CompositionPrincipes actifs
Olipudase alfa (produite dans une lignée cellulaire d'ovaire de hamster chinois [CHO]).
Excipients
Méthionine, phosphate de sodium dibasique heptahydraté, phosphate de sodium monobasique monohydraté, saccharose.
Un flacon de 4 mg contient 0,60 mg de sodium.
Un flacon de 20 mg contient 3,02 mg de sodium.
Indications/Possibilités d’emploiXenpozyme est indiqué en tant que traitement enzymatique substitutif des manifestations non neurologiques du déficit en sphingomyélinase acide (Acid Sphingomyelinase Deficiency, ASMD) de type A/B ou B chez les patients pédiatriques et adultes.
Posologie/Mode d’emploiLe traitement par Xenpozyme doit être supervisé par un professionnel de santé ayant l'expérience de la prise en charge de l'ASMD ou d'une maladie métabolique héréditaire comparable. Le professionnel de santé doit avoir accès à un traitement médical d'urgence approprié pour prendre en charge les réactions sévères potentielles telles que les réactions d'hypersensibilité systémiques graves. Le traitement par Xenpozyme doit toujours être instauré selon le schéma d'escalade de dose ci-dessous (voir tableaux 1 et 2) et suivi d'une dose d'entretien afin de limiter le risque de réactions associées à la perfusion, y compris les réactions de phase aiguë et les augmentations des transaminases hépatiques. En cas d'oubli de doses, voir ci-dessous.
La perfusion à domicile pour les patients, sous supervision d'un professionnel de santé, peut être envisagée pendant la phase d'entretien du traitement (voir ci-dessous).
Posologie
Le métabolisme rapide par Xenpozyme de la sphingomyéline (SM) accumulée génère des produits de dégradation pro-inflammatoires, qui peuvent induire des réactions associées à la perfusion et/ou des augmentations transitoires des enzymes hépatiques. Un schéma d'escalade de dose peut limiter la majorité de ces effets indésirables (voir «Données précliniques»).
La posologie de Xenpozyme est calculée à partir du poids corporel réel pour les patients avec un indice de masse corporelle (IMC) ≤30 ou à partir du poids corporel adapté pour les patients avec un IMC > 30 (voir la rubrique concernant les patients avec un IMC > 30).
Toutes les instructions pour la posologie et l'administration (voir ci-dessous), la préparation et la manipulation (voir «Remarques particulières» et «Préparation de la solution pour perfusion en fonction de la posologie») doivent être suivies afin d'éviter les erreurs posologiques, y compris le surdosage (voir «Surdosage»). Veuillez noter que l'escalade de dose chez les patients pédiatriques est différente de celle chez les adultes. En plus du schéma d'escalade de dose, chaque dose doit être administrée en utilisant un débit de perfusion échelonné (voir tableaux 4 et 5).
Adultes
Phase d'escalade de dose
La dose initiale recommandée de Xenpozyme est de 0,1 mg/kg* pour les adultes (voir également la sous-rubrique concernant les doses manquées pour des recommandations supplémentaires) et, par la suite, la dose doit être augmentée selon le schéma d'escalade de dose présenté dans le tableau 1:
Tableau 1: Schéma d'escalade de dose chez l'adulte
|
Patients adultes (≥18 ans)
| |
Première dose (Jour 1/Semaine 0)
|
0,1 mg/kg*
| |
Deuxième dose (Semaine 2)
|
0,3 mg/kg*
| |
Troisième dose (Semaine 4)
|
0,3 mg/kg*
| |
Quatrième dose (Semaine 6)
|
0,6 mg/kg*
| |
Cinquième dose (Semaine 8)
|
0,6 mg/kg*
| |
Sixième dose (Semaine 10)
|
1 mg/kg*
| |
Septième dose (Semaine 12)
|
2 mg/kg*
| |
Huitième dose (Semaine 14)
|
3 mg/kg* (dose d'entretien recommandée)
|
* Le poids corporel réel doit être utilisé pour les patients avec un IMC ≤ 30. Pour les patients avec un IMC > 30, un poids corporel adapté doit être utilisé comme décrit ci-dessous.
Phase d'entretien
La dose d'entretien recommandée de Xenpozyme est de 3 mg/kg* toutes les 2 semaines.
* Le poids corporel réel doit être utilisé pour les patients avec un IMC ≤30. Pour les patients avec un IMC > 30, un poids corporel adapté doit être utilisé comme décrit ci-dessous.
Insuffisance hépatique
Aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance hépatique (voir «Pharmacocinétique»).
Insuffisance rénale
Aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance rénale (voir «Pharmacocinétique»).
Patients âgés
Aucun ajustement posologique n'est recommandé chez les patients âgés de plus de 65 ans (voir «Pharmacocinétique»).
Enfants et adolescents
Phase d'escalade de dose
La dose initiale recommandée de Xenpozyme chez les patients pédiatriques est de 0,03 mg/kg*, et la dose doit ensuite être augmentée selon le schéma d'escalade de dose présenté dans le tableau 2:
Tableau 2: Schéma d'escalade de dose chez les patients pédiatriques
|
Patients pédiatriques (0 à < 18 ans)
| |
Première dose (Jour 1/Semaine 0)
|
0,03 mg/kg*
| |
Deuxième dose (Semaine 2)
|
0,1 mg/kg*
| |
Troisième dose (Semaine 4)
|
0,3 mg/kg*
| |
Quatrième dose (Semaine 6)
|
0,3 mg/kg*
| |
Cinquième dose (Semaine 8)
|
0,6 mg/kg*
| |
Sixième dose (Semaine 10)
|
0,6 mg/kg*
| |
Septième dose (Semaine 12)
|
1 mg/kg*
| |
Huitième dose (Semaine 14)
|
2 mg/kg*
| |
Neuvième dose (Semaine 16)
|
3 mg/kg* (dose d'entretien recommandée)
|
* Le poids corporel réel doit être utilisé pour les patients avec un IMC ≤ 30. Pour les patients avec un IMC > 30, un poids corporel adapté doit être utilisé comme décrit ci-dessous.
Phase d'entretien
La dose d'entretien recommandée de Xenpozyme est de 3 mg/kg* toutes les 2 semaines.
* Le poids corporel réel doit être utilisé pour les patients avec un IMC ≤ 30. Pour les patients avec un IMC > 30, un poids corporel adapté doit être utilisé comme décrit ci-dessous.
Groupes de patients particuliers
Patients avec un IMC > 30
Chez les patients adultes et pédiatriques ayant un indice de masse corporelle (IMC) > 30, le poids corporel adapté utilisé pour calculer la dose de Xenpozyme est estimé selon la méthode suivante (pour les phases d'escalade de dose et d'entretien).
Poids corporel (kg) à utiliser pour le calcul de la dose = 30 × (taille réelle en m)2
Exemple:
Pour un patient avec
un IMC de 38,
un poids corporel de 110 kg et
une taille de 1,70 m
La dose à administrer sera calculée en utilisant un poids corporel égal à 30 × 1,702 = 86,7 kg.
Doses manquées
Une dose est considérée comme manquée lorsqu'elle n'est pas administrée dans les 3 jours suivant la date prévue. En cas de dose manquée de Xenpozyme, la dose suivante doit être administrée dès que possible comme décrit au tableau 3. Par la suite, les administrations doivent être programmées toutes les 2 semaines à partir de la date de la dernière administration.
Tableau 3: Recommandation posologique en cas de doses manquées*
|
Nombre de perfusions manquées
|
Phase d'escalade de dose
|
Phase d'entretien
| |
Si une perfusion est manquée:
|
La dernière dose tolérée doit être administrée, avant la reprise de l'escalade de dose selon le schéma posologique chez les adultes (tableau 1) ou chez les patients pédiatriques (tableau 2).
|
La dose d'entretien doit être administrée et le calendrier de traitement ajusté en conséquence.
| |
Si 2 perfusions consécutives sont manquées:
|
Un niveau de dose inférieur à la dernière dose tolérée (en utilisant une dose minimale de 0,3 mg/kg) doit être administré, avant la reprise de l'escalade de dose conformément au tableau 1 ou au tableau 2.
|
Une dose inférieure à la dose d'entretien (c. à d. 2 mg/kg) doit être administrée. Puis, pour les perfusions suivantes, la dose d'entretien (3 mg/kg) doit être administrée toutes les 2 semaines.
| |
Si 3 perfusions consécutives ou plus sont manquées:
|
Chez les patients n'ayant pas achevé l'escalade de dose, il faudra procéder de la manière suivante:
·chez les patients adultes, reprise de l'escalade de dose à 0,1 mg/kg et poursuite conformément au tableau 1.
·chez les patients pédiatriques, reprise de l'escalade de dose à 0,03 mg/kg et poursuite conformément au tableau 2.
|
L'escalade de dose doit être reprise à 0,3 mg/kg et poursuivie conformément au tableau 1 ou au tableau 2.
Chez les patients qui ont manqué des perfusions d'entretien sur une plus longue période, qui pourrait voir une nouvelle accumulation de sphingomyéline, le médecin traitant devra:
·chez les patients adultes, envisager la reprise de la dose à 0,1 mg/kg et l'escalade de la dose conformément au tableau 1.
·chez les patients pédiatriques, envisager la reprise de la dose à 0,03 mg/kg et une escalade de dose conformément au tableau 2.
|
*Lors de la prochaine perfusion planifiée après une dose manquée, si la dose administrée est de 0,3 ou 0,6 mg/kg, cette dose doit être administrée deux fois conformément au tableau 1 ou au tableau 2.
Surveillance des taux de transaminases
Les taux de transaminases (alanine aminotransférase [ALAT] et aspartate aminotransférase [ASAT]) doivent être recueillis avant l'initiation du traitement et surveillés pendant toute la phase d'escalade de dose (voir «Mises en garde et précautions»). Si les taux de transaminases avant perfusion sont augmentés par rapport à la valeur initiale et > 2 fois la limite supérieure de la normale (LSN), la posologie de Xenpozyme peut être ajustée (répétition de la dose précédente ou réduction de la dose) ou le traitement peut être temporairement interrompu en fonction de l'élévation des transaminases. Si un patient nécessite un ajustement de posologie ou une interruption du traitement, la reprise du traitement doit suivre le schéma d'escalade de dose indiqué dans le tableau 1 et le tableau 2 pour les patients adultes et pédiatriques, respectivement, ainsi que les recommandations en cas de doses manquées (voir «Doses manquées»).
Mode d'administration
Xenpozyme est exclusivement réservé à une utilisation par voie intraveineuse. Les perfusions doivent être administrées de manière progressive, de préférence à l'aide d'une pompe à perfusion.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir «Remarques concernant la manipulation».
Après reconstitution et dilution, la solution est administrée par perfusion intraveineuse. Les débits de perfusion doivent être augmentés progressivement pendant la perfusion uniquement en l'absence de réactions associées à la perfusion (en cas de réactions associées à la perfusion, voir «Mises en garde et précautions»). Le débit de perfusion et la durée de la perfusion (+/- 5 min) pour chaque étape de perfusion sont détaillés dans le tableau 4 et le tableau 5. Lors de la détermination du débit de perfusion dans les tableaux 4 et 5, utiliser le niveau de dose indiqué dans le schéma d'escalade de dose, disponible soit dans le tableau 1 (patients adultes), soit dans le tableau 2 (patients pédiatriques).
Tableau 4: Débit de perfusion et durée de la perfusion chez les patients adultes
|
Dose* (mg/kg)
|
Débit de perfusion
Durée de la perfusion
|
Durée approximative de la perfusion
| |
|
Étape 1
|
Étape 2
|
Étape 3
|
Étape 4
|
| |
0,1
|
20 ml/h
pendant 20 min
|
60 ml/h
pendant 15 min
|
NA
|
NA
|
35 min
| |
0,3 à 3
|
3,33 ml/h
pendant 20 min
|
10 ml/h
pendant 20 min
|
20 ml/h
pendant 20 min
|
33,33 ml/h
pendant 160 min
|
220 min
|
h: heure; min: minute; NA: non applicable
* Niveau de dose selon le schéma d'escalade de dose dans le tableau 1
Tableau 5: Débit de perfusion et durée de la perfusion chez les patients pédiatriques
|
Dose* (mg/kg)
|
Débit de perfusion
Durée de la perfusion
|
Durée approximative de la perfusion
| |
Étape 1
|
Étape 2
|
Étape 3
|
Étape 4
| |
0,03
|
0,1 mg/kg/h pendant toute la durée de la perfusion
|
NA
|
NA
|
NA
|
18 min
| |
0,1
|
0,1 mg/kg/h pendant 20 min
|
à partir de 0,3 mg/kg/h
|
NA
|
NA
|
35 min
| |
0,3
|
0,1 mg/kg/h pendant 20 min
|
0,3 mg/kg/h pendant 20 min
|
à partir de 0,6 mg/kg/h
|
NA
|
60 min
| |
0,6
|
0,1 mg/kg/h pendant 20 min
|
0,3 mg/kg/h pendant 20 min
|
0,6 mg/kg/h pendant 20 min
|
à partir de 1 mg/kg/h
|
80 min
| |
1
|
100 min
| |
2
|
160 min
| |
3
|
220 min
|
h: heure; min: minute; NA: non applicable
* Niveau de dose selon le schéma d'escalade de dose dans le tableau 2
Les signes et les symptômes de Réactions Associées à la Perfusion (RAP), tels que céphalées, urticaire, fièvre, nausée et vomissements, et autres signes ou symptômes d'hypersensibilité doivent être surveillés pendant la perfusion. Selon la gravité des symptômes, il pourra être nécessaire de ralentir, d'interrompre ou d'arrêter la perfusion, et un traitement médical approprié pourra être instauré.
En cas de réaction d'hypersensibilité sévère et/ou de réaction anaphylactique, le traitement par Xenpozyme doit être arrêté immédiatement (voir «Mises en garde et précautions»).
À la fin de la perfusion (une fois la seringue ou la poche de perfusion vide), la ligne de perfusion doit être rincée avec une solution injectable de chlorure de sodium à 0,9% (9 mg/ml) en utilisant le même débit de perfusion que celui utilisé lors de la dernière partie de la perfusion.
Perfusion à domicile pendant la phase d'entretien
Une perfusion à domicile sous la supervision d'un professionnel de santé peut être envisagée pour les patients sous dose d'entretien qui tolèrent bien leurs perfusions. La décision de passer à des perfusions à domicile doit être prise après évaluation et recommandation par le médecin prescripteur.
Une assistance médicale appropriée, notamment un soignant formé aux mesures d'urgence, doit être disponible lors de l'administration de Xenpozyme. En cas de survenue de réactions anaphylactiques ou d'autres réactions aiguës, arrêter immédiatement la perfusion de Xenpozyme, instaurer un traitement médical approprié et consulter un médecin. En cas de réactions d'hypersensibilité sévères, les perfusions suivantes doivent impérativement avoir lieu dans un cadre clinique approprié disposant du matériel de réanimation nécessaire. La dose et les débits de perfusion doivent rester les mêmes que ceux utilisés dans le cadre clinique surveillé et ne doivent pas être modifiés sans une décision du médecin prescripteur. En cas de doses manquées ou de retard de perfusion, le médecin prescripteur doit être contacté, car il est possible que les perfusions suivantes aient lieu dans un cadre clinique surveillé.
Contre-indicationsHypersensibilité à l'olipudase alfa ou à l'un des excipients mettant en jeu le pronostic vital (réaction anaphylactique) (voir «Mises en garde et précautions»).
Mises en garde et précautionsTraçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro du lot du médicament administré doivent être clairement documentés.
Absence de passage de la barrière hémato-encéphalique
Xenpozyme n'est pas susceptible de traverser la barrière hémato-encéphalique ni de moduler les manifestations de la maladie au niveau du système nerveux central (SNC).
Réactions associées à la perfusion (RAP)
Au cours des études cliniques, environ 60 % des patients traités par Xenpozyme ont présenté des RAP. Ces RAP comprenaient des réactions d'hypersensibilité et des réactions de phase aiguë (voir «Effets indésirables»). Les RAP les plus fréquentes étaient: céphalées, urticaire, fièvre, nausée et vomissements (voir «Effets indésirables»). Les RAP sont généralement survenues entre le moment de la perfusion et jusqu'à 24 heures après la fin de la perfusion.
Des effets indésirables graves, incluant le décès, sont survenus après un surdosage pendant la phase d'escalade de dose (voir «Surdosage»). Pour prévenir le risque de telles réactions, il convient de respecter les recommandations posologiques des rubriques «Posologie/Mode d'emploi» ainsi que les instructions de préparation et de manipulation (voir «Remarques particulières» et «Préparation de la solution pour perfusion en fonction de la posologie»).
Hypersensibilité, y compris anaphylaxie
Des réactions d'hypersensibilité, dont l'anaphylaxie, ont été signalées chez des patients traités par Xenpozyme (voir «Effets indésirables»). Au cours des études cliniques, des réactions d'hypersensibilité sont survenues chez 9 (22,5 %) patients adultes et 9 (45%) patients pédiatriques, dont un patient pédiatrique qui a présenté une anaphylaxie.
Des réactions d'hypersensibilité légères à modérées telles que l'urticaire, l'érythème, le prurit, le rash cutané et l'angiœdème ont été signalées chez plus d'un patient adulte. Plusieurs patients pédiatriques ont présenté des réactions d'hypersensibilité légères à modérées telles que l'urticaire, l'érythème, le rash cutané et le prurit.
Les patients doivent être étroitement surveillés pendant la perfusion et pendant une période de temps appropriée après la perfusion, en fonction de l'évaluation clinique. Les patients doivent être informés des symptômes potentiels d'hypersensibilité/d'anaphylaxie et de la nécessité de solliciter des soins médicaux immédiats en cas d'apparition de symptômes. La conduite à tenir en cas de RAP doit être basée sur la sévérité des signes et des symptômes et peut inclure une interruption temporaire de la perfusion de Xenpozyme, une diminution du débit de perfusion et/ou un traitement médical approprié.
En cas de réaction d'hypersensibilité sévère ou d'anaphylaxie, Xenpozyme doit être arrêté immédiatement et un traitement médical approprié doit être mis en place. Le patient ayant présenté une anaphylaxie au cours de l'étude clinique a suivi un protocole de désensibilisation personnalisé qui lui a permis de reprendre le traitement à long terme par Xenpozyme à la dose d'entretien recommandée. Le médecin prescripteur doit évaluer les risques et les bénéfices d'une reprise du traitement par Xenpozyme après une anaphylaxie ou une réaction d'hypersensibilité sévère. Si une reprise de l'administration de Xenpozyme est envisagée après une anaphylaxie, le médecin prescripteur doit contacter le représentant local de Sanofi pour obtenir des conseils sur la réadministration. Chez ces patients, il convient de faire preuve d'une extrême prudence, en ayant à disposition des mesures de réanimation appropriées, en cas de reprise de l'administration de Xenpozyme.
En cas de RAP légères ou modérées, le débit de perfusion peut être ralenti ou la perfusion temporairement interrompue, la durée de chaque étape de perfusion peut être augmentée et/ou la dose de Xenpozyme peut être réduite. Si un patient nécessite une réduction de la dose, une nouvelle escalade de dose doit suivre le schéma d'escalade de dose indiqué dans le tableau 1 et le tableau 2 pour les patients adultes et pédiatriques, respectivement (voir «Posologie/Mode d'emploi»).
Les patients peuvent recevoir une prémédication par antihistaminiques, antipyrétiques et/ou glucocorticoïdes afin de prévenir ou réduire les réactions allergiques.
Immunogénicité
Des patients adultes et pédiatriques ont développé des anticorps anti-médicament (anti-drug antibodies, ADA) au cours des essais cliniques (voir «Effets indésirables»). Des RAP et des réactions d'hypersensibilité peuvent survenir indépendamment du développement d'ADA. La majorité des RAP et des réactions d'hypersensibilité étaient légères ou modérées et ont été prises en charge selon des pratiques cliniques standard.
La recherche des ADA de type IgE peut être envisagée pour les patients ayant présenté une réaction d'hypersensibilité sévère à l'olipudase alfa.
Tandis qu'aucune perte d'efficacité n'a été rapportée pendant les études cliniques, la recherche des ADA de type IgG peut être envisagée en cas de perte de réponse au traitement.
Augmentation transitoire des transaminases
Des augmentations transitoires des transaminases (ALAT ou ASAT) dans les 24 à 48 heures suivant les perfusions ont été rapportées pendant la phase d'escalade de dose de Xenpozyme chez 4 patients adultes et 7 patients pédiatriques (voir «Effets indésirables»). Lors de la perfusion suivante programmée, ces taux élevés de transaminases étaient généralement revenus aux taux observés avant la perfusion de Xenpozyme.
Les taux de transaminases (ALAT et ASAT) doivent être mesurés dans le mois précédant l'initiation du traitement par Xenpozyme (voir «Posologie/Mode d'emploi»). Lors de l'escalade de dose ou de la reprise du traitement après des doses manquées, les taux de transaminases doivent être mesurés dans les 72 heures précédant la prochaine perfusion de Xenpozyme planifiée. Si, pendant l'escalade de dose, le taux de transaminases initial ou avant perfusion est > 2 fois la limite supérieure de la normale (LSN), les taux de transaminases doivent également être mesurés dans les 72 heures suivant la fin de la perfusion. Si les taux de transaminases sont supérieurs à la valeur initiale et > 2 fois la LSN, la dose de Xenpozyme peut être ajustée (reprise de la dose antérieure ou réduction de la dose) ou le traitement peut être temporairement interrompu, en fonction de l'évaluation clinique.
Une fois la dose d'entretien recommandée atteinte, le dosage des transaminases peut être effectué dans le cadre de la prise en charge clinique de routine de l'ASMD.
Sodium
Ce médicament contient 0,60 mg de sodium par flacon de 4 mg ou 3,02 mg de sodium par flacon de 20 mg, ce qui équivaut à 0,03% et 0,15%, respectivement, de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte ou adolescent et à ≤0,08% et ≤0,38%, respectivement, de l'apport alimentaire quotidien maximal acceptable chez un enfant de moins de 16 ans.
InteractionsAucune étude n'a été réalisée concernant les études médicamenteuses. L'olipudase alfa étant une protéine humaine recombinante, aucune interaction médicamenteuse médiée par le cytochrome P450 n'est attendue.
Grossesse, allaitementFemmes en âge de procréer
Un test de grossesse peut être envisagé chez les femmes en âge de procréer avant le début du traitement afin d'exclure toute grossesse.
Il est conseillé aux femmes en âge de procréer d'utiliser une contraception efficace pendant le traitement et jusqu'à 14 jours après la dernière dose si Xenpozyme est arrêté.
Grossesse
À ce jour, on ne dispose que de données limitées sur l'utilisation de l'olipudase alfa chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir «Données précliniques»). Xenpozyme n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception, sauf si les bénéfices potentiels pour la mère sont supérieurs aux risques potentiels, y compris ceux pour le fœtus.
Allaitement
On ne sait pas si l'olipudase alfa est excrétée dans le lait maternel. L'olipudase alfa a été détectée dans le lait d'animaux en lactation (voir «Données précliniques»). Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement par Xenpozyme en prenant en compte le bénéfice de l'allaitement maternel pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'y a pas de données disponibles chez l'être humain concernant les effets de l'olipudase alfa sur la fertilité des hommes et des femmes. Les données des études effectuées chez l'animal ne montrent aucun effet nocif direct ou indirect sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesUne hypotension ayant été rapportée lors des études cliniques, Xenpozyme peut avoir une influence mineure sur l'aptitude à conduire des véhicules et l'utilisation de machines (voir «Effets indésirables»).
Effets indésirablesRésumé du profil de sécurité
Les effets indésirables graves rapportés chez les patients traités par Xenpozyme étaient un épisode d'extrasystoles dans le contexte d'antécédents de cardiomyopathie chez un (2,5%) patient adulte et une réaction anaphylactique, une urticaire, un rash, une hypersensibilité et une augmentation du taux d'alanine aminotransférase, chacun rapporté chez un (5%) patient pédiatrique. L'incidence des RAP sévères liées à une hypersensibilité était plus élevée chez les patients pédiatriques que chez les adultes. Chez un patient adulte, des effets indésirables récurrents, se manifestant sous la forme d'un rash cutané, ont conduit à une interruption durable du traitement.
Les effets indésirables médicamenteux les plus fréquemment rapportés (EI chez ≥10% des patients ayant reçu Xenpozyme) étaient les suivants: céphalées (31,7%), urticaire (26,7 %), fièvre (25 %), nausée (20%), douleurs abdominales (16,7 %), vomissements (16,7%), prurit (13,3 %), myalgie (13,3 %), rash (11,7 %), augmentation du taux de protéine C-réactive (11,7 %), douleur abdominale haute (10 %) et érythème (10 %).
L'analyse groupée de la sécurité d'emploi réalisée à partir de 4 études cliniques (une étude de tolérance chez des patients adultes, ASCEND, ASCEND-Peds et une étude d'extension chez des patients adultes et pédiatriques) incluait un total de 60 patients (40 patients adultes et 20 patients pédiatriques) traités par Xenpozyme à des doses allant jusqu'à 3 mg/kg toutes les 2 semaines.
Liste des effets indésirables
Les effets indésirables rapportés dans le cadre de l'analyse groupée de la sécurité d'emploi lors des études cliniques achevées sont présentés par classe de systèmes d'organes et par catégorie de fréquence: très fréquent (≥1/10), fréquent (≥1/100, < 1/10).
Tableau 6: Effets indésirables rapportés dans le cadre de l'analyse groupée de la sécurité d'emploi lors des études cliniques achevées
|
Classe de systèmes d'organes
|
Fréquence
| |
Très fréquent
|
Fréquent
| |
Affections du système immunitaire
|
|
Anaphylaxie et hypersensibilité
| |
Affections du système nerveux
|
Céphalées (31,7 %)
|
| |
Affections oculaires
|
|
Hyperémie oculaire, gêne oculaire, prurit de l'œil
| |
Affections cardiaques
|
|
Palpitations, tachycardie
| |
Affections vasculaires
|
|
Hypotension, bouffées de chaleur, bouffées congestives
| |
Affections respiratoires, thoraciques et médiastinales
|
|
Œdème pharyngé, gonflement pharyngé, sensation de gorge serrée, sibilances, irritation du larynx, dyspnée, irritation de la gorge
| |
Affections gastro-intestinales
|
Nausée (20 %), douleurs abdominales (15 %), vomissements (16,7 %)
Douleur abdominale haute (10 %)
|
Diarrhée, gêne abdominale, douleur gastro-intestinale
| |
Affections hépatobiliaires
|
|
Hépatalgie
| |
Affections de la peau et du tissu sous-cutané
|
Urticaire (21,7 %),
prurit (10 %), rash (11,7 %), érythème (10 %)
|
Angiœdème, érythème pigmenté fixe,rash papuleux, rash maculeux, rash maculopapuleux, rash érythémateux, rash prurigineux, rash morbilliforme, papule, macule
| |
Affections musculosquelettiques et du tissu conjonctif
|
Myalgie (11,7 %)
|
Douleur osseuse, arthralgie, dorsalgie
| |
Troubles généraux et anomalies au site d'administration
|
Fièvre (25 %)
|
Douleur, frissons, douleur au site du cathéter, réaction au site d'introduction du cathéter, prurit au site du cathéter, gonflement au site du cathéter, fatigue, asthénie
| |
Investigations
|
Protéine C-réactive augmentée (11,7 %)
|
Alanine aminotransférase augmentée, aspartate aminotransférase augmentée, ferritine sérique augmentée, protéine C-réactive anormale, température augmentée
|
Description de certains effets indésirables
Réactions associées à la perfusion (RAP), notamment réactions d'hypersensibilité/anaphylactiques
Des RAP ont été rapportées chez 57,5 % des patients adultes et chez 65% des patients pédiatriques. Les symptômes de RAP les plus fréquemment rapportés chez les patients adultes étaient: céphalées (25 %), nausée (17,5 %), urticaire (17,5 %), myalgie (12,5 %), arthralgie (10%), fièvre (10%), prurit (10 %), vomissements (7,5%), douleurs abdominales (7,5%), érythème (7,5 %) et fatigue (7,5 %). Les symptômes de RAP les plus fréquemment rapportés chez les patients pédiatriques étaient: fièvre (40%), urticaire (40 %), vomissements (30%), protéine C-réactive augmentée (20 %), céphalées (20%), nausée (20%), érythème (15 %), rash (15%), ferritine sérique augmentée (15 %), douleurs abdominales (10 %) et prurit (10 %). Les RAP sont généralement survenues entre le moment de la perfusion et jusqu'à 24 heures après la fin de la perfusion. La plupart des RAP ont été classifiées comme légères ou modérées.
Des RAP liées à l'hypersensibilité, dont l'anaphylaxie, sont survenues chez 30 % des patients, 22,5 % des patients adultes et 45% des patients pédiatriques, dans les études cliniques. Les symptômes de RAP liés à l'hypersensibilité les plus fréquemment rapportés étaient l'urticaire (25 %), le prurit (10 %), l'érythème (10 %) et le rash (8,3 %).
Lors des études cliniques, un patient pédiatrique a présenté une réaction anaphylactique sévère. En outre, indépendamment du programme d'études cliniques, un patient âgé de 16 mois atteint d'ASMD de type A, traité par Xenpozyme, a présenté 2 réactions anaphylactiques. Des anticorps anti-olipudase alfa de type IgE ont été détectés chez les deux patients.
Chez 2 patients adultes et 3 patients pédiatriques, les symptômes de RAP étaient associés à des modifications des paramètres biologiques (p.ex. protéine C-réactive, ferritine), indiquant une réaction de phase aiguë. Tous les événements peuvent être traités comme d'autres RAP.
Augmentation des transaminases
Des augmentations transitoires des transaminases (ALAT ou ASAT) dans les 24 à 48 heures suivant une perfusion ont été signalées chez quelques patients traités par Xenpozyme pendant la phase d'escalade de dose lors des études cliniques. Lors de la perfusion suivante planifiée, ces taux élevés de transaminases étaient généralement revenus aux taux antérieurs à la perfusion précédente.
Au total, après 52 semaines de traitement par Xenpozyme, le taux moyen d'ALAT avait diminué de 46,9 % et le taux moyen d'ASAT de 40,2%, par rapport aux taux initiaux. Chez les patients adultes, l'ensemble des 16 patients avec un taux initial d'ALAT élevé avaient normalisé leur ALAT et 10 des 12 patients avec un taux initial d'ASAT élevé avaient normalisé leur ASAT.
Immunogénicité
Dans l'ensemble, 19 patients adultes sur 40 (47,5 %) et 15 patients pédiatriques sur 20 (75 %) traités avec Xenpozyme ont développé des anticorps anti-médicament (anti-drug antibodies, ADA) pendant le traitement. Le délai médian de séroconversion à partir de la première perfusion de Xenpozyme était d'environ 52 semaines chez les patients adultes et d'environ 12 semaines chez les patients pédiatriques. La majorité des patients positifs aux ADA (16 patients adultes sur 19 et 10 patients pédiatriques sur 15) présentaient une faible réponse en ADA (titre maximal ≤400) ou sont revenus à un statut négatif aux ADA. Trois patients adultes et 4 patients pédiatriques positifs aux ADA présentaient une réponse en ADA intermédiaire (titre maximal dans la plage de 800 à 6400). Huit des 19 patients adultes positifs aux ADA et 9 des 15 patients pédiatriques positifs aux ADA ont développé des anticorps neutralisants (AcN) inhibant l'activité de l'olipudase alfa. Deux patients adultes et 3 patients pédiatriques ont développé des AcN à plusieurs points d'évaluation. Aucun des patients n'a développé d'AcN inhibant l'absorption cellulaire de l'olipudase alfa. Un patient pédiatrique a présenté une réaction anaphylactique et a développé des ADA de type IgE et de type IgG avec un titre maximal de 1600.
Aucun effet des ADA n'a été observé sur la pharmacocinétique et l'efficacité de Xenpozyme dans les populations adultes et pédiatriques. Le pourcentage de patients présentant des RAP apparues au cours du traitement (y compris des réactions d'hypersensibilité) était plus élevé chez les patients qui avaient développé des ADA apparus au cours du traitement que chez ceux qui n'en avaient pas développé (70,6 % contre 46,2 %). Les RAP étaient contrôlables et n'ont pas entraîné d'interruption du traitement.
Population pédiatrique
À l'exception d'une incidence de RAP liées à l'hypersensibilité plus élevée chez les patients pédiatriques que chez les adultes, le profil de sécurité d'emploi de Xenpozyme chez les patients pédiatriques et adultes était similaire.
Utilisation à long terme
La durée médiane d'exposition était de 4,95 ans (plage: 0,4 à 9,6 ans) chez les patients adultes et de 6,15 ans (plage: 4,3 à 8,2 ans) chez les patients pédiatriques. Dans l'ensemble, le profil des événements indésirables observés chez les patients adultes et pédiatriques dans le cadre d'une utilisation à long terme était en cohérence avec celui observé pendant la première année de traitement.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Un nombre limité de cas de surdosage de Xenpozyme a été rapporté chez des patients pédiatriques pendant l'escalade de dose. Certains de ces patients ont présenté des effets indésirables graves dans les 24 heures suivant le début du traitement, y compris le décès. Les principales observations cliniques comprenaient un arrêt respiratoire, une hypotension, des élévations marquées des tests hépatiques et des hémorragies gastro-intestinales.
Traitement
Il n'y a pas d'antidote spécifique connu en cas de surdosage de Xenpozyme. En cas de surdosage, la perfusion doit être arrêtée immédiatement et le patient doit être étroitement surveillé en milieu hospitalier afin de détecter le développement de RAP, y compris de réactions de phase aiguë. Pour la prise en charge des effets indésirables, voir «Mises en garde et précautions».
Propriétés/EffetsCode ATC
A16AB25
Mécanisme d'action
L'olipudase alfa (sphingomyélinase acide humaine recombinante) est une enzyme recombinante qui réduit l'accumulation de sphingomyéline (SM) dans les organes des patients atteints de déficit en sphingomyélinase acide (ASMD). Xenpozyme ne devrait pas traverser la barrière hémato-encéphalique ou moduler les manifestations de la maladie au niveau du système nerveux central.
Pharmacodynamique
Le taux de céramide et de lyso-sphingomyéline (une forme désacylée de la SM) a été utilisé pour évaluer l'activité pharmacodynamique de Xenpozyme chez des patients atteints d'ASMD.
Après administration répétée de Xenpozyme chez des patients adultes et pédiatriques, les concentrations plasmatiques de céramide augmentent provisoirement après chaque dose (après la perfusion), en diminuant progressivement au cours du traitement. Dans l'étude DFI12712/ASCEND, la variation en pourcentage de la moyenne des moindres carrés (MC) entre le début de l'étude et la Semaine 52 (erreur-type, E-T) de la concentration plasmatique de céramide avant la perfusion était de -36,4% (5,3) dans le groupe de traitement par Xenpozyme et de -0,2% (5,6) dans le groupe placebo. Chez les patients pédiatriques, les valeurs de la moyenne des MC de la concentration plasmatique de céramide avant la perfusion, entre le début de l'étude et la Semaine 52 (E-T: 5,1) ont diminué de 57%.
La concentration plasmatique de lyso-sphingomyéline a considérablement augmenté chez les patients adultes et pédiatriques atteints d'ASMD. Après l'administration répétée de Xenpozyme, les valeurs plasmatiques de lyso-sphingomyéline ont significativement diminué, reflétant une réduction de la teneur en sphingomyéline dans les tissus. Dans l'étude DFI12712/ASCEND, la variation en pourcentage de la moyenne des MC entre le début de l'étude et la Semaine 52 (E-T) de la concentration plasmatique de lyso-sphingomyéline avant la perfusion était de -77,7% (3,9) dans le groupe de traitement par Xenpozyme et de -5,0% (4,2) dans le groupe placebo. Chez les patients pédiatriques, les valeurs de la moyenne des MC de la concentration plasmatique de lyso-sphingomyéline avant la perfusion, entre le début de l'étude et la Semaine 52 (E-T: 1,3) ont diminué de 87,2%.
La teneur en sphingomyéline dans le foie, évaluée par histopathologie, entre l'inclusion dans l'étude et la Semaine 52 dans le groupe de patients adultes traités par Xenpozyme a diminué de 92,0% (E-T: 8,1) (contre une augmentation de +10,3% [E-T: 7,8] dans le groupe placebo).
Efficacité clinique
L'efficacité de Xenpozyme a été évaluée dans 3 études cliniques (étude ASCEND chez des patients adultes, étude ASCEND-Peds chez des patients pédiatriques et une étude d'extension chez des patients adultes et pédiatriques) portant sur un total de 61 patients atteints d'ASMD.
Étude clinique chez des patients adultes
L'étude ASCEND est une étude de phase II/III multicentrique, randomisée, en double aveugle, contrôlée par placebo, à doses répétées, conduite chez des patients adultes atteints d'ASMD de type A/B et B. Au total, 36 patients ont été randomisés selon un rapport 1:1 pour recevoir soit Xenpozyme, soit un placebo. Le traitement a été administré aux deux groupes en perfusion intraveineuse une fois toutes les 2 semaines. Chez les patients recevant Xenpozyme, la dose a été augmentée de 0,1 mg/kg jusqu'à une dose cible de 3 mg/kg. L'étude a été divisée en 2 périodes consécutives: une période d'analyse principale (PAP) randomisée, contrôlée par placebo, en double aveugle, d'une durée de 52 semaines, suivie d'une période d'extension du traitement (PET) d'une durée maximale de 4 ans.
Les patients randomisés dans le bras placebo de la PAP sont passés au traitement actif dans la PET pour atteindre progressivement la dose cible de 3 mg/kg, tandis que les patients du bras originel Xenpozyme ont continué le traitement.
Les patients inclus dans l'étude avaient une capacité de diffusion pulmonaire du monoxyde de carbone (DLco) ≤70% de la valeur normale prédite, un volume splénique ≥6 multiples de la normale (MN) mesuré par imagerie par résonance magnétique (IRM) et des scores ≥5 pour le score lié à la splénomégalie (splenomegaly related score, SRS). Dans l'ensemble, à l'inclusion, les caractéristiques démographiques et celles de la maladie étaient similaires entre les deux groupes de traitement. L'âge médian des patients était de 30 ans (intervalle: 18 à 66 ans). L'âge moyen (écart-type, ET) au moment du diagnostic de l'ASMD était de 18 (18,4) ans. À l'inclusion dans l'étude, des manifestations neurologiques ont été observées chez 9 patients adultes sur 36 (25%) présentant un diagnostic clinique compatible avec un ASMD de type A/B. Les 27 patients restants présentaient un diagnostic clinique compatible avec un ASMD de type B.
Cette étude comprenait 2 critères principaux d'évaluation de l'efficacité indépendants: la variation en pourcentage de la DLco (en % de la valeur normale prédite) et du volume splénique (en MN), mesuré par IRM, entre l'inclusion et la Semaine 52.
Les critères secondaires d'évaluation de l'efficacité incluaient la variation en pourcentage du volume hépatique (en MN) et la numération plaquettaire entre l'inclusion et la Semaine 52.
Des améliorations de la variation moyenne en pourcentage de la DLco (en % de la valeur prédite) (p = 0,0004) et du volume splénique (p < 0,0001), ainsi que du volume hépatique moyen (p < 0,0001) et de la numération plaquettaire (p = 0,0185) ont été observées dans le groupe Xenpozyme par rapport au groupe placebo pendant la période d'analyse principale de 52 semaines. Une amélioration significative de la variation moyenne en pourcentage de la DLco (en % de la valeur prédite), du volume splénique, du volume hépatique et de la numération plaquettaire a été constatée à la Semaine 26 du traitement, la première évaluation du critère d'évaluation après administration.
Les résultats de la PAP à la Semaine 52 sont détaillés dans le tableau 7.
Tableau 7: Valeurs moyennes (ET) des critères d'évaluation de l'efficacité à l'inclusion et variation en pourcentage de la moyenne des MC (E-T) entre l'inclusion et la Semaine 52
|
|
Placebo
(N = 18)
|
Xenpozyme
(N = 18)
|
Différence
(IC à 95%)
|
Valeur de p*
| |
Critères d'évaluation principaux
|
| |
DLco moyenne en % de la valeur prédite à l'inclusion dans l'étude
|
48,5 (10,8)
|
49,4 (11,0)
|
NA
|
NA
| |
Variation de la DLco en % de la valeur prédite entre l'inclusion dans l'étude et la Semaine 52
|
3 (3,4)
|
22 (3,3)
|
19 (4,8)
[9,3, 28,7]
|
0,0004
| |
Volume splénique moyen (MN) à l'inclusion dans l'étude
|
11,2 (3,8)
|
11,7 (4,9)
|
NA
|
NA
| |
Pourcentage de variation du volume splénique entre l'inclusion dans l'étude et la Semaine 52
|
0,5 (2,5)
|
-39,4 (2,4)
|
-39,9 (3,5)
[-47,1, -32,8]
|
< 0,0001
| |
Critères d'évaluation secondaires
|
| |
Volume hépatique moyen (MN) à l'inclusion dans l'étude
|
1,6 (0,5)
|
1,4 (0,3)
|
NA
|
NA
| |
Pourcentage de variation du volume hépatique entre l'inclusion dans l'étude et la Semaine 52
|
-1,5 (2,5)
|
-28,1 (2,5)
|
-26,6 (3,6)
[-33,9, -19,3]
|
< 0,0001
| |
Numération plaquettaire moyenne (109/l) à l'inclusion dans l'étude
|
115,6 (36,3)
|
107,2 (26,9)
|
NA
|
NA
| |
Pourcentage de variation de la numération plaquettaire entre l'inclusion dans l'étude et la Semaine 52
|
2,5 (4,2)
|
16,8 (4,0)
|
+14,3 (5,8)
[2,6, 26,1]
|
0,0185
|
* Statistiquement significatif après ajustement pour multiplicité
Dix-sept des 18 patients ayant reçu le placebo et 18 des 18 patients ayant reçu Xenpozyme pendant 52 semaines (PAP) ont respectivement démarré ou continué le traitement par Xenpozyme sur une période de temps allant jusqu'à 4 ans. Les effets durables de Xenpozyme sur les critères d'évaluation de l'efficacité jusqu'à la Semaine 104 sont présentés dans les figures 1 et 2 et dans le tableau 8.
Figure 1: Courbe des moyennes des MC (IC à 95%) de la variation en pourcentage de la DLco (% de la valeur prédite) entre l'inclusion et la Semaine 104 - Population en ITTm (intention de traiter modifiée)

Les barres verticales représentent les IC à 95% pour les moyennes des MC.
Les moyennes des MC et les IC à 95% sont basés sur un modèle mixte pour mesures répétées, utilisant des données jusqu'à la Semaine 104.
Les patients du groupe placebo/Xenpozyme ont reçu le placebo jusqu'à la Semaine 52 puis ont reçu Xenpozyme.
Figure 2: Courbe des moyennes des MC (IC à 95%) de la variation en pourcentage du volume splénique (MN) entre l'inclusion et la Semaine 104 - Population en ITTm (intention de traiter modifiée)
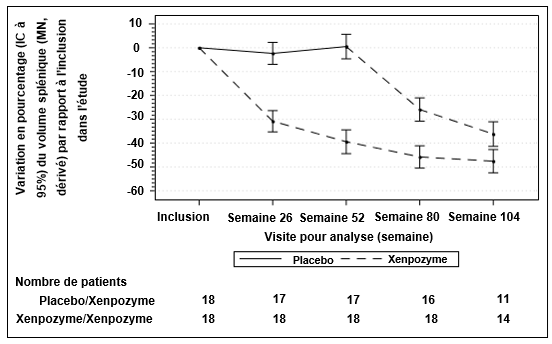
Les barres verticales représentent les IC à 95% pour les moyennes des MC.
Les moyennes des MC et les IC à 95% sont basés sur un modèle mixte pour mesures répétées, utilisant des données jusqu'à la Semaine 104.
Les patients du groupe placebo/Xenpozyme ont reçu le placebo jusqu'à la semaine 52 puis ont reçu Xenpozyme.
Tableau 8: Variation en pourcentage (E-T) de la moyenne des MC entre l'inclusion dans l'étude et la Semaine 104 du volume hépatique (MN) et de la numération plaquettaire (109/l) chez les patients traités avec Xenpozyme pendant 104 semaines
|
|
Groupe ayant reçu de l'olipudase alfa
| |
|
Semaine 52 (début de la PET)
|
Semaine 104
| |
N
Variation en pourcentage du volume hépatique (ET)
|
17
-27,8 (2,5)
|
14
-33,4 (2,2)
| |
N
Variation en pourcentage de la numération plaquettaire (ET)
|
18
16,6 (4,0)
|
13
24,9 (6,9)
|
N: Nombre de patients
Étude d'extension chez des patients adultes
Cinq patients adultes ayant participé à une étude en ouvert d'escalade de dose chez des patients atteints d'ASMD ont poursuivi le traitement dans une étude d'extension en ouvert et ont reçu Xenpozyme jusqu'à > 9 ans.
Des améliorations durables de la DLco en % de la valeur prédite, des volumes splénique et hépatique et de la numération plaquettaire, par rapport à l'inclusion dans l'étude, ont été observées chez les patients adultes au cours de l'étude (voir tableau 9).
Tableau 9: Variation moyenne en pourcentage (ET) des paramètres d'efficacité entre l'inclusion dans l'étude et le Mois 78
|
|
Mois 78
(N = 5)
| |
Variation en pourcentage de la DLco en % de la valeur prédite (ET)
|
55,3% (48,1)
| |
Variation en pourcentage du volume splénique (ET)
|
-59,5% (4,7)
| |
Variation en pourcentage du volume hépatique (ET)
|
-43,7% (16,7)
| |
Variation en pourcentage de la numération plaquettaire (ET)
|
38,5% (14,7)
|
N: Nombre de patients
Population pédiatrique
L'étude ASCEND-Peds (étude clinique de phase I/II) est une étude multicentrique, en ouvert, à doses répétées visant à évaluer la sécurité d'emploi et la tolérance de Xenpozyme administré pendant 64 semaines chez des patients pédiatriques âgés de < 18 ans atteints d'ASMD (type A/B et B). En outre, les critères d'évaluation exploratoires de l'efficacité liés à l'organomégalie, aux fonctions pulmonaire et hépatique, et à la croissance linéaire ont été évalués à la Semaine 52.
Au total, 20 patients (4 adolescents âgés de 12 à < 18 ans, 9 enfants âgés de 6 à < 12 ans et 7 nourrissons/jeunes enfants âgés de < 6 ans) ont été traités par une dose progressive de Xenpozyme via un schéma d'escalade de dose de 0,03 mg/kg à une dose cible de 3 mg/kg. Le traitement a été administré par perfusion intraveineuse une fois toutes les 2 semaines jusqu'à 64 semaines. Les patients inclus dans l'étude avaient un volume splénique ≥5 multiples de la normale mesuré par IRM. Les patients étaient âgés de 1,5 à 17,5 ans, les deux sexes étant également représentés. L'âge moyen (ET) au moment du diagnostic de l'ASMD était de 2,5 (2,5) ans. À l'inclusion dans l'étude, des manifestations neurologiques ont été observées chez 8 patients pédiatriques sur 20 (40%) présentant un diagnostic clinique compatible avec un ASMD de type A/B. Les 12 patients restants présentaient un diagnostic clinique compatible avec un ASMD de type B.
Le traitement par Xenpozyme a entraîné des améliorations de la variation moyenne en pourcentage de la DLco en % de la valeur prédite, des volumes splénique et hépatique, des numérations plaquettaires et de la progression de la croissance linéaire (telle que mesurée par les Z-scores de la taille) à la Semaine 52 par rapport à l'inclusion (voir tableau 10).
Tableau 10: Variation en pourcentage (E-T) de la moyenne des MC ou variation (ET) des paramètres d'efficacité entre l'inclusion et la Semaine 52 (toutes cohortes d'âge confondues)
|
|
Valeur à l'inclusion dans l'étude (N = 20)
|
Semaine 52 (N = 20)
| |
DLco moyenne en % de la valeur prédite (ET)
|
54,8 (14,2)
|
71,7 (14,8)
| |
Variation en pourcentage de la DLco en % de la valeur prédite*
|
|
32,9 (8,3)
| |
IC à 95%
|
|
13,4; 52,5
| |
Volume splénique moyen (MN) (ET)
|
19,0 (8,8)
|
9,3 (3,9)
| |
Variation en pourcentage du volume splénique (MN)
|
|
-49,2 (2,0)
| |
IC à 95%
|
|
-53,4, -45,0
| |
Volume hépatique moyen (MN) (ET)
|
2,7 (0,7)
|
1,5 (0,3)
| |
Variation en pourcentage du volume hépatique (MN)
|
|
-40,6 (1,7)
| |
IC à 95%
|
|
-44,1, -37,1
| |
Numération plaquettaire moyenne (109/l) (ET)
|
137,7 (62,3)
|
173,6 (60,5)
| |
Variation en pourcentage de la numération plaquettaire
|
|
34,0 (7,6)
| |
IC à 95%
|
|
17,9, 50,1
| |
Z-scores moyens de la taille (ET)
|
-2,1 (0,8)
|
-1,6 (0,8)
| |
Variation des Z-scores de la taille*
|
|
0,6 (0,4)
| |
IC à 95%
|
|
(0,38, 0,73)
|
* La DLco a été évaluée chez 9 patients pédiatriques âgés de ≥5 ans qui étaient capables d'effectuer le test. La variation de la taille (Z-score) a été évaluée chez 19 patients pédiatriques.
Les effets de Xenpozyme sur les volumes splénique et hépatique, la numération plaquettaire et la taille (Z-scores) ont été observés dans toutes les cohortes d'âge pédiatriques incluses dans l'étude.
Étude d'extension chez des patients pédiatriques
Vingt patients pédiatriques de l'étude ASCEND-Peds ont poursuivi le traitement dans une étude d'extension en ouvert et ont reçu Xenpozyme jusqu'à > 8 ans.
Des améliorations durables des paramètres d'efficacité (DLco en % de la valeur prédite, volumes splénique et hépatique, numération plaquettaire, Z-score de taille et âge osseux) ont été observées chez les patients pédiatriques au cours de l'étude jusqu'au Mois 48 (voir tableau 11).
Tableau 11: Variation en pourcentage de la moyenne des MC ou variation (ET) des paramètres d'efficacité entre l'inclusion dans l'étude et le Mois 48
|
|
Mois 48
| |
N
Variation en pourcentage de la DLco en % de la valeur prédite (ET)
|
5
60,3 (58,5)
| |
N
Variation en pourcentage du volume splénique (ET)
|
7
-69,1 (4,1)
| |
N
Variation en pourcentage du volume hépatique (ET)
|
7
-55,4 (11,0)
| |
N
Variation en pourcentage de la numération plaquettaire (ET)
|
5
35,8 (42,4)
| |
N
Variation des Z-scores de la taille (ET)
|
5
2,3 (0,8)
| |
N
Variation de l'âge osseux (mois) (ET)
|
7
18,5 (19,0)
|
N: Nombre de patients
PharmacocinétiqueLa pharmacocinétique de l'olipudase alfa a été évaluée chez 49 patients adultes atteints d'ASMD issus de toutes les études cliniques, recevant des administrations uniques ou multiples. À la dose de 3 mg/kg administrée une fois toutes les 2 semaines, la moyenne (coefficient de variation en pourcentage, CV %) de la concentration maximale (Cmax) et l'aire sous la courbe de la concentration en fonction du temps sur un intervalle d'administration (ASC0-τ) à l'état d'équilibre étaient, respectivement, de 30,2 µg/ml (17%) et 607 µg·h/ml (20%).
Absorption
Il n'y a pas d'absorption puisque Xenpozyme est administré par voie intraveineuse.
Distribution
Le volume de distribution moyen estimé (CV %) de l'olipudase alfa est de 13,1 l (18%).
Métabolisme
L'olipudase alfa est une enzyme humaine recombinante qui devrait être éliminée par dégradation protéolytique en petits peptides et acides aminés.
Élimination
La clairance moyenne (CV %) de l'olipudase alfa est de 0,331 l/h (22%). La demi-vie terminale moyenne (t½) est comprise entre 31,9 et 37,6 heures.
Linéarité/non-linéarité
L'olipudase alfa a démontré une pharmacocinétique linéaire à travers la plage de doses de 0,03 à 3 mg/kg. Après un protocole d'escalade de dose allant de 0,1 mg/kg à la dose d'entretien de 3 mg/kg, administrée une fois toutes les 2 semaines, une accumulation minime de l'olipudase alfa dans le plasma a été observée.
Cinétique pour certains groupes de patients
Aucune différence cliniquement significative dans la pharmacocinétique de l'olipudase alfa n'a été observée en fonction du sexe.
Insuffisance hépatique
L'olipudase alfa est une protéine recombinante qui devrait être éliminée par dégradation protéolytique. Par conséquent, une insuffisance hépatique ne devrait pas avoir d'effet sur la pharmacocinétique de l'olipudase alfa.
Insuffisance rénale
Quatre patients (11,1%) présentant une insuffisance rénale légère (60 ml/min ≤ clairance de la créatinine < 90 ml/min) ont été inclus dans l'étude ASCEND. Aucune différence cliniquement significative de la pharmacocinétique de l'olipudase alfa n'a été observée chez les patients atteints d'une insuffisance rénale légère. L'impact d'une insuffisance rénale modérée à sévère sur la pharmacocinétique de l'olipudase alfa n'est pas connu. L'olipudase alfa ne devrait pas être éliminée par excrétion rénale. Par conséquent, l'insuffisance rénale ne devrait pas avoir d'effet sur la pharmacocinétique de l'olipudase alfa.
Patients âgés
Une analyse de pharmacocinétique de population n'a pas mis en évidence de différence d'exposition chez les patients âgés (seuls 2 patients entre 65 et 75 ans ont été inclus dans les essais cliniques de Xenpozyme).
Enfants et adolescents
La pharmacocinétique de l'olipudase alfa a été évaluée chez 20 patients pédiatriques, dont 4 adolescents, 9 enfants et 7 jeunes enfants/nourrissons (tableau 12). Les expositions à l'olipudase alfa étaient plus faibles chez les patients pédiatriques que chez les patients adultes. Cependant, ces différences n'ont pas été considérées comme cliniquement significatives.
Tableau 12: Moyennes (CV %) des paramètres pharmacocinétiques de l'olipudase alfa après l'administration d'une dose de 3 mg/kg toutes les 2 semaines chez des patients adolescents, enfants et jeunes enfants/nourrissons atteints d'ASMD
|
Groupe d'âge
|
Âge (ans)
|
Cmax (µg/ml)
|
ASC0-τ (µg.h/ml)
| |
Adolescents (N = 4)
|
12, < 18
|
27,5 (8)
|
529 (7)
| |
Enfants (n = 9)
|
6, < 12
|
24,0 (10)
|
450 (15)
| |
Jeunes enfants/nourrissons (N = 7)
|
< 6
|
22,8 (8)
|
403 (11)
| |
Les statistiques descriptives représentent les estimations post-hoc des expositions à l'état d'équilibre à l'aide de l'analyse de pharmacocinétique de population.
ASC0-τ: aire sous la courbe de la concentration plasmatique en fonction du temps sur un intervalle d'administration; Cmax: concentration plasmatique maximale; N: nombre total de patients.
|
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité à dose unique et de toxicité à doses répétées menées chez des animaux de type sauvage (souris, rats, lapins, chiens et singes) à des niveaux de doses 10 fois supérieurs à la dose maximale recommandée chez l'homme (DMRH) et correspondant à une exposition jusqu'à 4 fois supérieure à celle de l'homme (sur la base de l'ASC) à la fréquence et à la dose thérapeutique d'entretien recommandées, ne mettent en évidence aucun risque particulier pour l'homme.
Toxicité en cas d'administration répétée
Chez des souris knockout pour la sphingomyélinase acide (acid sphingomyelinase knockout, ASMKO - modèle animal pour l'ASMD), une mortalité a été observée après l'administration par injection intraveineuse en bolus de doses uniques d'olipudase alfa ≥3,3 fois plus élevées que la DMRH. Cependant, des études de toxicité à doses répétées montrent que l'administration d'olipudase alfa selon un schéma d'escalade de dose n'a pas entraîné de mortalité liée au composé et a réduit la gravité d'autres résultats de toxicité jusqu'à la plus forte dose testée (30 mg/kg), étant 10 fois supérieure à la DMRH.
Génotoxicité
Aucune étude d'évaluation de la mutagénicité de l'olipudase alfa n'a été menée.
Carcinogénicité
Aucune étude d'évaluation du potentiel carcinogène de l'olipudase alfa n'a été menée.
Toxicité sur la reproduction
Une incidence accrue d'exencéphalie a été observée lorsque des souris gravides ont été traitées quotidiennement par olipudase alfa à des niveaux d'exposition correspondant à l'exposition chez l'homme à la dose thérapeutique d'entretien et à la fréquence recommandées. Cette incidence était légèrement plus élevée que les données historiques des témoins. La pertinence de cette observation chez l'homme est inconnue. L'administration intraveineuse quotidienne d'olipudase alfa à des lapines gravides n'a pas entrainé de malformations ou de variations fœtales à des expositions correspondant à 10,5 fois l'exposition (sur la base de l'ASC) chez l'humain à la dose thérapeutique d'entretien et à la fréquence recommandées.
Chez les souris qui ont reçu 3 mg/kg d'olipudase alfa le jour 7 du post-partum, l'olipudase alfa a été détectée dans le lait 2 jours après l'administration.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Médicament reconstitué
La stabilité physico-chimique de Xenpozyme après reconstitution avec de l'eau stérile pour préparations injectables a été démontrée jusqu'à 48 heures à une température comprise entre 2 °C et 8 °C ou à température ambiante (jusqu'à 25 °C). D'un point de vue microbiologique, le médicament reconstitué doit être utilisé immédiatement. Si la dilution n'est pas effectuée immédiatement, les durées et conditions de conservation pendant l'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C ou 6 heures à température ambiante (jusqu'à 25 °C).
Médicament dilué
La stabilité physico-chimique après dilution avec 9 mg/ml (0,9%) de solution injectable de chlorure de sodium entre 0,1 mg/ml et 3,5 mg/ml a été démontrée pendant 48 heures entre 2 °C et 8 °C ou à température ambiante (jusqu'à 25 °C).
D'un point de vue microbiologique, le médicament dilué doit être utilisé immédiatement. Si le médicament n'est pas utilisé immédiatement après dilution, les durées et conditions de conservation pendant l'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, suivie de 12 heures (durée de la perfusion incluse) à température ambiante (jusqu'à 25 °C).
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Xenpozyme est une poudre pour solution à diluer pour perfusion en flacon. Les flacons sont réservés à un usage unique.
Les perfusions doivent être administrées de manière progressive, de préférence à l'aide d'une pompe à perfusion.
Les instructions d'utilisation figurent à la fin de l'information professionnelle.
La poudre pour solution à diluer pour perfusion doit être reconstituée avec de l'eau stérile pour préparations injectables, diluée avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9%), puis administrée par perfusion intraveineuse.
Les étapes de reconstitution et de dilution doivent être réalisées dans des conditions aseptiques. Des dispositifs de filtration ne doivent jamais être utilisés pendant la préparation de la solution pour perfusion. Éviter la formation de mousse pendant les étapes de reconstitution et de dilution.
La solution diluée doit être filtrée à l'aide d'un filtre en ligne à faible liaison protéique de 0,2 μm pendant l'administration.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation locale.
Numéro d’autorisation67287 (Swissmedic)
PrésentationXenpozyme 4 mg, poudre pour solution à diluer pour perfusion
·Conditionnement avec 1 flacon à usage unique de 4 mg [A]
·Conditionnement avec 10 flacons à usage unique de 4 mg [A]
Xenpozyme 20 mg, poudre pour solution à diluer pour perfusion
·Conditionnement avec 1 flacon à usage unique de 20 mg [A]
·Conditionnement avec 10 flacons à usage unique de 20 mg [A]
Titulaire de l’autorisationsanofi-aventis (suisse) sa, 1214 Vernier/GE
Mise à jour de l’informationJuin 2025
Les informations suivantes sont réservées aux professionnels de la santé:
Préparation de la solution pour perfusion en fonction de la posologie
La poudre pour solution à diluer pour perfusion doit être reconstituée avec de l'eau stérile pour préparations injectables, diluée avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9%), puis administrée par perfusion intraveineuse.
Les étapes de reconstitution et de dilution doivent être réalisées dans des conditions aseptiques. Des dispositifs de filtration ne doivent jamais être utilisés pendant la préparation de la solution pour perfusion. Éviter la formation de mousse pendant les étapes de reconstitution et de dilution.
La solution diluée doit être filtrée à l'aide d'un filtre en ligne à faible liaison protéique de 0,2 μm pendant l'administration.
1) Déterminer le nombre de flacons à reconstituer en fonction du poids de chaque patient et de la dose prescrite.
Poids du patient (kg) × dose (mg/kg) = dose du patient (en mg). Exemple lors de l'utilisation de flacons de 20 mg: dose du patient (en mg) divisée par 20 mg/flacon = nombre de flacons à reconstituer. Si le nombre de flacons comprend une décimale, arrondir au nombre entier supérieur.
2) Sortir le nombre nécessaire de flacons du réfrigérateur et les mettre de côté pendant environ 20 à 30 minutes pour leur permettre d'atteindre la température ambiante.
3) Reconstituer chaque flacon en injectant les quantités suivantes:
·1,1 ml d'eau stérile pour préparations injectables dans le flacon de 4 mg
·5,1 ml d'eau stérile pour préparations injectables dans le flacon de 20 mgInjecter lentement, goutte à goutte, le long de la paroi interne du flacon.
4) Incliner et faire rouler doucement chaque flacon. Éviter de faire mousser. Chaque flacon contiendra une solution limpide, incolore, à 4 mg/ml.
5) Inspecter à l'œil nu la solution reconstituée dans les flacons pour vérifier l'absence de particules et de coloration anormale. La solution de Xenpozyme doit être limpide et incolore. Les flacons contenant des particules opaques ou une coloration anormale ne doivent pas être utilisés.
6) Prélever le volume de solution reconstituée correspondant à la dose prescrite à partir du nombre approprié de flacons et diluer avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9%), dans une seringue ou une poche de perfusion en fonction du volume de perfusion (voir tableau 1 pour le volume de perfusion total recommandé en fonction de l'âge et/ou du poids des patients).
Tableau 1: Volumes de perfusion recommandés
|
|
Poids corporel ≥3 kg à < 10 kg
|
Poids corporel ≥10 kg à < 20 kg
|
Poids corporel ≥20 kg (patients pédiatriques < 18 ans)
|
Patients adultes (≥18 ans)
| |
Dose (mg/kg)
|
Volume total de perfusion (ml)
|
Volume total de perfusion (ml)
|
Volume total de perfusion (ml)
|
Volume total de perfusion (ml)
| |
0,03
|
Volume variable en fonction du poids corporel
|
Volume variable en fonction du poids corporel
|
5
|
NA
| |
0,1
|
Volume variable en fonction du poids corporel
|
5
|
10
|
20
| |
0,3
|
5
|
10
|
20
|
100
| |
0,6
|
10
|
20
|
50
|
100
| |
1,0
|
20
|
50
|
100
|
100
| |
2,0
|
50
|
75
|
200
|
100
| |
3,0
|
50
|
100
|
250
|
100
|
·Pour les volumes finaux variables de perfusion calculés en fonction du poids des patients pédiatriques (voir tableau 1):
·préparer une solution pour perfusion à 0,1 mg/ml en ajoutant 0,25 ml (1 mg) de solution reconstituée préparée à l'étape 3) et 9,75 ml de solution injectable de chlorure de sodium à 9 mg/ml (0,9%) dans une seringue vide de 10 ml.
·Calculer le volume (ml) requis pour obtenir la dose adaptée au patient (mg).
Exemple: 0,3 mg ÷ 0,1 mg/ml = 3 ml
·Injecter le volume requis de la solution pour perfusion à 0,1 mg/ml dans une seringue stérile vide de la taille adaptée en fonction du volume de perfusion.
·Instructions de dilution pour obtenir un volume total tel que 5 ml ≤ volume total ≤20 ml à l'aide d'une seringue:
·Injecter lentement le volume requis de solution reconstituée le long de la paroi intérieure de la seringue vide.
·Ajouter lentement la quantité suffisante de solution injectable de chlorure de sodium à 9 mg/ml (0,9%) pour obtenir le volume de perfusion total requis (éviter la formation de mousse dans la seringue).
·Instructions de dilution pour un volume total ≥50 ml en utilisant une poche de perfusion:
·Poche de perfusion vide:
·Injecter lentement le volume nécessaire de solution reconstituée obtenue lors de l'étape 3) dans une poche de perfusion stérile de taille appropriée.
·Ajouter lentement la quantité nécessaire de solution injectable de chlorure de sodium à 9 mg/ml (0,9%) pour obtenir le volume de perfusion total requis (éviter la formation de mousse dans la poche).
·Poche de perfusion préremplie:
·Retirer de la poche de perfusion préremplie de solution injectable de chlorure de sodium à 9 mg/ml (0,9%) le volume de solution injectable approprié pour obtenir un volume final tel que spécifié dans le tableau 1.
·Ajouter lentement le volume requis de solution reconstituée préparée à l'étape 3) dans la poche de perfusion (éviter la formation de mousse dans la poche).
7) Retourner délicatement la seringue ou la poche de perfusion pour mélanger. Ne pas agiter. Puisqu'il s'agit d'une solution protéique, une légère floculation (apparition de fines fibres translucides) apparaît parfois après dilution.
8) La solution diluée doit être filtrée à l'aide d'un filtre en ligne à faible liaison protéique de 0,2 μm pendant l'administration.
9) Une fois la perfusion terminée, la ligne de perfusion doit être rincée avec une solution injectable de chlorure de sodium à 9 mg/ml (0,9%) en utilisant le même débit de perfusion que celui utilisé pour la dernière partie de la perfusion.
|