Propriétés/EffetsCode ATC
L04AA42
Mécanisme d'action
Le siponimod est un modulateur des récepteurs de la sphingosine 1phosphate (S1P). Le siponimod se lie sélectivement à deux des cinq récepteurs de S1P couplés à la protéine G (GPCR): S1P1 et S1P5. Le siponimod agit en tant qu'antagoniste fonctionnel sur les récepteurs de S1P1 des lymphocytes et empêche ainsi les lymphocytes de sortir des ganglions lymphatiques. Cela diminue la recirculation des lymphocytes T dans le système nerveux central (SNC) et limite ainsi l'inflammation centrale. Le siponimod passe la barrière hématoencéphalique. Il n'a aucune influence durable sur les lymphocytes T à mémoire effecteurs des tissus périphériques et dans le sang et n'influence pas l'activation des lymphocytes.
Pharmacodynamique
Dans les études animales, les effets directs du siponimod sur les cellules nerveuses ont été détectés, par le S1P1 sur les astrocytes et par le S1P5 sur les oligodendrocytes. Dans un modèle de souris de l'encéphalomyélite auto-immune expérimentale, un effet neuroprotecteur direct, indépendant des effets sur les lymphocytes, a été également mis en évidence pour le siponimod utilisé centralement (par perfusion intracérébroventriculaire).
Système immunitaire
Mayzent induit une réduction dose-dépendante du nombre de lymphocytes dans le sang périphérique dans les 6 heures qui suivent la première dose, ce qui est dû à la rétention réversible (séquestration) des lymphocytes dans les tissus lymphoïdes.
En cas d'administration quotidienne continue, le nombre de lymphocytes diminue en continu et atteint une valeur minimale médiane (IC à 90%) d'environ 0,560 (0,271 à 1,08) cellules/nl chez un patient type atteint de SEP-SP, présentant le génotype CYP2C9*1/*1 ou CYP2C9*1/*2, non originaire du Japon, ce qui correspond à une réduction de 20 à 30% par rapport à la valeur initiale. Lors d'une prise quotidienne, les faibles nombres de lymphocytes sont maintenus.
Chez une large majorité (90%) des patients atteints de SEP-SP, le nombre de lymphocytes revient à la normale dans les 10 jours suivant l'arrêt du traitement. Après l'arrêt du traitement par Mayzent, la réduction du nombre de lymphocytes périphériques peut persister pendant 3 à 4 semaines après la dernière dose.
Électrophysiologie cardiaque
Fréquence et rythme cardiaques
Mayzent entraîne une diminution temporaire de la fréquence cardiaque et de la conduction auriculoventriculaire au début du traitement (voir «Effets indésirables»). Ceci est lié à une activation des canaux potassiques rectifiants entrants couplés aux protéines G (G-protein-coupled inwardly rectifying potassium, GIRK) via la stimulation des récepteurs de S1P1, ce qui conduit à une hyperpolarisation cellulaire et une excitabilité réduite. En raison de son antagonisme fonctionnel sur les récepteurs de S1P1, la titration initiale du siponimod désensibilise les canaux GIRK jusqu'à ce que la dose d'entretien soit atteinte.
Potentiel d'allongement de l'intervalle QT
Les effets des doses thérapeutiques (2 mg) et suprathérapeutiques (10 mg) du siponimod sur la repolarisation cardiaque ont été étudiés dans une étude détaillée de l'intervalle QT. Les résultats n'indiquent pas un potentiel arythmogène en lien avec l'allongement de l'intervalle QT avec le siponimod, car ce dernier a augmenté l'intervalle QTcF moyen ajusté par rapport à la valeur initiale et corrigé par rapport au placebo (ΔΔQTcF) de plus de 5 ms, avec un effet moyen maximal de respectivement 7,8 ms (2 mg) et 7,2 ms (10 mg) 3 heures après l'administration. La limite supérieure de l'IC à 95% unilatéral pour l'intervalle ΔΔQTcF est restée à tout moment inférieure à 10 ms. L'analyse catégorielle n'a mis en évidence aucune valeur QTc conditionnée par le traitement supérieure à 480 ms, aucune augmentation de l'intervalle QTc de plus de 60 ms par rapport à la valeur initiale et aucune valeur de QT/QTc corrigée ou non corrigée dépassant 500 ms.
Fonction pulmonaire
L'administration unique ou multiple de Mayzent pendant 28 jours n'est pas associée à une augmentation cliniquement pertinente de la résistance respiratoire, mesurée par le volume expiratoire maximal en 1 seconde (VEMS) à 25 et à 75% du volume pulmonaire (DEM25-75%). Aux doses uniques non thérapeutiques (> 10 mg), on observe une légère tendance à une réduction du VEMS. Le traitement concomitant par Mayzent et le propranolol a entraîné une baisse minimale du VEMS par rapport au propranolol seul, les modifications avec le médicament seul ou avec l'association se situant dans la variabilité physiologique du VEMS et étant cliniquement non significatives.
Efficacité clinique
L'efficacité de Mayzent a été étudiée dans une étude de phase 3 dans laquelle une dose de 2 mg de Mayzent une fois par jour a été évaluée chez des patients atteints de SEP-SP. Une étude de détermination de la dose de phase 2 menée chez des patients atteints de SEP-RR a indiqué une réduction dose-dépendante des lésions inflammatoires à l'IRM et a montré que Mayzent à 2 mg produit un effet presque maximal.
Étude A2304 (EXPAND) pour la SEP-SP
L'étude A2304 était une étude de phase 3 randomisée, en double aveugle, contrôlée contre placebo, axée sur les événements et la durée de suivi, menée auprès de patients atteints de SEP-SP qui ont présenté au cours des 2 dernières années une progression détectable, en l'absence ou indépendamment de poussées, aucune indication de poussée au cours des 3 mois précédant le début de l'étude et un score EDSS (Expanded Disability Status Scale, échelle étendue du handicap) médian de 3,0 à 6,5 au début de l'étude.
Le score EDSS médian à l'inclusion était de 6,0. Les patients de plus de 61 ans n'ont pas été inclus dans l'étude. En ce qui concerne l'activité de la maladie, les paramètres caractéristiques de l'activité inflammatoire de la SEP-SP pouvaient être définis par des poussées ou par l'imagerie (à savoir, lésions en T1 rehaussées par agent de contraste ou lésions en T2 actives [nouvelles ou en croissance]).
Les patients ont été randomisés dans un ratio 2:1 pour recevoir Mayzent 2 mg une fois par jour ou un placebo. Les évaluations cliniques ont été réalisées à la sélection, ainsi que tous les 3 mois et au moment d'une poussée. Les évaluations de l'IRM ont été réalisées à la sélection et tous les 12 mois.
Le critère d'évaluation principal de l'étude était le délai jusqu'à une progression du handicap confirmée à 3 mois (confirmed disability progression, CDP), qui était déterminée par une augmentation d'au moins 1 point par rapport à la valeur initiale du score EDSS (augmentation de 0,5 point pour les patients ayant un score EDSS de 5,5 ou plus) à 3 mois. Les critères d'évaluation secondaires importants étaient le délai jusqu'à une aggravation confirmée à 3 mois d'au moins 20% par rapport à l'inclusion du résultat au test chronométré de marche sur une distance de 25 pieds (timed 25 foot walk test, T25FW) et la variation du volume des lésions en T2 par rapport à l'inclusion. D'autres critères d'évaluation secondaires étaient le délai jusqu'à une CDP à 6 mois, le pourcentage de variation du volume cérébral et les mesures de l'activité inflammatoire de la maladie (taux annualisé de poussées, lésions à l'IRM). La modification de la vitesse de traitement cognitif dans le test de substitution symboles/chiffres (Symbol Digit Modalities Test) était un critère exploratoire.
La durée de l'étude était variable d'un patient à l'autre (durée médiane de l'étude: 21 mois, plage de 1 jour à 37 mois).
1651 patients ont été randomisés dans cette étude pour recevoir soit Mayzent 2 mg (N = 1105) soit le placebo (N = 546); 82% des patients traités par Mayzent et 78% des patients sous placebo ont terminé l'étude. L'âge médian était de 49,0 ans, la durée médiane de la maladie de 16,0 ans et le score EDSS médian de 6,0 au début de l'étude; 64% des patients n'avaient eu aucune poussée au cours des 2 années précédant le début de l'étude et 76% n'avaient aucune lésion rehaussée par le gadolinium (Gd) lors de l'IRM initiale; 78% des patients avaient reçu auparavant un traitement pour la SEP.
Le délai jusqu'à l'apparition d'une progression du handicap confirmée à 3 et à 6 mois était significativement allongé sous siponimod, avec une réduction du risque de progression du handicap confirmée à 3 mois de 21% par rapport au placebo (hazard ratio [HR] 0,79, p = 0,0134) et une réduction du risque de progression du handicap confirmée à 6 mois de 26% par rapport au placebo (HR 0,74, p = 0,0058).
Les résultats de cette étude sont résumés dans le tableau 3 et dans les figures 1 et 2.
Tableau 3 Résultats cliniques et d'IRM de l'étude A2304
|
Critères d'évaluation
|
A2304 (EXPAND)
| |
Siponimod 2 mg
(n = 1099)
|
Placebo
(n = 546)
| |
Critères d'évaluation cliniques
| |
Critère d'évaluation principal d'efficacité: proportion de patients présentant une progression du handicap confirmée à 3 mois (critère d'évaluation principal)
|
26,3%
|
31,7%
| |
Réduction du risque1
|
21% (p = 0,0134)
| |
Proportion de patients présentant une augmentation confirmée à 3 mois de 20% du résultat au test de marche de 25 pieds
|
39,7%
|
41,4%
| |
Réduction du risque1
|
6% (p = 0,4398)
| |
Proportion de patients présentant une progression du handicap confirmée à 6 mois
|
19,9%
|
25,5%
| |
Réduction du risque1
|
26% [(p = 0,0058)]6
| |
Taux annualisé de poussées (TAP)
|
0,071
|
0,152
| |
Réduction du taux2
|
55% [(p < 0,0001)]6
| |
Critères d'évaluation IRM
| |
Modification du volume des lésions en T2 (mm3) par rapport à la valeur initiale3
|
+184 mm3
|
+879 mm3
| |
Différence de variation du volume des lésions en T2
|
-695 mm3 (p < 0,0001)7
| |
Pourcentage de variation du volume cérébral par rapport à la valeur initiale (IC à 95%)3
|
-0,497%
|
-0,649%
| |
Différence du pourcentage de variation du volume cérébral
|
0,152% [(p = 0,0002)]6
| |
Nombre cumulé moyen de lésions rehaussées par le Gd, pondérées en T1 (IC à 95%)4
|
0,081
|
0,596
| |
Réduction du taux
|
86% [(p < 0,0001)]6
| |
Proportion de patients présentant une aggravation de 4 points au test de substitution symboles/chiffres (Symbol Digit Modalities Test)5
|
16,0%
|
20,9%
| |
Réduction du risque1
|
25% [(p = 0,0163)]6
| |
1
D'après les modèles de Cox pour le délai jusqu'à la progression
2 D'après un modèle pour les événements récurrents
3 Moyenne au mois 12 et au mois 24
4 Jusqu'au mois 24
5 Confirmée à 6 mois
6 [Valeur p nominale pour les critères d'évaluation non inclus dans les tests hiérarchiques et non ajustés en fonction de la multiplicité]
7 Valeur p non confirmée; le procédé de test hiérarchique a été terminé avant l'atteinte du critère d'évaluation
|
Figure 1 Patients présentant une progression du handicap confirmée à 3 et à 6 mois, à l'aide des courbes de Kaplan-Meier du score EDSS (ensemble d'analyse complet, étude A2304)
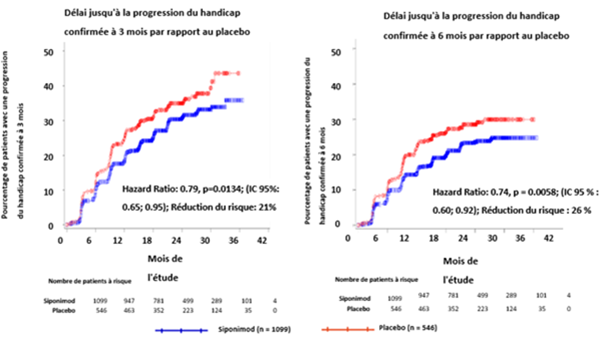
Les résultats de l'étude ont montré une réduction du risque cohérente concernant le délai jusqu'à la progression du handicap confirmée à 3 mois et à 6 mois avec Mayzent par rapport au placebo dans des sous-groupes définis selon le sexe, l'âge, le traitement précédent de la sclérose en plaques, l'activité des poussées avant l'étude, l'activité de la maladie par IRM à l'inclusion, la durée de la maladie et le niveau de handicap à l'inclusion.
Mayzent a montré un effet positif au test de substitution symboles/chiffres (Symbol Digit Modalities Test, SDMT). Pour Mayzent, la variation par rapport aux valeurs initiales a été stable ou meilleure et elle s'est dégradée pour le placebo avec une différence significative entre les groupes de 1,1 point au mois 12 (p = 0,0132) et de 2,3 points au mois 24 (p = 0,0002). Dans une étude exploratoire, Mayzent a réduit le risque d'une aggravation confirmée à 6 mois de 4 points du SDMT de 25% (p = 0,0163) par rapport au placebo. Une aggravation de 4 points s'est révélée être déjà cliniquement significative.
Dans le sous-groupe des patients (47,1%, n = 779) présentant une maladie active (définie comme les patients présentant une poussée dans les 2 années précédant l'étude et/ou la présence de lésions en T1 rehaussées par le Gd au début de l'étude), les caractéristiques à l'inclusion étaient similaires à celles de la population globale. L'âge médian était de 47 ans, la durée médiane de la maladie était de 15 ans et le score EDSS médiane au début de l'étude était de 6,0 (voir «Pharmacocinétique»).
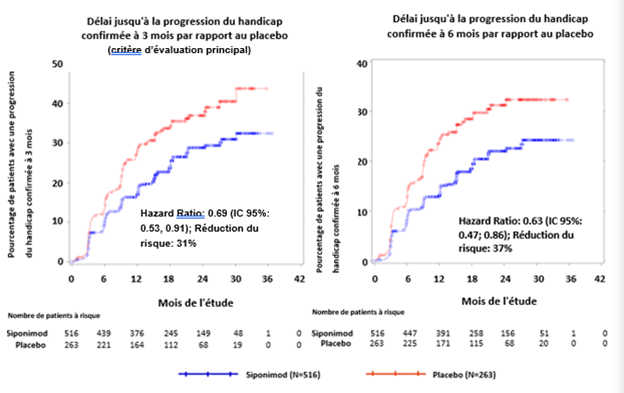
Le délai jusqu'à l'apparition d'une progression du handicap confirmée à 3 et à 6 mois a été significativement prolongé pour les patients présentant une maladie active traités par le siponimod, de 31% par rapport au placebo (hazard ratio [HR] 0,69; IC à 95%: 0,53; 0,91) et de 37% par rapport au placebo (HR 0,63; IC à 95%: 0,47; 0,86). Le TAP (poussées confirmées) a été réduit de 46% par rapport au placebo (rapport de TAP 0,54; IC à 95%: 0,39; 0,77). La réduction relative du nombre cumulé de lésions pondérées en T1, rehaussées par le Gd sur 24 mois a été de 85% (risque relatif 0,155; IC à 95%: 0,104; 0,231) par rapport au placebo. Les différences de modification du volume des lésions en T2 et du pourcentage de modification du volume cérébral (moyenne sur les mois 12 et 24) par rapport au placebo étaient de 1163 mm3 (IC à 95%: 1484, 843 mm3) et 0,141% (IC à 95% 0,020; 0,261%), respectivement.
Dans le sous-groupe de patients (n = 827) sans signe ni symptôme d'activité de la maladie (définie comme les patients sans poussée dans les 2 années précédant l'étude et sans présence de lésions en T1 rehaussées par agent de contraste au début de l'étude), les effets sur la progression du handicap confirmée à 3 et à 6 mois étaient faibles (la réduction du risque était de 7% et 13%, respectivement).
Figure 2 Patients présentant une progression du handicap confirmée à 3 et à 6 mois, à l'aide des courbes de Kaplan-Meier du score EDSS – sous-groupe présentant une maladie inflammatoire active (ensemble d'analyse complet, étude A2304)
|