques, fièvre, œdèmes, symptômes pseudo-grippaux, douleurs, frissons, augmentation de poids.
Rares: inflammations, énergie accrue, sensation anormale, inflammation de muqueuses, sensation de chaleur.
Investigations
Occasionnels: allongement du temps de thromboplastine partielle activée, détection d'auto-anticorps (y compris anticorps ADN double brin), élévation du taux sanguin de lactate-déshydrogénase.
Rares: analyses urinaires anormales.
Lésions, intoxications et complications liées aux procédures*
Occasionnels: blessure accidentelle, guérison difficile d'une plaie.
Rares: complications dues à l'utilisation.
Population pédiatrique
La fréquence et la sévérité des infections, des réactions d'hypersensibilité et des réactions au site d'injection étaient plus élevées chez les enfants que chez les adultes. À part cela, la fréquence et la nature des effets indésirables observés étaient similaires chez les patients pédiatriques et chez les adultes. Pendant la phase contrôlée de 32 semaines de l'étude sur l'AJIp, des infections ont été observées chez 24% des patients (contrôle 15%), des réactions d'hypersensibilité chez 1,5% des patients (contrôle 0%) et des réactions au site d'injection chez 37% des patients (contrôle 20%). Sur la totalité de la période d'étude, comprenant aussi la phase d'extension en ouvert d'une durée allant jusqu'à 4 ans, des infections sont apparues chez 80% des patients, dont 6% de cas sévères, et des réactions d'hypersensibilité sont survenues chez 6% des patients.
Réactions au site d'injection
Au cours des essais pivots contrôlés menés sur des adultes et des enfants, 13% au total des patients sous adalimumab ont présenté des réactions au site d'injection (érythèmes et/ou prurit, hémorragie, douleur ou tuméfaction) par rapport à 7% au total des patients sous placebo ou substance de comparaison active. Les réactions au site d'injection n'ont en général pas nécessité l'arrêt du médicament.
Infections
Au cours des études pivots contrôlées effectuées chez des adultes et des enfants, le taux d'infection a atteint 1,51 par année-patient chez les patients traités par l'adalimumab et 1,46 par année-patient chez les patients sous placebo ou contrôle actif. L'incidence des infections graves a été de 0,04 par annéepatient chez les patients traités par l'adalimumab et de 0,03 par année-patient chez les patients traités par placebo et par contrôle actif. Les infections étaient principalement des infections des voies respiratoires supérieures, des rhinopharyngites et des sinusites. La plus grande partie des patients ont poursuivi le traitement par l'adalimumab après guérison de l'infection.
Au cours des études contrôlées et ouvertes portant sur l'adalimumab chez l'adulte et l'enfant, des infections graves (ayant, rarement, une issue fatale) incluant des cas de tuberculose (miliaire et extra-pulmonaire) et des infections opportunistes invasives (comme histoplasmose disséminée ou extrapulmonaire, blastomycose, coccidioïdomycose, infection à Pneumocystis, candidose (muguet), aspergillose et listériose) ont été signalées. La plupart des cas de tuberculose sont survenus dans les huit premiers mois après le début du traitement et peuvent être le reflet d'une réactivation d'une maladie latente. L'incidence de la réactivation d'une tuberculose a été particulièrement élevée lors de l'administration de doses d'adalimumab supérieures à celles recommandées.
Tumeurs malignes
Pendant les périodes contrôlées (d'une durée d'au moins 12 semaines) des études pivots portant sur l'adalimumab chez des patients adultes atteints de polyarthrite rhumatoïde active modérée à sévère, d'arthrite psoriasique, de spondylarthrite ankylosante (maladie de Bechterew), de maladie de Crohn, de colite ulcéreuse et de psoriasis, on a observé un taux d'incidence (intervalle de confiance à 95%) des maladies malignes (à l'exception de lymphomes et du cancer de la peau non-mélanome) de 6,9 (4,4; 10,6) par 1000 années-patients chez les 5196 patients traités par l'adalimumab, contre un taux de 6,4 (3,5; 11,9) par 1000 années-patients chez les 3347 patients des groupes contrôles (la durée moyenne de traitement était de 4,0 mois pour les patients traités par l'adalimumab et de 3,9 mois pour les patients traités par la substance de contrôle). Le taux (intervalle de confiance de 95%) de cancer de la peau non-mélanome s'est élevé à 8,9 (6,1; 13,1) par 1000 années-patients chez les patients traités par l'adalimumab et à 3,2 (1,3; 7,7) par 1000 années-patients chez les patients du groupe contrôle. Parmi ces carcinomes de la peau, le carcinome épidermoïde a été observé avec un taux (intervalle de confiance de 95%) de 2,7 (1,4; 5,5) par 1000 années-patients chez les patients traités par l'adalimumab contre 0,6 (0,1; 4,6) par 1000 années-patients chez les patients du groupe contrôle. Le taux (intervalle de confiance à 95%) de lymphomes s'est élevé à 0,7 (0,2; 2,7) par 1000 années-patients chez les patients traités par l'adalimumab et à 0,6 (0,1; 4,6) par 1000 années-patients chez les patients du groupe contrôle.
Si l'on regroupe les études cliniques contrôlées et les études de prolongation ouvertes, toujours en cours et menées à terme, le taux d'incidence des tumeurs malignes (à l'exception des lymphomes et du cancer de la peau non-mélanome) s'élève à quelque 8,6 par 1000 années-patients. Le taux de cancers de la peau non-mélanome est d'environ 9,8 par 1000 années-patients et celui des lymphomes d'environ 1,3 par 1000 années-patients. Ces essais étaient d'une durée moyenne de presque 3,3 ans et incluaient 6279 patients traités pendant au moins 1 an par l'adalimumab ou ayant développé une tumeur maligne dans l'année suivant le début du traitement, ce qui correspond à 26'045 années-patients.
Les expériences post-marketing montrent que depuis janvier 2003, le taux d'incidence déclaré des tumeurs malignes (à l'exception des lymphomes et du cancer de la peau non-mélanome) s'élève à environ 1,7 par 1000 années-patients (principalement chez les patients atteints de polyarthrite rhumatoïde). Dans le cas des carcinomes de la peau non-mélanome et des lymphomes, les taux déclarés s'élèvent respectivement à environ 0,2 et 0,4 par 1000 années-patients (voir «Mises en garde et précautions»).
Auto-anticorps
Au cours des études 1-5 portant sur la polyarthrite rhumatoïde, des auto-anticorps ont été recherchés à plusieurs reprises dans des échantillons de sérum de patients. Au cours de ces études adéquates et bien contrôlées, 11,9% des patients traités par l'adalimumab et 8,1% des patients traités par le placebo et la substance de comparaison active ayant présenté un titre négatif d'anticorps antinucléaires avant traitement ont présenté un titre positif à la 24e semaine. Dans toutes les études portant sur la polyarthrite rhumatoïde, le rhumatisme psoriasique et la spondylarthrite ankylosante (maladie de Bechterew), deux patients sur les 3989 patients traités par l'adalimumab ont présenté les symptômes cliniques d'une nouvelle poussée de syndrome de type lupus. Après l'arrêt du traitement, l'état de santé des patients s'est amélioré. Aucun des patients n'a présenté de néphrite lupique ni de symptômes affectant le système nerveux central. Les effets d'un traitement au long cours par l'adalimumab sur l'apparition de maladies auto-immunes ne sont pas connus.
Psoriasis: rechute et détérioration
Sous inhibiteurs du TNF, y compris adalimumab, des cas de rechute de psoriasis, y compris de psoriasis pustuleux et palmo-plantaire, ainsi que des cas de détérioration d'un psoriasis existant ont été rapportés. Beaucoup de ces patients utilisaient concomitamment des immunosuppresseurs (p.ex. méthotrexate, corticostéroïdes). Une hospitalisation a été nécessaire pour certains d'entre eux. Le psoriasis s'est amélioré chez la plupart des patients après l'arrêt de l'inhibiteur du TNF. Chez certains patients, il y a eu une nouvelle exacerbation du psoriasis à la prise d'un autre inhibiteur du TNF. Lors de cas sévères ou si un traitement topique n'améliore pas l'affection ou même la détériore, il y a lieu d'envisager l'arrêt du traitement par AMGEVITA.
Foie: élévation des taux d'ALAT
Polyarthrite rhumatoïde et arthrite psoriasique:
Dans des études contrôlées de phase III portant sur l'adalimumab (40 mg s.c. toutes les deux semaines), une élévation des taux d'ALAT (≥3× la LSN) est survenue au cours de la phase d'observation entre la 4e et la 104e semaine chez 3,7% des patients traités par l'adalimumab et chez 1,6% des patients sous traitement de contrôle. Dans ces études, de nombreux patients utilisaient également des médicaments provoquant une élévation des enzymes hépatiques (p.ex. AINS, MTX).
Arthrite juvénile idiopathique polyarticulaire:
Dans une étude contrôlée de phase III portant sur l'adalimumab, on a enregistré au cours d'une durée d'observation de 32 semaines, parmi les patients sous méthotrexate, une augmentation des taux d'ALAT (à ≥3× la LSN) chez 5,3% des patients recevant l'adalimumab et chez 2,7% des patients recevant le traitement de contrôle. Les patients traités sans méthotrexate n'ont présenté aucune augmentation des taux d'ALAT.
Maladie de Crohn:
Dans les études contrôlées de phase III portant sur l'adalimumab (dose initiale de 160 mg le jour 1 et de 80 mg le jour 15, ou de 80 mg le jour 1 et de 40 mg le jour 15, puis de 40 mg toutes les deux semaines), une élévation des taux ALAT (≥3× la LSN) est survenue pendant la phase d'observation entre la 4e et la 52e semaine chez 0,9% des patients traités par l'adalimumab et chez 0,9% des patients sous traitement de contrôle.
Colite ulcéreuse:
Dans les études contrôlées de phase III portant sur l'adalimumab (dose initiale de 160 mg le jour 1, de 80 mg le jour 15, puis de 40 mg toutes les deux semaines), des augmentations du taux d'ALAT (≥3× la LSN) ont été constatées chez 1,5% des patients sous l'adalimumab et chez 1% des patients sous traitement de contrôle au cours de la période d'observation de 1 à 52 semaines.
Psoriasis en plaques:
Dans des études contrôlées de phase III portant sur l'adalimumab (dose initiale de 80 mg, puis de 40 mg toutes les deux semaines), une élévation des taux d'ALAT (≥3× la LSN) est survenue durant la phase d'observation entre la 12e et la 24e semaine chez 1,8% des patients traités par l'adalimumab et chez 1,8% des patients sous traitement de contrôle.
Spondylarthrite ankylosante:
Dans des études contrôlées de phase III portant sur l'adalimumab (40 mg toutes les deux semaines), une élévation des taux d'ALAT (≥3× la LSN) est survenue pendant la phase d'observation entre la 12e et la 24e semaine chez 2,4% des patients traités par l'adalimumab et chez 0,66% des patients sous traitement de contrôle.
Dans toutes les indications, les patients présentant une élévation des taux d'ALAT étaient asymptomatiques. En outre, dans la plupart des cas, l'élévation n'est survenue que de façon transitoire et a disparu au cours de la poursuite du traitement. Cependant, des réactions hépatiques sévères (y compris une insuffisance hépatique) ont été rapportées dans de très rares cas au cours des observations de l'utilisation chez des patients sous inhibiteurs du TNF, dont l'adalimumab.
Association avec l'azathioprine/6-mercaptopurine
Dans des études auprès de patients adultes atteints de maladie de Crohn, l'incidence d'effets indésirables à type de tumeurs malignes ou d'infections sévères était plus élevée sous l'association de l'adalimumab avec l'azathioprine/6-mercaptopurine que sous l'adalimumab seul.
Immunogénicité
La production d'anticorps dirigés contre l'adalimumab est associée à une clairance accrue et à une efficacité réduite de l'adalimumab. Il n'existe pas de rapport manifeste entre la présence d'anticorps neutralisants contre l'adalimumab et la survenue d'effets indésirables. L'immunogénicité au long cours de l'adalimumab est inconnue.
Les patients atteints de polyarthrite rhumatoïde qui ont participé aux études 1, 2 et 3 ont été examinés à plusieurs reprises quant au développement d'anticorps dirigés contre l'adalimumab au cours des périodes de traitement de 6 et de 12 mois. Des anticorps contre l'adalimumab ont été détectés chez 58 (5,5%) des 1053 patients ayant reçu de l'adalimumab dans les études pivots, par rapport à 2 (0,5%) des 370 patients ayant reçu un placebo. La fréquence était de 12,4% chez les patients sans co-administration de méthotrexate (étude 2), par rapport à 0,6% chez les patients avec une co-administration de méthotrexate. Les patients sous adalimumab seul administré toutes les deux semaines développent éventuellement plus souvent des anticorps que les patients recevant des administrations hebdomadaires d'adalimumab. La réponse ACR20 était plus faible chez les patients ayant développé des anticorps que chez les patients sans anticorps détectables dans le cadre d'un traitement avec la dose recommandée de 40 mg toutes les deux semaines en monothérapie.
Chez les patients atteints d'arthrite juvénile idiopathique polyarticulaire, des anticorps dirigés contre l'adalimumab ont été identifiés chez 27 (15,8%) des 171 patients sous adalimumab. La fréquence était de 22 (25,6%) patients sur 86 sans co-administration de méthotrexate, par rapport à 5 (5,9%) patients sur 85 recevant l'adalimumab en association avec le méthotrexate.
Chez les patients atteints d'arthrite psoriasique, des anticorps dirigés contre l'adalimumab ont été identifiés chez 38 (10%) des 376 patients sous adalimumab. La fréquence était de 13,5% (24 patients sur 178) dans le groupe sans co-administration de méthotrexate, par rapport à 7% (14 patients sur 198) dans le groupe recevant l'adalimumab en association avec le méthotrexate.
Chez les patients atteints de spondylarthrite ankylosante, des anticorps dirigés contre l'adalimumab ont été identifiés chez 17 (8,3%) des 204 patients sous adalimumab. La fréquence était de 8,6% (16 patients sur 185) dans le groupe sans co-administration de méthotrexate, par rapport à 5,3% (1 patient sur 19) dans le groupe recevant l'adalimumab en association avec le méthotrexate.
Chez les patients présentant une maladie de Crohn ou une colite ulcéreuse, des anticorps dirigés contre l'adalimumab ont été détectés au cours du traitement par l'adalimumab chez 7 (2,6%) des 269 patients atteints de la maladie de Crohn et chez 19 (3,9%) des 487 patients atteints de colite ulcéreuse.
Parmi les patients atteints de psoriasis en plaques qui ont reçu de l'adalimumab en monothérapie pendant une longue période et ont participé à une étude dans laquelle le traitement a été suspendu, puis repris, la proportion de patients ayant développé des anticorps contre l'adalimumab après la reprise du traitement (11 patients sur 482; 2,3%) était similaire à celle avant l'interruption du traitement (11 patients sur 590; 1,9%).
Vu que toute analyse sur l'immunogénicité n'est applicable qu'à l'agent actif examiné, les comparaisons des taux d'anticorps entre différents médicaments doivent être interprétées avec prudence.
Immunogénicité d'AMGEVITA dans les études de comparabilité
Des anticorps liants et neutralisants ont été détectés avec AMGEVITA et le médicament de référence à des fréquences comparables.
SurdosageAucune toxicité par dépassement de dose n'a été observée au cours des essais cliniques. La dose la plus élevée étudiée a été une application répétée par voie intraveineuse de 10 mg/kg.
Propriétés/EffetsCode ATC: L04AB04
L'adalimumab est un anticorps monoclonal humain fabriqué dans des cellules CHO par technologie de l'ADN recombinant. Il a été mis au point avec des chaînes lourdes et légères humaines par la méthode «phage display». On obtient ainsi un anticorps doté de régions variables des chaînes lourdes et légères sans séquence peptidique animale, ce qui permet une spécificité pour le facteur de nécrose tumorale (TNF) humain, et doté de régions constantes humaines IgG1 (chaîne lourde) et kappa (chaîne légère). L'adalimumab se lie avec une affinité et une spécificité élevées au facteur de nécrose tumorale soluble (TNF-α), mais pas à la lymphotoxine (TNF-β). Il comporte 1330 acides aminés et son poids moléculaire est de 148 kilodaltons environ.
AMGEVITA est un médicament biosimilaire.
Mécanisme d'action
L'adalimumab se lie spécifiquement au TNF et neutralise la fonction biologique du TNF par inhibition de son interaction avec les récepteurs du TNF p55 et p75 à la surface des cellules. Le TNF est une cytokine naturelle importante pour les réponses inflammatoires et immunitaires normales. Chez les patients atteints de polyarthrite rhumatoïde, de rhumatisme psoriasique ou de spondylarthrite ankylosante (maladie de Bechterew), on observe des concentrations élevées de TNF dans le liquide synovial, qui jouent un rôle important aussi bien dans le cadre de l'inflammation pathologique que dans le cadre de la destruction de l'articulation, signes caractéristiques de la polyarthrite rhumatoïde.
L'adalimumab module aussi des réactions biologiques induites ou régulées par le TNF, entre autres les modifications des concentrations en molécules d'adhésion responsables de la migration des leucocytes (ELAM-1, VCAM-1 et ICAM-1, avec une CI50 de 1-2× 10-10 M).
Pharmacodynamique
Après traitement par l'adalimumab, en comparaison avec les valeurs initiales, on a observé chez les patients atteints de polyarthrite rhumatoïde une régression rapide des valeurs des paramètres de la phase aiguë de l'inflammation (protéine C réactive [CRP]), vitesse de sédimentation des érythrocytes (VSE) et cytokines sériques (IL-6). La concentration sérique des métalloprotéinases matricielles (MMP-1 et MMP-3), qui entraînent le remodelage tissulaire responsable de la destruction du cartilage, a également diminué après administration d'adalimumab. On constate souvent, chez les patients atteints de polyarthrite rhumatoïde, de rhumatisme psoriasique ou de spondylarthrite ankylosante (maladie de Bechterew), une anémie légère à modérée, une diminution du nombre de lymphocytes et une augmentation des nombres de neutrophiles et de thrombocytes. Chez les patients traités par l'adalimumab, on observe généralement une amélioration de ces signes hématologiques d'une inflammation chronique.
Une régression rapide des taux de CRP après le traitement par l'adalimumab a été observée aussi chez les patients atteints de maladie de Crohn, de colite ulcéreuse, de la forme polyarticulaire de l'arthrite juvénile idiopathique. Chez les patients atteints de maladie de Crohn, une diminution (statistiquement non significative) des cellules exprimant des marqueurs d'inflammation dans le côlon, y compris une diminution significative de l'expression du TNF-α, a été observée.
Efficacité clinique
Polyarthrite rhumatoïde
L'adalimumab a été étudié chez plus de 3000 patients dans le cadre de toutes les études cliniques sur la polyarthrite rhumatoïde. Certains patients ont été traités sur une période de plus de 60 mois. L'efficacité et la tolérance de l'adalimumab dans le traitement de la polyarthrite rhumatoïde ont été étudiées dans cinq études randomisées, en double aveugle et bien contrôlées.
L'étude 1 a évalué 271 patients souffrant d'une polyarthrite rhumatoïde active modérée à sévère, ayant ≥18 ans ou plus, en échec d'un traitement par au moins un, mais pas plus de quatre antirhumatismaux de fond, pour lesquels on avait observé une efficacité insuffisante du méthotrexate à une dose de 12,5 à 25 mg (10 mg en cas d'intolérance au méthotrexate) par semaine, et pour lesquels la dose de méthotrexate était restée constante pendant l'étude, entre 10 et 25 mg par semaine. Les patients avaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. La polyarthrite rhumatoïde avait été diagnostiquée en appliquant les critères de l'American College of Rheumatology (ACR). Pendant 24 semaines, les patients ont reçu, toutes les deux semaines, des doses de 20, 40 ou 80 mg d'adalimumab ou un placebo.
L'étude 2 a évalué 544 patients présentant une polyarthrite rhumatoïde active modérée à sévère, ayant ≥18 ans ou plus, en échec d'un traitement comportant au moins un antirhumatismal de fond. Les patients présentaient ≥10 articulations enflées et ≥12 articulations sensibles à la pression et avaient également été diagnostiqués selon les critères de l'ACR. Pendant 26 semaines, les patients ont reçu par injection sous-cutanée 20 ou 40 mg d'adalimumab toutes les deux semaines, en alternance avec un placebo la semaine suivante, ou un placebo chaque semaine. Le placebo a été administré chaque semaine au même moment. Les patients ne suivaient aucun traitement d'appoint par agents antirhumatismaux de fond.
L'étude 3 a évalué 619 patients souffrant d'une polyarthrite rhumatoïde active modérée à sévère, ayant 18 ans ou plus, chez lesquels le méthotrexate à la dose de 12,5 à 25 mg (10 mg en cas d'intolérance au méthotrexate) par semaine, avait eu une efficacité insuffisante et pour lesquels la dose de méthotrexate était restée constante pendant l'étude, entre 12,5 et 25 mg par semaine. À la différence de l'étude 1, l'inclusion des patients de l'étude 3 ne supposait pas obligatoirement l'échec d'un traitement par antirhumatismaux de fond (méthotrexate exclus). Les patients avaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. La polyarthrite rhumatoïde avait été diagnostiquée en appliquant les critères de l'ACR. Cette étude comprenait trois groupes. Le groupe 1 a reçu une injection de placebo chaque semaine pendant 52 semaines. Le deuxième groupe a reçu 20 mg d'adalimumab chaque semaine pendant 52 semaines. Le troisième groupe a reçu 40 mg d'adalimumab toutes les deux semaines et une injection de placebo la semaine suivante. Enfin, 457 patients ont été inclus dans une phase d'extension en ouvert de 5 ans au maximum pendant laquelle ils ont reçu 40 mg d'adalimumab toutes les deux semaines.
L'étude 4 a évalué 636 patients souffrant de polyarthrite rhumatoïde active modérée à sévère, ayant 18 ans ou plus. Ces patients remplissaient les critères diagnostiques de la polyarthrite rhumatoïde de l'ACR depuis au moins trois mois et présentaient ≥6 articulations enflées et ≥9 articulations sensibles à la pression. Ces patients soit n'avaient jamais été traités par antirhumatismaux de fond soit pouvaient poursuivre leur traitement rhumatologique en cours, à condition qu'il ait été stable depuis au moins 28 jours. Les patients ont été randomisés dans le groupe de traitement 40 mg d'adalimumab ou placebo toutes les deux semaines, pendant 24 semaines.
Dans l'étude 5 concernant la polyarthrite rhumatoïde précoce, 525 patients adultes (≥18 ans) atteints d'une polyarthrite rhumatoïde active modérée à sévère précoce (durée de la maladie inférieure à 3 ans), et n'ayant pas été traités par le méthotrexate, ont été évalués. Dans cette étude, l'efficacité de l'adalimumab associé au méthotrexate a été comparée au méthotrexate en monothérapie en ce qui concerne la réduction des signes et des symptômes et du taux de progression des lésions articulaires lors de la polyarthrite rhumatoïde. Les patients ont été randomisés dans le groupe de traitement 40 mg d'adalimumab toutes les deux semaines associé au méthotrexate ou dans le groupe traité par méthotrexate toutes les deux semaines en monothérapie. La durée du traitement a été de 104 semaines.
Les résultats de ces cinq études sont exprimés sous forme du pourcentage de patients présentant une amélioration de leur polyarthrite rhumatoïde selon les critères de réponse de l'ACR. Le critère d'évaluation principal des études 1, 2 et 3 et le critère secondaire de l'étude 4 étaient le pourcentage de patients ayant atteint un taux de réponse ACR20 à la semaine 24 ou 26. Le critère d'évaluation principal de l'étude 5 portant sur la polyarthrite rhumatoïde précoce était le pourcentage de patients atteignant une réponse ACR50 à la semaine 52. Les études 3 et 5 ont défini comme critère d'évaluation principal supplémentaire le retard de progression de la maladie (constaté par radiographie) à la semaine 52. Dans l'étude 3, les changements de la qualité de vie ont été également étudiés comme critère d'évaluation principal.
Taux de réponse ACR
Le pourcentage de patients traités par l'adalimumab ayant atteint des taux de réponse ACR20, ACR50 et ACR70 a été similaire dans les études 1, 2, 3 et 4. Les résultats pour la dose de 40 mg d'adalimumab administrée toutes les deux semaines sont résumés dans le tableau 2.
Tableau 2: Taux de réponse ACR au cours des études contrôlées par placebo (en pourcentage des patients)
|
Taux de Réponse
|
Étude 1a*
|
Étude 2a*
|
Étude 3a*
|
Étude 4
| |
|
Placebo/
MTXc
n=60
|
Adalimumabb/
MTXc
n=63
|
Placebo
n=110
|
Adalimumabb
n=113
|
Placebo/
MTXc
n=200
|
Adalimumabb/
MTXc
n=207
|
Traitement
standard/
Placebo
n=318
|
Traitement
standard/
Adalimumab
n=318
| |
ACR 20
| |
6 mois
|
13,3%
|
65,1%
|
19,1%
|
46,0%
|
29,5%
|
63,3%
|
34,9%
|
53,0%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
24,0%
|
58,9%
|
NA
|
NA
| |
ACR 50
| |
6 mois
|
6,7%
|
52,4%
|
8,2%
|
22,1%
|
9,5%
|
39,1%
|
11,1%
|
29,2%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
9,5%
|
41,5%
|
NA
|
NA
| |
ACR 70
| |
6 mois
|
3,3%
|
23,8%
|
1,8%
|
12,4%
|
2,5%
|
20,8%
|
3,2%
|
14,9%
| |
12 mois
|
NA
|
NA
|
NA
|
NA
|
4,5%
|
23,2%
|
NA
|
NA
|
a Etude 1 au bout de 24 semaines, étude 2 au bout de 26 semaines et étude 3 au bout de 24 et 52 semaines.
b 40 mg d'adalimumab administrés toutes les deux semaines
c MTX=méthotrexate
* p<0,01 adalimumab contre placebo
NA=sans objet
Les patients ayant reçu, au cours de l'étude 2, 40 mg d'adalimumab par semaine, ont atteint au bout de 6 mois des taux de réponse ACR20, ACR50 et ACR70 statistiquement significatifs à hauteur de 53,4%, 35,0% et 18,4%.
Au cours des études 1-4, une amélioration de toutes les composantes individuelles des critères de réponse de l'ACR (nombre d'articulations sensibles à la pression et enflées, évaluation de l'activité de la maladie et des douleurs par le médecin et le patient, évaluation au moyen de l'index de handicap du HAQ et concentrations de CRP [mg/dl]) a été observée par comparaison avec le placebo au bout de 24 et 26 semaines. Au cours de l'étude 3, ces améliorations se sont aussi maintenues pendant 52 semaines. De plus, les taux de réponse ACR se sont maintenus jusqu'à la semaine 104 chez la majorité des patients ayant participé à la phase d'extension en ouvert. Les résultats à deux ans de l'étude montrent que chez 24% des patients traités par l'adalimumab, un effet clinique, défini comme un taux de réponse ACR70 maintenu pendant 6 mois, a pu être obtenu. Un effet clinique durable jusqu'à 5 ans a pu être démontré pendant les phases non contrôlées de l'étude III. Le taux de réponse ACR observé à la semaine 52 a pu être maintenu lorsque l'adalimumab a été administré sans interruption pendant 5 ans, avec un taux de réponse ACR20 de 75,5% dans le sous-groupe de 220 patients évalués après 5 ans. Le taux de réponse ACR70 après 5 ans était de 34,7%. Chez 25,7% des patients, la dose de méthotrexate administrée concomitamment a pu être réduite sans diminution de l'effet clinique; le même phénomène a été observé pour les corticostéroïdes chez 29,9% de ces mêmes patients.
La figure 1 ci-dessous illustre la persistance du taux de réponse ACR20 obtenu avec l'adalimumab au cours de l'étude 3. Dans le cadre de cette étude, 84,7% des patients ayant atteint un taux de réponse ACR20 à la semaine 24 l'ont maintenu jusqu'à la semaine 52.
Figure 1: Taux de réponse ACR 20 sur 52 semaines pendant l'étude 3
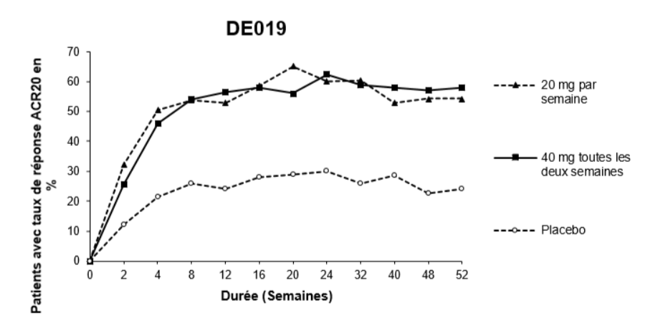
Au cours de l'étude 4, les taux de réponse ACR20 des patients traités par l'adalimumab plus le traitement standard ont été meilleurs de manière statistiquement significative que ceux des patients prenant un placebo plus le traitement standard (p<0,001).
Dans les quatre études, les patients traités par l'adalimumab ont atteint les taux de réponse ACR20, ACR50 et ACR70 plus rapidement et plus souvent que les patients recevant le placebo. Au cours de l'étude 1, une différence statistiquement significative a été observée pour les taux de réponse ACR20 à la semaine 1 (premier examen dans le cadre de l'étude) entre les patients traités par l'adalimumab (26,0%) et les patients recevant un placebo (5,0%). Des différences statistiquement significatives des taux de réponse ACR20 ont aussi été observées au cours des études 2, 3 et 4 à la semaine 2 (premier examen dans le cadre de l'étude) entre les patients traités par l'adalimumab (36,4%, 29,1% et 33,7%) et les patients recevant le placebo (7,3%, 13,0% et 8,6%). On a obtenu un tableau similaire au cours des quatre études pour le temps écoulé jusqu'à ce que les premiers taux de réponse ACR50 et ACR70 soient atteints.
Pour certains patients ne prenant pas concomitamment du méthotrexate, une augmentation de la fréquence d'administration de l'adalimumab à 40 mg par semaine pourrait apporter un bénéfice supplémentaire. Cette observation a été confirmée lors d'une étude en ouvert au long cours pendant laquelle la fréquence d'administration a été augmentée pour les patients ne répondant que partiellement au traitement, de 40 mg toutes les deux semaines à 40 mg par semaine.
Dans l'étude 5, la thérapie associant l'adalimumab et le méthotrexate chez des patients atteints de polyarthrite rhumatoïde précoce et n'ayant pas été traités par méthotrexate a entraîné un taux de réponse rapide et significativement plus élevé à la semaine 52 qu'avec le méthotrexate en monothérapie, avec un taux de réponse maintenu jusqu'à la semaine 104 (voir tableau 3).
Tableau 3: Taux de réponse dans l'étude 5 (en pourcentage du nombre de patients)
|
Taux de réponse*
|
MTX
n=257
|
Adalimumab/MTX
n=268
| |
ACR20
| |
Semaine 52
|
62,6%
|
72,8%
| |
Semaine 104
|
56,0%
|
69,4%
| |
ACR50
| |
Semaine 52
|
45,9%
|
61,6%
| |
Semaine 104
|
42,8%
|
59,0%
| |
ACR70
| |
Semaine 52
|
27,2%
|
45,5%
| |
Semaine 104
|
28,4%
|
46,6%
|
* p<0,05, adalimumab/méthotrexate comparé au méthotrexate pour ACR20
* p<0,001, adalimumab/méthotrexate comparé au méthotrexate pour ACR50 et 70
Tous les critères de réponse ACR ont montré une amélioration à la semaine 52 sous traitement par l'adalimumab/méthotrexate, qui s'est maintenue jusqu'à la semaine 104. Au cours de l'étude sur deux ans, 48,5% des patients ayant été traités par l'association adalimumab/méthotrexate ont atteint une réponse clinique majeure (ACR70 pendant six mois). En comparaison, 27,2% des patients traités par méthotrexate en monothérapie (p<0,001) ont atteint ces résultats.
Tableau 4: Taux de réponse DAS28 dans l'étude 5 sur la polyarthrite rhumatoïde précoce
|
Taux de réponse DAS28
|
MTX
n=257
|
Adalimumab/MTX
n=268
| |
Différence moyenne par rapport au début de l'étude
| |
Valeur initiale (valeur moyenne)
|
6,3
|
6,3
| |
Semaine 52 (valeur moyenne ± écart type)
|
-2,8 ± 1,4
|
-3,6 ± 1,3*
| |
Semaine 104 (valeur moyenne ± écart type)
|
-3,1 ± 1,4
|
-3,8 ± 1,3*
| |
Rémission (DAS28<2,6)
| |
Semaine 52 (pourcentage du nombre de patients)
|
20,6%
|
42,9%*
|
* p<0,001, adalimumab/méthotrexate comparé au méthotrexate
Réponse radiologique
Au cours de l'étude 3, dans laquelle les patients traités par l'adalimumab étaient en moyenne atteints de polyarthrite rhumatoïde depuis environ 11 ans, les lésions articulaires structurelles ont été évaluées par radiographie et exprimées en fonction du changement du score global de Sharp (SST) modifié et de ses composantes, le score d'érosion et le joint space narrowing score = JSN (score évaluant l'amincissement de l'espace articulaire). Une différence statistiquement significative a été observée au bout de 6 mois et s'est maintenue jusqu'à 12 mois pour le changement du score global de Sharp modifié ainsi que pour le score d'érosion. Au bout de 52 semaines, les patients traités par adalimumab/méthotrexate présentaient moins de modifications radiologiques que les patients traités par le méthotrexate seul. Cet effet de ralentissement de la progression des lésions structurelles a pu être maintenu pendant 5 ans.
Parmi les patients initialement traités par 40 mg d'adalimumab toutes les deux semaines, 55% ont subi un examen radiologique après 5 ans. Le ralentissement de la progression des lésions structurelles a pu être maintenu, et, chez 50% de ces patients, la progression des lésions structurelles a pu être complètement arrêtée, ce qui s'est manifesté par une modification du score TSS de zéro ou moins. Les patients ayant été traités par le méthotrexate pendant la phase en double aveugle de l'étude ont présenté une progression minimale des lésions structurelles lorsqu'ils ont été traités par l'adalimumab pendant la phase en ouvert de l'étude.
Tableau 5: Modification radiologique à 12 mois dans l'étude 3 avec traitement de fond par le méthotrexate
|
|
Placebo
n=200
|
Adalimumaba
n=207
|
Différence entre l'adalimumaba et le placebo
|
Valeur p
| |
Changement du score global de Sharp modifié (moyenne)
|
2,7
|
0,1
|
-2,6
|
=0,001b
| |
Modification des érosions (moyenne)
|
1,6
|
0,0
|
-1,6
|
=0,001
| |
Pas de nouvelles érosions (% de patients)
|
46,2
|
62,9
|
16,7
|
=0,001
| |
Modification du score JSN (moyenne)
|
1,0
|
0,1
|
-0,9
|
=0,002
|
a 40 mg, administrés toutes les deux semaines
b sur la base des valeurs moyennes, mesurées par le SST
Dans l'étude 5 portant sur la polyarthrite rhumatoïde précoce, les patients traités par l'adalimumab avaient une durée de la maladie moyenne inférieure à 9 mois et n'avaient auparavant pas été traités par le méthotrexate. Les lésions articulaires structurelles ont été évaluées par radiographie et exprimées en fonction du changement du score global de Sharp modifié. Les résultats après 52 semaines sont décrits dans le tableau 6. Un changement statistiquement significatif du score global de Sharp modifié et du score d'érosion a été constaté à la semaine 52 et a été maintenu jusqu'à la semaine 104.
Tableau 6: Différences radiologiques moyennes à la semaine 52 dans l'étude 5
|
|
MTX n=257
IC 95%
|
Adalimumab/MTX
n=268
IC 95%
|
Valeur p*
| |
Score global de Sharp
|
5,7 (4,2-7,3)
|
1,3 (0,5-2,1)
|
<0,001
| |
Score d'érosion
|
3,7 (2,7-4,7)
|
0,8 (0,4-1,2)
|
<0,001
| |
JSN Score
|
2,0 (1,2-2,8)
|
0,5 (0-0,1)
|
<0,001
|
* Comparaison entre adalimumab/méthotrexate et méthotrexate au moyen du test U de Mann-Whitney
Le pourcentage de patients sans progression radiologique de la maladie (accroissement par rapport aux valeurs initiales du score global de Sharp modifié ≤0,5) était significativement plus élevé sous adalimumab/méthotrexate en association comparé au méthotrexate en monothérapie à la semaine 52 (respectivement 63,8% et 37,4%, p<0,001) et à la semaine 104 (respectivement 61,2% et 33,5%, p<0,001).
Dans le cadre de la phase d'extension en ouvert de l'étude 3, 77% des patients à l'origine traités par l'adalimumab ont été évalués par radiographie au bout de deux ans. L'inhibition de la progression des lésions structurelles a été maintenue. 54% des patients étudiés ne présentaient pas de progression des lésions structurelles, ce qui s'est reflété par une modification du SST de 0 ou inférieure.
Qualité de vie et capacités fonctionnelles physiques
Pour l'évaluation de la qualité de vie en rapport avec l'état de santé, critère prescrit de l'étude 3 après 52 semaines, l'index de handicap contenu dans le Health Assessment Questionnaire (HAQ = questionnaire d'évaluation de l'état de santé) a été utilisé au cours des quatre études adéquates et bien contrôlées. Au cours des quatre études, des améliorations statistiquement significatives et plus fortes de l'index de handicap du HAQ ont été observées pour l'ensemble des doses/traitements par l'adalimumab par comparaison avec le placebo entre les valeurs initiales et les valeurs au bout de 6 mois. Au cours de l'étude 3, l'amélioration moyenne (IC) du HAQ entre avant le traitement et à la semaine 52 a été de -0,60 (-0,65, -0,55) pour les patients traités par adalimumab/méthotrexate et de -0,25 (-0,33, -0,17) chez les patients traités par placebo/méthotrexate (p<0,001). Chez 82% des patients traités par adalimumab/méthotrexate, et pour lesquels une amélioration de 0,5 ou plus au questionnaire HAQ a été obtenue à la semaine 52, cette amélioration a été maintenue jusqu'au mois 60 de la phase d'extension en ouvert.
Dans l'étude 5, l'étude contrôlée portant sur la polyarthrite rhumatoïde précoce par comparaison avec le méthotrexate, l'amélioration de l'index de handicap du HAQ et de la composante physique du SF 36 dans la semaine 52 était plus importante (p<0,001) sous l'association adalimumab/méthotrexate que sous le méthotrexate en monothérapie et a été maintenue jusqu'à la semaine 104.
Par ailleurs, la qualité de vie globale en rapport avec l'état de santé a été évaluée pour les quatre études adéquates et bien contrôlées à l'aide du Short Form Health Survey (SF 36 = analyse courte de l'état de santé). Au cours de ces quatre études, des améliorations statistiquement significatives plus fortes du score résumé pour les composantes physiques du questionnaire SF 36 ont été observées pour l'ensemble des doses/fréquences d'injection de l'adalimumab par comparaison avec le placebo entre les valeurs initiales et le mois 6, et ont été maintenues jusqu'à la semaine 52 au cours de l'étude 3. Les scores résumés des composantes mentales du questionnaire SF 36 au cours des études 2 et 4 ont été statistiquement significativement plus élevés au mois 6 pour l'adalimumab par comparaison avec le placebo. On a observé une amélioration statistiquement significative plus forte des scores de douleur et de vitalité du questionnaire SF 36 au cours des quatre études pour la dose de 40 mg d'adalimumab toutes les deux semaines par comparaison avec le placebo, entre les valeurs initiales et le mois 6. Ces résultats ont été renforcés par les scores atteints dans le cadre du Functional Assessment of Chronic Illness Therapy (FACIT = évaluation fonctionnelle du traitement de maladies chroniques), selon lesquels, pour les trois études analysées, on a eu une diminution statistiquement significative de la fatigue au mois 6, qui a été maintenue jusqu'à la semaine 52 au cours de l'étude 3. Le score SF 36 a été calculé jusqu'à la semaine 156 (3 ans) et l'amélioration a été maintenue pendant cette période pour les patients restés dans l'étude.
Arthrite juvénile idiopathique polyarticulaire (AJIp)
La sécurité et l'efficacité de l'adalimumab ont été évaluées dans une étude multicentrique randomisée, en double aveugle, par groupes parallèles, auprès de 171 enfants (de 4 à 17 ans) atteints d'AJI polyarticulaire. L'analyse a été effectuée après avoir stratifié les patients en deux groupes: ceux qui recevaient du méthotrexate (MTX) et ceux qui n'en recevaient pas. Les patients ont reçu des doses stables d'AINS et/ou de prednisone (≤0,2 mg/kg/jour ou au maximum 10 mg par jour). Dans la phase initiale en ouvert («open-label lead-in», OL LI), d'une durée de 16 semaines, tous les patients ont reçu toutes les deux semaines une dose d'adalimumab de 24 mg/m2 (sans dépasser une dose maximale de 40 mg). La répartition des patients est présentée dans le tableau 7.
Tableau 7: Répartition des patients par âge et par dose d'adalimumab administrée pendant la phase OL LI
|
Groupe d'âge
|
Nombre de patients au début de l'étude
n (%)
|
Dose minimale, moyenne et maximale
| |
4 à 7 ans
|
31 (18,1)
|
10, 20 et 25 mg
| |
8 à 12 ans
|
71 (41,5)
|
20, 25 et 40 mg
| |
13 à 17 ans
|
69 (40,4)
|
25, 40 et 40 mg
|
Les patients ayant atteint une réponse ACR30 pédiatrique à 16 semaines ont été randomisés pour la phase d'étude en double aveugle (double blind, DB), dans laquelle ils ont reçu toutes les 2 semaines l'adalimumab (24 mg/m2 jusqu'à une dose unique maximale de 40 mg) ou un placebo. Ce traitement a été poursuivi pour une durée maximale de 32 semaines ou jusqu'à la survenue d'une nouvelle poussée de la maladie. Les critères définissant une nouvelle poussée de la maladie ont englobé une aggravation de ≥30% par rapport au début de l'étude pour ≥3 des 6 critères ACR pédiatriques, une activité de la maladie dans ≥2 articulations et pas plus d'un critère sur 6 présentant une amélioration de >30%. Au bout de 32 semaines ou en présence d'une nouvelle poussée de la maladie, les patients étaient éligibles pour une participation à la phase d'extension en ouvert («open-label extension», OLE).
Au bout de 16 semaines (à l'issue de la phase OL LI), 94,1% (80 patients sur 85) du groupe sous adalimumab et MTX et 74,4% (64 patients sur 86) du groupe sous adalimumab en monothérapie avaient atteint une réponse ACR30. Les résultats de la période en double aveugle sont présentés dans le tableau 8.
Tableau 8: Réponse ACR30 pédiatrique dans l'étude sur l'AJI
Résultats d'efficacité
|
En double aveugle,
32 semaines
|
Adalimumab/MTX
(n=38)
|
Placebo/MTX
(n=37)
|
Adalimumab
(n=30)
|
Placebo
(n=28)
| |
Nouvelle poussée de la maladie après 32 semainesa (n/N)
|
36,8% (14/38)
|
64,9% (24/37)b
|
43,3% (13/30)
|
71,4% (20/28)c
| |
Temps moyen écoulé jusqu'à une nouvelle poussée de la maladie
|
>32 semaines
|
20 semaines
|
>32 semaines
|
14 semaines
|
a La réponse ACR30/50/70 pédiatrique à 48 semaines était significativement supérieure à celle des patients ayant reçu le placebo
b p=0,015
c p=0,031
Parmi les patients ayant atteint une réponse à 16 semaines (n=144), la réponse ACR30/50/70/90 pédiatrique s'est maintenue jusqu'à six ans dans la phase OLE chez ceux qui ont reçu de l'adalimumab pendant toute la durée de l'étude. 19 patients au total (11 qui avaient 4 à 12 ans au début de l'étude et 8 qui avaient 13 à 17 ans au début de l'étude) ont été traités pendant 6 ans ou plus.
Il est apparu que la réponse globale au traitement associant l'adalimumab et le MTX était supérieure et que moins de patients ayant reçu ce traitement ont développé des anticorps en comparaison avec le groupe sous adalimumab seul. Compte tenu de ces résultats, il est recommandé d'utiliser l'adalimumab en association avec le MTX. L'utilisation de l'adalimumab en monothérapie n'est recommandée que chez les patients pour lesquels le MTX est inapproprié (cf. «Posologie/Mode d'emploi»).
Arthrite psoriasique
L'efficacité de l'adalimumab a été étudiée chez 413 patients. Dans l'étude principale, 313 patients adultes atteints d'arthrite psoriasique modérée à sévère, n'ayant pas répondu suffisamment au traitement par anti-inflammatoires non stéroïdiens ont été traités. 158 (50,5%) des patients traités prenaient du méthotrexate au moment de la randomisation. L'adalimumab a été administré à la dose de 40 mg toutes les 2 semaines sur une période de 24 semaines. Après la fin des études, 383 patients ont été inclus dans une phase d'extension en ouvert pendant laquelle l'adalimumab a été administré toutes les deux semaines. 382 des patients inclus ont été traités par l'adalimumab au moins au début de cette étude d'extension. Concernant les patients évaluables à 48 et 144 semaines, voir plus bas.
Taux de réponse ACR et PASI
Le tableau 9 montre que l'adalimumab était supérieur au placebo dans tous les paramètres de progression de la maladie (p<0,001). Chez les patients atteints d'arthrite psoriasique traités par l'adalimumab, une efficacité clinique a été observée dès le premier contrôle (2 semaines); cette efficacité était significative après 12 semaines et s'est maintenue pendant les 24 semaines de traitement.
Les patients chez lesquels au moins 3% de la surface corporelle étaient atteints de psoriasis ont été évalués d'après le Psoriatic Area and Severity Index (PASI). Les lésions cutanées dues au psoriasis se sont améliorées chez ces patients par rapport au placebo, selon le PASI.
Le taux de réponse était comparable lors du traitement avec ou sans méthotrexate.
Les taux de réponse ACR se sont maintenus pendant la phase d'extension en ouvert jusqu'à 136 semaines.
Tableau 9: Taux de réponse ACR et PASI dans une étude contrôlée contre placebo chez des patients atteints d'arthrite psoriasique (en pourcentage du nombre de patients)
|
Taux de réponse*
|
Placebo
|
Adalimumab
| |
|
N=162
|
N=151
| |
ACR 20
| |
Semaine 12
|
14%
|
58%
| |
Semaine 24
|
15%
|
57%
| |
ACR 50
| |
Semaine 12
|
4%
|
36%
| |
Semaine 24
|
6%
|
39%
| |
ACR 70
| |
Semaine 12
|
1%
|
20%
| |
Semaine 24
|
1%
|
23%
| |
|
N=69
|
N=69
| |
PASI 50
| |
Semaine 12
|
15%
|
72%
| |
Semaine 24
|
12%
|
75%
| |
PASI 75
| |
Semaine 12
|
4%
|
49%
| |
Semaine 24
|
1%
|
59%
|
* p<0,001 pour toutes les comparaisons entre l'adalimumab et le placebo
Au cours des études portant sur le rhumatisme psoriasique, les modifications radiologiques ont été évaluées. Des radiographies des mains, des poignets et des pieds ont été réalisées au début de l'étude (Baseline) et à la semaine 24 de la période en double aveugle pendant laquelle les patients ont reçu soit l'adalimumab, soit un placebo, ainsi qu'à la semaine 48, pendant laquelle tous les patients recevaient l'adalimumab. Un score de Sharp global modifié (mTSS) incluant les articulations interphalangiennes distales (différent du score total de Sharp pour la polyarthrite rhumatoïde) a été utilisé.
Comparativement au placebo, l'adalimumab a réduit la vitesse de progression des lésions articulaires périphériques. Une modification du score de Sharp global modifié de 0,8 ± 2,5 (moyenne ± écart type) par rapport à la valeur initiale a été observée dans le groupe placebo (à la semaine 24), contre 0,0 ± 1,9 dans le groupe adalimumab (à la semaine 48, n=133); p<0,001).
84% des patients qui ont été traités par l'adalimumab et qui n'ont présenté aucune progression radiologique entre l'inclusion et la semaine 48 (n=102) n'ont pas non plus présenté de progression radiologique jusqu'à la semaine 144.
L'évaluation de l'index de handicap du HAQ et du questionnaire Short Form Health Survey (SF 36) a montré, dans le groupe de patients traités par l'adalimumab, une amélioration statistiquement significative des capacités fonctionnelles physiques à la semaine 24 comparativement aux patients traités par le placebo.
L'amélioration des capacités fonctionnelles physiques s'est maintenue au cours de la phase d'extension en ouvert de l'étude, jusqu'à la semaine 136.
Maladie de Crohn
La sécurité et l'efficacité d'une dose multiple d'adalimumab ont été étudiées chez plus de 1500 patients atteints d'une forme active modérée à sévère de la maladie de Crohn (indice d'activité de la maladie de Crohn [CDAI = Crohn's Disease Activity Index] ≥220 et ≤450) dans le cadre d'études randomisées, contrôlées contre placebo et en double aveugle. L'administration concomitante d'aminosalicylates, de corticostéroïdes ou d'immunomodulateurs à doses constantes était permise, et l'un de ces médicaments au moins a été administré à 80% des patients.
L'induction d'une rémission clinique (définie par un CDAI <150) a été évaluée dans le cadre de deux études (CLASSIC I et GAIN). 299 patients n'ayant pas reçu de traitement par antagonistes du TNF au préalable, répartis par randomisation dans quatre groupes de traitement, ont participé à l'étude CLASSIC I; un placebo a été administré au groupe placebo à la semaine 0 et 2, le groupe 160/80 a reçu 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, le groupe 80/40 a été traité par 80 mg à la semaine 0 et 40 mg à la semaine 2, et le groupe 40/20 a été traité par 40 mg à la semaine 0 et 20 mg à la semaine 2. Dans l'étude GAIN, 325 patients qui ne répondaient plus à l'infliximab ou ne le toléraient pas, ont été randomisés pour recevoir soit 160 mg d'adalimumab à la semaine 0 et 80 mg à la semaine 2, soit un placebo à la semaine 0 et 2.
Le maintien de la rémission clinique a été évalué dans le cadre de l'étude CHARM. Celle-ci portait sur 854 patients qui ont reçu en ouvert 80 mg d'adalimumab à la semaine 0 et 40 mg d'adalimumab à la semaine 2. À la semaine 4, les patients ont été randomisés et ont reçu soit 40 mg d'adalimumab toutes les deux semaines, soit 40 mg d'adalimumab toutes les semaines, soit un placebo. L'étude a duré au total 56 semaines. Les patients présentant une réponse clinique (diminution du CDAI ≥70) à la semaine 4 ont été stratifiés et analysés (séparément de ceux qui ne présentaient aucune réponse clinique à la semaine 4). Une réduction progressive des corticostéroïdes était permise après la semaine 8.
Résultats cliniques
En comparaison avec le placebo, un pourcentage statistiquement significativement plus élevé de patients traités par 160/80 mg d'adalimumab dans le cadre des études CLASSIC I et GAIN a affiché une rémission clinique à la semaine 4, indépendamment du fait que les patients aient suivi au préalable un traitement par des antagonistes du TNF ou aient déjà été exposés à un traitement par l'infliximab (voir tableau 10).
Tableau 10: Induction d'une rémission et d'une réponse cliniques (en pourcentage de patients)
|
|
CLASSIC I: Patients n'ayant pas été traités par l'infliximab
|
GAIN: Patients ayant été traités par l'infliximab
| |
Placebo
N=74
|
Adalimumab
160/80 mg
N=76
|
Placebo
N=166
|
Adalimumab
160/80 mg
N=159
| |
Semaine 4
| |
Rémission clinique
|
12%
|
36%*
|
7%
|
21%*
| |
Réponse clinique
(CR-100)
|
24%
|
50%**
|
25%
|
38%**
| |
Réponse clinique
(CR-70)
|
34%
|
58%**
|
34%
|
52%**
|
Toutes les valeurs p sont des comparaisons appariées des pourcentages pour l'adalimumab vs placebo.
* p<0,001
** p<0,01
58% (499/854) des patients participant à l'étude CHARM ont montré une réponse clinique à la semaine 4 et ont été évalués dans le cadre de l'analyse primaire. 48% des patients qui présentaient une réponse clinique à la semaine 4 avaient déjà reçu au préalable une autre thérapie anti-TNF. Aux semaines 26 et 56, un pourcentage statistiquement significativement plus élevé des groupes recevant une thérapie d'entretien par l'adalimumab et ayant affiché une réponse clinique dans la semaine 4 a atteint une rémission clinique, par comparaison avec le groupe bénéficiant d'une thérapie d'entretien par placebo. Par ailleurs, toujours par comparaison avec le groupe ayant reçu une thérapie d'entretien par placebo, on a observé que dans les groupes ayant été traités par une thérapie d'entretien par l'adalimumab et ayant bénéficié au début du traitement simultanément de corticostéroïdes, le pourcentage de patients avec une rémission clinique et en mesure d'interrompre l'administration de corticostéroïdes pendant au moins 90 jours aux semaines 26 et 56 était statistiquement significativement plus élevé (voir tableau 11).
Une analyse post hoc a montré une diminution statistiquement significative des hospitalisations et opérations intra-abdominales liées à la maladie sous adalimumab par rapport au placebo pendant la phase en double aveugle.
Tableau 11: Maintien de la rémission et de la réponse cliniques (en pourcentage de patients)
|
|
Placebo
|
40 mg d'adalimumab
toutes les deux semaines
|
40 mg d'adalimumab
toutes les semaines
| |
Semaine 26
|
N=170
|
N=172
|
N=157
| |
Rémission clinique
|
17%
|
40%*
|
47%*
| |
Réponse clinique (CR-100)
|
27%
|
52%*
|
52%*
| |
Réponse clinique (CR-70)
|
28%
|
54%*
|
56%*
| |
Patients présentant une rémission sans stéroïdes ≥90 joursa
|
3% (2/66)
|
19% (11/58)**
|
15% (11/74)**
| |
Semaine 56
|
N=170
|
N=172
|
N=157
| |
Rémission clinique
|
12%
|
36%*
|
41%*
| |
Réponse clinique (CR-100)
|
17%
|
41%*
|
48%*
| |
Réponse clinique (CR-70)
|
18%
|
43%*
|
49%*
| |
Patients présentant une rémission sans stéroïdes ≥90 joursa
|
5% (3/66)
|
29% (17/58)*
|
20% (15/74)**
|
* p<0,001 pour adalimumab vs placebo (comparaisons appariées des pourcentages)
** p<0,002 pour adalimumab vs placebo (comparaisons appariées des pourcentages)
a Parmi ceux ayant reçu initialement des corticostéroïdes
Les résultats de rémission clinique présentés dans le tableau 11 sont restés relativement constants, en dépit d'une exposition préalable aux antagonistes du TNF.
Parmi les patients qui ont affiché une réponse à la semaine 4 et atteint une rémission dans le courant de l'étude, les patients issus des groupes de thérapie d'entretien par l'adalimumab ont pu maintenir cette rémission significativement plus longtemps que les patients issus du groupe de thérapie d'entretien par placebo (voir figure 2).
Figure 2: Jours de rémission clinique chez les patients qui ont atteint une rémission clinique durant l'étude CHARM (Intent-to-Treat-Population)
Parmi les patients qui n'affichaient aucune réponse à la semaine 4, 43% des patients des groupes de thérapie d'entretien par l'adalimumab jusqu'à la semaine 12, contre un taux de 30% chez les patients du groupe de thérapie d'entretien par placebo. Ces résultats permettent de conclure que la poursuite d'une thérapie d'entretien jusqu'à la semaine 12 peut présenter un avantage pour certains patients qui n'avaient affiché aucune réponse à la semaine 4. La poursuite de la thérapie au-delà des 12 semaines n'a conduit à aucune autre augmentation significative des réponses (voir «Posologie/Mode d'emploi»).
Dans l'étude CLASSIC I, 117/276 patients et dans les études GAIN et CHARM, 272/777 patients ont poursuivi le traitement par l'adalimumab pendant au moins 3 ans dans une étude ouverte de suivi. Respectivement 88 et 189 patients étaient toujours en rémission clinique après 3 ans. Le taux de réponse clinique (CR-100) s'est maintenu chez respectivement 102 et 233 patients.
Dans l'étude M05-769 (EXTEND) endoscopique, randomisée et contrôlée contre placebo, 135 patients ont été examinés avec comme critère principal la guérison de la muqueuse (définie comme la disparition des ulcérations de la muqueuse). Après une phase d'induction de l'adalimumab de 4 semaines, les patients ont été randomisés. À la semaine 12, 27,4% des patients traités par l'adalimumab présentaient une guérison de la muqueuse contre 13,1% de ceux sous placebo (p=0,056); à la semaine 52, 24,2% des patients sous adalimumab présentaient une guérison de la muqueuse contre 0% de ceux sous placebo (p<0,001).
Résultats de la perspective des patients/Patient-Reported Outcomes
Le résultat total issu du «questionnaire portant sur les maladies inflammatoires de l'intestin» (inflammatory bowel disease questionnaire = IBDQ), questionnaire spécialement conçu pour la maladie, à la semaine 4, a indiqué une amélioration statistiquement significative, en comparaison au placebo, chez les patients qui avaient été randomisés pour bénéficier d'un traitement par l'adalimumab 160/80 mg dans le cadre des études CLASSIC I et GAIN. En comparaison avec le groupe placebo, les groupes traités par l'adalimumab dans le cadre de l'étude CHARM ont affiché une amélioration statistiquement significative du score total IBDQ par rapport aux valeurs initiales observées dans les semaines 26 et 56.
Qualité de vie et capacités fonctionnelles physiques
La qualité de vie en rapport avec l'état de santé et les capacités fonctionnelles physiques ont été évaluées dans l'étude sur l'arthrite psoriasique à l'aide du Health Assessment Questionnaire (HAQ). Les patients traités par l'adalimumab ont montré, par rapport aux patients traités par placebo, des améliorations statistiquement significatives et plus fortes de l'index de handicap du HAQ entre les valeurs initiales et la semaine 24.
Les résultats du Short Form Health Survey (SF 36) renforcent ces résultats avec un Physical Component Summary (PCS) Score ainsi que des Pain and Vitality Domain Scores statistiquement significatifs.
Colite ulcéreuse
La sécurité et l'efficacité ont été examinées chez des patients adultes atteints de colite ulcéreuse active modérée à sévère (score Mayo de 6 à 12, avec sous-score endoscopique de 2 à 3) dans deux études randomisées, en double aveugle, avec contrôle contre placebo. Les patients pouvaient prendre en même temps une médication permanente sous forme d'aminosalicylates, de glucocorticoïdes et/ou d'immunomodulateurs.
L'induction d'une rémission clinique (définie comme un score Mayo ≤2 sans sous-score >1) a été examinée chez 390 patients naïfs d'inhibiteurs du TNF. Ces patients ont été traités aux semaines 0 et 2 par un placebo, par 160 mg et 80 mg d'adalimumab ou par 80 mg et 40 mg d'adalimumab, puis aux semaines 4 et 6 par un placebo ou par 40 mg d'adalimumab. Tous les patients sont ensuite passés à un traitement d'entretien par 40 mg d'adalimumab toutes les 2 semaines.
À la semaine 8, on a pu constater une rémission clinique chez 18% des patients traités avec une dose d'induction de 160 mg/80 mg d'adalimumab, vs 9% des patients ayant reçu le placebo (p=0,031). Aucune supériorité statistiquement significative de l'adalimumab n'a été observée avec la dose d'induction de 80 mg/40 mg (10%, p=0,833).
L'efficacité au cours de la phase d'induction et de la phase d'entretien (52 semaines au total) a été évaluée chez 248 patients traités par 160 mg/80 mg/40 mg toutes les 2 semaines versus 246 patients sous placebo. Une rémission à 8 et à 52 semaines a été constatée chez 16,5% (p=0,019) et 17,3% (p=0,004) des patients sous adalimumab vs 9,3% et 8,5% des patients sous placebo. Les taux de réponse persistante, de rémission et de guérison de la muqueuse sont résumés dans le tableau 12.
Tableau 12: Taux de réponse persistante, de rémission et de guérison de la muqueuse dans l'étude UC II, pourcentage de patients (intervalle de confiance à 95%)
|
|
Placebo et intervalle de confiance à 95%
|
40 mg d'adalimumab toutes les deux semaines et intervalle de confiance à 95%
| |
Semaines 8 et 52
| |
Réponse persistante
|
12% (IC: 8,1 à 16,3)
|
24%** (IC: 18,5 à 29,1)
| |
Rémission persistante
|
4% (IC: 1,6 à 6,5)
|
8%* (IC: 5,0 à 11,9)
| |
Guérison durable de la muqueuse
|
11% (IC: 6,7 à 14,4)
|
19%* (IC: 13,7 à 23,4)
|
a Intervalle de confiance pour la proportion, sur la base d'une approximation de la distribution binomiale par loi normale
La rémission clinique signifie un score Mayo ≤2 sans aucun sous-score >1
* p <0,05 pour adalimumab versus placebo
** p <0,001 pour adalimumab versus placebo
La guérison de la muqueuse signifie un sous-score endoscopique de 0 ou de 1
Une réponse signifie un score Mayo réduit de ≥3 points et inférieur de ≥30% au score initial ainsi qu'un sous-score d'hémorragies rectales de 0 ou de 1 ou réduit de ≥1 point versus valeur initiale.
Sur les 125 patients ayant atteint une réponse à 8 semaines, 59 (47%) étaient encore répondeurs à 52 semaines, 36 (29%) étaient en rémission, 51 (41%) présentaient une guérison de la muqueuse et 18 (20% des 90 patients répondeurs à 8 semaines et initialement sous corticostéroïdes) étaient en rémission sans utilisation de corticostéroïdes depuis ≥90 jours.
Une réduction statistiquement significative des taux d'hospitalisations de toutes causes et des taux d'hospitalisations imputables à la colite ulcéreuse a été observée dans l'analyse incluant les deux études UC.
Le traitement anti-TNF par l'infliximab avait échoué auparavant chez presque 40% des patients de l'étude UC II. L'efficacité de l'adalimumab était plus faible chez ces patients que chez les patients naïfs de traitements par anti-TNF. Dans ce sous-groupe, une rémission à 52 semaines a été atteinte dans 10% des patients sous adalimumab vs 3% sous placebo (p=0,039).
Les patients des études UC I et II ont pu continuer à être traités dans le cadre de l'étude à long terme en ouvert (UC-III). 3 ans après le traitement par l'adalimumab, 75% (301/402) étaient encore en rémission clinique d'après le score Mayo partiel.
Une amélioration de la qualité de vie, évaluée à l'aide du score IBDQ total (Inflammatory Bowel Disease Questionnaire) spécifique de la maladie, était atteinte à 52 semaines vs placebo (p=0,007).
Spondylarthrite ankylosante (maladie de Bechterew)
L'efficacité de 40 mg d'adalimumab toutes les deux semaines en injection sous-cutanée a été étudiée dans deux études randomisées, en double aveugle, contrôlées contre placebo pendant 24 semaines chez 393 patients atteints de spondylarthrite ankylosante active (score d'activité de la maladie [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)]) >4 (valeurs initiales moyennes de 6,3 aussi bien dans le groupe adalimumab que dans le groupe placebo) n'ayant pas répondu suffisamment aux traitements conventionnels. 79 patients (20,1%) ont reçu simultanément un agent antirhumatismal de fond et 37 patients (9,4%) des glucocorticoïdes. La période en aveugle fut suivie d'une phase d'extension en ouvert pendant laquelle les patients ont reçu 40 mg d'adalimumab par injection sous-cutanée toutes les deux semaines pendant une durée maximale de 28 semaines supplémentaires.
Dans l'étude de plus grande ampleur avec 315 patients, les résultats ont indiqué des améliorations statistiquement significatives des signes et des symptômes de la spondylarthrite ankylosante chez les patients ayant reçu de l'adalimumab, par rapport aux patients sous placebo. Une réponse significative a été constatée pour la première fois après 2 semaines de traitement et s'est maintenue jusqu'à la semaine 24.
Les taux de réponse selon l'Assessment in Ankylosing Spondylitis (ASAS) 20/50/70 ont été atteints à la semaine 12 chez 58%, 38% et 23% des patients sous adalimumab contre 21%, 10% et 5% des patients sous placebo (p<0,001 adalimumab versus placebo). Une réponse relativement comparable a été constatée à la semaine 24.
Comme constaté avec le BASDAI, le traitement par l'adalimumab a entraîné une amélioration des signes et symptômes. Chez 45% des patients traités par l'adalimumab, une réduction d'au moins 50% des valeurs initiales du BASDAI a été atteinte à la semaine 12, comparé aux 16% des patients traités par le placebo (p<0,01). Des résultats comparables ont été constatés à la semaine 24.
De plus, la diminution moyenne des taux initiaux de protéine C réactive (CRP) à la semaine 12 était plus importante avec le traitement par l'adalimumab (-1,3 mg/dl) qu'avec le placebo (-0,1 mg/dl), (p<0,001).
Des résultats comparables (non tous statistiquement significatifs) ont été constatés dans une étude plus réduite, randomisée, en double aveugle, contrôlée contre placebo, chez 82 patients adultes atteints de spondylarthrite ankylosante active.
Dans les études portant sur la spondylarthrite ankylosante, les résultats rapportés par les patients ont été évalués à l'aide du Generic Health Status Questionnaire Short Form-36 (SF 36) et du Disease Specific Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL). Des améliorations significatives plus importantes de l'ASQoL et de la composante physique du SF 36 ont été observées à la semaine 12 chez les patients traités par l'adalimumab par rapport aux patients du groupe placebo, et se sont maintenues jusqu'à la semaine 24.
Psoriasis
L'efficacité et la sécurité de l'adalimumab ont été examinées au cours d'études randomisées, en double aveugle, contrôlées, menées chez plus de 1600 patients atteints de psoriasis en plaques chronique modéré à sévère, ayant 18 ans ou plus et candidats à un traitement systémique ou une photothérapie.
Au cours de l'étude 1, 1212 patients atteints de psoriasis en plaques chronique sur ≥10% de leur surface corporelle (Body Surface Area, BSA) et présentant un score PASI (Psoriatic Area and Severity Index) ≥12, ont été évalués sur trois périodes de traitement. Pendant la période A, les patients ont reçu par voie sous-cutanée un placebo ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1. Les patients ayant obtenu au bout de 16 semaines de traitement au minimum une réponse PASI 75 (définie par une amélioration d'au moins 75% du score PASI par rapport à la valeur initiale) ont poursuivi leur traitement en ouvert pendant la période B supplémentaire. Ils ont reçu 40 mg d'adalimumab toutes les deux semaines. Après 17 semaines de traitement en ouvert, les patients ayant conservé au minimum une réponse PASI 75 à la semaine 33 et ayant reçu un traitement actif pendant la période A, sont passés à la période de traitement C. Ils ont reçu durant 19 semaines supplémentaires 40 mg d'adalimumab ou un placebo toutes les 2 semaines. Le score PASI initial moyen était de 18,9 pour tous les groupes de traitement. Le score PGA (Physician's Global Assessment) initial pour tous les groupes allait de «modéré» (52,6%) à «sévère» (41,3%) et «très sévère» (6,1%).
L'étude 2 a comparé l'efficacité et la sécurité de l'adalimumab versus méthotrexate et placebo chez 271 patients atteints de psoriasis en plaques chronique avec un BSA de 10% et un score PASI ≥10. Pendant 16 semaines, les patients ont reçu un placebo, du méthotrexate (7,5-20 mg) ou une dose initiale de 80 mg d'adalimumab par voie sous-cutanée à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1. Le score PASI initial moyen était de 19,7 pour tous les groupes de traitement. Le score PGA initial allait pour tous les groupes de «léger» (0,4%) à «modéré» (47,8%), «sévère» (45,6%) et «très sévère» (6,3%).
1469 patients provenant des études de phase II et III ont été inclus dans une étude d'extension ouverte à 3 périodes, comportant une période de poursuite du traitement (104-252 semaines), une période d'interruption du traitement (jusqu'à une récidive ou au maximum de 52 semaines), puis une période de reprise du traitement (16 semaines).
Dans l'étude 3, 148 patients atteints de psoriasis en plaques chronique avec un BSA ≥5% ont été évalués pendant au moins 1 an. Les patients ont reçu un placebo ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab toutes les deux semaines à partir de la semaine 1, ou une dose initiale de 80 mg d'adalimumab à la semaine 0, suivie de 40 mg d'adalimumab chaque semaine à partir de la semaine 1.
Résultats cliniques
Le critère d'évaluation principal des études 1, 2 et 3 était le pourcentage de patients ayant atteint à la semaine 16 (études 1 et 2) ou à la semaine 12 (étude 3) une réduction de leur score PASI d'au moins 75% par rapport à la valeur initiale (PASI 75). Les études 1–3 menées sur le psoriasis ont également porté entre autres sur le score PGA et d'autres valeurs PASI.
Étude 1: en plus du critère d'évaluation principal ci-dessus, l'étude 1 avait comme second critère d'évaluation principal la perte d'une réponse appropriée à la semaine 33 et pendant ou avant la semaine 52. La perte d'une réponse appropriée était définie comme une réponse <pasi50 par rapport à la valeur initiale avec un minimum d'augmentation de 6 points du score PASI par rapport à la semaine 33.
Des données contrôlées portant sur un traitement par l'adalimumab versus placebo sont disponibles pour une durée de traitement de 52 semaines. Dans une étude comparative contrôlée contre placebo, menée auprès des patients ayant obtenu une disparition prolongée des troubles jusqu'à la semaine 33 sous adalimumab, 95,1% des patients ayant poursuivi le traitement par l'adalimumab sont restés sans récidive jusqu'à la semaine 52, contre 71,6% des patients sous placebo (c.-à-d. après l'arrêt de l'administration de l'adalimumab à la semaine 33).
Parmi les patients présentant une diminution de réponse appropriée après la re-randomisation dans le groupe placebo et ayant ensuite été inclus dans l'étude d'extension en ouvert, 38% (25/66) et 55% (36/66) ont retrouvé une réponse PASI 75 après respectivement 12 et 24 semaines de thérapie active.
Cette réponse tardive après récidive est probablement liée à une évolution sévère du psoriasis dans ce sous-groupe de patients.
Dans les études 1 et 2 sur le psoriasis, plus de patients traités par l'adalimumab que de patients sous placebo ont atteint à la semaine 16 une réduction d'au moins 75% par rapport au score PASI initial. D'autres paramètres cliniques significatifs incluant le score PASI 100 (p.ex. disparition complète des signes cutanés du psoriasis) et le score PGA «clair ou minime» ont été également améliorés par rapport au placebo.
Étude 2: Dans l'étude 2 sur le psoriasis, les patients recevant l'adalimumab ont présenté de meilleurs résultats pour les scores PASI 75, PASI 100 et PGA «clair ou minime» que les patients sous méthotrexate.
Tableau 13: Étude 1 sur le psoriasis - Efficacité à la semaine 16 (% de patients)
|
|
Placebo
N=398
|
40 mg d'adalimumab 1 sem./2
N=814
| |
≥PASI 75
|
6,5
|
70,9a
| |
PASI 100
|
0,8
|
20,0a
| |
PGA: clair/minimal
|
4,3
|
62,2a
|
a p<0,001, adalimumab vs placebo
Tableau 14: Étude 2 sur le psoriasis - Efficacité à la semaine 16 (% de patients)
|
|
Placebo
N=53
|
MTX
N=110
|
40 mg d'adalimumab 1 sem./2
N=108
| |
≥PASI 75
|
18,9
|
35,5
|
79,6a, b
| |
PASI 100
|
1,9
|
7,3
|
16,7a, b
| |
PGA: clair/minime
|
11,3
|
30,0
|
73,1 a, b
|
a p<0,001, adalimumab vs placebo
b p<0,001 adalimumab vs méthotrexate
Étude de prolongation: au total, 233 patients qui avaient obtenu une réponse PASI-75 à la semaine 16 et à la semaine 33 et avaient reçu dans l'étude 1 sur le psoriasis un traitement continu par l'adalimumab pendant 52 semaines ont poursuivi le traitement par l'adalimumab dans l'étude de prolongation ouverte. La réponse PASI-75 ou la réponse PGA définie comme un score PGA «clair» ou «minime» ont été respectivement de 74,7% et 59,0% chez ces patients après 108 semaines supplémentaires de traitement en ouvert (au total 160 semaines). Dans une analyse de Non Responder Imputation (NRI), dans laquelle tous les patients qui avaient interrompu leur participation à l'étude en raison d'effets indésirables ou d'absence d'efficacité ou chez lesquels la dose avait été augmentée, ont été considérés comme non-répondeurs, la réponse PASI 75 et la réponse PGA définie comme un score PGA «clair» ou «minime» ont été respectivement de 69,6% et 55,7% chez ces patients après 108 semaines supplémentaires de poursuite du traitement en ouvert (au total 160 semaines).
Dans l'étude de prolongation, 347 patients ayant présenté une réponse durable ont participé à une évaluation de l'interruption du traitement et de la reprise du traitement. Pendant la période d'interruption du traitement, les symptômes psoriasiques sont réapparus chez 54,2% (188/347) des patients en moyenne en l'espace d'environ 5 mois (diminution du score PGA à «modéré» ou plus grave). Aucun de ces patients n'a connu de phénomène de rebond pendant la période d'interruption. Au total, 76,5% (218/285) des patients ayant participé à la période ultérieure de reprise du traitement ont présenté une réponse PGA définie comme un score PGA «clair» ou «minime» 16 semaines après la reprise du traitement, qu'ils aient eu une récidive pendant l'arrêt (69,1% [123/178] ou non 88,8% [95/107]). Le profil de sécurité observé pendant la période de reprise du traitement a été similaire à celui observé avant l'interruption du traitement.
Étude 3: Les résultats de l'étude 3 sur le psoriasis confirment l'efficacité démontrée dans les études 1 et 2. Les patients de l'étude 1 ayant présenté une réponse PASI 75 et ayant été re-randomisés à la semaine 33 dans le groupe adalimumab, ont présenté avant ou à la semaine 52 une diminution de la réponse appropriée inférieure à celle présentée par les patients re-randomisés dans le groupe placebo (4,9% vs 28,4%, p<0,001).
Qualité de vie
Les résultats rapportés par les patients (Patient reported Outcomes, PRO) ont été évalués à l'aide de divers paramètres. La qualité de vie a été déterminée au cours des études 1 et 2 à l'aide de l'indice spécifique de la maladie DLQI (Dermatology Life Quality Index). Dans l'étude 1, les patients sous adalimumab ont présenté aux semaines 4 et 16 des améliorations du score total DLQI, du degré de la maladie, des douleurs et du prurit en comparaison avec les patients sous placebo. Les résultats DLQI ont été maintenus jusqu'à la semaine 52.
Dans l'étude 2, les patients sous adalimumab ont présenté à la semaine 16 des améliorations du score total DLQI, du degré de la maladie et du prurit en comparaison avec les patients sous placebo ou sous méthotrexate, ainsi que des améliorations cliniquement significatives des douleurs comparativement aux patients sous placebo.
La qualité de vie générale en rapport avec l'état de santé a été définie à l'aide du questionnaire Short Form Health Survey (SF 36) dans l'étude 1. Les patients sous adalimumab ont présenté une amélioration significativement plus importante des scores résumés physique (Physical Component Summary, PCS) et mental (Mental Component Summary, MCS) du SF 36.
Étude sur la polyarthrite rhumatoïde (PR) comparant AMGEVITA et le médicament de référence
L'efficacité et la sécurité d'AMGEVITA ont été évaluées comparativement au médicament de référence au cours de l'étude comparative randomisée, en double aveugle, contrôlée contre traitement actif (étude 1 RA-ABP), menée auprès de 526 patients ≥18 ans atteints de polyarthrite rhumatoïde active modérée à sévère et présentant une réponse insuffisante au méthotrexate. Les patients ont reçu 40 mg d'AMGEVITA ou la même quantité du médicament de référence par voie sous-cutanée toutes les deux semaines pendant jusqu'à 22 semaines.
Le critère d'évaluation principal, la réponse ACR20, s'est situé dans la plage d'équivalence prédéfinie et a montré l'équivalence clinique entre AMGEVITA et le médicament de référence.
Le profil de sécurité général d'AMGEVITA ressemble à celui du médicament de référence.
Étude sur le psoriasis en plaques (Ps) comparant AMGEVITA et le médicament de référence
L'efficacité et la sécurité d'AMGEVITA ont été évaluées comparativement au médicament de référence au cours d'une étude randomisée, en double aveugle, contrôlée contre traitement actif (étude 1 Ps-ABP) menée auprès de 350 patients ≥18 ans atteints de psoriasis en plaques modéré à sévère chez lesquels une thérapie systémique ou une photothérapie était envisagée. Les patients ont reçu soit AMGEVITA, soit le médicament de référence, à la dose initiale ou de charge de 80 mg par voie sous-cutanée à la semaine 1/au jour 1, puis 40 mg par voie sous-cutanée toutes les deux semaines, en commençant une semaine après l'administration de la dose initiale. L'amélioration du score PASI (en pourcentage) par rapport à la valeur initiale à la semaine 16 s'est située dans la plage d'équivalence prédéfinie et a donc démontré l'équivalence clinique entre AMGEVITA et le médicament de référence.
PharmacocinétiqueAbsorption
Après l'administration sous-cutanée d'une dose unique de 40 mg, on observe une absorption lente du principe actif adalimumab. Les concentrations sériques maximales ont été atteintes environ cinq jours après administration. La biodisponibilité absolue moyenne de l'adalimumab calculée à partir de trois études après administration d'une dose unique de 40 mg par voie sous-cutanée s'est élevée à 64%. Les concentrations, après administration d'une dose unique par voie intraveineuse, comprises entre 0,25 et 10 mg/kg, étaient approximativement proportionnelles à la dose.
Après administration sous-cutanée de 40 mg d'adalimumab toutes les deux semaines à des patients atteints de polyarthrite rhumatoïde, on peut prévoir l'accumulation d'adalimumab, en fonction de la demi-vie, à des concentrations moyennes à l'état d'équilibre d'environ 5 µg/ml (sans administration concomitante de méthotrexate) et de 8 à 9 µg/ml (avec administration concomitante de méthotrexate). À l'état d'équilibre, les concentrations sériques minimales d'adalimumab après administration sous-cutanée de 20, 40 et 80 mg toutes les deux semaines ou chaque semaine augmentent de manière proportionnelle à la dose.
Pendant la période d'induction, on obtient, avec une dose d'attaque de 160 mg d'adalimumab à la semaine 0, suivie de 80 mg d'adalimumab à la semaine 2, un taux sérique minimal d'adalimumab d'environ 12 mcg/ml chez les patients atteints de la maladie de Crohn. Un taux minimal moyen d'environ 7 mcg/ml a été observé chez les patients atteints de la maladie de Crohn qui ont reçu une dose d'entretien de 40 mg d'adalimumab toutes les deux semaines.
Chez les patients atteints de colite ulcéreuse qui ont reçu une dose d'induction de 160 mg d'adalimumab à la semaine 0, puis de 80 mg à la semaine 2, la concentration sérique minimale d'adalimumab était d'environ 12 μg/ml pendant la période d'induction. Pendant la phase d'entretien, la concentration minimale moyenne à l'état d'équilibre était de 8 μg/ml.
Distribution
Après l'administration sous-cutanée d'une dose unique de 40 mg, on observe une distribution lente du principe actif adalimumab.
Après administration unique par voie intraveineuse de doses comprises entre 0,25 et 10 mg/kg, le volume de distribution (Vss) s'est élevé à des valeurs comprises entre 4,7 et 6,0 litres, qui correspondent à une distribution homogène entre les liquides vasculaires et extravasculaires. Les concentrations d'adalimumab dans le liquide synovial observées chez différents patients atteints de polyarthrite rhumatoïde étaient comprises entre 31% et 96% des concentrations sériques.
Élimination
Après l'administration par voie intraveineuse de doses uniques comprises entre 0,25 et 10 mg/kg, la clairance a atteint normalement des valeurs inférieures à 12 ml/heure. La demi-vie moyenne s'est élevée à environ deux semaines. La clairance normalisée en fonction du poids corporel est similaire entre les patients juvéniles et les patients adultes.
Au cours d'études au long cours pendant lesquelles le médicament a été administré pendant plus de deux ans, aucun signe de modification de la clairance n'a été observé.
Cinétique pour certains groupes de patients
Les évaluations pharmacocinétiques des groupes de patients regroupant les données de plus de 1300 patients adultes atteints de polyarthrite rhumatoïde et de 171 patients atteints d'arthrite juvénile idiopathique (âgés de 4 à 17 ans) ont révélé une clairance apparente plus élevée en rapport avec un poids corporel plus élevé: La clairance est également accrue en présence d'anticorps dirigés contre l'adalimumab. La cinétique de l'adalimumab chez les patients ayant une fonction rénale ou hépatique limitée n'a pas été étudiée.
Enfants et adolescents
Arthrite juvénile idiopathique polyarticulaire
Après l'administration sous-cutanée de 24 mg/m2 (sans dépasser une dose maximale de 40 mg) toutes les deux semaines chez des patients atteints d'arthrite juvénile idiopathique polyarticulaire, les concentrations minimales moyennes à l'état d'équilibre étaient comparables à celles observées chez les adultes recevant la dose de 40 mg (adalimumab en monothérapie: 5,6 ± 5,6 µg/ml (102% CV) et 10,9 ± 5,2 µg/ml (47,7% CV) lors d'un traitement en association avec le méthotrexate; les valeurs ont été saisies entre la semaine 20 et la semaine 48).
Interactions avec d'autres médicaments
Chez 21 patients suivant un traitement stable par le méthotrexate, aucune modification statistiquement significative des profils de concentration sérique du méthotrexate n'a été observée après l'administration d'adalimumab. À l'inverse, le méthotrexate, après administration unique ou multiple, a abaissé la clairance apparente de l'adalimumab de 29% ou 44% (voir «Interactions»).
Données précliniquesSur la base d'études de toxicité après administration unique ou répétée ainsi que d'études de génotoxicité, les données précliniques n'ont pas mis en évidence de danger particulier pour l'homme.
Une étude de toxicité sur le développement périnatal et embryonnaire, menée sur des singes cynomolgus, n'a révélé quasiment aucun effet nocif pour la gestation et le développement de l'embryon. On a observé chez les fœtus des animaux femelles traités par 30 et 100 mg/kg un retard de l'ossification plus fréquent que dans le groupe de contrôle. L'administration répétée d'adalimumab a provoqué une accumulation, et le niveau d'exposition des animaux femelles traités était nettement plus élevé que ne pouvait le laisser penser le traitement.
En raison du fait qu'il n'existe aucun modèle adapté d'anticorps ayant une réactivité croisée limitée avec le TNF des rongeurs et du développement d'anticorps neutralisants chez les rongeurs, aucune étude de cancérogénicité ni d'évaluation de la fertilité et de la toxicité postnatale de l'adalimumab n'a été entreprise.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Influence sur les méthodes de diagnostic
Aucune influence de l'adalimumab sur des méthodes de diagnostic n'a été observée.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
La préparation ne contient pas d'agent conservateur. Pour des raisons microbiologiques, la solution injectable prête à l'emploi doit être utilisée immédiatement après ouverture.
Remarques concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver AMGEVITA dans l'emballage d'origine pour protéger le contenu de la lumière.
Conserver AMGEVITA hors de la portée des enfants.
La seringue préremplie ou le stylo prérempli peuvent être conservés à une température de 25 °C maximum pour une durée allant jusqu'à 14 jours. La seringue préremplie ou le stylo prérempli doivent être conservés à l'abri de la lumière et doivent être éliminés s'ils n'ont pas été utilisés dans cette période de 14 jours, et ce même s'ils ont été remis au réfrigérateur entre-temps.
Remarques concernant la manipulation
AMGEVITA solution injectable doit être utilisé sous la direction ou la supervision d'un médecin. Après une formation adaptée aux méthodes d'injection par voie sous-cutanée, les patients peuvent s'injecter eux-mêmes la solution injectable AMGEVITA lorsque le médecin estime que cela est opportun et lorsque le suivi médical nécessaire est assuré.
La solution injectable est limpide et incolore à légèrement jaunâtre.
Vous trouverez des informations détaillées concernant l'utilisation dans la notice d'emballage.
Il est recommandé d'éliminer le médicament non utilisé ou les déchets conformément à la réglementation locale.
Numéro d’autorisation66979, 67204 (Swissmedic).
PrésentationAMGEVITA 40 mg solution injectable en seringue préremplie avec protection d'aiguille: 1 ou 2 seringue(s) préremplie(s) (B).
AMGEVITA 40 mg solution injectable en stylo prérempli: 1 ou 2 stylo(s) prérempli(s) (B).
Titulaire de l’autorisationAmgen Switzerland AG, Risch.
Domicile: 6343 Rotkreuz.
Mise à jour de l’informationJuillet 2019.
Version#100719
|