Propriétés/EffetsCode ATC
S01LA06
Mécanisme d'action
Une transmission accrue du signal par la voie VEGF-A (facteur de croissance vasculaire endothélial A) est associée à une angiogenèse oculaire pathologique et à un œdème rétinien. Le brolucizumab se lie avec une grande affinité aux isoformes VEGF-A (p.ex. VEGF110, VEGF121 et VEGF165) et empêche ainsi le VEGF-A de se fixer à ses récepteurs VEGFR-1 et VEGFR-2. En inhibant la liaison au VEGF-A, le brolucizumab supprime la prolifération des cellules endothéliales, réduisant ainsi la néovascularisation pathologique et la perméabilité vasculaire.
Pharmacodynamie
La dégénérescence maculaire liée à l'âge (DMLA) néovasculaire (humide) est caractérisée par une néovascularisation choroïdienne (NVC) pathologique. La perte de sang et de liquide due à la NVC peut entraîner un épaississement ou un œdème de la rétine ou un saignement sous/intrarétinien, entraînant une perte d'acuité visuelle.
Dans les études HAWK et HARRIER, les paramètres anatomiques connexes faisaient partie de l'évaluation de l'activité de la maladie, qui a servi de base aux décisions thérapeutiques. Une réduction de l'épaisseur centrale de la rétine (ECR) et la présence de liquide intrarétinien/sous-rétinien (LIR/LSR) ou de liquide épithélial pigmentaire sous-rétinien (sous-EPR) ont été observées chez des patients traités par Beovu dès les 4 semaines suivant le début du traitement et jusqu'aux semaines 48 et 96.
Dans ces études, une réduction de la taille des lésions de NVC a été observée chez les patients traités avec Beovu dès 12 semaines après le début du traitement et aux semaines 48 et 96 après le début du traitement.
Efficacité clinique
L'innocuité et l'efficacité de Beovu ont été évaluées dans deux études de phase III (HAWK et HARRIER) randomisées, multicentriques, en double aveugle avec contrôle actif, menées auprès de patients atteints de DMLA néovasculaire. Au total, 1'817 patients ont été traités dans ces essais pendant deux ans (1'088 par Beovu et 729 par aflibercept). Les patients étaient âgés de 50 à 97 ans, avec une valeur moyenne de 76 ans.
Dans l'étude HAWK, les patients ont été randomisés selon un rapport 1:1:1 et affectés à l'un des schémas posologiques suivants:
1.Beovu 3 mg administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
2.Beovu 6 mg, administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
3.Aflibercept 2 mg, administré toutes les 8 semaines («q8w») après les 3 premières doses mensuelles.
Dans l'étude HARRIER, les patients ont été randomisés selon un rapport 1:1 et affectés à l'un des schémas posologiques suivants:
1.Beovu 6 mg, administré toutes les 12 ou 8 semaines («q12w/q8w») après les 3 premières doses mensuelles.
2.Aflibercept 2 mg, administré toutes les 8 semaines («q8w») après les 3 premières doses mensuelles.
Dans les deux études, les patients traités par le brolucizumab ont été traités toutes les 12 semaines après les 3 premières doses mensuelles (semaines 0, 4 et 8) avec la possibilité de passer à un intervalle de traitement de 8 semaines en fonction de l'activité de la maladie. L'activité de la maladie a été évaluée par un médecin au cours des 12 premières semaines (semaines 16 et 20) et à chacune des 12 semaines de traitement prévues. Les patients qui présentaient une activité pathologique au cours de l'une de ces visites (p.ex. diminution de l'acuité visuelle, augmentation de l'épaisseur centrale de la rétine (ECR) ou présence de liquides rétiniens (LIR/LSR, sous-EPR) ont reçu un traitement toutes les huit semaines.
Résultats
Le critère d'efficacité primaire des essais était la variation de la meilleure acuité visuelle corrigée (MAVC) par rapport à l'acuité visuelle initiale à la semaine 48, telle que mesurée par les tableaux des lettres ETDRS, dans le but principal de démontrer la non-infériorité de Beovu par rapport à l'aflibercept. Les deux études ont démontré que Beovu (administré selon un schéma thérapeutique de 12/8 semaines) n'était pas inférieur à l'aflibercept à la dose de 2 mg (administré toutes les 8 semaines) en termes d'efficacité.
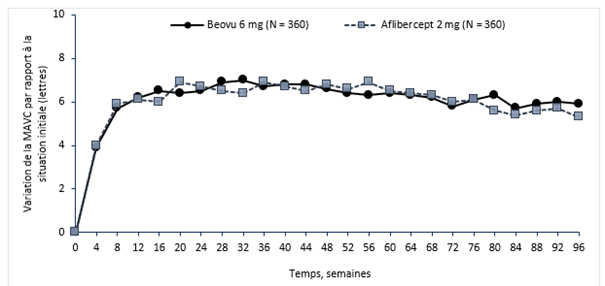
Dans l'étude HAWK, les patients des groupes Beovu 6 mg et aflibercept ont atteint, à la semaine 48, une variation moyenne de respectivement +6,6 lettres et +6,8 lettres (p <0,0001), par rapport à la situation initiale. La variation moyenne par rapport à la situation initiale dans le groupe recevant 3 mg de Beovu était de +6,1 lettres (p = 0,0003). La proportion de patients ayant gagné au moins 15 lettres d'acuité visuelle par rapport à la situation initiale était de 33,6% dans le groupe brolucizumab contre 25,4% dans le groupe aflibercept. La proportion de patients ayant perdu 15 lettres ou plus d'acuité visuelle par rapport à la situation initiale était de 6,4% dans le groupe recevant 6 mg de brolucizumab contre 5,5% dans le groupe recevant l'aflibercept.
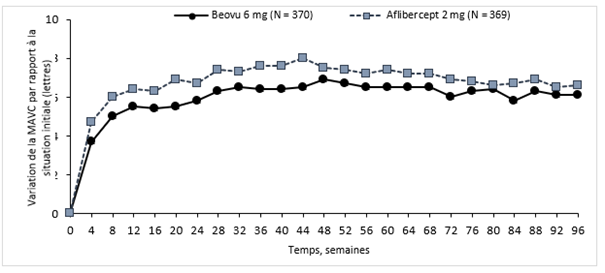
Dans l'étude HARRIER, les patients des groupes Beovu et aflibercept ont atteint, à la semaine 48, une variation moyenne de respectivement +6,9 lettres et +7,6 lettres (p <0,0001) par rapport à la situation initiale. La proportion de patients ayant gagné au moins 15 lettres d'acuité visuelle par rapport à la situation initiale était de 29,3% dans le groupe brolucizumab contre 29,9% dans le groupe aflibercept. La proportion de patients ayant perdu 15 lettres ou plus d'acuité visuelle par rapport à la situation initiale était de 3,8% dans le groupe recevant 6 mg de brolucizumab contre 4,8% dans le groupe recevant l'aflibercept.
Le gain d'acuité visuelle observée la première année a été maintenu la deuxième année.
Figure 0-1: Variation moyenne de l'acuité visuelle par rapport à la situation initiale jusqu'à la semaine 96 dans les études HAWK et HARRIER
Étude HAWK
Étude HARRIER
Dans les études HAWK et HARRIER, respectivement 56% et 51% des patients traités par Beovu 6 mg à 12 semaines d'intervalle ont atteint ce gain d'acuité visuelle à la semaine 48 (variation moyenne par rapport à la situation initiale), et respectivement 45% et 39% des patients à la semaine 96.
Parmi les patients considérés comme adaptés à cet intervalle de traitement au cours du premier intervalle de 12 semaines de traitement, respectivement 85% et 82% ont maintenu l'intervalle de 12 semaines jusqu'à la semaine 48. Chez respectivement 82% et 75% des patients traités à la semaine 48 avec l'intervalle de traitement de 12 semaines, l'intervalle de traitement de 12 semaines a été maintenu de la semaine 48 à la semaine 96.
Les effets du traitement dans les sous-groupes évaluables (p.ex. âge, sexe, origine ethnique, acuité visuelle en situation initiale, épaisseur de la rétine en situation initiale, type de lésion, taille de la lésion, état hydrique) dans les deux études concordaient en grande partie avec les résultats dans la population totale.
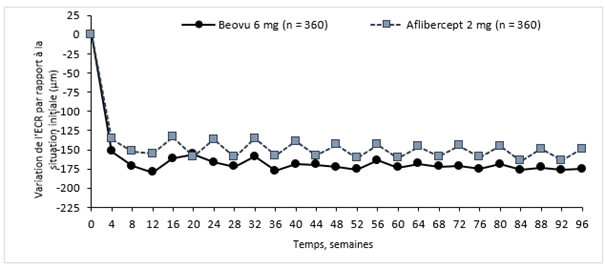
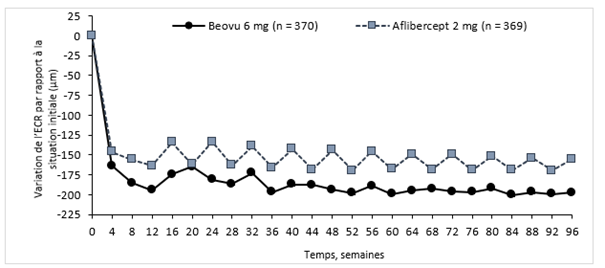
L'activité de la maladie a été évaluée sur la base des variations de l'acuité visuelle ou de critères morphologiques, notamment l'épaisseur centrale de la rétine (ECR) et la présence de liquides rétiniens (LIR/LSR, sous-EPR). À la semaine 16, lorsque l'activité de la maladie a été évaluée pour la première fois pour déterminer l'intervalle de traitement, le nombre de patients ayant montré une activité de la maladie était statistiquement moins élevé dans le groupe sous Beovu à la dose de 6 mg que dans le groupe sous aflibercept à la dose de 2 mg (24% contre 35% dans HAWK, p = 0,0013; 23% contre 32% dans HARRIER, p = 0,0021). L'activité de la maladie a été évaluée tout au long des études. Les critères morphologiques de l'activité de la maladie étaient plus faibles au cours des semaines 48 et 96 dans le groupe Beovu comparativement au groupe aflibercept (Tableau 0-2).
Tableau 0-1: Évaluation de l'activité de la maladie dans les études HAWK et HARRIER jusqu'à la semaine 96
|
|
|
Étude HAWK
|
|
Étude HARRIER
|
| |
Résultats d'efficacité (critères d'évaluation secondaires préétablis)
|
À la semaine
|
Beovu 6 mg
(N=360)
|
Aflibercept
2 mg
(N=360)
|
Différence (IC à 95%) brolucizumab et aflibercept
|
Beovu 6 mg
(N=360)
|
Aflibercept
2 mg
(N=369)
|
Différence (IC à 95%) brolucizumab et aflibercept
| |
Variation moyenne de l'ECR par rapport à la situation initiale (µm)
|
16 c)
|
-161,4
(ET = 6,2)
|
-133,6
(ET = 6,2)
|
-27,8
(-45,1, -10,5)
p = 0,0008 a)
|
-174,4
(ET = 6,7)
|
-134,2
(ET = 6,7)
|
-40,2
(-58,9, -21,6)
p <0,0001 a)
| |
48
|
-172.8
(ET = 6,7)
|
-143,7
(ET = 6,7)
|
-29,0
(-47,6, -10,4)
p = 0,0012 a)
|
-193,8
(ET = 6,8)
|
-143,9
(ET = 6,8)
|
-49,9
(-68,9, -30,9)
p <0,0001 a)
| |
96
|
-174.8
(ET = 7,3)
|
-148,7
(ET = 7,3)
|
-26,0
(-46,2, -5,9)
p = 0,0115 b)
|
-197,7
(ET= 7,0)
|
-155,1
(ET= 7,0)
|
-42,6
(-62,0, -23,3)
p <0,0001 b)
|
ECR: épaisseur centrale de la rétine, LIR/LSR: liquide intrarétinien/liquide sous-rétinien, EPR: épithélium pigmentaire rétinien
a) Critère d'évaluation secondaire dans l'étude HARRIER, analyse de confirmation dans l'étude HAWK. Valeurs p unilatérales pour la supériorité du brolucizumab
b) Critères d'évaluation secondaire dans les études HAWK et HARRIER; valeurs de p bilatéral
c) Jusqu'à la semaine 16, l'exposition au traitement était identique, permettant une comparaison coordonnée de Beovu avec l'aflibercept.
Figure 0-2: Variation de l'épaisseur centrale de la rétine, en situation initiale jusqu'à la semaine 96 dans les études HAWK et HARRIER
Étude HAWK
Étude HARRIER
Dans les deux études, le traitement par Beovu a entraîné des modifications cliniquement significatives par rapport à la situation initiale en ce qui concerne le critère d'évaluation secondaire de l'efficacité préspécifié des «résultats rapportés par les patients» enregistrés au moyen du questionnaire NEI VFQ-25 sur la santé oculaire de l'Institut national des yeux des États-Unis. L'ampleur de ces modifications était comparable à celle des études publiées et correspondait à une augmentation de 15 lettres dans la meilleure acuité visuelle corrigée (MACV). Le bénéfice des résultats rapportés par les patients a été maintenu au cours de la deuxième année.
Aucune différence cliniquement significative n'a été observée entre Beovu et l'aflibercept en ce qui concerne les variations du score total NEI VFQ-25 et des sous-échelles entre la situation initiale et la semaine 48 (vision générale, douleur oculaire, vision de près, vision de loin, activité sociale, bien-être mental, difficultés à exercer des rôles sociaux, dépendance envers autrui, conduite automobile, vision des couleurs et vision périphérique).
|