CompositionPrincipes actifs
Siponimod (sous forme d'acide fumarique de siponimod).
Excipients
Noyau des comprimés: lactose monohydraté, cellulose microcristalline, crospovidone, dibéhénate de glycérol et silice colloïdale anhydre.
Un comprimé à 0,25 mg contient 62,2 mg de lactose monohydraté.
Un comprimé à 1 mg contient 61,4 mg de lactose monohydraté
Un comprimé à 2 mg contient 60,3 mg de lactose monohydraté.
Enrobage: poly(alcool vinylique), dioxyde de titane (E171), oxyde de fer rouge (E172), talc, lécithine de soja (E322), gomme xanthane, oxyde de fer noir (E172, seulement les comprimés à 0,25 mg et à 1 mg), oxyde de fer jaune (E172, seulement les comprimés à 2 mg).
Indications/Possibilités d’emploiMayzent est indiqué dans le traitement des patients adultes atteints de sclérose en plaques secondairement progressive (SEP-SP) avec activité inflammatoire détectée par des poussées cliniques ou à l'imagerie.
Posologie/Mode d’emploiLe traitement par Mayzent doit être initié et surveillé par un neurologue expérimenté dans le traitement de patients atteints de SEP.
Avant l'introduction du traitement par Mayzent, le génotype CYP2C9 du patient doit être déterminé. Mayzent ne doit pas être utilisé chez les patients présentant un génotype CYP2C9*3/*3 (voir «Contreindications», «Mises en garde et précautions» et «Pharmacocinétique»).
Pour les patients présentant un génotype CYP2C9*2/*3 ou *1/*3, la dose d'entretien recommandée est de 1 mg une fois par jour (un comprimé à 1 mg ou quatre comprimés à 0,25 mg) (voir «Mises en garde et précautions» et «Pharmacocinétique»).
En raison du manque de données, aucune recommandation posologique ne peut être donnée pour d'autres allèles de CYP2C9 rares pour lesquels on peut supposer une activité de CYP2C9 réduite ou absente, en particulier, par exemple, les allèles CYP2C9 *5, *6, *8 et *11, (voir aussi les rubriques «Contre-indications», «Mises en garde et précautions», «Interactions» et «Pharmacocinétique»).
La dose d'entretien recommandée de Mayzent pour les patients présentant tous les autres génotypes CYP2C9 est de 2 mg.
Mayzent est pris une fois par jour au cours ou en dehors des repas. Les comprimés pelliculés de Mayzent doivent être pris entiers avec de l'eau.
Avant le début du traitement
Évaluation ophtalmologique
Un examen ophtalmologique pour l'évaluation du fond de l'œil, macula y comprise, doit être exécuté (voir «Mises en garde et précautions»).
Évaluation dermatologique
Un examen dermatologique doit être exécuté. Les lésions cutanées suspectes doivent être clarifiées sans délai (voir «Mises en garde et précautions»).
Instauration du traitement
Le traitement doit être initié au moyen d'une boîte de début de traitement, qui couvre 5 jours.
Les patients atteints de certaines affections cardiaques préexistantes doivent être surveillés pendant les 6 premières heures suivant la première dose de Mayzent en vue de détecter tout signe et symptôme de bradycardie (voir «Mises en garde et précautions»).
La titration de la dose commence par 0,25 mg une fois par jour aux jours 1 et 2, suivi de 0,5 mg une fois par jour au jour 3, 0,75 mg au jour 4 et 1,25 mg au jour 5, de sorte que le patient atteint sa dose d'entretien prescrite de Mayzent en 6 jours (voir tableau 1).
Pendant les 6 premiers jours de traitement, la dose quotidienne recommandée doit être prise une fois par jour le matin avec un repas ou indépendamment de celui-ci.
Tableau 1: Schéma de titration de la dose pour atteindre la dose d'entretien de Mayzent
|
Titration
|
Dose de titration
|
Schéma de titration
|
Emballage
| |
Jour 1
|
0,25 mg
|
1 × 0,25 mg
|
| |
Jour 2
|
0,25 mg
|
1 × 0,25 mg
|
| |
Jour 3
|
0,5 mg
|
2 × 0,25 mg
|
DÉBUT DE TRAITEMENT
| |
Jour 4
|
0,75 mg
|
3 × 0,25 mg
|
| |
Jour 5
|
1,25 mg
|
5 × 0,25 mg
|
| |
Jour 6
|
2 mg°
|
1 × 2 mg°
|
ENTRETIEN pour les génotypes CYP2C9 *1/*1, *1/*2 ou *2/*2
|
° La dose d'entretien recommandée est de 1 mg (1 × 1 mg ou 4 × 0,25 mg) par jour pour les patients ayant le génotype CYP2C9*2/*3 ou *1/*3. La sécurité du patient n'est pas affectée par une exposition excédentaire de 0,25 mg le jour 5.
Doses omises au début du traitement
Si une dose de titration est omise un jour pendant les 6 premiers jours de traitement, celui-ci doit être recommencé avec une nouvelle boîte de début de traitement.
Dose omise après le jour 6
Si une dose est omise, la dose prescrite doit être prise à l'horaire habituel suivant. La dose suivante ne doit pas être doublée pour compenser un comprimé oublié.
Reprise du traitement d'entretien après interruption du traitement
Lorsque le traitement d'entretien par Mayzent est interrompu pendant 4 doses quotidiennes successives ou plus, le traitement doit être repris au moyen d'une nouvelle boîte de début de traitement (voir «Instauration du traitement»). Les interruptions de traitement à raison de 4 doses quotidiennes successives manquantes au maximum n'exigent pas une nouvelle titration et le traitement doit être poursuivi avec la dose d'entretien.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Le siponimod ne doit pas être utilisé chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) (voir «Contre-indications»). Bien qu'aucun ajustement posologique ne soit nécessaire chez les patients présentant une insuffisance hépatique légère à modérée, la prudence est de rigueur en début de traitement chez ces patients (voir «Mises en garde et précautions», «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique de Mayzent n'est nécessaire pour les patients atteints d'insuffisance rénale.
Enfants et adolescents (moins de 18 ans)
Aucune étude n'a été menée pour les enfants et les adolescents.
Patients âgés
Mayzent n'a pas été étudié chez les patients de plus de 65 ans. Les patients ayant participé aux études cliniques étaient âgés de 61 ans au maximum. Chez les patients âgés, Mayzent doit être utilisé avec prudence, car on ne dispose pas de données suffisantes concernant la sécurité et l'efficacité.
Contre-indications·Hypersensibilité au principe actif, le siponimod, aux arachides, au soja ou à l'un des excipients selon la composition
·Syndrome d'immunodéficience
·Antécédents de leucoencéphalopathie multifocale progressive (LEMP) ou de méningite à cryptocoque
·Maladies malignes actives
·Insuffisance hépatique sévère (classe C de Child-Pugh)
·Patients ayant eu, au cours des 6 derniers mois, un infarctus du myocarde (IM), une angine de poitrine instable, un accident vasculaire cérébral/un accident ischémique transitoire (AIT), une insuffisance cardiaque décompensée ayant nécessité un traitement hospitalier, ou une insuffisance cardiaque de classe III-IV selon la New York Heart Association (NYHA)
·Patients avec un bloc AV du 2e degré de type Mobitz II connu à l'anamnèse, un bloc AV de 3e degré, un bloc sino-auriculaire ou maladie du sinus, lorsqu'ils ne portent pas de stimulateur cardiaque (voir «Mises en garde et précautions»)
·Patients homozygotes pour le génotype CYP2C9*3 (CYP2C9*3/*3)
·Pendant la grossesse ou chez les femmes en âge de procréer qui n'utilisent pas de méthode contraceptive fiable
Mises en garde et précautionsInfections
Un effet pharmacodynamique central de Mayzent consiste en la réduction dépendante de la dose du nombre de lymphocytes périphériques de 20 à 30% de la valeur de référence. Cet effet est attribué à la rétention réversible (séquestration) des lymphocytes dans les tissus lymphoïdes (voir «Mécanisme d'action/Pharmacodynamique»).
L'effet de Mayzent sur le système immunitaire peut augmenter le risque d'infection.
Avant l'initiation du traitement par Mayzent, un hémogramme complet actuel (à savoir, datant de moins de 6 mois ou effectué à l'arrêt du traitement précédent) doit être disponible. Il est en outre recommandé de contrôler l'hémogramme complet 3 à 4 mois après le début du traitement et ensuite au moins une fois par an ainsi qu'en cas de signes d'infection. En cas de numération lymphocytaire totale confirmée < 0,2 × 109/l, la dose doit être réduite à 1 mg, car dans les études cliniques, la dose de siponimod a été réduite chez les patients ayant une numération lymphocytaire totale < 0,2 × 109/l. En cas de numération lymphocytaire totale confirmée < 0,2 × 109/l chez des patients recevant déjà une dose de 1 mg de siponimod, le traitement doit être interrompu jusqu'à ce que la valeur atteigne 0,6 × 109/l. Une nouvelle initiation de traitement par siponimod peut alors être envisagée.
L'instauration du traitement par Mayzent doit être retardée chez les patients souffrant d'une infection active grave, jusqu'à disparition de l'infection. Étant donné que les effets pharmacodynamiques résiduels, par exemple la réduction du nombre de lymphocytes périphériques, peuvent se maintenir pendant 3 à 4 semaines après l'arrêt de Mayzent, il convient de poursuivre la surveillance des infections pendant cette période (voir «Arrêt du traitement»).
Les patients qui reçoivent Mayzent, doivent être informés de signaler immédiatement à leur médecin tout symptôme d'infection. Des mesures diagnostiques et thérapeutiques efficaces doivent être appliquées chez les patients présentant des symptômes d'infection pendant le traitement. Une interruption du traitement par Mayzent doit être envisagée lorsque le patient développe une infection sévère.
Des cas de méningite à cryptocoque (MC) en lien avec l'utilisation de Mayzent ont été rapportés. Des cas de MC ont également été mentionnés pour un autre modulateur des récepteurs de la sphingosine-1phosphate (S1P). Les médecins doivent être attentifs aux symptômes ou signes cliniques de la MC. Un examen diagnostique doit être immédiatement effectué chez les patients présentant de tels symptômes et signes. Le traitement par Mayzent doit dans le même temps être interrompu jusqu'à exclusion d'une MC. Si une MC est diagnostiquée, un traitement approprié doit être initié immédiatement. Une reprise ultérieure du traitement par Mayzent est contre-indiquée dans de tels cas (voir rubrique «Contre-indications»).
Des cas d'infection par le virus de l'herpès, y compris des cas de méningite ou de méningoencéphalite causés par le virus de la varicelle-zona (VVZ) ont été rapportés avec Mayzent. Les patients sans antécédent de varicelle confirmé par un médecin ou sans documentation d'une vaccination complète contre le virus de la varicelle-zona (VVZ) doivent être testés pour dépister les anticorps contre le VVZ avant de débuter le traitement par Mayzent (voir «Vaccination»).
Leucoencéphalopathie multifocale progressive
Chez les patients atteints de SEP traités par des modulateurs des récepteurs de S1P, des cas de leucoencéphalopathie multifocale progressive (LEMP) sont survenus. La LEMP est une infection virale opportuniste du cerveau causée par le virus JC (VJC), survenant généralement uniquement chez des patients dont le système immunitaire a été affaibli et menant en règle générale au décès ou à un handicap grave. Des cas de LEMP sont survenus chez des patients traités par modulateurs du récepteur S1P qui n'avaient pas été antérieurement traités par le natalizumab (connu pour son lien avec la LEMP), qui ne prenaient aucun autre médicament immunosuppresseur ou immunomodulateur et qui ne souffraient d'aucune pathologie systémique entraînant une altération de la fonction du système immunitaire. La plupart des cas de LEMP liés aux modulateurs des récepteurs de S1P sont survenus chez des patients traités depuis au moins 2 ans. Le lien entre le risque de LEMP et la durée du traitement n'est pas connu.
Aux premiers signes ou symptômes évocateurs d'une LEMP, Mayzent doit être arrêté et une évaluation diagnostique appropriée doit être effectuée. Les symptômes typiques associés à la LEMP sont multiformes, progressent sur plusieurs jours à plusieurs semaines et incluent une faiblesse progressive d'un côté du corps ou une lourdeur des extrémités, des troubles de la vision et des modifications du processus de pensée, de la mémoire et de l'orientation entraînant une confusion et des modifications de la personnalité. Des signes peuvent être présents à l'IRM avant l'apparition des signes ou symptômes cliniques. Des cas de LEMP diagnostiqués sur la base des observations à l'IRM et de la détection de l'ADN du VJC dans le LCR en l'absence de signes cliniques ou de symptômes spécifiques de la LEMP ont été rapportés chez des patients traités par des médicaments contre la SEP associés à un risque de LEMP, y compris des modulateurs du récepteur de S1P. Des symptômes d'une LEMP sont apparus ensuite chez un grand nombre de ces patients.
Une surveillance à l'IRM de signes évocateurs d'une LEMP peut donc être utile. En cas d'observations suspectes, des examens supplémentaires doivent être réalisés afin de permettre un diagnostic précoce de la LEMP. Après l'arrêt d'un autre médicament contre la SEP pour lequel un lien a été établi avec la LEMP, une mortalité et une morbidité liées à la LEMP plus faibles ont été rapportées chez les patients atteints de LEMP et asymptomatiques dans un premier temps comparativement aux patients atteints de LEMP présentant des signes cliniques et des symptômes caractéristiques au moment du diagnostic. On ignore si ces différences sont dues à la détection précoce et à l'arrêt du traitement contre la SEP ou à des différences d'évolution de la pathologie chez ces patients. En cas de confirmation d'une LEMP, le traitement par Mayzent doit être arrêté définitivement.
Un syndrome inflammatoire de reconstitution immunitaire (en anglais Immune reconstitution inflammatory syndrome, IRIS) a été rapporté chez des patients traités par des modulateurs des récepteurs de S1P chez lesquels une LEMP est survenue et qui ont ensuite arrêté le traitement. L'IRIS se manifeste par une dégradation de l'état clinique du patient pouvant survenir rapidement; il peut entraîner des complications neurologiques graves ou mener au décès et il s'accompagne souvent de modifications caractéristiques à l'IRM. L'apparition d'un IRIS chez des patients atteints d'une LEMP a eu lieu le plus souvent quelques mois après l'arrêt du modulateur du récepteur de S1P. Il convient de surveiller l'apparition d'un IRIS et de traiter l'inflammation qui l'accompagne de manière appropriée.
Vaccination
Pour les patients n'ayant pas d'anticorps, une vaccination complète avec le vaccin contre la varicelle est recommandée avant de débuter le traitement par Mayzent, ce qui décale d'un mois l'initiation du traitement par Mayzent pour permettre le développement de l'effet complet du vaccin (voir «Effets indésirables»).
Vaccins vivants atténués
L'utilisation de vaccins vivants atténués (p. ex., vaccin contre la varicelle et vaccin contre la fièvre jaune) peut être associée à un risque d'infection et doit donc être évitée pendant le traitement par Mayzent et pendant 4 semaines après la fin du traitement par Mayzent (voir «Interactions»).
Autres types de vaccins
Les autres types de vaccins peuvent être moins efficaces lorsqu'ils sont administrés pendant le traitement par Mayzent. Une interruption du traitement de 1 semaine avant à 4 semaines après la vaccination prévue est recommandée. La décision de poursuivre ou d'interrompre le traitement par Mayzent doit se fonder sur l'évaluation du rapport bénéfice/risque au cas par cas (voir «Fin du traitement par le siponimod» et «Interactions»).
En cas d'interruption du traitement par le siponimod en raison d'une vaccination, un retour possible de l'activité de la maladie doit être pris en considération (voir «Fin du traitement par le siponimod»).
Traitement concomitant avec des thérapies antinéoplasiques, immunomodulatrices ou immunosuppressives
La prudence s'impose en cas d'utilisation concomitante de thérapies antinéoplasiques, immunomodulatrices ou immunosuppressives (y compris corticostéroïdes) car il existe un risque d'effets additifs sur le système immunitaire pendant une telle thérapie (voir «Interactions»).
Œdème maculaire
Dans l'étude clinique de phase 3 (A2304), un œdème maculaire (voir «Effets indésirables») avec ou sans troubles visuels a été signalé plus fréquemment sous siponimod (1,8%) que sous placebo (0,2%). La plupart des cas sont survenus dans les 3 à 4 premiers mois de traitement. Pour cette raison, un examen ophtalmologique est recommandé pour tous les patients avant le début du traitement, ainsi que 3 à 4 mois après le début du traitement. Étant donné que des œdèmes maculaires sont apparus également lors d'un traitement à long terme, les patients sous traitement par Mayzent doivent signaler immédiatement l'apparition de troubles visuels. Un examen du fond d'œil, y compris de la macula, est recommandé.
Les patients ayant des antécédents de diabète, d'uvéite, ou d'affections sous-jacentes/simultanées de la rétine présentent un risque accru d'œdème maculaire. Il est recommandé que ces patients soient examinés régulièrement par un ophtalmologue pendant le traitement par Mayzent.
La poursuite du traitement par Mayzent chez les patients atteints d'un œdème maculaire n'a pas été étudiée. Il est recommandé d'arrêter le siponimod lorsque le patient développe un œdème maculaire. La décision d'arrêt ou de poursuite du traitement par Mayzent doit être prise en tenant compte du bénéfice potentiel et des risques pour chaque patient.
Bradycardie et bradyarythmie
Fréquence cardiaque
En raison du risque de troubles sévères du rythme cardiaque ou de bradycardie importante, Mayzent ne doit pas être utilisé chez les patients présentant les affections suivantes:
·antécédents d'arrêt cardiaque datant de plus de 6 mois avant le traitement par Mayzent,
·maladie cérébrovasculaire,
·bradycardie symptomatique ou syncopes répétées à l'anamnèse,
·hypertension non contrôlée ou
·apnée du sommeil sévère non traitée.
Si un traitement est envisagé, il convient de prendre l'avis d'un cardiologue avant le début du traitement afin d'établir la stratégie de surveillance la plus appropriée.
Chez ces patients, un traitement par le siponimod ne doit être envisagé que lorsque le bénéfice attendu est supérieur aux risques possibles (voir informations ci-dessous).
Une étude approfondie de l'intervalle QT n'a montré aucun effet direct significatif sur l'allongement de l'intervalle QT et Mayzent ne s'accompagne pas d'un potentiel arythmogène dans le cadre d'un allongement de l'intervalle QT. L'initiation d'un traitement par Mayzent peut entraîner une réduction de la fréquence cardiaque et un allongement indirect de l'intervalle QT pendant la phase de titration. Mayzent n'a pas été étudié chez les patients présentant un allongement significatif de l'intervalle QT (QTc > 500 msec) ou sous médication allongeant l'intervalle QT. Lorsqu'un traitement par Mayzent est envisagé chez des patients présentant un allongement significatif de l'intervalle QT préexistant ou sous traitement par un médicament allongeant l'intervalle QT ayant des propriétés arythmogènes connues, un cardiologue doit être consulté avant l'initiation du traitement afin d'établir la meilleure stratégie de surveillance pendant l'initiation du traitement.
Mayzent n'a pas été étudié chez les patients présentant des arythmies qui nécessitent un traitement par des antiarythmiques de la classe Ia (par exemple, quinidine, procaïnamide) ou de la classe III (par exemple, amiodarone, sotalol). Les antiarythmiques de la classe Ia et de la classe III ont été associés à des cas de torsade de pointes chez les patients présentant une bradycardie. Vu que l'initiation du traitement par Mayzent entraîne une réduction de la fréquence cardiaque, Mayzent ne doit pas être administré avec ces médicaments pendant l'initiation du traitement.
On ne dispose que d'une expérience limitée avec les patients qui sont traités de façon concomitante par des inhibiteurs calciques abaissant la fréquence cardiaque (comme vérapamil ou diltiazem) ou d'autres substances actives qui peuvent réduire la fréquence cardiaque (par exemple, ivabradine ou digoxine). Dans les études cliniques menées sur Mayzent, les patients n'ont pas reçu ces médicaments. L'utilisation concomitante de ces principes actifs pendant l'initiation de Mayzent peut être associée à une bradycardie sévère et à un bloc cardiaque. En raison des effets additifs possibles sur la fréquence cardiaque, le traitement par Mayzent ne doit pas être initié chez les patients qui sont traités de manière concomitante par ces principes actifs (voir «Interactions»).
En cas de prise des principes actifs susmentionnés, si un traitement concomitant par Mayzent est néanmoins envisagé, un cardiologue doit être consulté avant l'initiation du traitement par Mayzent afin d'envisager un passage à un médicament ne réduisant pas la fréquence cardiaque ou d'établir une surveillance appropriée lors de l'initiation du traitement.
Étant donné que l'initiation du traitement par Mayzent entraîne une réduction temporaire de la fréquence cardiaque (voir «Effets indésirables»), il convient d'adopter en début de traitement, un schéma de titration de dose qui permet d'atteindre la dose d'entretien de Mayzent au jour 6 (voir «Posologie/Mode d'emploi»).
Après la première dose de la titration, la réduction de la fréquence cardiaque commence dans un délai d'une heure et la diminution du jour 1 est à son maximum après environ 3 à 4 heures. En cas de poursuite de la titration de la dose, on observe d'autres réductions de la fréquence cardiaque au cours des jours suivants, la réduction maximale de la valeur initiale du jour 1 étant atteinte au jour 5 à 6. La réduction quotidienne la plus élevée de la fréquence cardiaque moyenne horaire absolue après administration est observée au jour 1, le pouls diminuant en moyenne de 5 à 6 battements par minute (bpm). Les jours suivants, la réduction après l'administration est moins marquée. En cas de poursuite de l'administration, la fréquence cardiaque augmente après le jour 6 et atteint la valeur du placebo en 10 jours après le début du traitement.
Des fréquences cardiaques de moins de 40 bpm ont rarement été observées. Les patients chez lesquels une bradycardie est survenue étaient en général asymptomatiques. Certains patients ont présenté des symptômes légers à modérés, tels des vertiges ou de la fatigue, qui ont disparu dans les 24 heures sans intervention (voir «Effets indésirables»). Si nécessaire, la chute de la fréquence cardiaque induite par le siponimod peut être annulée par administration parentérale d'atropine ou d'isoprénaline.
Temps de conduction auriculoventriculaire
L'initiation du traitement par Mayzent a été associée à des retards temporaires de la conduction auriculoventriculaire qui suivent un schéma temporel similaire à celui de la réduction de la fréquence cardiaque observée pendant la titration de la dose. Les retards de la conduction auriculoventriculaire se manifestent dans la plupart des cas sous la forme d'un bloc auriculoventriculaire (AV) de 1er degré (allongement de l'intervalle PR à l'électrocardiogramme). Au moment de l'initiation du traitement par Mayzent, un bloc AV de 2e degré, le plus souvent de type Mobitz I (Wenckebach), a été observé chez moins de 1,7% des patients des études cliniques. Les anomalies de la conduction ont été en général temporaires, asymptomatiques, résolues en 24 heures et n'ont pas nécessité l'arrêt du traitement par Mayzent.
Recommandation pour l'initiation du traitement
Par prudence, les patients atteints des affections cardiaques suivantes doivent être surveillés pendant une période de 6 heures après l'administration de la première dose de Mayzent en vue de détecter tout signe et symptôme de bradycardie:
·bradycardie sinusale (fréquence cardiaque (FC) < 55 bpm),
·antécédents connus de bloc auriculoventriculaire (bloc AV) de 1er ou 2e degré (type Mobitz I),
·antécédents connus d'infarctus du myocarde ou antécédents connus d'insuffisance cardiaque, si non contre-indiqué.
Il est recommandé de réaliser un électrocardiogramme (ECG) avant l'administration et à la fin de la période d'observation. Si des symptômes liés à une bradyarythmie ou à des troubles de la conduction se présentent après l'administration ou si l'ECG montre, 6 heures après l'administration, un bloc AV de 2e degré nouveau ou accru ou un intervalle QTc ≥500 msec, un traitement approprié doit être instauré et la surveillance poursuivie jusqu'à la résolution des symptômes/anomalies. Lorsqu'un traitement pharmacologique est nécessaire, la surveillance doit être poursuivie pendant la nuit et la surveillance de 6 heures doit être répétée après la deuxième dose.
Les effets bradyarythmiques sont marqués lorsque Mayzent est administré en plus d'un traitement par bêtabloquant.
Chez les patients recevant un bêtabloquant à dose stable, la fréquence cardiaque au repos doit être déterminée avant l'initiation du traitement par Mayzent. Lorsque la fréquence cardiaque au repos sous traitement en cours par bêtabloquant se situe à > 50 bpm, Mayzent peut être instauré. Lorsque la fréquence cardiaque au repos se situe à ≤50 bpm, le traitement par bêtabloquant doit être interrompu jusqu'à ce que la fréquence cardiaque s'élève à > 50 bpm. Le traitement par Mayzent peut alors être commencé et le traitement par bêtabloquant peut être repris une fois que la dose d'entretien souhaitée de Mayzent est atteinte (voir «Interactions»).
Dose oubliée en début de traitement et reprise du traitement après interruption de celui-ci
Si une des doses de la titration est omise pendant les 6 premiers jours de traitement ou si 4 doses quotidiennes successives ou plus sont omises pendant le traitement d'entretien, les recommandations initiales de titration et de surveillance s'appliquent (voir «Posologie/Mode d'emploi»).
Fonction hépatique
Avant le début du traitement par Mayzent, il convient de disposer des valeurs des transaminases et de la bilirubine actuelles (à savoir obtenues au cours des 6 derniers mois). Dans l'étude A2304, des taux d'alanine aminotransférase (ALAT) ou d'aspartate aminotransférase (ASAT) correspondant à trois fois la limite supérieure de la normale (LSN) ont été observés chez 5,6% des patients traités par Mayzent 2 mg contre 1,5% des patients sous placebo (voir «Effets indésirables»). Dans les études cliniques, Mayzent a été arrêté lorsque l'augmentation était supérieure à trois fois la LSN et que le patient présentait des symptômes liés à la fonction hépatique, ou lorsque l'augmentation était supérieure à 5 fois la LSN.
Chez les patients développant des symptômes indiquant une insuffisance hépatique, comme des nausées inexpliquées, des vomissements, des douleurs abdominales, une fatigue, une perte d'appétit, un rash avec éosinophilie ou ictère et/ou urines foncées pendant le traitement, les enzymes hépatiques doivent être mesurées et Mayzent arrêté si une atteinte hépatique significative est confirmée. Une reprise du traitement est conditionnée par la mise en évidence d'une autre cause de l'atteinte hépatique et par la mise en balance du bénéfice de la reprise du traitement pour le patient et des risques d'une nouvelle survenue des troubles de la fonction hépatique.
Bien que l'on ne dispose d'aucune donnée permettant d'établir que les patients atteints d'une affection hépatique préexistante présentent un risque accru de développer une augmentation des paramètres de la fonction hépatique (valeurs de TFH) lors de l'utilisation de Mayzent, la prudence est recommandée lors de l'administration de Mayzent à des patients ayant des antécédents de pathologie hépatique sévère.
Néoplasies cutanées
Des carcinomes basocellulaires (CBC) et autres néoplasies cutanées telles que le mélanome malin, le cancer épidermoïde, le sarcome de Kaposi et le carcinome à cellules de Merkel ont été rapportés chez des patients ayant été traités par des modulateurs des récepteurs de S1P. Dans l'étude A2304, le carcinome basocellulaire (CBC) a été le néoplasme le plus fréquent et a été rapporté avec une fréquence similaire dans le groupe de traitement qui recevait le siponimod 2 mg (1,1%, 12 patients) et dans le groupe placebo (1,3%, 7 patients). La fréquence des carcinomes épidermoïdes (CE) dans l'étude A2304 était identique chez les patients traités par Mayzent et ceux ayant reçu le placebo (0,2%). Dans une étude à long terme, une légère augmentation de l'incidence de CBC et de CE a été observée en cas de prise prolongée.
Chez tous les patients, notamment ceux présentant un risque élevé de néoplasies cutanées malignes (par exemple, antécédent de mélanome malin), mais aussi ceux ne présentant pas de risque élevé de néoplasies cutanées malignes, des examens dermatologiques réguliers devront être effectués avant le début d'un traitement par Mayzent, puis au cours du traitement. Les lésions cutanées suspectes doivent être clarifiées sans délai. Les patients traités par le siponimod doivent être informés d'éviter une exposition non protégée aux rayons du soleil (par exemple, antécédent de mélanome malin). Les patients sous traitement par Mayzent ne doivent pas suivre de photothérapie par UV-B, ni de photochimiothérapie par PUVA.
Symptômes/signes neurologiques ou psychiatriques inattendus
De rares cas de syndrome d'encéphalopathie postérieure réversible (SEPR) ont été rapportés pour un autre modulateur des récepteurs de la sphingosine 1phosphate (S1P). De tels événements n'ont pas été signalés pour Mayzent dans le cadre du programme de développement. Si un patient sous traitement par Mayzent devait toutefois développer des symptômes/signes neurologiques ou psychiatriques inattendus (par exemple, déficits cognitifs, modifications du comportement, troubles de la vision corticaux ou autres symptômes/signes neurologiques corticaux ou symptômes/signes évocateurs d'une élévation de la pression intracrânienne) ou une altération neurologique accélérée, le médecin doit procéder sans tarder à un examen physique et neurologique complet et envisager une imagerie par résonance magnétique (IRM).
Traitement préalable par des immunosuppresseurs ou immunomodulateurs
Lors du passage depuis d'autres thérapies modifiant la maladie, la demi-vie et le mécanisme d'action de l'autre traitement doivent être pris en compte pour éviter un effet additif sur le système immunitaire et minimiser le risque d'une réactivation de maladie. La détermination du nombre de lymphocytes périphériques (hémogramme complet) est recommandée avant l'initiation de Mayzent pour s'assurer que les effets immunitaires du traitement précédent (par exemple, cytopénie) sont résolus.
Effets sur la tension artérielle
Les patients présentant une hypertension non contrôlée par voie médicamenteuse ont été exclus d'une participation aux études cliniques avant l'autorisation. Le siponimod doit être utilisé avec une grande prudence chez les patients présentant une hypertension non contrôlée.
Dans l'étude A2304, l'hypertension a été signalée plus fréquemment chez les patients atteints de SEP-SP sous siponimod (12,6%) que sous placebo (9,0%). Le traitement par le siponimod a entraîné une élévation des tensions artérielles systolique et diastolique; les valeurs de tension artérielle ont augmenté peu de temps après l'initiation du traitement et ont atteint un maximum après environ 6 mois de traitement (systolique 3 mmHg, diastolique 1,2 mmHg) pour rester stables par la suite. L'effet a persisté en cas de poursuite du traitement.
La tension artérielle doit être surveillée régulièrement pendant le traitement par le siponimod et une éventuelle hypertension artérielle doit être traitée.
Génotype du CYP2C9
Avant le début du traitement par Mayzent, le génotype du CYP2C9 du patient doit être déterminé afin d'établir son statut de métabolisation par le CYP2C9 (voir aussi «Contre-indications», «Posologie/Mode d'emploi» et «Pharmacocinétique»). Les patients porteurs homozygotes du génotype CYP2C9*3 (CYP2C9*3/*3) (environ 0,3 à 0,4% de la population caucasienne, plus rare dans d'autres groupes ethniques) ne doivent pas être traités par Mayzent, car l'utilisation de Mayzent chez ces patients entraîne des taux plasmatiques de siponimod nettement augmentés (voir aussi «Pharmacocinétique» et «Contre-indications»).
La dose d'entretien recommandée de Mayzent chez les porteurs du génotype CYP2C9*2/*3 (1,4 à 1,7% de la population) et du génotype CYP2C9*1/*3 (9 à 12% de la population) est de 1 mg une fois par jour pour éviter une exposition élevée au siponimod (voir aussi «Posologie/Mode d'emploi» et «Pharmacocinétique»).
Les effets d'autres variantes que *2 et *3 sur la pharmacocinétique du siponimod n'ont pas encore été étudiés. Bien qu'aucune étude de l'influence des allèles plus rares CYP2C9*5, *6, *8 et *11 sur le métabolisme du siponimod n'ait été réalisée, on ne peut exclure une augmentation des taux de siponimod en raison de la diminution ou de la perte de l'activité enzymatique chez les porteurs de ces polymorphismes du CYP2C9 (voir aussi «Interactions» et «Pharmacocinétique»). La fréquence totale des quatre allèles *5, *6, *8 et *11 s'élève à 10% chez les personnes africaines/d'origine africaine, à 2% chez les personnes hispaniques et à < 0,4% chez les personnes caucasiennes et asiatiques. Compte tenu des données insuffisantes disponibles, aucune recommandation ne peut être faite pour un ajustement posologique pour ces génotypes.
Aucun symptôme clinique spécifique et significatif d'une toxicité aiguë n'a été observé chez des porteurs hétérozygotes des allèles CYP2C9*2 et *3, qui sont caractérisés par une réduction du métabolisme du CYP2C9, recevant dans des études cliniques une dose non ajustée de 2 mg de siponimod. Après une exposition prolongée, une légère augmentation de la fréquence des œdèmes maculaires est survenue (voir aussi «Interactions» et «Pharmacocinétique»). On ne sait toutefois pas avec certitude si ces différences sont liées à une augmentation de l'exposition au siponimod.
Femmes en âge de procréer
En raison du risque pour le fœtus, le siponimod est contre-indiqué pendant la grossesse et chez les femmes en âge de procréer qui n'utilisent pas de méthode contraceptive fiable. Avant le début du traitement, les femmes en âge de procréer doivent être informées du risque pour le fœtus, présenter un test de grossesse négatif et utiliser une méthode contraceptive fiable pendant le traitement et pendant au moins 10 jours après l'arrêt du traitement (voir «Contre-indications», «Grossesse, Allaitement»).
Fin du traitement par le siponimod
Une aggravation sévère de la maladie, notamment un retour de l'activité de la maladie, a été signalée dans de rares cas après l'arrêt d'un autre modulateur des récepteurs de S1P. La possibilité d'une aggravation sévère de la maladie après l'arrêt du traitement par le siponimod doit être prise en compte. Après l'arrêt du siponimod, les patients doivent être surveillés afin d'identifier tout signe d'une éventuelle aggravation sévère ou d'un retour d'une activité importante de la maladie. Si nécessaire, un traitement approprié doit être initié. Après l'arrêt du traitement, Mayzent peut encore être retrouvé dans le sang pendant 10 jours. L'instauration d'autres traitements pendant cette période entraîne une exposition concomitante au siponimod.
Surveillez les patients atteints de LEMP après l'arrêt de Mayzent afin de repérer l'apparition d'un syndrome inflammatoire de reconstitution immunitaire (LEMP-IRIS) (voir mise en garde «Leuco-encéphalopathie multifocale progressive»).
Chez une large majorité (90%) des patients atteints de SEP-SP, le nombre de lymphocytes revient à la normale dans les 10 jours suivant l'arrêt du traitement. Les effets pharmacodynamiques résiduels, tels que la réduction du nombre de lymphocytes périphériques, peuvent persister pendant 3 à 4 semaines après la dernière dose. L'utilisation d'immunosuppresseurs pendant cette période peut conduire à un effet additif sur le système immunitaire et il convient donc d'être prudent pendant 3 à 4 semaines après la dernière dose.
Effet sur les examens hématologiques
Étant donné que le siponimod réduit le nombre de lymphocytes dans le sang par redistribution dans les organes lymphoïdes secondaires, le nombre de lymphocytes dans le sang périphérique ne peut pas être utilisé pour évaluer le statut des sous-groupes de lymphocytes chez les patients sous traitement par siponimod. Le nombre de lymphocytes circulants étant réduit, les examens de laboratoire des cellules mononucléées circulantes exigent de plus grandes quantités de sang.
Composants particuliers
Les comprimés contiennent des phospholipides issus de graines de soja. Les patients qui sont hypersensibles à l'arachide ou au soja ne doivent pas prendre ce médicament (voir «Contre-indications»).
Les comprimés contiennent du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
InteractionsInteractions pharmacocinétiques
Potentiel d'autres médicaments à influencer la pharmacocinétique du siponimod
Le siponimod est essentiellement métabolisé par le cytochrome P450 2C9 (CYP2C9) (79,3%) et, dans une moindre mesure, par le cytochrome P450 3A4 (CYP3A4) (18,5%). Le CYP2C9 est une enzyme polymorphe et le génotype influence la mesure dans laquelle les deux voies métaboliques oxydatives contribuent chacune à l'élimination totale. La modélisation pharmacocinétique basée sur la physiologie (PBPK) renvoie à une inhibition et une induction différenciées des voies de signalisation de CYP3A4 en fonction du génotype du CYP2C9. Ainsi, en présence de substances qui peuvent influencer le CYP3A ou le CYP2C9, les effets médicamenteux («drug-drug interactions», DDI) dépendent vraisemblablement du génotype du CYP2C9 (voir «Mises en garde et précautions» et «Pharmacocinétique»).
Les génotypes CYP2C9*5, *6, *8 et *11 sont aussi associés à une perte partielle à complète de l'activité enzymatique du CYP2C9. Aucune étude pharmacocinétique de ces polymorphismes n'a été réalisée à ce jour. Cependant, chez les porteurs de ces génotypes, des concentrations plus élevées d'autres substrats du CYP2C9, comme la phénytoïne ou la warfarine, ont été observées avec la nécessité d'ajuster la posologie de ces substrats (voir aussi «Mises en garde et précautions» et «Pharmacocinétique»).
Inhibiteurs du CYP2C9 et du CYP3A4
En raison d'une augmentation significative de l'exposition au siponimod, l'utilisation concomitante du siponimod et de médicaments qui engendrent une inhibition modérée de CYP2C9 et une inhibition modérée ou puissante du CYP3A4 n'est pas recommandée. Ce traitement concomitant peut dans ce contexte consister en un inhibiteur modéré double des CYP2C9/CYP3A4 (par exemple, le fluconazole) ou en un inhibiteur modéré du CYP2C9 en association avec un inhibiteur modéré ou puissant du CYP3A4 distinct.
L'utilisation concomitante de fluconazole (inhibiteur modéré double des CYP2C9/CYP3A4) à 200 mg par jour à l'état d'équilibre et d'une dose unique de siponimod à 4 mg chez des sujets sains ayant un génotype CYP2C9*1/*1 a conduit à un doublement de l'aire sous la courbe (ASC) du siponimod. Conformément à l'évaluation du potentiel d'interaction de substances actives de médicaments au moyen d'une modélisation pharmacocinétique basée sur la physiologie (PBPK), on s'attend pour les génotypes CYP2C9*1/*1, *1/*2, *1/*3 et *2/*3 à une augmentation au plus d'un facteur 2 de l'ASC du siponimod avec tous les types d'inhibiteurs du CYP3A4 et du CYP2C9. Chez les patients CYP2C9*2/*2, on s'attend à une augmentation de 2,7 fois de l'ASC du siponimod en présence d'inhibiteurs modérés des CYP2C9/CYP3A4. Actuellement, aucune donnée n'est disponible concernant les interactions avec les inhibiteurs du CYP2C9 et du CYP3A4 pour d'autres génotypes du CYP2C9 associés à une diminution ou une absence d'activité du CYP2C9.
Inducteurs du CYP2C9 et du CYP3A4
En raison d'une réduction cliniquement pertinente de l'exposition au siponimod, il convient d'être prudent lors de l'utilisation concomitante de Mayzent avec des médicaments inducteurs puissants du CYP3A4/modérés du CYP2C9. Ce traitement concomitant peut consister en un inducteur double modéré du CYP2C9/puissant du CYP3A4 (par exemple, rifampicine ou carbamazépine) ou un inducteur modéré du CYP2C9 en association avec un inducteur puissant du CYP3A4 distinct.
Il convient aussi d'être prudent en cas d'utilisation concomitante de Mayzent avec des inducteurs modérés du CYP3A4 (par exemple, modafinil) ou des inducteurs puissants du CYP3A4 chez des patients ayant un génotype CYP2C9*1/*3 ou *2/*3, pour lesquels un ajustement posologique est recommandé (voir «Recommandations posologiques»). Aucune donnée concernant les interactions avec les inducteurs du CYP2C9 et du CYP3A4 n'est actuellement disponible pour d'autres génotypes du CYP2C9 associés à une réduction ou une absence d'activité du CYP2C9.
Selon les études cliniques des effets de médicaments et les tests in silico (pharmacocinétique basée sur la physiologie), on s'attend à ce que les inducteurs puissants du CYP3A4/modérés du CYP2C9 (par exemple, carbamazépine) et les inducteurs modérés du CYP3A4 (par exemple, modafinil) réduisent l'exposition au siponimod de respectivement jusqu'à 76% et 51%.
L'administration concomitante de siponimod 2 mg par jour et de rifampicine 600 mg par jour (inducteur puissant du CYP3A4 et modéré du CYP2C9) a réduit l'ASCtau,ss et la Cmax,ss du siponimod de respectivement 57% et 45% chez les patients ayant un génotype CYP2C9*1/*1.
Le siponimod n'est pas un substrat des transporteurs d'efflux Pgp, BCRP ou MRP. On suppose donc que les médicaments qui influencent l'activité de ces transporteurs n'ont pas d'effet sur la pharmacocinétique du siponimod.
L'absorption du siponimod dans les hépatocytes a lieu uniquement par diffusion passive. Par conséquent, des interactions entre le siponimod et les transporteurs hépatiques d'absorption (OATP, OCT, OAT) ne sont pas attendues.
Potentiel du siponimod à influencer la pharmacocinétique d'autres médicaments
Les études in vitro montrent que le siponimod et ses métabolites (M17 et M3), dans les concentrations thérapeutiquement pertinentes, inhibent peu ou pas du tout l'activité des enzymes CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5) ou les activent peu ou pas du tout (CYP1A2, CYP2B6, CYP2C9 et CYP3A4/5).
Sur la base de données in vitro, aucun effet inhibiteur du siponimod et de ses métabolites (M17 et M3) n'est attendu sur l'absorption des médicaments et/ou principes actifs biologiques utilisés simultanément qui sont transportés par OATP1B1, OATP1B3, OAT1, OAT3, OCT1 ou OCT2. On estime que, aux concentrations thérapeutiques, il n'y a aucune inhibition de l'efflux des médicaments et/ou principes actifs biologiques utilisés simultanément qui sont transportés par BCRP, BSEP, MATE1, MATE2K ou par Pgp.
Interactions pharmacodynamiques
Traitements antinéoplasiques, immunomodulateurs ou immunosuppresseurs
Mayzent n'a pas été étudié en association avec des traitements antinéoplasiques, immunomodulateurs ou immunosuppresseurs. En cas d'administration concomitante, la prudence s'impose, car il existe un risque d'effets additifs sur le système immunitaire pendant un tel traitement et dans les semaines qui suivent la fin de l'administration d'un de ces médicaments (voir «Mises en garde et précautions»).
Lors du passage depuis d'autres thérapies modifiant la maladie, la demi-vie et le mécanisme d'action de l'autre traitement doivent être pris en compte pour éviter un effet additif sur le système immunitaire et minimiser le risque d'une réactivation de la maladie.
En raison des propriétés et de la durée des effets immunosuppresseurs de l'alemtuzumab décrits dans l'information sur le produit, l'initiation d'un traitement par Mayzent après l'alemtuzumab n'est pas recommandée, à moins que le bénéfice du traitement par Mayzent soit nettement supérieur aux risques pour le patient.
La prise de Mayzent peut être immédiatement commencée après l'arrêt de l'interféron bêta ou de l'acétate de glatiramère.
Antiarythmiques, médicaments allongeant l'intervalle QT et médicaments pouvant réduire la fréquence cardiaque
Pendant l'initiation du traitement, en raison des effets additifs potentiels sur la fréquence cardiaque, Mayzent ne doit pas être utilisé de façon concomitante chez les patients qui prennent des antiarythmiques de la classe Ia (par exemple, quinidine, procaïnamide), de la classe III (par exemple, amiodarone, sotalol), des médicaments allongeant l'intervalle QT ayant des propriétés arythmogènes connues, des inhibiteurs calciques réduisant la fréquence cardiaque (tels que vérapamil ou diltiazem) ou d'autres principes actifs qui peuvent réduire la fréquence cardiaque (par exemple, ivabradine ou digoxine). Lorsqu'un traitement par Mayzent est envisagé, L'avis d'un cardiologue doit être sollicité avant le début du traitement (voir «Mises en garde et précautions»).
Bêtabloquants
Lorsque le siponimod est initié chez des patients qui prennent des bêtabloquants, la prudence est de rigueur en raison des effets additifs sur la diminution de la fréquence cardiaque (voir «Mises en garde et précautions»). Un traitement par bêtabloquant peut être initié chez des patients qui reçoivent Mayzent à dose stable.
L'effet chronotrope négatif de l'administration concomitante du siponimod et du propranolol a été évalué dans une étude spécifique sur la pharmacodynamique et la sécurité. Lorsque le propranolol a été administré en plus du siponimod à l'état d'équilibre pharmacocinétique, les effets chronotropes négatifs ont été moins prononcés (plus faibles qu'additifs) que lors de l'administration du siponimod en plus du propranolol à l'état d'équilibre pharmacocinétique (effet additif sur la fréquence cardiaque).
Vaccination
Vaccins vivants atténués
L'utilisation de vaccins vivants atténués (par exemple, vaccin contre la varicelle et vaccin contre la fièvre jaune) peut être associée à un risque d'infection et doit donc être évitée pendant la prise de Mayzent et pendant 4 semaines après la fin du traitement par Mayzent (voir «Mises en garde et précautions»).
Autres types de vaccins
Pendant le traitement par Mayzent et pendant 4 semaines après le traitement, l'efficacité des vaccinations peut être affectée. L'efficacité d'une vaccination n'est pas considérée comme perturbée lorsque le traitement par le siponimod est interrompu pendant 1 semaine avant et 4 semaines après la vaccination (voir «Reprise du traitement d'entretien après interruption du traitement»). Dans une étude spécifique de phase I incluant des volontaires sains, après un traitement préalable par le siponimod d'une durée maximale de 10 jours, la poursuite de l'administration ou une interruption plus courte du traitement de 10 jours avant à 14 jours après la vaccination a entraîné une diminution d'environ 15 à 30% des taux de réponse à un vaccin grippal quadrivalent comparativement au placebo, tandis que le traitement concomitant par le siponimod n'a pas modifié de manière significative les taux de réponse à une vaccination PPV-23 comparativement au placebo (voir «Mises en garde et précautions»).
Contraceptifs oraux
L'efficacité des contraceptifs oraux étudiés (association d'éthinylestradiol et de lévonorgestrel) a été maintenue sous traitement par le siponimod. Le siponimod n'a présenté aucune influence sur la pharmacodynamique des contraceptifs (estradiol, progestérone; FSH, LH, taille du follicule, score de Hoogland, SHBG). Par comparaison avec l'administration seule de contraceptifs oraux, l'administration concomitante du siponimod a augmenté de 1,29 fois l'aire sous la courbe pendant l'administration (ASCtau) du lévonorgestrel (geometric mean ratio (GMR, rapport de moyenne géométrique): 1,29, IC à 90%: 1,24–1,34) et la concentration plasmatique maximale à l'équilibre (Cmax,ss) de 1,18 fois (GMR: 1,18, IC à 90%: 1,11–1,26). Le siponimod n'influence pas la pharmacocinétique de l'éthinylestradiol (ASCtau GMR: 1,00, IC à 90%: 0,96–1,05; Cmax,ss GMR: 1,02, IC à 90%: 0,96–1,08).
Aucune étude d'interaction avec des contraceptifs oraux qui contiennent d'autres gestagènes n'a été réalisée.
Grossesse, allaitementFemmes en âge de procréer/contraception chez les femmes
Mayzent est contre-indiqué chez les femmes en âge de procréer qui n'utilisent pas de méthodes contraceptives fiables (voir «Contre-indications»).
Il convient donc d'informer les femmes en âge de procréer du fait que les études animales ont montré que le siponimod est nuisible au fœtus en développement (voir «Données précliniques»). Pour les patientes en âge de procréer, il faut disposer d'un test de grossesse négatif avant de commencer le traitement par le siponimod. Les patientes doivent utiliser des méthodes contraceptives efficaces lors de l'utilisation de Mayzent et pendant au moins dix jours après la fin du traitement par Mayzent (méthodes qui entraînent moins de 1% de grossesses) (voir «Mises en garde et précautions»).
Si le traitement par le siponimod est interrompu dans le but de planifier une grossesse, un retour éventuel de l'activité pathologique doit être envisagé.
Grossesse
On ne dispose d'aucune donnée sur l'utilisation de Mayzent chez les femmes enceintes, permettant de démontrer un risque lié au médicament d'événement indésirable du développement.
Les études animales ont montré des embryo- et fœtotoxicités engendrées par le siponimod chez des rats et des lapins, ainsi que des effets tératogènes chez des rats, y compris des cas de mort embryofoetale et d'anomalies squelettiques ou viscérales, lors d'une exposition comparable à l'exposition humaine à une dose quotidienne de 2 mg (voir «Données précliniques»). En outre, les expériences cliniques avec un autre modulateur des récepteurs de S1P lors d'une utilisation pendant la grossesse ont montré un risque d'anomalies congénitales graves 2 fois plus élevé en comparaison avec le taux observé dans la population générale.
Par conséquent, le siponimod est contre-indiqué pendant la grossesse (voir «Contre-indications»). Le siponimod doit être arrêté au moins 10 jours avant la planification d'une grossesse (voir «Mises en garde et précautions»). En cas de grossesse pendant le traitement, le siponimod doit être arrêté. Un conseil médical doit être donné concernant le risque d'effets néfastes du traitement sur le fœtus et une échographie doit être réalisée.
Allaitement
On ignore si le siponimod ou de ses métabolites principaux passent dans le lait maternel chez les humains. Chez le rat, le siponimod et ses métabolites sont excrétés dans le lait. Le siponimod ne doit pas être utilisé pendant l'allaitement.
Fertilité
L'effet du siponimod sur la fertilité chez l'homme n'a pas été étudié. Le siponimod n'affecte pas les organes reproducteurs mâles des rats et des singes ni les paramètres de fertilité des rats.
Effet sur l’aptitude à la conduite et l’utilisation de machinesMayzent n'a aucune influence ou a une influence négligeable sur l'aptitude à la conduite et sur l'utilisation de machines.
Toutefois, au début du traitement par le siponimod, des vertiges et des bradyarythmies peuvent survenir. C'est pourquoi, pendant les premiers jours de traitement par le siponimod, les patients ne doivent pas conduire de véhicule ni utiliser de machines (voir «Mises en garde et précautions»).
Effets indésirablesDans l'étude clinique de phase 3, A2304, 1651 patients atteints de SEP-SP ont été randomisés selon un ratio 2:1 dans le groupe Mayzent, 2 mg une fois par jour, ou le groupe placebo. La durée médiane du traitement s'élevait à 18 mois (plage de 0 à 37 mois). Au moment de l'autorisation, les données de sécurité à long terme étaient très limitées. Les effets indésirables les plus fréquents sous siponimod 2 mg sont les céphalées (15,2%) et l'hypertension (12,6%).
Les effets indésirables mis en évidence dans les études cliniques ont été définis essentiellement sur la base des expériences découlant de l'étude pivot A2304 (tableau 2) et sont classés par classes de systèmes d'organes selon la classification MedDRA.
Pour chaque classe de systèmes d'organes, les effets indésirables médicamenteux sont répertoriés en fonction de la fréquence, les effets indésirables médicamenteux les plus fréquents étant nommés en premier. En outre, la catégorie de fréquence respective pour chaque effet indésirable médicamenteux se base sur les définitions de fréquence suivantes selon la convention (CIOMS III): très fréquent (≥1/10); fréquent (≥1/100 à < 1/10); occasionnel (≥1/1000 à < 1/100); rare (≥1/10 000 à < 1/1000); très rare (< 1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 2 Tableau récapitulatif des effets indésirables
|
Infections et infestations
| |
Fréquent
|
Zona
| |
Occasionnel
|
Méningite à cryptocoque*#
| |
Rare
|
Leucoencéphalopathie multifocale progressive*#
| |
Tumeurs bénignes, malignes et non précisées (incl kystes et polypes)
| |
Fréquent
|
Naevus mélanocytaire#
Carcinome basocellulaire*#
| |
Occasionnel
|
Carcinome épidermoïde*#
Mélanome malin
| |
Affections hématologiques et du système lymphatique
| |
Fréquent
|
Lymphopénie
| |
Affections du système nerveux
| |
Très fréquent
|
Céphalées
| |
Fréquent
|
Vertiges
Convulsions
Tremblements
| |
Affections oculaires
| |
Fréquent
|
Œdème maculaire
| |
Affections cardiaques
| |
Fréquent
|
Bradycardie
Bloc auriculoventriculaire (1er et 2e degré)
| |
Affections vasculaires
| |
Très fréquent
|
Hypertension#
| |
Affections gastro-intestinales
| |
Fréquent
|
Nausées, diarrhée
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Fréquent
|
Douleurs dans les extrémités
| |
Troubles généraux et anomalies au site d'administration
| |
Fréquent
|
Œdème périphérique
Asthénie
| |
Investigations
| |
Très fréquent
|
Élévation des valeurs aux tests de la fonction hépatique
| |
Fréquent
|
Diminution des valeurs aux tests de la fonction pulmonaire
|
# voir également «Mises en garde et précautions»
* Effets indésirables des médicaments issus de l'extension en ouvert de l'étude de phase 3 A2304
Description d'effets indésirables sélectionnés du médicament
Infections
Dans l'étude A2304, le taux total d'infections chez les patients atteints de sclérose en plaques secondairement progressive (SEP-SP) était comparable entre les patients sous siponimod et ceux sous placebo (49,0% vs 49,1%). Cependant, une augmentation des infections de type zona a été rapportée sous siponimod (2,5%) par comparaison avec le placebo (0,7%). Aucune autre augmentation du taux d'incidence (TI) des infections par le virus varicelle-zona n'a été observée en cas d'exposition chronique. Des cas de méningite ou de méningo-encéphalite causés par le virus varicelle-zona ont également été rapportés sous traitement par Mayzent (voir «Mises en garde et précautions»).
Des cas de leucoencéphalopathie multifocale progressive (LEMP) et de méningite à cryptocoque ont été rapportés sous Mayzent (voir «Mises en garde et précautions» et «Contre-indications»).
Œdème maculaire
Un œdème maculaire a été rapporté plus fréquemment chez les patients sous siponimod (1,8%) que chez ceux sous placebo (0,2%). Bien que la plupart des cas soient survenus dans les 3 à 4 mois après l'initiation du siponimod, des cas ont également été signalés chez des patients traités par le siponimod depuis plus de 6 à 12 mois (voir «Mises en garde et précautions»). Certains patients ont présenté une vision floue ou une réduction de l'acuité visuelle, d'autres par contre étaient asymptomatiques et n'ont été diagnostiqués qu'au cours d'un examen ophtalmologique de routine. Après l'arrêt du médicament, l'œdème maculaire a généralement régressé ou s'est résorbé spontanément. Le risque de récidive suite à une nouvelle exposition n'a pas été étudié.
Bradycardie
L'initiation du traitement par le siponimod entraîne une réduction temporaire de la fréquence cardiaque et peut être associée en outre, à un retard de la conduction auriculoventriculaire (voir «Mises en garde et précautions»). Une bradycardie a été rapportée chez 6,2% des patients traités par le siponimod contre 3,1% des patients sous placebo, et un bloc AV a été observé chez 1,7% des patients traités par le siponimod contre 0,7% des patients sous placebo.
La baisse maximale de la fréquence cardiaque se manifeste au cours des 6 premières heures après la prise de la dose.
Une réduction temporaire, dépendante de la dose, de la fréquence cardiaque a été observée pendant la phase de titration initiale et s'est stabilisée aux doses ≥5 mg. Les événements bradyarythmiques (blocs AV et pauses sinusales) ont été détectés avec une plus haute incidence sous traitement par le siponimod que sous placebo.
La plupart des blocs AV et des pauses sinusales se sont produits à des doses supérieures à la dose thérapeutique de 2 mg. L'incidence a été nettement plus élevée lorsqu'aucune titration de la dose n'a été effectuée.
La chute de la fréquence cardiaque provoquée par le siponimod peut être inversée par l'atropine ou l'isoprénaline.
Tests de la fonction hépatique
Des élévations des taux d'enzymes hépatiques (le plus souvent, élévation de l'ALAT) ont été rapportées chez des patients atteints de SEP traités par le siponimod. Dans l'étude A2304 menée auprès de patients atteints de SEP-SP, une augmentation des paramètres hépatiques a été observée plus fréquemment chez les patients sous siponimod (11,3%) que chez les patients sous placebo (3,1%), essentiellement en raison des augmentations des transaminases hépatiques (ALAT/ASAT/GGT). La plupart des élévations se sont produites dans les 6 mois suivant le début du traitement. Les taux d'ALAT sont revenus à la normale dans un délai d'environ 1 mois après l'arrêt du siponimod (voir «Mises en garde et précautions»).
Tension artérielle
Dans l'étude clinique de phase 3 menée auprès de patients atteints de SEP-SP, une hypertension a été plus fréquemment signalée chez les patients sous siponimod (12,6%) que chez ceux sous placebo (9,0%) (voir «Mises en garde et précautions»).
Convulsions
Dans l'étude A2304 menée auprès de patients atteints de SEP-SP, des convulsions ont été plus fréquemment signalées chez les patients sous siponimod (1,7%) que chez ceux sous placebo (0,4%). On ne sait pas si ces événements doivent être attribués aux effets de la SEP, du siponimod ou à une association des deux.
Effets sur les voies respiratoires
Lors du traitement par le siponimod, de faibles réductions des valeurs du volume expiratoire maximal en 1 seconde (VEMS) et de la capacité de diffusion pulmonaire du monoxyde de carbone (DLCO) ont été observées. Après le mois 3 et le mois 6 de traitement dans l'étude A2304 menée auprès de patients atteints de SEP-SP, la variation moyenne par rapport à la valeur initiale était de 0,1 litre (l) dans le groupe siponimod à chaque échéance, alors qu'aucune variation n'a été observée dans le groupe placebo. Ces observations étaient légèrement plus élevées (variation moyenne du VEMS d'environ 0,15 l par rapport à la valeur initiale) chez les patients atteints d'affections respiratoires comme la bronchopneumopathie chronique obstructive (BPCO) ou l'asthme et traités par le siponimod. En cas de traitement à long terme, cette réduction n'a pas entraîné d'événements indésirables cliniquement significatifs et n'a pas été associée à une augmentation des signalements de toux ou de dyspnée.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes volontaires sains ont reçu le siponimod à une dose unique (0,1 à 75 mg) ou en doses multiples (0,25 à 20 mg). La dose unique maximale tolérée a été définie à 25 mg sur la base de l'apparition d'une bradycardie symptomatique après une dose unique de 75 mg. La dose multiple étudiée la plus élevée de 20 mg pendant 28 jours a été bien tolérée (9 participants ont reçu le dernier jour la posologie de 100 mg et 5 participants ont reçu, par erreur, jusqu'à 200 mg par jour pendant 3 à 4 jours). Certains des 9 participants ont présenté des élévations légères à modérées des paramètres hépatiques, tout en étant asymptomatiques.
Un patient (avec des antécédents de dépression) a pris 84 mg de siponimod. À part une légère élévation des transaminases hépatiques, aucun autre événement indésirable en raison du surdosage n'est apparu chez ce patient.
Lorsque le surdosage est une première exposition à Mayzent ou survient pendant la phase de titration de Mayzent, il est important de surveiller les signes et symptômes de bradycardie, la surveillance pouvant se poursuivre pendant toute la nuit. Des mesures régulières du pouls et de la tension artérielle sont nécessaires et un électrocardiogramme doit être effectué (voir «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Il n'existe aucun antidote spécifique pour le siponimod. Ni la dialyse ni l'échange plasmatique ne conduisent à une élimination significative du siponimod de l'organisme.
Propriétés/EffetsCode ATC
L04AE03
Groupe pharmacothérapeutique: modulateurs des récepteurs de la sphingosine 1phosphate (S1P)
Mécanisme d'action
Le siponimod est un modulateur des récepteurs de la sphingosine 1phosphate (S1P). Le siponimod se lie sélectivement à deux des cinq récepteurs de S1P couplés à la protéine G (GPCR): S1P1 et S1P5. Le siponimod agit en tant qu'antagoniste fonctionnel sur les récepteurs de S1P1 des lymphocytes et empêche ainsi les lymphocytes de sortir des ganglions lymphatiques. Cela diminue la recirculation des lymphocytes T dans le système nerveux central (SNC) et limite ainsi l'inflammation centrale. Le siponimod passe la barrière hématoencéphalique. Il n'a aucune influence durable sur les lymphocytes T à mémoire effecteurs des tissus périphériques et dans le sang et n'influence pas l'activation des lymphocytes.
Pharmacodynamique
Dans les études animales, les effets directs du siponimod sur les cellules nerveuses ont été détectés, par le S1P1 sur les astrocytes et par le S1P5 sur les oligodendrocytes. Dans un modèle de souris de l'encéphalomyélite auto-immune expérimentale, un effet neuroprotecteur direct, indépendant des effets sur les lymphocytes, a été également mis en évidence pour le siponimod utilisé centralement (par perfusion intracérébroventriculaire).
Système immunitaire
Mayzent induit une réduction dose-dépendante du nombre de lymphocytes dans le sang périphérique dans les 6 heures qui suivent la première dose, ce qui est dû à la rétention réversible (séquestration) des lymphocytes dans les tissus lymphoïdes.
En cas d'administration quotidienne continue, le nombre de lymphocytes diminue en continu et atteint une valeur minimale médiane (IC à 90%) d'environ 0,560 (0,271 à 1,08) cellules/nl chez un patient type atteint de SEP-SP, présentant le génotype CYP2C9*1/*1 ou CYP2C9*1/*2, non originaire du Japon, ce qui correspond à une réduction de 20 à 30% par rapport à la valeur initiale. Lors d'une prise quotidienne, les faibles nombres de lymphocytes sont maintenus.
Chez une large majorité (90%) des patients atteints de SEP-SP, le nombre de lymphocytes revient à la normale dans les 10 jours suivant l'arrêt du traitement. Après l'arrêt du traitement par Mayzent, la réduction du nombre de lymphocytes périphériques peut persister pendant 3 à 4 semaines après la dernière dose.
Électrophysiologie cardiaque
Fréquence et rythme cardiaques
Mayzent entraîne une diminution temporaire de la fréquence cardiaque et de la conduction auriculoventriculaire au début du traitement (voir «Effets indésirables»). Ceci est lié à une activation des canaux potassiques rectifiants entrants couplés aux protéines G (G-protein-coupled inwardly rectifying potassium, GIRK) via la stimulation des récepteurs de S1P1, ce qui conduit à une hyperpolarisation cellulaire et une excitabilité réduite. En raison de son antagonisme fonctionnel sur les récepteurs de S1P1, la titration initiale du siponimod désensibilise les canaux GIRK jusqu'à ce que la dose d'entretien soit atteinte.
Potentiel d'allongement de l'intervalle QT
Les effets des doses thérapeutiques (2 mg) et suprathérapeutiques (10 mg) du siponimod sur la repolarisation cardiaque ont été étudiés dans une étude détaillée de l'intervalle QT. Les résultats n'indiquent pas un potentiel arythmogène en lien avec l'allongement de l'intervalle QT avec le siponimod, car ce dernier a augmenté l'intervalle QTcF moyen ajusté par rapport à la valeur initiale et corrigé par rapport au placebo (ΔΔQTcF) de plus de 5 ms, avec un effet moyen maximal de respectivement 7,8 ms (2 mg) et 7,2 ms (10 mg) 3 heures après l'administration. La limite supérieure de l'IC à 95% unilatéral pour l'intervalle ΔΔQTcF est restée à tout moment inférieure à 10 ms. L'analyse catégorielle n'a mis en évidence aucune valeur QTc conditionnée par le traitement supérieure à 480 ms, aucune augmentation de l'intervalle QTc de plus de 60 ms par rapport à la valeur initiale et aucune valeur de QT/QTc corrigée ou non corrigée dépassant 500 ms.
Fonction pulmonaire
L'administration unique ou multiple de Mayzent pendant 28 jours n'est pas associée à une augmentation cliniquement pertinente de la résistance respiratoire, mesurée par le volume expiratoire maximal en 1 seconde (VEMS) à 25 et à 75% du volume pulmonaire (DEM25-75%). Aux doses uniques non thérapeutiques (> 10 mg), on observe une légère tendance à une réduction du VEMS. Le traitement concomitant par Mayzent et le propranolol a entraîné une baisse minimale du VEMS par rapport au propranolol seul, les modifications avec le médicament seul ou avec l'association se situant dans la variabilité physiologique du VEMS et étant cliniquement non significatives.
Efficacité clinique
L'efficacité de Mayzent a été étudiée dans une étude de phase 3 dans laquelle une dose de 2 mg de Mayzent une fois par jour a été évaluée chez des patients atteints de SEP-SP. Une étude de détermination de la dose de phase 2 menée chez des patients atteints de SEP-RR a indiqué une réduction dose-dépendante des lésions inflammatoires à l'IRM et a montré que Mayzent à 2 mg produit un effet presque maximal.
Étude A2304 (EXPAND) pour la SEP-SP
L'étude A2304 était une étude de phase 3 randomisée, en double aveugle, contrôlée contre placebo, axée sur les événements et la durée de suivi, menée auprès de patients atteints de SEP-SP qui ont présenté au cours des 2 dernières années une progression détectable, en l'absence ou indépendamment de poussées, aucune indication de poussée au cours des 3 mois précédant le début de l'étude et un score EDSS (Expanded Disability Status Scale, échelle étendue du handicap) médian de 3,0 à 6,5 au début de l'étude.
Le score EDSS médian à l'inclusion était de 6,0. Les patients de plus de 61 ans n'ont pas été inclus dans l'étude. En ce qui concerne l'activité de la maladie, les paramètres caractéristiques de l'activité inflammatoire de la SEP-SP pouvaient être définis par des poussées ou par l'imagerie (à savoir, lésions en T1 rehaussées par agent de contraste ou lésions en T2 actives [nouvelles ou en croissance]).
Les patients ont été randomisés selon un ratio de 2:1 pour recevoir Mayzent 2 mg une fois par jour ou un placebo. Les évaluations cliniques ont été réalisées à la sélection, ainsi que tous les 3 mois et au moment d'une poussée. Les évaluations par IRM ont été réalisées à la sélection et tous les 12 mois.
Le critère d'évaluation principal de l'étude était le délai jusqu'à une progression du handicap confirmée à 3 mois (confirmed disability progression, CDP), qui était déterminée par une augmentation d'au moins 1 point par rapport à la valeur initiale du score EDSS (augmentation de 0,5 point pour les patients ayant un score EDSS de 5,5 ou plus) à 3 mois. Les critères d'évaluation secondaires importants étaient le délai jusqu'à une détérioration confirmée à 3 mois d'au moins 20% par rapport à l'inclusion du résultat au test chronométré de marche sur une distance de 25 pieds (timed 25 foot walk test, T25FW) et la variation du volume des lésions en T2 par rapport à l'inclusion. D'autres critères d'évaluation secondaires étaient le délai jusqu'à une CDP à 6 mois, le pourcentage de variation du volume cérébral et les mesures de l'activité inflammatoire de la maladie (taux annualisé de poussées, lésions à l'IRM). La modification de la vitesse de traitement cognitif dans le test de substitution symboles/chiffres (Symbol Digit Modalities Test) était un critère exploratoire.
La durée de l'étude a été variable d'un patient à l'autre (durée médiane de l'étude: 21 mois, plage de 1 jour à 37 mois).
1651 patients ont été randomisés dans cette étude pour recevoir soit Mayzent 2 mg (N = 1105) soit le placebo (N = 546); 82% des patients traités par Mayzent et 78% des patients sous placebo ont terminé l'étude. L'âge médian était de 49,0 ans, la durée médiane de la maladie de 16,0 ans et le score EDSS médian de 6,0 au début de l'étude; 64% des patients n'avaient eu aucune poussée au cours des 2 années précédant le début de l'étude et 76% n'avaient aucune lésion rehaussée par le gadolinium (Gd) lors de l'IRM initiale; 78% des patients avaient reçu auparavant un traitement pour la SEP.
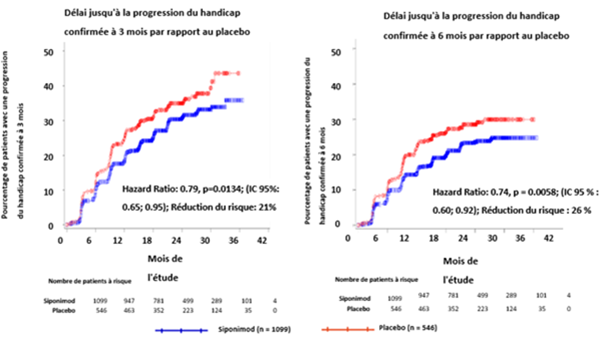
Le délai jusqu'à l'apparition d'une progression du handicap confirmée à 3 et à 6 mois était significativement allongé sous siponimod, avec une réduction du risque de progression du handicap confirmée à 3 mois de 21% par rapport au placebo (hazard ratio [HR] 0,79, p = 0,0134) et une réduction du risque de progression du handicap confirmée à 6 mois de 26% par rapport au placebo (HR 0,74, p = 0,0058).
Les résultats de cette étude sont résumés dans le tableau 3 et dans les figures 1 et 2.
Tableau 3 Résultats cliniques et d'IRM de l'étude A2304
|
Critères d'évaluation
|
A2304 (EXPAND)
| |
Siponimod 2 mg
(n = 1099)
|
Placebo
(n = 546)
| |
Critères d'évaluation cliniques
| |
Critère d'évaluation principal d'efficacité: proportion de patients présentant une progression du handicap confirmée à 3 mois (critère d'évaluation principal)
|
26,3%
|
31,7%
| |
Réduction du risque1
|
21% (p = 0,0134)
| |
Proportion de patients présentant une augmentation confirmée à 3 mois de 20% du résultat au test de marche de 25 pieds
|
39,7%
|
41,4%
| |
Réduction du risque1
|
6% (p = 0,4398)
| |
Proportion de patients présentant une progression du handicap confirmée à 6 mois
|
19,9%
|
25,5%
| |
Réduction du risque1
|
26% [(p = 0,0058)]6
| |
Taux annualisé de poussées (TAP)
|
0,071
|
0,152
| |
Réduction du taux2
|
55% [(p < 0,0001)]6
| |
Critères d'évaluation IRM
| |
Modification du volume des lésions en T2 (mm3) par rapport à la valeur initiale3
|
+184 mm3
|
+879 mm3
| |
Différence de variation du volume des lésions en T2
|
-695 mm3 (p < 0,0001)7
| |
Pourcentage de variation du volume cérébral par rapport à la valeur initiale (IC à 95%)3
|
-0,497%
|
-0,649%
| |
Différence du pourcentage de variation du volume cérébral
|
0,152% [(p = 0,0002)]6
| |
Nombre cumulé moyen de lésions rehaussées par le Gd, pondérées en T1 (IC à 95%)4
|
0,081
|
0,596
| |
Réduction du taux
|
86% [(p < 0,0001)]6
| |
Proportion de patients présentant une aggravation de 4 points au test de substitution symboles/chiffres (Symbol Digit Modalities Test)5
|
16,0%
|
20,9%
| |
Réduction du risque1
|
25% [(p = 0,0163)]6
| |
1
D'après les modèles de Cox pour le délai jusqu'à la progression
2 D'après un modèle pour les événements récurrents
3 Moyenne au mois 12 et au mois 24
4 Jusqu'au mois 24
5 Confirmée à 6 mois
6 [Valeur p nominale pour les critères d'évaluation non inclus dans les tests hiérarchiques et non ajustés en fonction de la multiplicité]
7 Valeur p non confirmée; le procédé de test hiérarchique a été terminé avant l'atteinte du critère d'évaluation
|
Figure 1 Patients présentant une progression du handicap confirmée à 3 et à 6 mois, à l'aide des courbes de Kaplan-Meier du score EDSS (ensemble d'analyse complet, étude A2304)
Les résultats de l'étude ont montré une réduction du risque cohérente concernant le délai jusqu'à la progression du handicap confirmée à 3 mois et à 6 mois avec Mayzent par rapport au placebo dans des sous-groupes définis selon le sexe, l'âge, le traitement précédent de la sclérose en plaques, l'activité des poussées avant l'étude, l'activité de la maladie par IRM à l'inclusion, la durée de la maladie et le niveau de handicap à l'inclusion.
Mayzent a montré un effet positif au test de substitution symboles/chiffres (Symbol Digit Modalities Test, SDMT). Pour Mayzent, la variation par rapport aux valeurs initiales a été stable ou meilleure et elle s'est dégradée pour le placebo avec une différence significative entre les groupes de 1,1 point au mois 12 (p = 0,0132) et de 2,3 points au mois 24 (p = 0,0002). Dans une étude exploratoire, Mayzent a réduit le risque d'une détérioration confirmée de 4 points du SDMT à 6 mois de 25% (p = 0,0163) par rapport au placebo. Une détérioration de 4 points s'est révélée être déjà cliniquement significative.
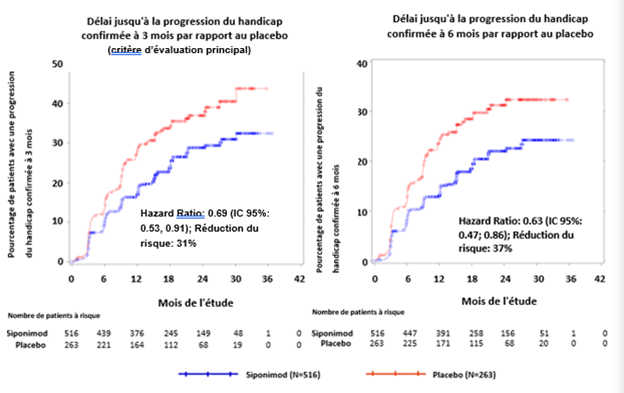
Dans le sous-groupe des patients (47,1%, n = 779) présentant une maladie active (définie comme les patients présentant une poussée dans les 2 années précédant l'étude et/ou la présence de lésions en T1 rehaussées par le Gd au début de l'étude), les caractéristiques à l'inclusion étaient similaires à celles de la population globale. L'âge médian était de 47 ans, la durée médiane de la maladie était de 15 ans et le score EDSS médian au début de l'étude était de 6,0 (voir «Pharmacocinétique»).
Le délai jusqu'à l'apparition d'une progression du handicap confirmée à 3 et à 6 mois a été significativement prolongé pour les patients présentant une maladie active traités par le siponimod, de 31% par rapport au placebo (hazard ratio [HR] 0,69; IC à 95%: 0,53; 0,91) et de 37% par rapport au placebo (HR 0,63; IC à 95%: 0,47; 0,86). Le TAP (poussées confirmées) a été réduit de 46% par rapport au placebo (rapport de TAP 0,54; IC à 95%: 0,39; 0,77). La réduction relative du nombre cumulé de lésions pondérées en T1, rehaussées par le Gd sur 24 mois a été de 85% (risque relatif 0,155; IC à 95%: 0,104; 0,231) par rapport au placebo. Les différences de modification du volume des lésions en T2 et du pourcentage de modification du volume cérébral (moyenne sur les mois 12 et 24) par rapport au placebo ont été de 1163 mm3 (IC à 95%: 1484, 843 mm3) et 0,141% (IC à 95% 0,020; 0,261%), respectivement.
Dans le sous-groupe de patients (n = 827) sans signe ni symptôme d'activité de la maladie (définie comme les patients sans poussée dans les 2 années précédant l'étude et sans présence de lésions en T1 rehaussées par agent de contraste au début de l'étude), les effets sur la progression du handicap confirmée à 3 et à 6 mois ont été faibles (la réduction du risque a été de 7% et 13%, respectivement).
Figure 2 Patients présentant une progression du handicap confirmée à 3 et à 6 mois, à l'aide des courbes de Kaplan-Meier du score EDSS – sous-groupe présentant une maladie inflammatoire active (ensemble d'analyse complet, étude A2304)
PharmacocinétiqueAbsorption
Le temps (Tmax) nécessaire pour atteindre les concentrations plasmatiques maximales (Cmax) après plusieurs administrations orales de siponimod est d'environ 4 heures (plage de 2 à 12 heures). La biodisponibilité orale absolue du siponimod est d'environ 84%. Pour 2 mg de siponimod administrés une fois par jour pendant 10 jours, on mesure au jour 10, une Cmax moyenne de 30,4 ng/ml et une ASCtau moyenne de 558 h*ng/ml. L'état d'équilibre est atteint après environ 6 jours d'une administration de doses répétées de siponimod une fois par jour.
La prise de nourriture n'a eu aucune influence sur l'exposition systémique au siponimod (Cmax et ASC). Dès lors, Mayzent peut être pris indépendamment de l'heure des repas.
Distribution
Le siponimod est distribué dans les tissus corporels avec un volume moyen de distribution de 124 l. La proportion de siponimod dans le plasma par rapport au sang complet est de 68% chez l'homme. Les études animales montrent que le siponimod passe aisément la barrière hématoencéphalique. Chez les personnes saines et chez les insuffisants hépatiques et rénaux, la liaison du siponimod aux protéines est > 99,9%.
Métabolisme
Le siponimod est largement métabolisé, principalement par le CYP2C9 (79,3%), suivi du CYP3A4 (18,5%).
L'activité pharmacologique des métabolites principaux M3 et M17 ne contribue vraisemblablement pas à l'effet clinique ni à la sécurité du siponimod chez l'homme.
Élimination
On estime pour les patients atteints de SEP une clairance systémique apparente (CL/F) de 3,11 l/h. La demi-vie d'élimination pertinente d'un point de vue pharmacocinétique est d'environ 30 heures.
Le siponimod est éliminé de la circulation systémique essentiellement par le métabolisme et l'excrétion ultérieure de la bile/selles. Environ 86,7% de la dose de siponimod est excrétée par les selles, dont 9,2% sous forme inchangée. Seules de faibles quantités sont excrétées par les urines (3,6%). On n'a pas détecté de siponimod inchangé dans les urines.
La demi-vie moyenne des métabolites du siponimod M17 et M3 après administration orale est respectivement d'environ 155 h et 30 h.
Linéarité/non-linéarité
La concentration du siponimod augmente de manière presque proportionnelle à la dose après plusieurs administrations de 0,3 mg à 20 mg de siponimod une fois par jour.
La concentration plasmatique à l'état d'équilibre est atteinte après environ 6 jours d'administration une fois par jour et les taux à l'état d'équilibre sont environ 2 à 3 fois plus élevés que les taux après la dose initiale. Un schéma de titration de dose est utilisé pour atteindre graduellement la dose thérapeutique clinique de 2 mg de siponimod après 6 jours et 4 jours d'administration supplémentaires sont nécessaires pour atteindre les concentrations plasmatiques de l'état d'équilibre.
Cinétique pour certains groupes de patients
Génotype du CYP2C9
Le génotype du CYP2C9 a une influence significative sur le métabolisme du siponimod.
Le traitement par Mayzent est contre-indiqué chez les patients homozygotes pour l'allèle CYP2C9*3 (génotype CYP2C9*3/*3) (voir «Contre-indications», «Mises en garde et précautions» et «Interactions»). L'utilisation de Mayzent chez ces patients entraîne des concentrations plasmatiques de siponimod nettement plus élevées. La dose d'entretien recommandée de Mayzent est de 1 mg par jour chez les patients ayant le génotype CYP2C9*2/*3 ou *1/*3, afin d'éviter une exposition plus élevée au siponimod (voir «Posologie/Mode d'emploi»).
Il existe d'autres polymorphismes moins fréquents du CYP2C9. La pharmacocinétique du siponimod chez les porteurs de ces génotypes n'a pas été étudiée. Quelques-uns de ces polymorphismes, en particulier les allèles *5, *6, *8 et *11, sont aussi associés à une diminution ou une absence de la fonction enzymatique (voir «Mises en garde et précautions» et «Interactions»). Après une dose unique de 0,25 mg de siponimod, l'ASCinf et l'ASClast de sujets ayant les génotypes CYP2C9*2/*3 et CYP2C9*3/*3 étaient environ 2 à 4 fois plus élevées, alors que par comparaison aux forts métaboliseurs (CYP2C9*1/*1), on n'a obtenu qu'une faible augmentation de 21% et 16%, respectivement. La demi-vie moyenne est prolongée chez les porteurs de CYP2C9*2/*3 et de CYP2C9*3/*3 (51 h et 126 h, respectivement).
Après administration orale multiple de siponimod à des patients atteints de SEP-SP forts métaboliseurs par CYP2C9 (CYP2C9*1/*1 et CYP2C9*1/*2), la clairance systémique apparente (Cl/F) a été estimée à 3,11 l/h. Pour les sujets ayant les génotypes CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3 et CYP2C9*3/*3, la Cl/F est respectivement de 2,5, 1,9, 1,6 et 0,9 l/h. L'augmentation résultante de l'ASC du siponimod chez les sujets portant les génotypes CYP2C9*2/*2, CYP2C9*1/*3, CYP2C9*2/*3 et CYP2C9*3/*3 par rapport à ceux ayant le génotype CYP2C9*1/*1 a été respectivement de 25, 61, 91 et 285%. Vu que la clairance apparente estimée pour les sujets ayant le génotype CYP2C9*1/*2 est comparable à celle des sujets ayant le génotype CYP2C9*1/*1, on s'attend à une exposition similaire au siponimod pour les deux génotypes.
Patients présentant une insuffisance rénale
Aucun ajustement de la dose de siponimod n'est nécessaire chez les patients atteints d'une insuffisance rénale légère, modérée ou sévère. La demi-vie moyenne et la Cmax du siponimod (total et non lié) ont été comparables entre les patients présentant une insuffisance rénale sévère et les sujets sains. Les ASC du siponimod total et non lié n'ont été que légèrement augmentées par rapport aux sujets sains (de 23 à 33%). Les effets d'une maladie rénale au stade terminal et d'une hémodialyse sur la pharmacocinétique du siponimod n'ont pas été étudiés. En raison de la forte liaison aux protéines plasmatiques (> 99,9%) du siponimod, on ne s'attend pas à ce que l'hémodialyse modifie la concentration du siponimod total ou non lié, et aucun ajustement posologique n'est attendu sur la base de ce constat.
Patients présentant une insuffisance hépatique
Aucun ajustement de la dose de siponimod n'est nécessaire chez les patients atteints d'une insuffisance hépatique. L'ASC du siponimod non lié est plus élevée de 15% à 50% chez les patients présentant une insuffisance hépatique modérée à sévère par rapport aux sujets sains pour la dose étudiée de 0,25 mg. La demi-vie moyenne du siponimod est restée inchangée chez les patients présentant une insuffisance hépatique.
Patients âgés
Les études cliniques ont inclus des patients dont l'âge maximal était de 61 ans (voir «Posologie/Mode d'emploi»).
Sexe
Le sexe n'a aucune influence sur la pharmacocinétique du siponimod.
Couleur de peau/origine ethnique
En cas d'administration unique, aucun signe de différence ethnique concernant les paramètres PK entre des sujets sains japonais et caucasiens n'a été relevé, ce qui indique que l'appartenance ethnique n'a aucune influence sur la pharmacocinétique du siponimod.
Données précliniquesLe profil de sécurité préclinique du siponimod a été étudié dans des études de toxicité avec doses uniques et multiples sur des souris (13 semaines au maximum), des rats (26 semaines au maximum) et des singes (52 semaines au maximum). Les toxicités limitant la dose selon les espèces animales étaient la néphrotoxicité chez la souris, un impact sur la prise de poids chez le rat ainsi que des effets indésirables sur le SNC, et des effets gastrointestinaux chez le singe. Les organes les plus affectés, identifiés par histopathologie chez les rongeurs et par les effets toxiques étaient entre autres les poumons, le foie, la thyroïde, les reins et l'utérus/vagin. Chez certains singes, des effets sur les muscles et la peau ont été observés.
La valeur NOAEL (no observed adverse effect levels, dose sans effet nocif observable) a été fixée à respectivement 50 et 15 mg/kg/jour pour les rats mâles et femelles, et à 10 mg/kg/jour pour les singes des deux sexes. Les marges de sécurité selon l'ASC pour les effets systémiques (facteur 171) ont été calculées sur la base d'une dose d'entretien de 2 mg/jour.
Génotoxicité et carcinogénicité
Les tests de génotoxicité in vitro (mutations bactériennes, test du micronoyau et test d'aberration chromosomique avec des lymphocytes humains) et une étude de micronoyau in vivo sur des rats n'ont mis en évidence aucun potentiel génotoxique du siponimod.
Dans les investigations de carcinogénicité, des lymphomes, hémoangiomes et hémangiosarcomes induits par le siponimod ont été identifiés chez la souris, des adénomes folliculaires et carcinomes de la thyroïde chez des rats mâles. Ces tumeurs ont été considérées comme étant spécifiques à la souris ou attribuées à des adaptations métaboliques du foie chez les rats. On ne connait pas l'importance clinique chez l'homme.
L'exposition au siponimod dans le plasma (ASC) à la dose la plus faible testée chez la souris est d'environ 29 fois celle observée chez l'homme à la dose recommandée chez l'homme de 2 mg.
Fertilité et toxicité pour la reproduction
Le siponimod n'a aucun effet sur les paramètres de fertilité chez les rats mâles comme femelles aux doses testées les plus élevées. Sur la base de l'exposition systémique chez l'homme (ASC) à une dose quotidienne de 2 mg, cela correspond à une marge de sécurité d'environ 16 fois. Une administration chronique n'a révélé aucun effet sur les organes reproducteurs des rats et des singes.
Les études de reproduction et de développement sur des rates et des lapines gravides ont montré que le siponimod engendre une embryotoxicité et une fœtotoxicité chez les rats et les lapins, ainsi qu'un effet tératogène chez les rats. Après une exposition prénatale au siponimod à une dose similaire à la dose recommandée chez l'homme de 2 mg/jour, une incidence accrue de pertes après implantation et d'anomalies fœtales (externes, urogénitales et sur le squelette) a été observée chez le rat, ainsi que des morts embryofœtales, des fausses couches et des anomalies fœtales (squelette et organes internes) chez le lapin.
L'exposition des rats et des lapins n'entraînant aucune malformation ni mort embryofœtale se situait sous l'exposition clinique. Cela indique qu'à la dose d'entretien de 2 mg/jour, il n'existe aucune marge de sécurité concernant les effets sur le développement embryofœtal et pré/postnatal. Chez les rates allaitantes ayant reçu une dose orale unique de 10 mg/kg, le siponimod et ses métabolites sont passés dans le lait maternel.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Après l'ouverture, conserver 3 mois à une température ne dépassant pas 25 °C.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2–8 °C).
Conserver dans l'emballage d'origine.
Conserver le médicament hors de portée des enfants.
Remarques concernant la manipulation
Aucune exigence particulière.
Mesures particulières d'élimination
Le médicament non utilisé ou le matériel résiduel doit être éliminé conformément à la règlementation nationale.
Numéro d’autorisation67230 (Swissmedic)
PrésentationBoîte de début de traitement de 12 comprimés pelliculés à 0,25 mg (B)
Boîte de 120 comprimés pelliculés à 0,25 mg (B)
Boîte de 28 comprimés pelliculés à 1 mg (B)
Boîtes de 28 comprimés pelliculés à 2 mg (B)
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz.
Mise à jour de l’informationJuin 2025
|