CompositionPrincipes actifs
Mépolizumab.
Excipients
Saccharose, phosphate monohydrogéné de sodium heptahydraté, acide citrique monohydraté, polysorbate 80, édétate de sodium, eau pour préparations injectables.
Teneur tonale en sodium: 0,72 mg/ml.
Indications/Possibilités d’emploiAsthme sévère à éosinophiles
Nucala est indiqué en tant que médicament complémentaire chez les adultes et les adolescents à partir de 12 ans qui souffrent d'un asthme sévère à éosinophiles, caractérisé par les critères suivants:
·au moins deux exacerbations au cours des 12 derniers mois sous traitement standard actuel (corticostéroïdes à inhaler à haute dose et un traitement d'entretien complémentaire) et/ou nécessité de traitement par des corticostéroïdes systémiques.
·taux sanguin d'éosinophiles ≥0,15 G/L* (soit ≥150 cellules/μl) lors de l'initiation du traitement ou ≥0,3 G/L (soit ≥300 cellules/μl) au cours des 12 derniers mois.
* Pour des informations plus détaillées sur la population de patients, voir la rubrique «Propriétés/Effets», «Efficacité clinique et sécurité»
Rhinosinusite chronique avec polypes nasaux (RSCaPN)
Nucala est indiqué en tant que médicament complémentaire aux corticostéroïdes par voie nasale chez les adultes à partir de 18 ans qui souffrent d'une rhinosinusite chronique avec polypes nasaux (RSCaPN) sévère, insuffisamment contrôlée par des corticostéroïdes systémiques intermittents et/ou une chirurgie.
Granulomatose éosinophilique avec polyangéite (GEPA)
Nucala est indiqué en tant que médicament complémentaire chez les adultes à partir de 18 ans qui souffrent d'une granulomatose éosinophilique avec polyangéite (GEPA), caractérisée par les critères suivants:
·GEPA récidivante ou résistante au traitement
·Stabilisation préalable de la maladie au moyen de corticostéroïdes systémiques
·Traitement de maintien nécessaire avec des corticostéroïdes systémiques et éventuellement des immunosuppresseurs économiseurs de stéroïdes
Syndrome hyperéosinophilique (SHE)
Nucala est indiqué comme médicament complémentaire pour le traitement des adultes et des adolescents à partir de 12 ans atteints de syndrome hyperéosinophilique sans fusion FIP1L1-PDGFRα (SHE F/P négatif) (voir rubrique «Propriétés/Effets»).
Le traitement par Nucala doit rester réservé aux médecins expérimentés dans le traitement de l'asthme sévère, de la RSCaPN, de la GEPA ou du SHE.
Posologie/Mode d’emploiPosologie
Asthme sévère à éosinophiles
Adultes et adolescents à partir de 12 ans
La dose recommandée est de 100 mg de Nucala, à administrer par voie sous-cutanée une fois toutes les 4 semaines.
Enfants de moins de 12 ans
La sécurité et l'efficacité de Nucala chez les enfants de moins de 12 ans n'ont pas été évaluées dans le cadre d'études contrôlées.
Durée du traitement
Le succès thérapeutique doit être évalué au plus tard après 8 administrations de Nucala pour décider si le traitement doit être poursuivi ou non. L'évaluation de la réponse au traitement complémentaire inclut une évaluation soigneuse du contrôle de l'asthme, du besoin de corticostéroïdes systémiques et de la fréquence des exacerbations avant et pendant le traitement. Si la réponse est satisfaisante, Nucala est destiné à un traitement à long terme. Le bénéfice et la nécessité de poursuivre le traitement doivent être réévalués au moins une fois par an par le médecin sur la base de son appréciation de la sévérité de la maladie et du contrôle des exacerbations.
Rhinosinusite chronique avec polypes nasaux (RSCaPN)
Adultes à partir de 18 ans
La dose recommandée est de 100 mg de Nucala, à administrer par voie sous-cutanée une fois toutes les 4 semaines.
Enfants et adolescents de moins de 18 ans
La sécurité et l'efficacité de Nucala chez les enfants et les adolescents de moins de 18 ans atteints de RSCaPN n'ont pas été étudiées.
Granulomatose éosinophilique avec polyangéite (GEPA)
Aucune étude clinique de détermination de la dose spécifique à la GEPA n'a été réalisée (voir rubrique Propriétés / Effets, étude MEA115921).
Il convient de respecter une distance d'au moins 5 cm entre deux sites d'injection (cf. «Remarques particulières», Remarques concernant la manipulation).
Adultes à partir de 18 ans
La dose recommandée est de 300 mg de Nucala, à administrer par voie sous-cutanée une fois toutes les 4 semaines.
Enfants et adolescents de moins de 18 ans
La sécurité et l'efficacité de Nucala chez les enfants et les adolescents de moins de 18 ans souffrant de GEPA n'ont pas été étudiées.
Durée du traitement
Si la réponse est satisfaisante, Nucala est destiné à un traitement à long terme. La nécessité de poursuivre le traitement doit être réévaluée par le médecin au minimum une fois par an, selon un rythme déterminé en fonction de la gravité de la maladie et de l'amélioration du contrôle des symptômes.
La nécessité de poursuivre le traitement doit être évaluée également chez les patients qui développent des manifestations de granulomatose éosinophilique avec polyangéite menaçant le pronostic vital compte tenu du fait que, Nucala n'a pas été étudié dans cette population.
Syndrome hyperéosinophilique (SHE)
Il convient de respecter une distance d'au moins 5 cm entre deux sites d'injection (cf. «Remarques particulières», Remarques concernant la manipulation).
Adultes et adolescents à partir de 12 ans
La dose recommandée est de 300 mg de Nucala, à administrer par voie sous-cutanée une fois toutes les 4 semaines.
Enfants de moins de 12 ans
La sécurité et l'efficacité de Nucala chez les enfants et les adolescents de moins de 12 ans n'ont pas été étudiées.
Durée du traitement
Si la réponse est satisfaisante, Nucala est destiné à un traitement à long terme. La nécessité de poursuivre le traitement doit être réévaluée par le médecin au minimum une fois par an, selon un rythme déterminé en fonction de la gravité de la maladie du patient et de l'amélioration du contrôle des symptômes.
La nécessité de poursuivre le traitement doit être évaluée également chez les patients qui développent des manifestations de syndrome hyperéosinophilique menaçant le pronostic vital compte tenu du fait que, Nucala n'a pas été étudié dans cette population.
Populations particulières de patients
Patients âgés (>65 ans)
Aucun ajustement de la posologie n'est nécessaire chez les patients âgés (cf. «Pharmacocinétique»).
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale (cf. «Pharmacocinétique»).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique (cf. «Pharmacocinétique»).
Mode d'administration (Remarques concernant la manipulation, cf. «Remarques particulières»)
Nucala stylo prérempli et seringue préremplie ne doivent être utilisés qu'en injection sous-cutanée (voir Mode d'emploi séparé).
Nucala peut être administré par le patient lui-même ou par un aidant si le médecin le juge opportun et que le patient ou l'aidant a été formé à la technique d'injection correspondante.
En cas d'auto-injection, les sites d'injection sont l'abdomen et la cuisse. Un aidant peut également faire l'injection dans le haut du bras.
Les injections ne doivent pas être administrées dans des zones cutanées sensibles, blessées, rouges ou indurées.
Des instructions détaillées concernant l'utilisation du stylo prérempli ou de la seringue préremplie figurent dans le mode d'emploi joint à l'emballage.
L'administration de Nucala peut provoquer des réactions d'hypersensibilité locale ou systémique. Les patients doivent être informés que de telles réactions sont possibles et qu'elles peuvent exiger un traitement médical immédiat. Après chaque administration, les patients doivent être attentifs pendant au moins 30 minutes à l'apparition de signes et symptômes d'une réaction d'hypersensibilité.
Contre-indicationsHypersensibilité au mépolizumab ou à l'un des excipients.
Mises en garde et précautionsNucala ne doit pas être utilisé pour le traitement d'une exacerbation d'asthme aiguë.
Des effets indésirables liés à l'asthme ou des exacerbations peuvent se produire au cours du traitement par Nucala. Les patients doivent être instruits de consulter un médecin si les symptômes d'asthme restent mal contrôlés ou s'aggravent après le début du traitement par Nucala.
Un arrêt abrupt de l'utilisation de corticostéroïdes après le début du traitement par Nucala n'est pas recommandé. Au besoin, les doses de corticostéroïdes doivent être réduites progressivement sous surveillance médicale.
Réactions d'hypersensibilité et réactions liées à l'administration
Des réactions systémiques de type immédiat ou de type retardé – y compris réactions d'hypersensibilité (par exemple anaphylaxie, urticaire, angio-œdème, rash, bronchospasme, hypotension) – ont été observées après l'administration de Nucala. Ces réactions se produisent généralement dans les premières heures suivant l'administration, mais parfois aussi de façon retardée (après plusieurs jours).
Maladies parasitaires
Les éosinophiles peuvent participer à la réponse immunitaire à certains helminthes. Chez les patients présentant une helminthiase, celle-ci doit être traitée avant l'initiation du traitement par le mépolizumab. Si une helminthiase survient chez des patients sous mépolizumab et ne répond pas à un traitement antihelminthique, il faut envisager une interruption temporaire du traitement par le mépolizumab.
Granulomatose éosinophilique avec polyangéite menaçant un organe ou le pronostic vital
Nucala n'a pas été étudié chez les patients avec des manifestations de granulomatose éosinophilique avec polyangéite menaçant un organe ou le pronostic vital (cf. «Posologie/Mode d'emploi»).
Syndrome hyperéosinophilique menaçant le pronostic vital
Nucala n'a pas été étudié chez les patients avec des manifestations de syndrome hyperéosinophilique menaçant le pronostic vital (cf. «Posologie/Mode d'emploi»)
Ce médicament contient moins de 1 mmol de sodium (23 mg) par injection de 100 mg (=1 ml), c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsAucune étude n'a été effectuée pour examiner les interactions du mépolizumab avec d'autres médicaments.
On ne dispose d'aucune expérience clinique concernant la réponse immunitaire aux vaccinations pendant un traitement par Nucala.
Grossesse, allaitementGrossesse
Les données disponibles sur l'utilisation du mépolizumab pendant la grossesse sont limitées ou inexistantes.
Les expérimentations animales n'ont fourni aucun indice de toxicité pour la reproduction (cf. «Données précliniques»).
Nucala ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
Chez l'homme, on ne dispose pas de données concernant le passage du mépolizumab dans le lait maternel. Chez des macaques crabiers (Macaca fascicularis), le mépolizumab a cependant été retrouvé dans le lait à des concentrations inférieures à 0,5% de la concentration plasmatique correspondante.
Il faut donc cesser soit l'allaitement soit le traitement par Nucala après avoir évalué les avantages de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère.
Fertilité
On ne dispose pas de données concernant la fertilité chez l'être humain. Les expérimentations animales n'ont révélé aucun effet indésirable d'un traitement anti-IL5 sur la fertilité (cf. «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude spécifique n'a été effectuée pour examiner l'influence de Nucala sur l'aptitude à conduire des véhicules et à utiliser des machines.
Effets indésirablesAsthme sévère à éosinophiles
Résumé du profil de sécurité
Dans les études cliniques auprès de patients atteints d'asthme sévère à éosinophiles, les effets indésirables le plus souvent rapportés au cours du traitement étaient des céphalées, des réactions au site d'injection et des douleurs dorsales. Le profil de sécurité était similaire dans tous les groupes de traitement, sauf pour les réactions au site d'injection, qui étaient plus fréquentes dans le groupe recevant les injections s.c. de mépolizumab 100 mg que dans le groupe recevant un placebo (8% versus 3%). Les réactions au site d'injection se sont généralement produites au début du traitement (jusqu'à la troisième injection); les rapports concernant les injections par la suite sont moins nombreux.
Liste des effets indésirables
La sécurité de Nucala a été évaluée chez un nombre total de 1327 adultes et adolescents à partir de 12 ans atteints d'asthme sévère à éosinophiles. Ces personnes ont reçu le médicament par voie sous-cutanée (s.c.) ou intraveineuse (i.v.) dans le cadre d'études cliniques de 24 à 52 semaines. Le tableau ci-dessous indique les effets indésirables observés chez les patients traités par 100 mg de mépolizumab s.c. dans les deux études contrôlées versus placebo (n = 263).
Les effets indésirables sont classifiés par classes de systèmes d'organes selon MedDRA et par fréquence en respectant la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 à <1/10), occasionnels (≥1/1000 à <1/100), rares (≥1/10 000 à <1/1000), très rares (<1/10 000) et de fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Infections et infestations
Fréquents: pharyngite, infections des voies respiratoires inférieures, infections des voies urinaires.
Occasionnels: Herpès zoster**
Affections du système nerveux
Très fréquents: céphalées (20%).
Affections respiratoires, thoraciques et médiastinales
Fréquents: congestion nasale.
Affections gastro-intestinales
Fréquents: douleurs dans l'abdomen supérieur.
Affections de la peau et du tissu sous-cutané
Fréquents: eczéma.
Affections musculosquelettiques et du tissu conjonctif
Fréquents: douleurs dorsales.
Troubles généraux et anomalies au site d'administration
Fréquents: fièvre, réactions au site d'injection*.
* Les symptômes les plus fréquents en rapport avec l'injection sous-cutanée englobent des douleurs, un érythème, une tuméfaction, des démangeaisons et une sensation de brûlure.
** Issu des rapports spontanés après la mise sur le marché
Le profil de sécurité de Nucala chez les patients souffrant d'asthme sévère (n=998) traités dans le cadre d'études de prolongation en ouvert sur une durée médiane de 2,8 ans (intervalle de 4 semaines à 4,5 ans) n'a pas révélé d'effets indésirables supplémentaires par rapport aux études contrôlées contre placebo. Les effets indésirables les plus fréquents étaient des céphalées (22%), des douleurs dorsales (14%) et des réactions au site d'injection (8%).
RSCaPN
Dans le cadre d'une étude randomisée de 52 semaines, en double aveugle et contrôlée contre placebo, portant sur des sujets souffrant de RSCaPN (100 mg de mépolizumab n = 206, placebo n = 201), aucun autre effet indésirable n'a été constaté par rapport aux études portant sur des patients souffrant d'asthme sévère.
GEPA
Dans le cadre d'une étude en double aveugle contrôlée contre placebo portant sur des patients souffrant de GEPA (300 mg de mépolizumab n = 68, placebo n = 68), aucun autre effet indésirable n'a été constaté par rapport aux études portant sur des patients souffrant d'asthme sévère.
SHE
Dans une étude randomisée, en double aveugle, contrôlée contre placebo, d'une durée de 32 semaines, menée chez des patients atteints de SHE (300 mg de mépolizumab n = 54, placebo n = 54), davantage d'infections sont apparues sous mépolizumab que sous placebo (37/54 vs 28/54). La majorité de ces cas étaient des infections des voies respiratoires supérieures. Le profil de sécurité du mépolizumab chez des patients atteints de SHE (n=102) dans l'étude ouverte de prolongation de 20 semaines a été similaire au profil de sécurité observé dans l'étude pivot contrôlée contre placebo. Aucun autre effet indésirable n'a été constaté par rapport aux études sur l'asthme sévère.
Effets indésirables identifiés après la mise sur le marché
Affections du système immunitaire
Rares: réactions d'hypersensibilité, y compris anaphylaxie
Affections musculosquelettiques et du tissu conjonctif
Fréquents: arthralgie
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageOn ne dispose d'aucune expérience clinique concernant le surdosage de mépolizumab.
Aucune toxicité dose-dépendante n'a été détectable chez des patients avec maladie à éosinophiles ayant reçu des doses intraveineuses allant jusqu'à 1500 mg dans le cadre d'une étude clinique.
Traitement
Aucun traitement spécifique n'est disponible en cas de surdosage de mépolizumab. Lors d'un surdosage, le patient doit recevoir le traitement de soutien approprié dans son cas et être surveillé en conséquence.
La marche à suivre dépendra des exigences cliniques ou des recommandations éventuellement disponibles du centre d'information toxicologique.
Propriétés/EffetsCode ATC
R03DX09
Mécanisme d'action
Le mépolizumab est un anticorps monoclonal humanisé (IgG1, kappa) qui est dirigé avec une haute affinité spécifiquement contre l'interleukine 5 (IL-5) humaine. L'IL-5 est la cytokine la plus importante pour la croissance, la différenciation, le recrutement, l'activation et la survie des éosinophiles. Le mépolizumab en concentrations d'ordre nanomolaire inhibe les effets biologiques de l'IL-5 en empêchant celle-ci de se lier à la chaîne alpha du complexe récepteur d'IL-5 exprimé à la surface cellulaire des éosinophiles. Ainsi, le mépolizumab bloque la voie de signalisation de l'IL-5 et réduit de cette manière la production et la survie des éosinophiles.
Pharmacodynamique
Dans les études cliniques, une diminution du taux des éosinophiles dans le sang a été observée après un traitement par le mépolizumab. L'ampleur et la durée de cette diminution étaient dose-dépendantes après l'administration sous-cutanée de doses de 12,5 à 125 mg.
Chez des patients souffrant d'asthme sévère l'administration sous-cutanée de 100 mg toutes les 4 semaines pendant une période de 32 semaines a permis de diminuer le taux sanguin d'éosinophiles à une valeur (moyenne géométrique) de 40 cellules/µl. Cela correspond à une réduction de 84% (moyenne géométrique) par rapport au placebo. Cette réduction dans cet ordre de grandeur a déjà été observée au cours des 4 premières semaines de traitement. Le degré de cette réduction du taux sanguin d'éosinophiles s'est maintenu chez les patients atteints d'asthme sévère (n=998) qui avaient été traités dans les études de prolongation en ouvert pendant une durée médiane de 2,8 ans (intervalle de 4 semaines à 4,5 ans).
Après une administration sous-cutanée de 100 mg toutes les 4 semaines pendant une période de 52 semaines chez des patients atteints de RSCaPN, le taux sanguin d'éosinophiles a baissé à 60 cellules/µl (moyenne géométrique). Cela correspond à une réduction de 83% (moyenne géométrique) versus placebo. Cette réduction a été observée au cours des 4 premières semaines de traitement et s'est maintenue pendant la durée du traitement.
Après une administration sous-cutanée de 300 mg toutes les 4 semaines pendant une période de 52 semaines chez des patients avec GEPA, le taux sanguin d'éosinophiles avait baissé à 38 cellules/µl (moyenne géométrique). Cela correspond à une réduction de 83% (moyenne géométrique) versus placebo.
Après une administration sous-cutanée de 300 mg toutes les 4 semaines pendant une période de 32 semaines chez des patients atteints de SHE, le taux sanguin d'éosinophiles avait baissé à 70 cellules/µl (moyenne géométrique). Cela correspond à une réduction de 92% (moyenne géométrique) par rapport au placebo. Une réduction de cet ordre de grandeur s'est maintenue pendant encore 20 semaines chez les patients qui ont poursuivi le traitement par mépolizumab durant la phase ouverte de prolongation de l'étude.
Immunogénicité
En raison du potentiel immunogène des médicaments à base de protéines ou de peptides, les patients peuvent développer des anticorps contre le mépolizumab après le traitement.
Sur 260 patients atteints d'asthme sévère ayant reçu au moins une dose sous-cutanée de 100 mg, 15 patients (6%) ont développé des anticorps contre le mépolizumab.
Le profil d'immunogénicité de Nucala chez les patients souffrant d'asthme sévère (n=998) qui avaient été traités dans les études de prolongation en ouvert pendant une durée médiane de 2,8 ans (intervalle de 4 semaines à 4,5 ans) était similaire à celui observé dans les études contrôlées contre placebo.
Sur 196 patients atteints de RSCaPN ayant reçu au moins une dose sous-cutanée de mépolizumab de 100 mg, 6 patients (3%) ont développé des anticorps contre le mépolizumab.
Au total, 1 patient sur 68 (1%) souffrant de GEPA ayant reçu au moins une dose sous-cutanée de 300 mg de mépolizumab a développé des anticorps contre le mépolizumab.
Au total, 1 patient sur les 53 (2%) atteints de SHE et traités avec au moins une dose sous-cutanée de 300 mg de mépolizumab a développé des anticorps dirigés contre le mépolizumab.
Toutes indications confondues, des anticorps neutralisants ont été trouvés chez un adulte (souffrant d'asthme sévère).
Chez la majorité des patients, la présence d'anticorps anti-mépolizumab n'a pas eu d'influence notable sur la PC ou la PD du mépolizumab; aucun indice suggérant un rapport des titres d'anticorps avec des modifications du taux d'éosinophiles n'a été trouvé.
Efficacité clinique
Asthme sévère à éosinophiles
L'efficacité du mépolizumab dans le traitement de l'asthme sévère à éosinophiles a été évaluée dans le cadre de 3 études cliniques randomisées, en double aveugle, par groupes parallèles, de 24 à 52 semaines, auprès de patients à partir de 12 ans. Ces études étaient conçues pour évaluer l'efficacité du mépolizumab administré sous forme d'injection sous-cutanée ou intraveineuse à intervalles de quatre semaines chez des patients dont l'asthme n'était pas contrôlé malgré un traitement standard en cours (par exemple corticostéroïdes à inhaler [CSI], association de CSI et d'agonistes bêta2-adrénergiques à longue durée d'action [LABA], antagonistes des leucotriènes et agonistes bêta2-adrénergiques à courte durée d'action [SABA]). Les exacerbations de l'asthme cliniquement importantes sont définies comme suit dans MEA112997 et MEA115588: aggravation des symptômes d'asthme, exigeant une corticothérapie orale/systémique et/ou une hospitalisation ou une admission dans un service d'urgence.
Études contrôlées versus placebo
Étude de recherche de dose MEA112997 (étude DREAM)
Les résultats de l'étude MEA112997 – une étude multicentrique de 52 semaines, randomisée, en double aveugle, avec contrôle versus placebo, par groupes parallèles, auprès de 616 patients – montrent que le mépolizumab (75 mg, 250 mg ou 750 mg) administré par voie intraveineuse a provoqué une réduction significative des exacerbations de l'asthme versus placebo. Aucune différence significative d'efficacité n'a été constatée entre les 3 posologies étudiées (voir le Tableau 1).
Tableau 1: Fréquence des exacerbations cliniquement importantes à 52 semaines dans la population en intention de traitement
|
|
Mépolizumab IV
|
Placebo
| |
75 mg
n = 153
|
250 mg
n = 152
|
750 mg
n = 156
|
n = 155
| |
Exacerbations par an
|
1,24
|
1,46
|
1,15
|
2,40
| |
Pourcentage de réduction
|
48%
|
39%
|
52%
|
| |
Rapport (IC à 95%)
|
0,52 (0,39; 0,69)
|
0,61 (0,46; 0,81)
|
0,48 (0,36; 0,64)
|
| |
Valeur de p
|
<0,001
|
<0,001
|
<0,001
|
-
|
Les résultats de cette étude suggèrent qu'un taux sanguin d'éosinophiles ≥150 cellules/µl lors des examens préalables et ≥300 cellules/µl dans les 12 mois précédents peut être utilisé comme marqueur biologique pour prédire quels patients peuvent profiter d'un traitement par le mépolizumab. Dans le groupe sous placebo, un tiers des patients présentant ces caractéristiques est cependant aussi resté exempt d'exacerbations pendant la période de traitement d'un an.
Les patients sélectionnés selon une valeur seuil plus élevée du taux sanguin d'éosinophiles ont présenté une plus forte réduction de la fréquence d'exacerbations. Il n'a cependant pas été vérifié dans quelle mesure des patients ayant profité du traitement complémentaire par le mépolizumab ont été exclus par la définition d'une valeur seuil élevée.
Tableau 2: Réduction estimée du taux d'exacerbations cliniquement significatives en fonction des valeurs seuils du taux sanguin d'éosinophiles à l'inclusion
|
Étude
|
Taux sanguin d'éosinophiles à l'inclusion
|
Rapport (IC à 95%)
| |
MEA112997
|
150 cellules/µl
|
0,70 (0,53; 0,93)
| |
|
300 cellules/µl
|
0,52 (0,41; 0,65)
| |
|
500 cellules/µl
|
0,42 (0,32; 0,54)
| |
MEA115588
|
150 cellules/µl
|
0,61 (0,45; 0,82)
| |
|
300 cellules/µl
|
0,49 (0,38; 0,63)
| |
|
500 cellules/µl
|
0,42 (0,31; 0,55)
|
Les résultats de cette étude ont servi de base pour définir les doses à évaluer dans les études ultérieures sur l'administration du mépolizumab par voie sous-cutanée. Nucala ne doit pas être administré par voie intraveineuse. Ce médicament doit être administré exclusivement par voie sous-cutanée.
Réduction des exacerbations (étude MEA115588), étude MENSA
L'étude MEA115588 (Mepolizumab as adjunctive therapy in patients with Severe Asthma) était une étude multicentrique randomisée, en double aveugle, avec contrôle versus placebo, par groupes parallèles, pour évaluer l'efficacité et la sécurité du mépolizumab en tant que médicament complémentaire chez 576 patients atteints d'asthme sévère à éosinophiles.
Les patients étaient âgés pour la plupart d'au moins 18 ans, avaient subi au moins deux exacerbations de l'asthme au cours des 12 derniers mois et leur asthme n'était pas contrôlé malgré leur pharmacothérapie anti-asthmatique en cours (corticostéroïdes à inhaler [CSI] à haute dose en association avec au moins un autre médicament approprié pour le contrôle de l'asthme, par exemple un agoniste bêta2-adrénergique à longue durée d'action [LABA] ou un antagoniste des leucotriènes). Pendant l'étude, les patients pouvaient utiliser des corticostéroïdes oraux et ont continué à recevoir leurs médicaments habituels contre l'asthme.
Un asthme sévère à éosinophiles était défini comme un taux d'éosinophiles ≥150 cellules/μl dans le sang périphérique au cours des 6 semaines précédant la randomisation (première dose) ou un taux sanguin ≥300 cellules/μl pendant l'année précédant la randomisation. Pendant 32 semaines, les patients ont reçu toutes les quatre semaines une injection de 100 mg de mépolizumab par voie sous-cutanée (s.c.), une injection de 75 mg de mépolizumab par voie intraveineuse (i.v.) ou un placebo. Les résultats du critère primaire de l'étude – la réduction de la fréquence des exacerbations cliniquement importantes de l'asthme – étaient statistiquement significatifs (p <0,001).
Le Tableau 3 donne un aperçu des résultats du critère primaire et des critères secondaires de l'étude MEA115588.
Tableau 3: Résultats du critère primaire et des critères secondaires à 32 semaines dans la population en intention de traitement (étude MEA115588)
|
|
Mépolizumab
(100 mg s.c.)
n = 194
|
Placebo
n = 191
| |
Critère primaire
| |
Fréquence des exacerbations cliniquement importantes
| |
Exacerbations par an
|
0,83
|
1,74
| |
Pourcentage de réduction
Rapport (IC à 95%)
|
53%
0,47 (0,35; 0,64)
|
-
| |
Valeur de p
|
<0,001
|
| |
Critères secondaires
| |
Fréquence des exacerbations ayant exigé une hospitalisation ou un traitement d'urgence
| |
Exacerbations par an
|
0,08
|
0,20
| |
Pourcentage de réduction
Rapport (IC à 95%)
|
61%
0,39 (0,18; 0,83)
|
-
| |
Valeur de p
|
0,015
|
| |
Fréquence des exacerbations ayant exigé une hospitalisation
| |
Exacerbations par an
|
0,03
|
0,10
| |
Pourcentage de réduction
Rapport (IC à 95%)
|
69%
0,31 (0,11; 0,91)
|
-
| |
Valeur p
|
0,034
|
| |
VEMS (ml) à 32 semaines avant l'administration d'un bronchodilatateur
| |
Variation moyenne versus valeur initiale (erreur type)
|
183 (31,1)
|
86 (31,4)
| |
Différence (mépolizumab vs placebo)
|
98
|
| |
IC à 95%
|
(11, 184)
|
| |
Valeur de p
|
0,028
|
| |
St. George's Respiratory Questionnaire (SGRQ) à 32 semaines
| |
Variation moyenne versus valeur initiale (erreur type)
|
-16,0 (1,13)
|
-9,0 (1,16)
| |
Différence (mépolizumab vs placebo)
|
-7,0
|
| |
IC à 95%
|
(-10,2; -3,8)
|
| |
Valeur de p
|
<0,001
|
|
Étude sur la réduction des corticostéroïdes oraux (étude SIRIUS)
L'étude MEA115575 a évalué l'effet du mépolizumab (100 mg par voie s.c.) pour réduire le besoin de corticostéroïdes oraux (CSO) dans le traitement d'entretien, en maintenant le contrôle de l'asthme chez des patients atteints d'asthme sévère à éosinophiles exigeant une corticothérapie systémique. Les patients, dont les taux d'éosinophiles dans le sang périphérique avaient été ≥300/µl dans les 12 mois précédant le screening/≥150/µl avant le début du traitement, ont reçu du mépolizumab ou un placebo à intervalles de quatre semaines pendant la phase de traitement. Au cours de la phase de réduction des CSO (semaines 4 à 20), la dose de CSO a été réduite toutes les 4 semaines tant que le contrôle de l'asthme restait maintenu. Les patients ont poursuivi leur traitement anti-asthmatique préexistant (corticostéroïdes à inhaler [CSI] à haute dose en association avec au moins un autre médicament pour le contrôle de l'asthme, par exemple agoniste bêta2-adrénergique à longue durée d'action [LABA] ou antagoniste des leucotriènes) pendant l'étude.
La population de l'étude, de 135 patients au total, présentait les caractéristiques suivantes: âge moyen de 50 ans, 55% de sexe féminin; 48% prenaient des corticostéroïdes oraux depuis au moins 5 ans et utilisaient une dose moyenne équivalente à environ 13 mg de prednisolone par jour au début de l'étude.
L'étude avait pour critère primaire la réduction de la dose journalière du CSO (semaines 20 à 24) obtenue sans perte du contrôle de l'asthme sous mépolizumab versus placebo (voir le Tableau 4).
Tableau 4: Résultats du critère primaire et des critères secondaires dans la population en intention de traitement de l'étude MEA115575
|
|
Mépolizumab
(100 mg s.c.)
n = 69
|
Placebo
n = 66
| |
Critère primaire
| |
Réduction (%) de la dose de CSO après 20 à 24 semaines par rapport à la dose initiale
| |
90% à 100%
|
16 (23%)
|
7 (11%)
| |
75% à <90%
|
12 (17%)
|
5 (8%)
| |
50% à <75%
|
9 (13%)
|
10 (15%)
| |
>0% à <50%
|
7 (10%)
|
7 (11%)
| |
Aucune réduction de la dose de CSO/contrôle insuffisant de l'asthme/arrêt prématuré du traitement
|
25 (36%)
|
37 (56%)
| |
Odds Ratio (IC à 95%)
|
2,39 (1,25; 4,56)
|
| |
Valeur de p
|
0,008
|
| |
Critères secondaires
| |
Réduction (%) de la dose journalière de CSO
| |
Réduction d'au moins 50%
|
37 (54%)
|
22 (33%)
| |
Odds Ratio (IC à 95%)
|
2,26 (1,10; 4,65)
|
| |
Valeur de p
|
0,027
|
| |
Réduction (%) de la dose journalière de CSO
| |
À ≤5 mg par jour
|
37 (54%)
|
21 (32%)
| |
Odds Ratio (IC à 95%)
|
2,45 (1,12; 5,37)
|
| |
Valeur de p
|
0,025
|
| |
Réduction (%) de la dose journalière de CSO
| |
À zéro
|
10 (14%)
|
5 (8%)
| |
Odds Ratio (IC à 95%)
|
1,67 (0,49; 5,75)
|
| |
Valeur de p
|
0,414
|
| |
Réduction médiane (%) de la dose journalière de CSO
| |
Réduction médiane (%) versus dose initiale (IC à 95%)
|
50,0 (20,0; 75,0)
|
0,0 (-20,0; 33,3)
| |
Différence médiane (IC à 95 %)
|
-30,0 (-66,7; 0,0)
|
| |
Valeur de p
|
0,007
|
|
À part cela, la qualité de vie liée à la santé a été évaluée à l'aide du SGRQ. À 24 semaines, une amélioration significative du score SGRQ moyen était constatable sous Nucala versus placebo: -5,8 (IC à 95%: -10,6; -1,0; p = 0,019). À 24 semaines, le pourcentage de patients ayant atteint une réduction cliniquement importante du score SGRQ (définie comme une réduction d'au moins 4 unités par rapport au score initial) était plus élevé sous Nucala (58%, 40/69) que sous placebo (41%, 27/66).
Le profil d'efficacité à long terme de Nucala chez les patients souffrant d'asthme sévère (n=998) qui avaient été traités dans les études de prolongation en ouvert MEA115666, MEA115661 et 201312 pendant une durée médiane de 2,8 ans (intervalle de 4 semaines à 4,5 ans) correspondait, en général, à celui des trois études contrôlées contre placebo.
Rhinosinusite chronique avec polypes nasaux (RSCaPN)
L'étude 205687 était une étude de 52 semaines, randomisée, en double aveugle et contrôlée contre placebo, dans laquelle 407 patients atteints de RSCaPN âgés de plus de 18 ans ont été examinés.
Les patients inclus dans cette étude devaient présenter un score VAS (échelle visuelle analogique) des symptômes d'obstruction nasale >5 (au max. 10 points), un score VAS total des symptômes >7 (au max. 10 points) et un score endoscopique bilatéral des polypes nasaux de 5 (au max. 8 points possibles; avec un score minimal de 2 par narine). En outre, les patients devaient avoir subi au moins une chirurgie des polypes nasaux au cours des 10 années précédentes.
Les patients ont reçu une dose de 100 mg de mépolizumab ou un placebo, administrés par voie sous-cutanée toutes les 4 semaines en complément de leur traitement de fond par corticostéroïdes intranasaux.
Les données démographiques et les propriétés à l'inclusion des patients ayant participé à l'étude 205687 sont présentés ci-dessous dans le Tableau 5:
Tableau 5: Données démographiques et propriétés à l'inclusion de la RSCaPN
|
|
N = 407
| |
Âge (années) des patients, moyenne (ET)
|
49 (13)
| |
Sexe féminin, n (%)
|
143 (35)
| |
Origine européenne, n (%)
|
379 (93)
| |
Durée (ans) de la RSCaPN, moyenne (ET)
|
11,4 (8,39)
| |
Patients ayant subi ≥1 chirurgie antérieure, n (%)
|
407 (100)
| |
Patients ayant subi ≥3 chirurgies antérieures, n (%)
|
124 (30)
| |
Utilisation de CSO à cause des PN (≥1 cure) dans les 12 derniers mois, n (%)
|
197 (48)
| |
Score endoscopique total des PNa b c, moyenne (ET), score max. = 8
|
5,5 (1,29)
| |
Score VASa d d'obstruction nasale, moyenne (ET), score max.= 10
|
9,0 (0,83)
| |
Score VAS total des symptômesa d, moyenne (ET), score max. = 10
|
9,1 (0,74)
| |
Score SNOT-22 totale, moyenne (ET), intervalle 0-110
|
64,1 (18,32)
| |
Score VAS composite des symptômesa, moyenne (ET), score max. = 10
|
9,0 (0,82)
| |
Score VASa,d de perte d'odorat, moyenne (ET), score max. = 10
|
9,7 (0,72)
| |
Asthme, n (%)
|
289 (71)
| |
AERD, n (%)
|
108 (27)
| |
Moyenne géométrique du taux d'éosinophiles à l'inclusion, cellules/µl (IC à 95%)
|
390 (360, 420)
|
RSCaPN = rhinosinusite chronique avec polypes nasaux, ET = écart-type, CSO = corticostéroïde oral, PN = polypes nasaux, VAS = échelle visuelle analogique, SNOT-22 = Sino-Nasal-Outcome-Test, AERD = maladie respiratoire exacerbée par l'aspirine
a Des scores plus élevés indiquent une sévérité plus importante de la maladie.
b Tels qu'évalués de manière aveugle par des investigateurs indépendants.
c Le score de PN est la somme des scores des deux narines (sur une échelle allant de 0 à 8), ceux-ci ayant été évalués pour chaque narine (0=absence de polypes; 1=petits polypes dans le méat nasal moyen, ne dépassant pas le dessous du bord inférieur du cornet moyen; 2=polypes atteignant le dessous du bord inférieur du cornet moyen; 3=grands polypes dépassant le bord inférieur du cornet inférieur ou polypes situés au centre du cornet moyen; 4=grands polypes provoquant une congestion/obstruction presque complète du méat nasal inférieur).
d Évalués tous les jours par les patients à l'aide d'une échelle allant de 0 à 10 (0=absence; 10=extrêmement grave).
e Le SNOT-22 est un test évaluant la qualité de vie liée à la santé et comprend 22 items dans 6 domaines de symptômes et de conséquences liés à la RSCaPN (nasaux, non nasaux, oreille/visage, sommeil, fatigue, conséquences émotionnelles). Des scores plus élevés indiquent une plus mauvaise qualité de vie liée à la santé.
Les critères d'évaluation co-primaires étaient la variation du score endoscopique total des PN à la semaine 52 par rapport à l'inclusion et la variation du score VAS moyen d'obstruction nasale aux semaines 49-52 par rapport à l'inclusion.
Les patients ayant reçu le mépolizumab présentaient par rapport au placebo des améliorations (réductions) significativement plus importantes du score endoscopique total des PN à la semaine 52 et du score VAS d'obstruction nasale aux semaines 49-52 (voir Tableau 6).
Tableau 6: Analyses des critères d'évaluation co-primaires (population en intention de traitement)
|
|
Placebo
(N=201)
|
Mépolizumab
100 mg s.c.
(N=206)
| |
Score endoscopique total à la semaine 52 a
| |
Score médian à l'inclusion (min, max)
|
6,0 (0, 8)
|
5,0 (2, 8)
| |
Variation médiane par rapport à l'inclusion
|
0,0
|
-1,0
| |
Valeur de p b
|
|
<0,001
| |
Différence ajustée des médianes entre les traitements (IC à 95%) c
|
|
-0,73 (-1,11, -0,34)
| |
Amélioration ≥1 point, n (%)
|
57 (28)
|
104 (50)
| |
Amélioration ≥2 points, n (%)
|
26 (13)
|
74 (36)
| |
Score VAS d'obstruction nasale (semaines 49 à 52) a
| |
Score médian à l'inclusion (min, max)
|
9,14 (5,31, 10,00)
|
9,01 (6,54, 10,00)
| |
Variation médiane par rapport à l'inclusion
|
-0,82
|
-4,41
| |
Valeur de p b
|
|
<0,001
| |
Différence ajustée des médianes entre les traitements (IC à 95%) c
|
|
-3,14 (-4,09, -2,18)
| |
Amélioration >1 point, n (%)
|
100 (50)
|
146 (71)
| |
Amélioration ≥3 points, n (%)
|
73 (36)
|
124 (60)
|
a Les participants à l'étude ayant subi une chirurgie nasale/sinusoplastie avant le rendez-vous de l'étude se sont vus attribuer le score le plus mauvais observé avant la chirurgie nasale/sinusoplastie. Les participants sortis de l'étude sans chirurgie nasale/sinusoplastie se sont vus attribuer le score le plus mauvais observé avant l'abandon de l'étude.
b Sur la base du test de la somme des rangs de Wilcoxon.
c Régression quantile avec comme covariables le groupe de traitement, la région géographique, le score à l'inclusion et le taux sanguin d'éosinophiles (log(s)) à l'inclusion.
Figure 1: Score endoscopique total des polypes nasaux (lecture centralisée) des répondeurs par contrôle
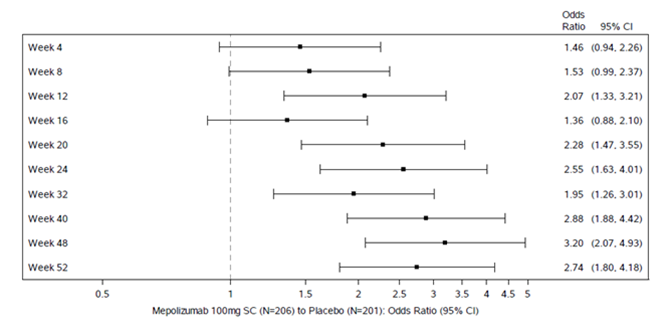
Tous les critères d'évaluation secondaires étaient statistiquement significatifs et confortaient les critères d'évaluation co-primaires. Le principal critère d'évaluation secondaire était le délai jusqu'à la première chirurgie des PN jusqu'à la semaine 52 (voir Figure 1). Les données sur les autres critères d'évaluation secondaires sont présentées dans le Tableau 7.
Délai jusqu'à la première chirurgie des PN
Pendant toute la période de traitement de 52 semaines, les patients du groupe mépolizumab ont eu une probabilité moindre de devoir subir une chirurgie des PN que les patients du groupe placebo (la chirurgie a été définie comme toute intervention impliquant des instruments entraînant une incision ou l'ablation de tissus [polypectomie] dans la cavité nasale).
Jusqu'à la semaine 52, 18 patients (9%) du groupe mépolizumab avaient subi une chirurgie des PN, contre 46 patients (23%) du groupe placebo.
Le délai jusqu'à la première chirurgie des PN était plus long chez les patients ayant reçu le mépolizumab que chez ceux sous placebo. Chez les patients traités par le mépolizumab, le risque de subir une intervention chirurgicale pendant la période de traitement était de 57% inférieur à celui observé chez les patients ayant reçu le placebo et était ainsi significativement plus faible (Hazard Ratio: 0,43; IC à 95% 0,25, 0,76; p ajusté/non ajusté =0,003); une analyse post-hoc a montré une réduction de 61% de la probabilité d'une intervention chirurgicale (Odds Ratio: 0,39, IC à 95%: 0,21, 0,72; p= 0,003).
Figure 2: Courbe de Kaplan-Meier pour le délai jusqu'à la première chirurgie des polypes nasaux
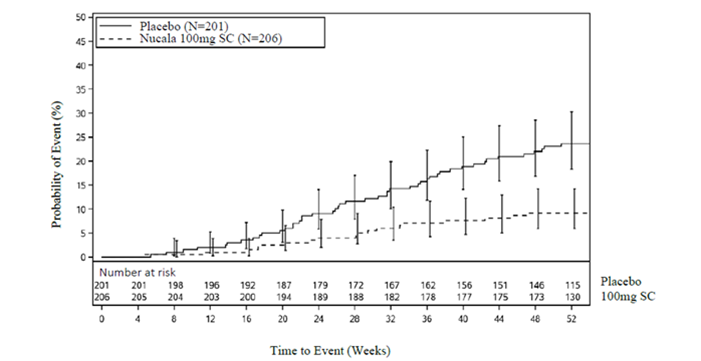
Tableau 7: Résultats des autres critères d'évaluation secondaires dans la population en intention de traitement
|
|
Placebo
(N=201)
|
Mépolizumab
(N=206)
| |
Score VAS total (semaines 49-52) a
| |
Score médian à l'inclusion (min, max)
|
9,20 (7,21, 10,00)
|
9,12 (7,17, 10,00)
| |
Variation médiane par rapport à l'inclusion
|
-0,90
|
-4,48
| |
Valeur de p ajustée/non ajustée b,c
|
|
<0,001/0,003
| |
Différence ajustée des médianes entre les traitements (IC à 95%) d
|
40
|
-3,18 (-4,10, -2,26)
| |
Amélioration ≥2,5 points (%)
|
|
64
| |
Score SNOT-22 total à la semaine 52 a, g
| |
N
|
198
|
205
| |
Score médian à l'inclusion (min, max)
|
64,0 (19, 110)
|
64,0 (17, 105)
| |
Variation médiane par rapport à l'inclusion
|
-14,0
|
-30,0
| |
Valeur de p ajustée/non ajustée b,c
|
|
<0,001/0,003
| |
Différence ajustée des médianes entre les traitements (IC à 95%) d
|
32
|
-16,49 (-23,57, -9,42)
| |
Amélioration ≥28 points (%)g
|
|
54
| |
Patients nécessitant des stéroïdes systémiques jusqu'à la semaine 52 à cause de polypes nasaux
| |
Nombre de patients avec ≥1 cure
|
74 (37)
|
52 (25)
| |
Odds Ratio par rapport au placebo (IC à 95%)
|
|
0,58 (0,36, 0,92)
| |
Valeur de p ajustée/non ajustée c, e
|
|
0,020/0,020
| |
Score VAS composite – symptômes nasaux (semaines 49-52) a,f
| |
Score médian à l'inclusion (min, max)
|
9,18 (6,03, 10,00)
|
9,11 (4,91, 10,00)
| |
Variation médiane par rapport à l'inclusion
|
-0,89
|
-3,96
| |
Valeur de p ajustée/non ajustée b,c
|
|
<0,001/0,020
| |
Différence ajustée des médianes entre les traitements (IC à 95%) d
|
40
|
-2,68 (-3,44, -1,91)
| |
Amélioration ≥2 points (%)h
|
|
66
| |
Score VAS de perte d'odorat (semaines 49-52) a
| |
Score médian à l'inclusion (min, max)
|
9,97 (6,69, 10,00)
|
9,97 (0,94, 10,00)
| |
Variation médiane par rapport à l'inclusion
|
0,00
|
-0,53
| |
Valeur de p ajustée/non ajustée b,c
|
|
<0,001/0,020
| |
Différence ajustée des médianes entre les traitements (IC à 95%) d
|
19
|
-0,37 (-0,65, -0,08)
| |
Amélioration ≥3 points (%)h
|
|
36
|
a Les patients ayant subi une chirurgie nasale/sinusoplastie avant le rendez-vous de l'étude se sont vus attribuer le score le plus mauvais observé avant la chirurgie nasale/sinusoplastie. Les participants sortis de l'étude sans chirurgie nasale/sinusoplastie se sont vus attribuer le score le plus mauvais observé avant l'abandon de l'étude.
b Sur la base du test de la somme des rangs de Wilcoxon.
c Multiplicité contrôlée à l'aide de tests des critères d'évaluation secondaires selon une hiérarchie prédéfinie.
d Régression quantile avec comme covariables le groupe de traitement, la région géographique, le score à l'inclusion et le taux sanguin d'éosinophiles (log(s)) à l'inclusion.
e Analyse ayant utilisé un modèle de régression logistique avec comme covariables le groupe de traitement, la région géographique, le nombre de cures de CSO pour les PN au cours des 12 derniers mois (0,1, >1 comme nombre ordinal), le score endoscopique total des PN à l'inclusion (lecture centralisée), le score VAS d'obstruction nasale et le taux sanguin d'éosinophiles (log(s)) à l'inclusion.
f Score VAS composite pour l'obstruction nasale, l'écoulement nasal, le mucus pharyngé et la perte d'odorat.
g Une amélioration a été observée dans les 6 domaines des symptômes et conséquences liés à la RSCaPN.
h La valeur limite de l'amélioration de chaque critère d'évaluation a été déterminée comme variation intra-individuelle judicieuse.
Critères d'évaluation du sous-groupe de patients atteints d'un asthme comorbide
Chez 289 (71%) patients atteints d'un asthme comorbide, des analyses prédéfinies chez les patients ayant reçu 100 mg de mépolizumab ont montré des améliorations des critères d'évaluation co-primaires par rapport au placebo, qui concordaient avec celles de la population totale.
Granulomatose éosinophilique avec polyangéite (GEPA)
MEA115921 était une étude d'une durée de 52 semaines, randomisée, en double aveugle, contrôlée contre placebo portant sur 136 patients à partir de 18 ans souffrant d'une GEPA récidivante ou réfractaire au traitement, sous dose stable de corticostéroïdes oraux (CSO; ≥7,5 à ≤50 mg/jour de prednisolone/prednisone). 53% (n = 72) des participants ont reçu simultanément des immunosuppresseurs à une dose stable.
Les patients ont reçu une dose de 300 mg de mépolizumab ou de placebo par voie sous-cutanée toutes les quatre semaines en complément de leur traitement de base par la prednisolone/prednisone avec ou sans immunosuppresseurs. La dose de corticostéroïde oral a été progressivement réduite à la discrétion du médecin investigateur.
Les critères d'évaluation primaire étaient la durée globale cumulative de rémission, la rémission étant définie par un Birmingham Vasculitis Activity Score (BVAS) de 0 (pas de vascularite active) plus une dose de prednisolone/prednisone ≤4 mg/jour, ainsi que le pourcentage de participants en rémission au bout de 36 et 48 semaines de traitement.
Rémission
Par rapport au groupe sous placebo, les participants sous 300 mg de mépolizumab ont obtenu une durée de rémission globale significativement plus longue. Par ailleurs, le pourcentage de participants en rémission sous 300 mg de mépolizumab à la semaine 36 et à la semaine 48 était significativement plus élevé que sous placebo (Tableau 8).
Tableau 8: Analyses des deux critères d'évaluation primaires (population ITT)
|
|
Nombre (%) de participants
| |
Placebo n = 68
|
Mépolizumab 300 mg n = 68
| |
Durée de rémission globale pendant 52 semaines
| |
0 semaine
|
55 (81)
|
32 (47)
| |
>0 à <12 semaines
|
8 (12)
|
8 (12)
| |
12 à <24 semaines
|
3 (4)
|
9 (13)
| |
24 à <36 semaines
|
0
|
10 (15)
| |
≥36 semaines
|
2 (3)
|
9 (13)
| |
Odds Ratio (mépolizumab/placebo)
|
|
5,91
| |
IC à 95 %
|
----
|
2,68; 13,03
| |
Valeur de p
|
----
|
<0,001
| |
Participants en rémission aux semaines 36 et 48
|
2 (3)
|
22 (32)
| |
Odds Ratio (mépolizumab / placebo)
|
|
16,74
| |
IC à 95%
|
----
|
3,61; 77,56
| |
Valeur de p
|
----
|
<0,001
|
Un Odds Ratio >1 parle en faveur du mépolizumab
Les participants sous 300 mg de mépolizumab ont obtenu une durée de rémission globale significativement plus longue (p <0,001), et le pourcentage de participants en rémission sous 300 mg de mépolizumab selon la définition de la rémission appliquée pour le critère d'évaluation secondaire (BVAS = 0 plus prednisolone/prednisone ≤7,5 mg/jour) était supérieur à celui sous placebo (p <0,001) tant à la semaine 36 qu'à la semaine 48.
Récidive
Par rapport au placebo, la durée jusqu'à la première récidive (définie comme une aggravation liée à une vascularite, de l'asthme ou des symptômes sinonasaux nécessitant une augmentation de la dose de corticostéroïdes ou d'immunosuppresseurs ou une hospitalisation) était significativement plus longue chez les participants sous 300 mg de mépolizumab (p <0,001). En outre, le taux annualisé de récidives était inférieur de 50% chez les patients sous mépolizumab par rapport à ceux sous placebo: 1,14 vs 2,27.
Réduction de la dose de corticostéroïdes oraux
Par rapport aux participants sous placebo, les participants sous 300 mg de mépolizumab ont reçu une dose quotidienne moyenne de corticostéroïdes oraux inférieure (p <0,001) durant les semaines 48 à 52. Dans le groupe sous 300 mg de mépolizumab, 12 participants (18%) ont pu complètement arrêter, de manière progressive, le traitement par les corticostéroïdes oraux contre 2 participants (3%) dans le groupe sous placebo.
Syndrome hyperéosinophilique (SHE)
L'étude 200622 était une étude de 32 semaines randomisée, contrôlée contre placebo, en double aveugle, dans laquelle 108 patients atteints de SHE âgés de ≥12 ans ont été examinés. Les patients atteints de SHE secondaire non hématologique (par ex. hypersensibilité aux médicaments, infection parasitaire, infection par le VIH, tumeur maligne non hématologique) ou de SEH F/P positif ont été exclus de l'étude. Les patients ont reçu par voie sous-cutanée 300 mg de mépolizumab ou un placebo toutes les 4 semaines, en plus de leur traitement stable pour le SHE. Un des 4 adolescents inclus a reçu 300 mg de mépolizumab, les autres 3 ont reçu le placebo, pendant 32 semaines pour chacun d'entre eux. Le traitement standard du SHE pouvait englober des CSO, des immunosuppresseurs ou des cytotoxiques. Les participants à l'étude avaient souffert d'au moins deux poussées de SHE au cours des 12 derniers mois et présentaient lors de la sélection un taux d'éosinophiles sanguins ≥1000 cellules/µl.
Le critère d'évaluation primaire de l'étude 200622 était le pourcentage de participants à l'étude qui ont souffert d'une poussée de SHE pendant la période de traitement de 32 semaines. Une poussée de SHE était définie comme une aggravation des signes et symptômes cliniques de SHE ou comme une élévation des éosinophiles (à deux moments au minimum) ayant nécessité une augmentation des CSO ou une augmentation de la dose ou l'administration d'un traitement adjuvant cytotoxique ou immunosuppresseur du SHE.
L'analyse primaire a comparé les patients des groupes du mépolizumab et du placebo qui avaient subi une poussée de SHE ou qui avaient quitté l'étude. Par rapport au groupe sous placebo, le nombre de patients du groupe ayant reçu 300 mg de mépolizumab qui ont subi une poussée de SHE ou qui ont quitté l'étude a été réduit de 50% pendant la période de traitement de 32 semaines; 28% versus 56% (OR 0,28, IC à 95% 0,12–0,64) (voir Tableau 9).
Les critères d'évaluation secondaires étaient le temps écoulé jusqu'à la première poussée de SHE, le pourcentage de participants à l'étude qui ont souffert d'une poussée de SHE entre la semaine 20 et la semaine 32, le taux de poussées de SHE et la variation du degré de gravité de la fatigue par rapport à la valeur initiale. Tous les critères d'évaluation secondaires ont été statistiquement significatifs et ont corroboré le critère d'évaluation primaire (voir Figure 3 et Tableau 10).
Tableau 9: Résultats obtenus pour le critère d'évaluation primaire / résultats de l'analyse primaire dans la population en intention de traitement (étude 200622)
|
|
Mépolizumab
N = 54
|
Placebo
N = 54
| |
Pourcentage de patients qui ont subi une poussée de SHE
| |
Participants à l'étude avec ≥1 poussée de SHE ou ayant arrêté l'étude (%)
|
15 (28)
|
30 (56)
| |
Participants à l'étude avec ≥1 poussée de SHE (%)
|
14 (26)
|
28 (52)
| |
Participants à l'étude sans poussée de SHE qui ont arrêté l'étude (%)
|
1 (2)
|
2 (4)
| |
Odds Ratio (IC à 95%)
|
0,28 (0,12–0,64)
|
| |
Valeur p au CMH
|
0,002
|
|
CMH = Cochran-Mantel-Haenszel
Temps écoulé jusqu'à la première poussée
Chez les participants à l'étude sous 300 mg de mépolizumab, le temps écoulé jusqu'à la première poussée de SHE a été significativement plus long que chez les patients sous placebo. Le risque d'une première poussée de SHE pendant la période de traitement a été inférieur de 66% chez les patients sous mépolizumab que chez les patients sous placebo (Hazard Ratio: 0,34, IC à 95% 0,18–0,67, p = 0,002).
Figure 3: Courbe de Kaplan-Meier pour le temps écoulé jusqu'à la première poussée de SHE
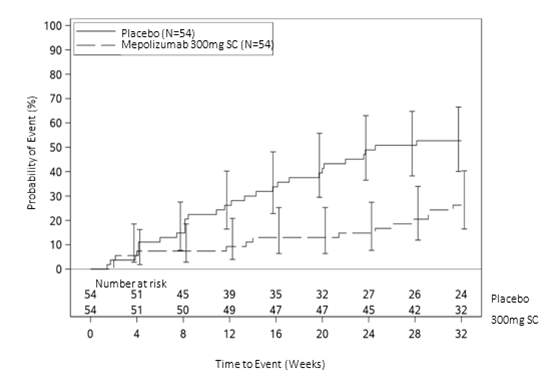
Tableau 10 Résultats pour d'autres critères d'évaluation secondaires dans la population en intention de traitement (étude 200622)
|
|
Mépolizumab
N = 54
|
Placebo
N = 54
| |
Poussées de SHE entre la semaine 20 et la semaine 32 incluse
| |
Participants à l'étude avec ≥1 poussée de SHE ou ayant quitté l'étude (%)
|
9 (17)
|
19 (35)
| |
Odds Ratio (IC à 95%)
|
0,33 (0,13–0,85)
| |
Valeur p CMH (non corrigée/corrigée)a
|
0,02/0,02
| |
Taux des poussées de SHE
| |
Taux annuel moyen estimé
|
0,50
|
1,46
| |
Rapport des taux (IC à 95%)
|
0,34 (0,19–0,63)
| |
Valeur p de Wilcoxon (ajustée/ajustée)a
|
0,002/0,02
| |
Variation du degré de gravité de la fatigue par rapport à la valeur initiale, sur la base de l'item 3 du Brief Fatigue Inventory (BFI) (manifestation la plus grave de la fatigue au cours des dernières 24 heures) à la semaine 32b
| |
Variation médiane de l'item 3 du BFI
|
-0,66
|
0,32
| |
Comparaison des valeurs p (mépolizumab vs placebo) (ajustée/ajustée) a
|
0,036/0,036
|
|
a Valeurs p ajustées, sur la base d'une hiérarchie de critères préalablement établie.
b Les patients dont les données manquaient ont été inclus en utilisant la valeur observée la plus mauvaise.
CMH = Cochran-Mantel-Haenszel
Phase ouverte de prolongation pour le SHE
Après la fin de l'étude 200622, des patients appropriés, dont 4 adolescents, ont poursuivi avec l'étude ouverte de prolongation de 20 semaines 205203, dans laquelle le profil de sécurité à long terme a été examiné et des données additionnelles ont été obtenues sur le bénéfice clinique du mépolizumab au-delà de 32 semaines chez les patients atteints de SHE.
L'effet du traitement par le mépolizumab sur la réduction des poussées de SHE observé dans l'étude 200622 s'est maintenu chez les patients qui ont poursuivi le traitement par le mépolizumab dans l'étude 205203: dans cette étude, 94% (47/50) des patients n'ont pas présenté de poussées.
Durant les semaines 16 à 20, 28% de tous les patients qui avaient reçu une dose moyenne de CSO > 0 mg/jour (prednisone ou équivalent) pendant les semaines 0 à 4, ont réussi à atteindre une réduction de ≥50% de leur dose journalière moyenne de CSO. Les données d'efficacité de cette étude suggèrent que le bénéfice clinique du mépolizumab se maintient sur 52 semaines et permet une réduction du traitement par CSO chez les patients atteints de SHE.
PharmacocinétiqueAdministré par voie sous-cutanée à des patients atteints d'asthme modéré à sévère, le mépolizumab a présenté aux doses de 12,5 mg à 250 mg une pharmacocinétique proportionnelle à la dose. La pharmacocinétique du mépolizumab concordait chez les patients souffrant d'asthme, de RSCaPN, de GEPA ou de SHE.
L'exposition systémique après administration sous-cutanée de 300 mg de mépolizumab était environ le triple de celle observée sous 100 mg.
Dans une étude pharmacocinétique comparative, menée chez des volontaires sains, la pharmacocinétique du mépolizumab s'est avérée comparable pour les différentes formulations (récemment préparées) après administration d'une dose sous-cutanée unique de 100 mg.
Absorption
Après l'administration sous-cutanée à des sujets sains ou à des patients asthmatiques, le mépolizumab a été résorbé lentement; il a atteint son pic de concentration plasmatique (Tmax) en l'espace d'un temps médian de 4 à 8 jours.
Après l'administration d'une dose sous-cutanée unique dans l'abdomen, la cuisse ou le haut du bras chez des sujets sains, la biodisponibilité absolue du mépolizumab était de 64%, 71% et 75% respectivement. Chez les patients asthmatiques, la biodisponibilité absolue du mépolizumab administré par voie sous-cutanée dans le haut du bras était de 74%. Après l'administration de doses sous-cutanées espacées de quatre semaines, le steady-state est atteint en l'espace de 16 semaines. Après l'administration de doses sous-cutanées répétées à intervalles de quatre semaines, l'état d'équilibre est atteint avec un rapport d'accumulation de 2. La pharmacocinétique du mépolizumab est indépendante du temps.
Distribution
Après administration d'une dose intraveineuse unique à des patients atteints d'asthme léger, le mépolizumab atteint un volume de distribution moyen de 55 à 85 ml/kg.
Métabolisme
Le mépolizumab est un anticorps monoclonal humanisé de type IgG1. Il est dégradé par des enzymes protéolytiques présentes non seulement dans le tissu hépatique, mais dans le corps entier.
Élimination
Après l'administration d'une dose intraveineuse unique chez des patients asthmatiques, la clairance systémique (CL) moyenne était de 1,9 à 3,3 ml/jour/kg, avec une demi-vie terminale moyenne d'environ 20 jours. Après l'administration sous-cutanée de mépolizumab, la demi-vie terminale moyenne (t1/2) était comprise entre 16 et 22 jours.
Cinétique pour certains groupes de patients
Patients âgés (>65 ans)
Aucune étude formelle n'a été effectuée auprès de patients âgés. L'analyse pharmacocinétique de population n'a toutefois fourni aucun indice d'une influence de l'âge (12 à 82 ans) sur la pharmacocinétique du mépolizumab.
Troubles de la fonction rénale
Aucune étude formelle n'a été effectuée pour évaluer l'influence d'une insuffisance rénale sur la pharmacocinétique du mépolizumab. Sur la base des analyses pharmacocinétiques de population, aucune adaptation posologique n'est nécessaire chez les patients dont la clairance de la créatinine est comprise entre 50 et 80 ml/min. Les données disponibles sur les patients présentant une clairance de la créatinine inférieure à 50 ml/min sont limitées.
Troubles de la fonction hépatique
Aucune étude formelle n'a été effectuée pour évaluer l'influence d'une insuffisance hépatique sur la pharmacocinétique du mépolizumab. Étant donné que le mépolizumab est dégradé par des enzymes protéolytiques ubiquitaires dans l'organisme, donc non limitées au tissu hépatique, l'élimination du mépolizumab ne devrait guère être influencée par une insuffisance hépatique.
Données précliniquesLes données précliniques obtenues dans des études conventionnelles de pharmacologie, de sécurité et de toxicité avec administration de doses répétées chez des singes ne suggèrent aucun risque particulier pour l'être humain. L'administration intraveineuse et sous-cutanée chez les singes a entraîné une réduction du taux d'éosinophiles dans le sang périphérique et dans les poumons, mais n'a conduit à aucun constat toxicologiquement important.
Les éosinophiles jouent manifestement un rôle dans les réactions du système immunitaire à certains parasites. Chez la souris, les études avec administration d'anticorps anti-IL-5 ou avec induction génétique d'un manque d'IL-5 ou d'éosinophiles n'ont révélé aucune fragilisation des défenses contre les parasites.
Le mépolizumab étant un anticorps monoclonal, aucune étude de mutagénicité ou de carcinogénicité n'a été effectuée.
Toxicité sur la reproduction
Gestation
Chez des singes, le mépolizumab n'a influencé ni la gestation, ni le développement embryo-fœtal ou postnatal (y compris fonction immunitaire). Aucun examen n'a été effectué pour détecter des malformations internes ou squelettiques. Les données obtenues chez des macaques crabiers (Macaca fascicularis) montrent que le mépolizumab peut traverser la barrière placentaire. Plusieurs mois après leur naissance, les petits présentaient encore des concentrations de mépolizumab ~1,2 à 2,4 fois plus élevées que leurs mères. Leur système immunitaire n'en a pas été affecté.
Fertilité
Aucun trouble de la fertilité n'a été observé dans l'étude de toxicité sur la fertilité et sur la reproduction générale menée chez la souris avec un anticorps analogue inhibant l'IL-5 de la souris.
Cette étude n'incluait pas d'évaluation sur la portée ni d'évaluation fonctionnelle sur la progéniture.
Remarques particulièresIncompatibilités
Aucune connue.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C) et à l'abri de la lumière dans l'emballage d'origine jusqu'à l'utilisation. Ne pas congeler.
La durée de conservation hors du réfrigérateur ne doit pas excéder 7 jours si le stylo prérempli ou la seringue préremplie a été conservé(e) à l'abri de la lumière et à une température ne dépassant 30°C. Ils doivent être jetés s'ils ont été conservés plus de 7 jours hors du réfrigérateur.
Le stylo prérempli et la seringue préremplie ne doivent plus être utilisés s'ils sont restés plus de 8 heures en dehors de leur emballage.
Conserver hors de la portée des enfants.
Remarques concernant la manipulation
Des instructions détaillées concernant l'utilisation du stylo prérempli ou de la seringue préremplie figurent dans le mode d'emploi joint à l'emballage.
Le stylo prérempli et la seringue préremplie sont destinés à un usage unique.
Élimination
Tout médicament non utilisé ou déchet doit être évacué conformément à la règlementation nationale.
Numéro d’autorisationNucala stylo prérempli 67350 (Swissmedic)
Nucala seringue préremplie 67351 (Swissmedic)
PrésentationStylo prérempli:
Emballage de 1 stylo prérempli de 100 mg/ml (B)
Seringue préremplie:
Emballage de 1 seringue préremplie de 100 mg/ml (B)
Titulaire de l’autorisationGlaxoSmithKline AG, 6340 Baar
Mise à jour de l’informationOctobre 2025
|