CompositionPrincipe actif
Semaglutidum
Excipients
Natrii salcaprozas corresp. natrium 22.9 mg, povidonum K 90, cellulosum microcristallinum, magnesii stearas
Indications/Possibilités d’emploiRybelsus est indiqué chez les adultes pour le traitement du diabète de type 2 insuffisamment contrôlé en complément d'un régime alimentaire et d'une activité physique:
·En monothérapie, quand l'utilisation de la metformine est considérée comme inappropriée en raison de contre-indications ou d'une intolérance
·En association avec d'autres médicaments hypoglycémiants.
Voir «Efficacité clinique» concernant les résultats des associations étudiées dans les essais cliniques et la sécurité cardiovasculaire.
Posologie/Mode d’emploiPosologie usuelle
La dose initiale de Rybelsus est de 3 mg une fois par jour. Après un mois de traitement, la dose devra être augmentée à une dose d'entretien de 7 mg une fois par jour. Si après au moins un mois de traitement avec la dose de 7 mg l'effet hypoglycémiant est insuffisant, la dose peut être augmentée à une dose d'entretien maximale de 14 mg une fois par jour.
Les patients traités par Rybelsus 14 mg une fois par jour peuvent passer à une injection sous-cutanée de 0.5 mg une fois par semaine (Ozempic). Les patients peuvent commencer avec Ozempic le jour après la dernière dose de Rybelsus. Les patients traités par Ozempic 0.5 mg en injection sous-cutanée une fois par semaine peuvent passer à Rybelsus 7 mg ou 14 mg une fois par jour. Les patients peuvent commencer avec Rybelsus dans un intervalle de 7 jours après la dernière injection d'Ozempic. Il n'existe aucune dose de Rybelsus équivalant à Ozempic 1 mg.
Lorsque Rybelsus est utilisé en association à la metformine et/ou à un inhibiteur du co-transporteur de sodium-glucose de type 2 (iSGLT2) ou à une thiazolidinedione, le traitement par metformine et/ou iSGLT2/thiazolidinedione peut être poursuivi à la même dose.
Lorsque Rybelsus est utilisé en association à une sulfonylurée ou de l'insuline, une diminution de la dose de la sulfonylurée ou de l'insuline pourra être envisagée afin de réduire le risque d'hypoglycémie (voir «Mises en garde et précautions»).
Instructions posologiques particulières
Patients âgés (≥65 ans)
Aucun ajustement de la dose n'est nécessaire chez les personnes âgées (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance rénale (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Rybelsus chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été étudiées.
Rybelsus est un comprimé pour administration par voie orale une fois par jour.
Mode d'administration
Rybelsus doit être pris à jeun. Rybelsus doit être avalé entier avec de l'eau (jusqu'à un demi-verre d'eau équivalent à 120 ml). Les comprimés ne doivent pas être écrasés ni mâchés. Les patients doivent attendre au moins 30 minutes avant de manger, de boire ou de prendre d'autres médicaments. Si le délai est inférieur à 30 minutes, l'absorption de sémaglutide peut être diminuée.
Si une dose est oubliée, elle ne doit pas être compensée. La dose suivante doit être prise le lendemain.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsRybelsus ne doit pas être utilisé chez les patients diabétiques de type 1 ou pour le traitement d'une acidocétose diabétique. Sur la base de cas rapportés chez des patients sous insulinothérapie, l'arrêt ou la diminution trop rapide de la dose d'insuline au moment de l'instauration du traitement par des agonistes des récepteurs du GLP-1 peut induire une acidocétose diabétique.
Effets gastro-intestinaux indésirables et déshydratation
L'utilisation d'agonistes des récepteurs du GLP-1 peut être associée à des réactions gastro-intestinales indésirables pouvant entraîner une déshydratation qui, dans de rares cas, est susceptible de détériorer la fonction rénale.
Pancréatite aiguë
Des cas de pancréatite aiguë ont été observés lors de l'utilisation d'agonistes des récepteurs du GLP-1. Les patients doivent être informés des symptômes caractéristiques de la pancréatite aiguë. En cas de suspicion de pancréatite, Rybelsus devra être arrêté, et si la pancréatite aiguë est confirmée, Rybelsus ne devra pas être réadministré. Il convient d'être prudent chez les patients ayant des antécédents de pancréatite.
En l'absence d'autres signes et symptômes de pancréatite aiguë, une augmentation isolée des enzymes pancréatiques n'indique pas nécessairement une pancréatite aiguë.
Hypoglycémie
Les patients traités par Rybelsus en association à une sulfonylurée ou à une insuline peuvent présenter une augmentation du risque d'hypoglycémie. Le risque d'hypoglycémie peut être diminué en réduisant la dose de la sulfonylurée ou de l'insuline lors de l'initiation du traitement par Rybelsus.
Risque de tumeurs des cellules C de la thyroïde
Les études précliniques avec des agonistes des récepteurs du GLP-1 chez les rongeurs suggèrent que les agonistes des récepteurs du GLP-1 peuvent être associés à un risque accru d'hyperplasie focale des cellules C de la thyroïde et des tumeurs des cellules C (voir «Données précliniques»).
On ignore si les agonistes des récepteurs GLP-1 sont associés aux tumeurs des cellules C de la thyroïde chez les humains, y compris au carcinome médullaire de la thyroïde (CMT). Les patients présentant un CMT et les patients présentant un syndrome de néoplasie endocrinienne multiple de type 2 (MEN 2) dans l'anamnèse n'ont pas été traités par le sémaglutide dans les études cliniques. Avant le traitement par Rybelsus, il est donc nécessaire d'évaluer attentivement les avantages et les risques dans ce groupe spécifique.
La valeur clinique de la surveillance systématique des taux sériques de calcitonine n'a pas été établie.
Rétinopathie diabétique
Une amélioration rapide du contrôle glycémique a été associée à une aggravation temporaire de la rétinopathie diabétique. Un contrôle glycémique à long terme réduit le risque de rétinopathie diabétique. Les patients présentant des antécédents de rétinopathie diabétique doivent faire l'objet d'un suivi attentif et doivent être traités sur place selon les recommandations cliniques.
Insuffisance cardiaque
Il n'existe pas d'expérience thérapeutique chez les patients présentant une insuffisance cardiaque congestive de classe IV NYHA (New York Heart Association), le sémaglutide n'est donc pas recommandé chez ces patients.
Patients ayant subi une chirurgie bariatrique
Il n'y a pas d'expérience clinique du sémaglutide chez les patients ayant eu une chirurgie bariatrique.
Effets (indésirables) gastro-intestinaux
Après la commercialisation, chez des patients traités par des agonistes des récepteurs du GLP-1, une insuffisance rénale aiguë et une aggravation d'une insuffisance rénale chronique pouvant parfois nécessiter une hémodialyse, ont été rapportées. Certains de ces événements étaient annoncés chez des patients ne présentant aucune affection rénale sous-jacente. La plupart des événements annoncés survenaient chez des patients qui présentaient déjà des nausées, vomissements, diarrhées et une déshydratation. La fonction rénale doit être surveillée lors de l'instauration ou de l'ajustement d'un traitement par Rybelsus chez des patients rapportant de sévères réactions indésirables gastro-intestinales.
Ce médicament contient 22.9 mg de sodium par comprimé, ce qui équivaut à 1 % de l'apport quotidien maximum recommandé par l'OMS de 2 g de sodium pour un adulte.
InteractionsDes études in vitro ont montré un très faible potentiel d'inhibition du sémaglutide ou d'induction des enzymes CYP et d'inhibition des transporteurs de médicaments.
Le sémaglutide retarde la vidange gastrique, ce qui est susceptible d'influencer le taux d'absorption des médicaments administrés par voie orale de façon concomitante.
Aucune interaction médicamenteuse cliniquement significative n'a été observée entre le sémaglutide et les médicaments évalués. Aucun ajustement de la dose n'est par conséquent nécessaire en cas d'administration concomitante de Rybelsus.
Il est important que les patients traités par Rybelsus et par d'autres médicaments administrés par voie orale suivent les instructions de dosage figurant dans la «Posologie/Mode d'emploi».
Effets de Rybelsus sur d'autres médicaments
Lévothyroxine
L'exposition totale (AUC) de la thyroxine (ajustée en fonction des taux endogènes) a été augmentée de 33 % après administration d'une dose unique de 600 µg de lévothyroxine en même temps que le sémaglutide. L'exposition maximale (Cmax) était inchangée. La surveillance des paramètres thyroïdiens doit être envisagée lors d'un traitement concomitant des patients par Rybelsus et lévothyroxine.
Rosuvastatine
L'exposition totale (AUC) de la rosuvastatine a été augmentée de 41 % et l'exposition maximale (Cmax) de 10 %. Étant donné la large marge thérapeutique de la rosuvastatine, l'ampleur des modifications de l'exposition n'est pas considérée comme cliniquement significative.
Metformine
L'exposition totale (AUC) de la metformine a été augmentée de 32 % et l'exposition maximale (Cmax) était inchangée. Étant donné la large marge thérapeutique de la metformine, l'ampleur des modifications de l'exposition n'est pas considérée comme cliniquement significative.
Furosémide
L'exposition totale (AUC) du furosémide a été augmentée de 28 % et l'exposition maximale (Cmax) diminuée de 34 %. Étant donné la large marge thérapeutique du furosémide, l'importance des modifications de l'exposition n'est pas considérée comme cliniquement significative.
Contraceptifs oraux
Le sémaglutide n'a pas modifié l'exposition (AUC ou Cmax) en cas d'administration concomitante d'un contraceptif oral (contenant de l'éthinylestradiol et du lévonorgestrel).
Warfarine et autres dérivés de la coumarine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la R- et S-warfarine après une dose unique de warfarine. Aussi l'effet pharmacodynamique de la warfarine tel que mesuré par le rapport normalisé international (INR) n'a pas été affecté de manière cliniquement significative. Toutefois, des cas de diminution de l'INR ont été rapportés lors de l'utilisation concomitante d'acénocoumarol et de sémaglutide. Lors de l'instauration du traitement par le sémaglutide chez des patients sous warfarine ou autres dérivés de la coumarine, il est donc recommandé de surveiller fréquemment l'INR.
Digoxine
Le sémaglutide n'a pas modifié l'exposition (AUC ou Cmax) de la digoxine.
Lisinopril
Le sémaglutide n'a pas modifié l'exposition (AUC ou Cmax) du lisinopril.
Effets d'autres médicaments sur Rybelsus
Oméprazole
Aucune modification cliniquement significative de l'AUC ou de la Cmax du sémaglutide n'a été observée lors d'une prise avec de l'oméprazole (p.ex. inhibiteurs de la pompe à protons qui augmentent le pH de l'estomac).
Interactions avec les aliments
La prise concomitante d'aliments réduit l'exposition au sémaglutide (voir «Posologie/Mode d'emploi»).
Grossesse, allaitementGrossesse
Il existe des données limitées sur l'utilisation du sémaglutide chez la femme enceinte.
Les expérimentations animales ont révélé une toxicité de reproduction (voir «Données précliniques»).
Le sémaglutide ne doit donc pas être utilisé pendant la grossesse. L'utilisation d'une contraception pendant le traitement par Rybelsus est recommandée chez les femmes en âge de procréer. En cas de projet de grossesse ou en cas de grossesse, le traitement par Rybelsus doit être interrompu. Le traitement par Rybelsus doit être arrêté au moins 2 mois avant un projet de grossesse en raison de sa longue demi-vie.
Allaitement
Aucune concentration mesurable de sémaglutide n'a été trouvée dans le lait maternel des femmes qui allaitent. Le salcaprozate de sodium était présent dans le lait maternel et certains de ses métabolites ont été excrétés dans le lait maternel à de faibles concentrations. Un risque pour l'enfant allaité ne pouvant être exclu, Rybelsus ne doit pas être utilisé pendant l'allaitement.
Fertilité
L'effet du sémaglutide sur la fertilité humaine est inconnu. Le sémaglutide n'a pas affecté la fertilité des rats mâles. Chez le rat femelle, une prolongation de la durée de l'œstrus et une légère réduction du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesDes vertiges peuvent survenir, surtout initialement, pendant la phase de titration de la dose et peuvent influencer l'aptitude à conduire et à utiliser des machines.
Lorsqu'il est utilisé en association à une sulfonylurée ou à une insuline, les patients doivent être informés qu'ils doivent prendre des précautions pour éviter une hypoglycémie lors de la conduite de véhicules ou de l'utilisation de machines (voir «Mises en garde et précautions»).
Effets indésirablesRésumé du profil de sécurité
Lors de 10 essais de phase 3a, 5 707 patients ont été exposés au sémaglutide seul ou en association à d'autres hypoglycémiants. La durée du traitement allait de 26 à 78 semaines.
Les effets indésirables les plus fréquemment rapportés pendant les essais cliniques étaient les affections gastro-intestinales, incluant nausées, diarrhées et vomissements. En général, ces réactions étaient d'intensité légère ou modérée et de courte durée.
Liste tabulée des effets indésirables
Le tableau 1 répertorie les effets indésirables rapportés dans des essais de phase 3 (pour plus d'informations, voir «Propriétés/Effets») et dans des données post-commercialisation chez les patients atteints de diabète sucré de type 2. La fréquence des effets indésirables (sauf complications de la rétinopathie diabétique, voir note de bas de page dans le tableau 1) repose sur les données groupées des essais de phase 3a, excluant l'essai d'évaluation des résultats cardiovasculaires.
Les effets indésirables sont indiqués ci-dessous par classe de systèmes d'organes de la classification MedDRA et par fréquence selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (<1/10, ≥1/100); «occasionnels» (<1/100, ≥1/1000), «rares» (<1/1000, ≥1/10 000), «très rares» (<1/10 000), «fréquence inconnue» (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1: Fréquence des effets indésirables du sémaglutide par voie orale
|
Classe de systèmes d'organes MedDRA
|
Très fréquents
|
Fréquents
|
Occasionnels
|
Rares
|
Fréquence inconnue
| |
Affections du système immunitaire
|
|
|
Hypersensibilitéc
|
Réaction anaphylactique
|
| |
Troubles du métabolisme et de la nutrition
|
Hypoglycémie en cas d'utilisation avec de l'insuline ou une sulfonylurée*
|
Hypoglycémie en cas d'utilisation avec d'autres antidiabétiques oraux*
Diminution de l'appétit
|
|
|
| |
Affections du système nerveux
|
|
Vertiges
|
Dysgueusie
|
|
| |
Affections oculaires
|
|
Complications de la rétinopathie diabétiqueb
|
|
|
| |
Affections cardiaques
|
|
|
Augmentation de la fréquence cardiaque
|
|
| |
Affections gastro-intestinales
|
Nausées
Diarrhée
|
Vomissements
Douleur abdominale
Distension abdominale
Constipation
Dyspepsie
Gastrite
Reflux gastro-œsophagien
Flatulences
|
Éructation
Retard de la vidange gastrique
|
Pancréatite aiguë
|
Obstruction intestinaled,e
| |
Affections hépatobiliaires
|
|
Lipase augmentée
Amylase augmentée
|
Cholélithiase
Cholécystite
|
|
| |
Troubles généraux et anomalies au site d'administration
|
|
Épuisement
|
|
|
| |
Investigations
|
|
|
Perte de poids
|
|
| |
* Hypoglycémie de grade 2 (ADA 2018, <3.0 mmol/l ou <54 mg/dl)
b Les complications de la rétinopathie diabétique englobent: photocoagulation rétinienne, traitement par des agents intravitreux, hémorragie vitreuse, cécité diabétique (occasionnel). La fréquence est basée sur les critères d'évaluation cardiovasculaires avec le sémaglutide s.c., mais on ne peut exclure que le risque de complications de la rétinopathie diabétique identifié s'applique également au sémaglutide p.o.
c Terme groupé couvrant également les effets indésirables liés à l'hypersensibilitié tels que les éruptions cutanées et l'urticaire.
d D'après les rapports post-commercialisation.
e Terme groupé couvrant les événements indésirables obstruction intestinale, iléus, obstruction de l’intestine grêle.
|
Description d'effets indésirables spécifiques et informations complémentaires
Hypoglycémie
Très fréquent – Hypoglycémie en cas d'utilisation avec de l'insuline (24 %) ou une sulfonylurée (11 %).
Fréquent – Hypoglycémie en cas d'utilisation avec d'autres antidiabétiques oraux*
Les hypoglycémies sévères ont principalement été observées lorsque Rybelsus était associé à une sulfonylurée (<0.1 % des patients, <0.001 événement/patient-année) ou à de l'insuline (1.1 % des patients, 0.013 événement/patient-année). Peu d'épisodes d'hypoglycémie (0.1 % des patients, 0.001 événement/patient-année) ont été observés lors de l'administration de Rybelsus en association avec d'autres antidiabétiques oraux que les sulfonylurées.
Affections gastro-intestinales
Très fréquents – Nausées (15 %), diarrhées (10 %)
Fréquents – Vomissements
Des nausées sont survenues chez 15 % des patients, des diarrhées chez 10 % et des vomissements chez 7 % des patients lorsqu'ils étaient traités par Rybelsus. La plupart de ces événements étaient d'intensité légère à modérée et de courte durée. Les événements ont entraîné un arrêt du traitement chez 4 % des patients. Les événements étaient plus fréquemment rapportés pendant les premiers mois de traitement.
Rare – Pancréatites aiguës
Des cas de pancréatites aiguës confirmées par adjudication ont été rapportés dans les essais cliniques de phase 3a pour le sémaglutide (< 0.1 %) et le comparateur (0.2 %). Dans l'essai d'évaluation des résultats cardiovasculaires, la fréquence des pancréatites aiguës confirmées par adjudication était de 0.1 % pour le sémaglutide et de 0.2 % pour le placebo (voir «Mises en garde et précautions»).
Arrêt du traitement à cause d'un effet indésirable
L'incidence de l'arrêt du traitement à cause d'effets indésirables était de 9 % chez les patients traités par Rybelsus. Les effets indésirables entraînant le plus fréquemment un arrêt du traitement étaient de nature gastro-intestinale.
Augmentation de la fréquence cardiaque
Une augmentation modérée de 2 battements par minute a été observée dans les essais de phase 3 sous Rybelsus.
Affections oculaires
Fréquent – Complications de la rétinopathie diabétique
Dans un essai clinique de 2 ans portant sur 3 297 patients diabétiques de type 2 présentant un risque cardiovasculaire élevé, traités par sémaglutide s.c., les complications de la rétinopathie diabétique étaient un critère d'évaluation. Lors de cet essai, des complications liées à la rétinopathie diabétique sont survenus chez plus de patients traités par sémaglutide s.c. (3.0 %) que chez ceux sous placebo (1.8 %). Plus de 80 % des patients présentant une complication de la rétinopathie diabétique avaient une rétinopathie diabétique documentée avant le début du traitement. Chez les patients qui n'avaient pas d'antécédents documentés de rétinopathie diabétique, le nombre d'événements sous sémaglutide s.c. et placebo ont été rapportés dans des proportions similaires.
Lors d'essais cliniques d'une durée allant jusqu'à 18 mois, évaluant Rybelsus et portant sur 6 352 patients diabétiques de type 2, les effets indésirables liés à la rétinopathie diabétique sont survenus dans des proportions similaires sous sémaglutide (4.2 %) et sous comparateurs (3.8 %).
Immunogénicité
Compte tenu des propriétés immunogènes potentielles des médicaments contenant des protéines ou des peptides, les patients traités par sémaglutide peuvent développer des anticorps. La proportion de sujets testés positifs aux anticorps anti-sémaglutide à tout moment après le début du traitement était de 14 (0.5 %) et 7 d'entre eux (soit 0.2 % de la population totale) avaient développé des anticorps avec un effet neutralisant sur le GLP-1 natif. L'activité neutralisante des anticorps est à ce jour encore incertaine.
Effets indésirables identifiés après la mise sur le marché
Affections du rein et des voies urinaires: insuffisance rénale aiguë (voir «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes effets d'un surdosage de sémaglutide observés dans les études cliniques peuvent être associés à des troubles gastro-intestinaux. En cas de surdosage, un traitement de soutien approprié doit être initié en fonction des signes cliniques et des symptômes du patient.
Une période d'observation prolongée et un traitement des symptômes peuvent être nécessaires, en tenant compte de la longue demi-vie du sémaglutide d'environ 1 semaine (voir «Pharmacocinétique»). Il n'existe aucun antidote spécifique à un surdosage de sémaglutide.
Propriétés/EffetsCode ATC
A10BJ06
Mécanisme d'action
Le sémaglutide est un analogue du GLP-1 humain qui participe à la régulation de l'homéostasie du glucose. Compte tenu de sa forte liaison à l'albumine, la clairance rénale du sémaglutide est retardée. De plus, le sémaglutide est moins sensible à la dégradation par l'enzyme DPP-4 du fait de sa structure modifiée par rapport au GLP-1 natif.
Le sémaglutide agit comme agoniste du récepteur cible du GLP-1 natif. Les récepteurs du GLP-1 sont exprimés dans le pancréas, le cerveau, le cœur, le système vasculaire, le système immunitaire et les reins. La stimulation des récepteurs du GLP-1 par le sémaglutide stimule la sécrétion d'insuline et réduit la sécrétion de glucagon de façon glucose-dépendante. Cela entraîne également un retard de la vidange gastrique en début de phase postprandiale.
Le sémaglutide réduit le poids corporel et la graisse corporelle en diminuant les apports énergétiques. Le mécanisme entraîne une réduction générale de l'appétit, comprenant une augmentation de la satiété et une diminution de la faim. La résistance insulinique est réduite. Ceci est probablement dû à la réduction du poids corporel.
Pharmacodynamique
Rybelsus réduit la glycémie à jeun ainsi que la glycémie mesurée par le patient. L'effet est rapide. Chez les patients diabétiques de type 2, la glycémie à jeun diminue dans la première semaine.
Toutes les évaluations pharmacodynamiques ont été effectuées au bout de 12 semaines de traitement (incluant l'augmentation de la dose) à l'état d'équilibre avec 1 mg de sémaglutide en injection une fois par semaine.
Glycémie à jeun et augmentations postprandiales
Le sémaglutide réduit les concentrations à jeun et postprandiales de glucose. Chez les patients diabétiques de type 2, le traitement par sémaglutide a entraîné une réduction de la glycémie aussi bien en termes de variation absolue par rapport à l'inclusion que de réduction relative par rapport au placebo des glycémies à jeun (1.6 mmol/l/29 mg/dl; réduction de 22 %), des glycémies postprandiales à 2 heures (4.1 mmol/l74 mg/dl; réduction de 37 %), de la glycémie moyenne sur 24 heures (1.7 mmol/l/30 mg/dl; réduction de 22 %) et des variations des pics de glycémie postprandiale sur 3 repas (0.6-1.1 mmol/l//11–20 mg/dl) par rapport au placebo.
Fonction bêta-cellulaire et sécrétion d'insuline
Le sémaglutide améliore la fonction bêta-cellulaire. En comparaison avec le placebo, le sémaglutide a amélioré la réponse insulinique de la première et deuxième phases en la multipliant par trois et par deux, respectivement, et augmenté la capacité de sécrétion maximale des cellules bêta après un test de stimulation à l'arginine chez des patients diabétiques de type 2. De plus, le traitement par sémaglutide a augmenté les concentrations d'insuline à jeun en comparaison avec le placebo.
Sécrétion de glucagon
Le sémaglutide réduit les concentrations à jeun et postprandiales de glucagon. Chez les patients diabétiques de type 2, le sémaglutide a entraîné les réductions relatives suivantes du glucagon en comparaison avec le placebo: glucagon à jeun (8–21 %), réponse postprandiale du glucagon (14–15 %) et concentration moyenne de glucagon sur 24 heures (12 %).
Sécrétion de glucagon et d'insuline glucose-dépendante
Le sémaglutide a abaissé les concentrations élevées de glucose sanguin en stimulant la sécrétion d'insuline et en réduisant la sécrétion de glucagon de façon glucose-dépendante. Avec le sémaglutide, le taux de sécrétion d'insuline chez les patients diabétiques de type 2 était comparable à celui des sujets sains.
Pendant l'hypoglycémie induite et en comparaison avec le placebo, le sémaglutide n'a pas modifié la réponse contre-régulatoire d'augmentation du glucagon et n'a pas limité la baisse du peptide C chez les patients diabétiques de type 2.
Vidange gastrique
Le sémaglutide a entraîné un léger retard de la vidange gastrique au début de la phase postprandiale, réduisant ainsi la vitesse d'apparition du glucose dans la circulation après le repas.
Poids et composition corporelle
Par rapport aux comparateurs (placebo, sitagliptine, empagliflozine et liraglutide), le traitement par Rybelsus visait une réduction de poids supérieure. La perte de poids était essentiellement due à la perte de tissu adipeux, qui était trois fois plus importante que la perte de masse musculaire.
Appétit, apport énergétique et choix des aliments
En comparaison avec le placebo, le sémaglutide a diminué de 18 à 35 % l'apport énergétique de 3 repas ad libitum consécutifs. Cette constatation a été étayée par une suppression de l'appétit induite par le sémaglutide à jeun et en phase postprandiale, un meilleur contrôle de l'alimentation, une diminution des envies alimentaires et une préférence relativement réduite pour les aliments à haute teneur en graisse.
Lipides à jeun et postprandiaux
En comparaison avec le placebo, le sémaglutide a diminué les concentrations de triglycérides et de cholestérol VLDL (lipoprotéines de très basse densité) à jeun de respectivement 12 % et 21 %. La réponse postprandiale des triglycérides et du cholestérol VLDL à un repas à haute teneur en graisse a été réduite de >40 %.
Électrophysiologie cardiaque (QTc)
L'effet du sémaglutide sur la repolarisation cardiaque a été testé lors d'un essai QTc approfondi. A un niveau d'exposition moyen qui était 4 fois supérieur à la dose maximale recommandée de Rybelsus, le sémaglutide n'a pas prolongé les intervalles QTc dans une mesure cliniquement significative.
Efficacité clinique
L'efficacité et la sécurité de Rybelsus ont été évaluées lors de huit essais contrôlés randomisés de phase 3a. Dans sept essais, le critère principal était l'évaluation de l'efficacité glycémique, et dans autre un essai, le critère principal portait sur l'évaluation de la sécurité cardiovasculaire.
Les essais de phase 3a incluaient 8 842 patients diabétiques de type 2 randomisés (5 169 étaient traités par Rybelsus), parmi lesquels 1 165 présentaient une insuffisance rénale modérée. Il s'agissait de comparer l'efficacité de Rybelsus par rapport au placebo, à l'empagliflozine, à la sitagliptine, au liraglutide et au dulaglutide.
Lors de tous les essais, le traitement par Rybelsus a entraîné une amélioration cliniquement significative de l'HbA1c, de la glycémie à jeun et du poids corporel pour une durée pouvant aller jusqu'à 78 semaines.
L'efficacité de Rybelsus n'a pas été influencée par l'âge, le sexe, la race, l'origine ethnique, le poids corporel, l'IMC, l'ancienneté du diabète, les affections du tractus gastro-intestinal supérieur ou le niveau d'atteinte de la fonction rénale à l'inclusion.
PIONEER 1 – Monothérapie
Lors d'un essai clinique en double aveugle de 26 semaines, 703 patients diabétiques de type 2 insuffisamment contrôlés par le régime alimentaire et l'activité physique ont été randomisés dans des groupes recevant 3 mg, 7 mg ou 14 mg de Rybelsus ou un placebo une fois par jour.
Tableau 2: Résultats de l'essai de 26 semaines comparant une monothérapie par Rybelsus à un placebo (PIONEER 1)
|
|
Rybelsus
7 mg
|
Rybelsus
14 mg
|
Placebo
| |
Population (N)1
|
175
|
175
|
178
| |
HbA1c (%)
| |
Inclusion2
|
8.0
|
8.0
|
7.9
| |
Variation entre l'inclusion et la semaine 263
|
-1.3
|
-1.5
|
-0.1
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-1.2 [-1.5; -1.0]§
|
-1,4 [-1.7; -1.2]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
72#
|
80#
|
34
| |
Poids corporel (kg)
| |
Inclusion2
|
89.0
|
88.1
|
88.6
| |
Variation entre l'inclusion et la semaine 263
|
-2.5
|
-4.1
|
-1.5
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-1.0 [-1.8; -0.2]§
|
-2.6 [-3.4; -1.8]§
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Les résultats qui ont été saisis après l'arrêt du produit de l'essai ou l'instauration d'un médicament de secours ne sont pas pris en compte.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous placebo (p<0.05)
|
PIONEER 2 – Rybelsus vs. empagliflozine, tous les deux en association avec la metformine
Lors d'un essai clinique en ouvert de 52 semaines, 822 patients diabétiques de type 2 ont été randomisés dans des groupes recevant 14 mg de Rybelsus une fois par jour ou 25 mg d'empagliflozine une fois par jour, tous les deux en association avec la metformine.
Le traitement par Rybelsus 14 mg une fois par jour a réduit l'HbA1c de -1.4 % à la semaine 26. La réduction était statistiquement significativement supérieure à celle obtenue sous empagliflozine, avec une différence de traitement estimée de -0.5 % [-0.7; -0.4]IC à 95%.
Tableau 3: Résultats de l'essai de 52 semaines comparant Rybelsus à l'empagliflozine (PIONEER 2)
|
|
Rybelsus
14 mg
|
Empagliflozine
25 mg
| |
Population (N)1
|
411
|
410
| |
HbA1c (%)
| |
Inclusion2
|
8.1
|
8.1
| |
Variation entre l'inclusion et la semaine 523
|
-1.3
|
-0.8
| |
Différence par rapport au traitement par empagliflozine3 [IC à 95 %]
|
-0.5 [-0.7; -0.4]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
72#
|
48
| |
Poids corporel (kg)
| |
Inclusion2
|
91.9
|
91.3
| |
Variation entre l'inclusion et la semaine 523
|
-4.7
|
-3.8
| |
Différence par rapport au traitement par empagliflozine3 [IC à 95 %]
|
-0.9 [-1.6; -0.2]§
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Les résultats qui ont été saisis après l'arrêt du traitement à l'étude ou après l'instauration d'un médicament de secours ne sont pas pris en compte.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous l'empagliflozine (p<0.05)
|
PIONEER 3 – Rybelsus versus sitagliptine, respectivement en association avec metformine ou metformine plus sulfonylurée
Lors d'un essai clinique en double aveugle et double placebo de 78 semaines, 1 864 patients diabétiques de type 2 ont été randomisés dans des groupes recevant 3 mg, 7 mg ou 14 mg de Rybelsus ou 100 mg de sitagliptine une fois par jour, tous en association avec la metformine seule ou avec la metformine plus une sulfonylurée.
Le traitement par Rybelsus 7 mg et 14 mg une fois par jour a démontré une réduction de l'HbA1c de respectivement 1.1 % et 1.4 % dans la semaine 26; cette réduction était statistiquement significativement supérieure à celle obtenue sous sitagliptine, avec une différence de traitement de -0.3 % [-0.4; -0.2]IC à 95 % et -0.6 % [-0.7; -0.5]IC à 95 %.
Les réductions de l'HbA1c et du poids corporel se sont maintenues pendant toute la durée de l'étude de 78 semaines (Tableau 4).
Tableau 4: Résultats de l'essai de 78 semaines comparant Rybelsus à la sitagliptine (PIONEER 3)
|
|
Rybelsus
7 mg
|
Rybelsus
14 mg
|
Sitagliptine
100 mg
| |
Population (N)1
|
465
|
465
|
467
| |
HbA1c (%)
| |
Inclusion2
|
8.4
|
8.3
|
8.3
| |
Variation entre l'inclusion et la semaine 783
|
-0.7
|
-1.1
|
-0.4
| |
Différence par rapport au traitement par sitagliptine3 [IC à 95 %]
|
-0.3 [-1.6; -0.2]§
|
-0.7 [-0.8; -0.5]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
50#
|
52#
|
39
| |
Poids corporel (kg)
| |
Inclusion2
|
91.3
|
91.2
|
90.9
| |
Variation entre l'inclusion et la semaine 783
|
-2.7
|
-3.5
|
-1.1
| |
Différence par rapport au traitement par sitagliptine3 [IC à 95 %]
|
-1.6 [-2.2; -0.9]§
|
-2.4 [-3.0; -1.7]§
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Les résultats qui ont été saisis après l'arrêt du traitement à l'étude ou après l'instauration d'un médicament de secours ne sont pas pris en compte.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle obtenue sous sitagliptine (p<0.05)
|
PIONEER 4 – Rybelsus versus liraglutide et placebo, respectivement en association avec metformine ou metformine plus inhibiteur du SGLT2
Lors d'un essai clinique en double aveugle et double placebo de 52 semaines, 711 patients diabétiques de type 2 ont été randomisés dans des groupes recevant 14 mg de Rybelsus, 1.8 mg de liraglutide en injection s.c. ou un placebo une fois par jour, tous en association avec la metformine ou avec la metformine plus un inhibiteur du SGLT2.
Le traitement par Rybelsus 14 mg une fois par jour a réduit l'HbA1c de 1.3 % dans la semaine 26; La réduction était statistiquement significativement supérieure à celle obtenue sous placebo et sous liraglutide, avec une différence de traitement estimée de -1.2 % [-1.4; -1.0]IC à 95% et -0.2 % [-0.3; -0.1]IC à 95 %.
Tableau 5: Résultats de l'essai de 52 semaines comparant Rybelsus au liraglutide et à un placebo (PIONEER 4)
|
|
Rybelsus
14 mg
|
Liraglutide
1.8 mg
|
Placebo
| |
Population (N)1
|
285
|
284
|
142
| |
HbA1c (%)
| |
Inclusion2
|
8.0
|
8.0
|
7.9
| |
Variation entre l'inclusion et la semaine 523
|
-1.2
|
-0.9
|
0.2
| |
Différence par rapport au traitement par liraglutide3 [IC à 95 %]
|
-0.3 [-0.4; -0.1]§
|
-
|
-
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-1.4 [-1.6; -1.2]§
|
-
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
69*
|
63
|
18
| |
Poids corporel (kg)
| |
Inclusion2
|
92.9
|
95.5
|
93.2
| |
Variation entre l'inclusion et la semaine 523
|
-5.0
|
-3.1
|
-1.2
| |
Différence par rapport au traitement par liraglutide3 [IC à 95 %]
|
-1.8 [-2.6; -1.0]§
|
-
|
-
| |
Différence par rapport au placebo3[IC à 95 %]
|
-3.8 [-4.8; -2.7]§
|
-
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Les résultats qui ont été saisis après l'arrêt du traitement à l'étude ou après l'instauration d'un médicament de secours ne sont pas pris en compte
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous liraglutide (p<0.05)
|
PIONEER 5 – Rybelsus versus placebo, respectivement en association avec une insuline basale seule, la metformine plus une insuline basale ou la metformine et/ou une sulfonylurée, chez des patients présentant une insuffisance rénale modérée
Lors d'un essai clinique en double aveugle de 26 semaines, 324 patients diabétiques de type 2 présentant une insuffisance rénale modérée (Débit de Filtration Glomérulaire estimé 30-59 ml/min/1.73 m2), qui étaient sous traitement antidiabétique stable, ont en outre été randomisés dans des groupes recevant 14 mg de Rybelsus ou un placebo une fois par jour.
Le profil d'efficacité et de sécurité chez des patients diabétiques de type 2 présentant une insuffisance rénale modérée de Rybelsus correspondait à celui généralement décrit pour les agonistes des récepteurs du GLP-1
Tableau 6: Résultats de l'essai de 26 semaines comparant Rybelsus à un placebo chez des patients diabétiques de type 2 présentant une insuffisance rénale modérée (PIONEER 5)
|
|
Rybelsus
14 mg
|
Placebo
| |
Population (N)1
|
163
|
161
| |
HbA1c (%)
| |
Inclusion2
|
8.0
|
7.9
| |
Variation entre l'inclusion et la semaine 263
|
-1.1
|
-0.1
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-1.0 [-1.2; -0.8]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
64#
|
21
| |
Poids corporel (kg)
| |
Inclusion2
|
91.3
|
90.4
| |
Variation entre l'inclusion et la semaine 263
|
-3.7
|
-1.1
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-2.7 [-3.5; -1.9]§
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Les résultats qui ont été saisis après l'arrêt du traitement à l'étude ou après l'instauration d'un médicament de secours ne sont pas pris en compte.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous placebo (p<0.05)
|
PIONEER 7 – Rybelsus versus sitagliptine, respectivement en association avec metformine, des inhibiteurs du SGLT2, une sulfonylurée ou une thiazolidinedione (essai d'ajustement flexible de la dose)
Lors d'un essai clinique en ouvert de 52 semaines, 504 patients diabétiques de type 2 ont été randomisés dans des groupes recevant Rybelsus (ajustement flexible de la dose de 3 mg, 7 mg et 14 mg une fois par jour) ou 100 mg de sitagliptine une fois par jour, tous en association avec 1 ou 2 hypoglycémiants oraux (metformine, inhibiteurs du SGLT2, sulfonylurée ou thiazolidinedione). La dose de Rybelsus était ajustée toutes les 8 semaines en fonction de la réponse glycémique et de la tolérance du patient. La dose de 100 mg de sitagliptine était fixe. L'efficacité et la sécurité de Rybelsus étaient évaluées à la semaine 52.
À la semaine 52, la proportion de patients traités par Rybelsus 3 mg, 7 mg et 14 mg était respectivement d'environ 9 %, 30 % et 60 %.
Tableau 7: Résultats de l'essai d'ajustement à dose flexible de 52 semaines comparant Rybelsus à la sitagliptine (PIONEER 7)
|
|
Rybelsus
Dose flexible
|
Sitagliptine
100 mg
| |
Population (N)1
|
253
|
251
| |
HbA1c (%)
| |
Inclusion2
|
8.3
|
8.3
| |
Variation entre l'inclusion et la semaine 523
|
-1.4
|
-0.7
| |
Différence par rapport au traitement par sitagliptine3 [IC à 95 %]
|
-0.7 [-0.9; -0.5]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
63#
|
28
| |
Poids corporel (kg)
|
88.9
|
88.4
| |
Inclusion2
|
-2.9
|
-0.8
| |
Variation entre l'inclusion et la semaine 523
|
-2.2 [-2.9; -1.5]§
|
-
| |
Différence par rapport au traitement par sitagliptine3 [IC à 95 %]
|
28#
|
13
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous sitagliptine (p<0.05)
|
PIONEER 8 – Rybelsus versus placebo, respectivement en association avec une insuline avec ou sans metformine
Lors d'un essai clinique en double aveugle de 52 semaines, 731 patients diabétiques de type 2 insuffisamment contrôlés sous insuline (basale, basale/en bolus ou prémélangée) avec ou sans metformine, ont été randomisés dans des groupes recevant 3 mg, 7 mg ou 14 mg de Rybelsus ou un placebo une fois par jour.
Le traitement par Rybelsus 7 mg et 14 mg une fois par jour a démontré une réduction de l'HbA1c dans la semaine 26; cette réduction était statistiquement significativement supérieure à celle obtenue sous placebo, avec une différence de traitement de -1.0 % [-1.2; -0.8]IC à 95 % et -1.4 % [-1.6; -1.2]IC à 95 %.
Tableau 8: Résultats de l'essai de 52 semaines comparant Rybelsus à un placebo en association à une insuline (PIONEER 8)
|
|
Rybelsus
7 mg
|
Rybelsus
14 mg
|
Placebo
| |
Population (N)1
|
182
|
181
|
184
| |
HbA1c (%)
| |
Inclusion2
|
8.2
|
8.2
|
8.2
| |
Variation entre l'inclusion et la semaine 523
|
-0.8
|
-1.2
|
0.0
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-0.9 [-1.1; -0.6]§
|
-1.3 [-1.5; -1.0]§
|
-
| |
Patients (%) ayant atteint une HbA1c <7.0 %2
|
47#
|
64#
|
10
| |
Poids corporel (kg)
| |
Inclusion2
|
87.1
|
84.6
|
86.0
| |
Variation entre l'inclusion et la semaine 523
|
-2.9
|
-4.3
|
0.6
| |
Différence par rapport au placebo3 [IC à 95 %]
|
-3.5 [-4.5; -2.6]§
|
-4.9 [-5.9; -3.9]§
|
-
| |
1
Full Analysis Set: tous les patients randomisés
2 Moyenne/proportion observée
3 Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région. Evaluée à l'aide d'un modèle de mélange pour mesures répétées, ajustée en fonction de la valeur de base, du traitement de base et de la région.
§ Statistiquement significatif (p<0.05)
# La probabilité d'atteindre l'objectif était statistiquement significativement supérieure sous Rybelsus à celle observée sous placebo (p<0.05)
|
Sécurité cardiovasculaire
Les effets cardiovasculaires du sémaglutide oral ont été examinés dans l'étude de résultats cardiovasculaire PIONEER 6. Des données supplémentaires sur la sécurité cardiovasculaire de sémaglutide en administration sous-cutanée ont été collectées dans l'étude de résultats cardiovasculaire SUSTAIN 6.
PIONEER 6
Lors de cet essai en double aveugle, 3183 patients diabétiques de type 2 à haut risque cardiovasculaire (2695 [85 %] patients atteints d'une maladie cardiovasculaire préexistante et 488 [15 %] patients présentant des facteurs de risque cardiovasculaire sans maladie cardiovasculaire préexistante) ont été randomisés dans des groupes recevant 14 mg de Rybelsus une fois par jour ou un placebo en plus du traitement antihyperglycémiant. La période d'observation médiane était de 16 mois. Le traitement pouvait être intensifié dans les deux bras, conformément aux directives de traitement en vigueur.
Le critère principal était le délai de survenue depuis la randomisation du premier événement cardiovasculaire majeur (MACE, mortalité cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal.). Le risque cardiovasculaire chez les patients traités par sémaglutide était numériquement réduit.
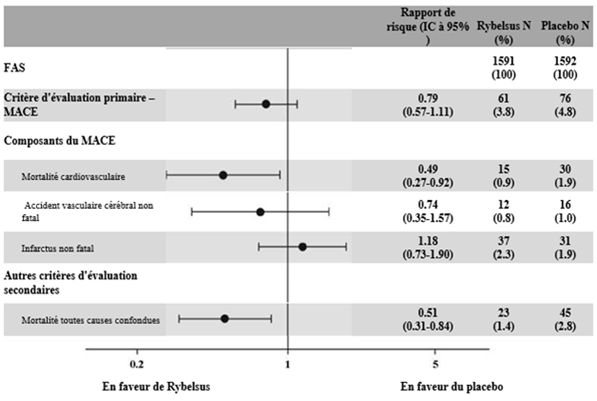
Figure 7: Graphique en forêt: effet du traitement sur le critère composite principal MACE, ses composantes et la mortalité toutes causes confondues (PIONEER 6)
Cet effet reposait principalement sur la diminution de la mortalité cardiovasculaire.
SUSTAIN 6
Lors de cet essai en double aveugle de 104 semaines, 3 297 patients diabétiques de type 2 à haut risque cardiovasculaire (2 735 [83 %] patients atteints d'une maladie cardiovasculaire préexistante et 562 [27 %] patients présentant des facteurs de risque cardiovasculaire sans maladie cardiovasculaire préexistante) ont été randomisés dans des groupes recevant 0.5 mg de sémaglutide s.c., 1 mg de sémaglutide s.c. ou un placebo en plus du traitement antihyperglycémiant. La période d'observation médiane était de 2 ans.
Le critère principal était le délai de survenue depuis la randomisation du premier événement cardiovasculaire majeur (MACE, décès cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal). Le risque cardiovasculaire chez les patients traités par sémaglutide sur deux ans en moyenne était réduit.
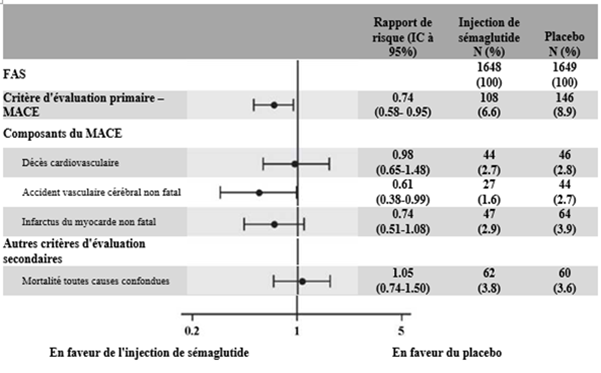
Figure 8: Graphique en forêt: effet du traitement sur le critère composite principal MACE, ses composantes et la mortalité toutes causes confondues (SUSTAIN 6)
La réduction du risque cardiovasculaire reposait essentiellement sur une diminution du nombre d'accidents vasculaires cérébraux non fatals. Malgré les résultats de l'étude PIONEER 6 portant sur le sémaglutide oral, le traitement par sémaglutide s.c. n'a démontré aucun effet positif sur la mortalité cardiovasculaire.
PharmacocinétiqueAbsorption
Le sémaglutide administré par voie orale a une faible biodisponibilité absolue et une absorption variable. L'administration quotidienne selon la posologie recommandée associée à une longue demi-vie réduit la fluctuation quotidienne de l'exposition.
Le sémaglutide est coformulé avec du salcaprozate de sodium, ce qui simplifie l'absorption du sémaglutide après administration orale. L'absorption du sémaglutide a lieu essentiellement dans l'estomac.
Les caractéristiques pharmacocinétiques du sémaglutide ont été largement étudiées chez des sujets sains et des patients diabétiques de type 2. Après administration orale, la concentration plasmatique maximale du sémaglutide a été atteinte 1 heure après la prise de la dose. L'exposition à l'état d'équilibre a été atteinte après 4 à 5 semaines d'administration une fois par jour. Des analyses pharmacocinétiques de la population ont démontré des concentrations moyennes à l'état d'équilibre d'environ 6.7 nmol/l et 14.6 nmol/l avec Rybelsus 7 mg et 14 mg chez les patients diabétiques de type 2. L'exposition systémique au sémaglutide a augmenté de manière proportionnelle à la dose.
L'absorption du sémaglutide est diminuée s'il est pris avec des aliments ou de grands volumes d'eau. Une période de jeûne post-dose plus longue entraîne une absorption plus élevée.
La biodisponibilité absolue estimée du sémaglutide est d'environ 1 % après administration orale.
Distribution
Le volume de distribution absolu estimé du sémaglutide chez des patients diabétiques de type 2 était d'environ 8 l. Le sémaglutide était fortement lié aux protéines plasmatiques (>99 %).
Métabolisme
Le sémaglutide est métabolisé par clivage protéolytique de la chaîne peptidique et bêta-oxydation séquentielle de la chaîne latérale des acides gras.
Élimination
L'élimination des matières associées au sémaglutide s'est faite principalement par l'urine et les fèces. Approximativement 3 % de la dose a été excrétée sous la forme de sémaglutide intact dans l'urine.
Avec une demi-vie d'élimination d'environ 1 semaine, le sémaglutide restera présent dans la circulation sanguine pendant approximativement 5 semaines après la dernière dose. La clairance du sémaglutide chez les patients diabétiques de type 2 est d'environ 0.04 l/h.
Niveau d'exposition moyen au sémaglutide en administration orale et sous-cutanée
L'exposition moyenne avec 0.5 mg de sémaglutide s.c. correspond à environ 90 % de celle avec 14 mg de Rybelsus dans des analyses pharmacocinétiques de la population. L'exposition moyenne avec 7 ou 14 mg de Rybelsus correspond à environ 60 % de celle avec 0.5 mg de sémaglutide s.c.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
L'insuffisance hépatique n'a eu aucun impact cliniquement significatif sur la pharmacocinétique du sémaglutide. La pharmacocinétique du sémaglutide a été évaluée chez des patients présentant une insuffisance hépatique légère, modérée ou sévère en comparaison avec des sujets présentant une fonction hépatique normale dans le cadre d'une étude utilisant des doses de sémaglutide administrées une fois par jour pendant 10 jours consécutifs.
Troubles de la fonction rénale
L'insuffisance rénale n'a pas affecté la pharmacocinétique du sémaglutide de manière cliniquement significative. La pharmacocinétique du sémaglutide a été évaluée chez des patients présentant une insuffisance rénale légère, modérée ou sévère et chez des patients dialysés atteints d'une maladie rénale en stade terminal comparativement à des sujets dont la fonction rénale était normale dans le cadre d'une étude utilisant des doses de sémaglutide administrées une fois par jour pendant 10 jours consécutifs. Ces résultats ont également été observés chez des patients diabétiques de type 2 et insuffisants rénaux dans des études cliniques de phase 3a (analyse pharmacocinétique de la population).
Patients âgés
L'âge n'a aucun effet sur la pharmacocinétique du sémaglutide, selon les données des essais cliniques portant sur des patients jusqu'à l'âge de 92 ans.
Enfants et adolescents
Le sémaglutide n'a pas été étudié chez les enfants et les adolescents.
Sexe
Le sexe n'a eu aucun effet cliniquement significatif sur la pharmacocinétique du sémaglutide.
Race et origine ethnique
La race (blanc, noir ou afro-américain, asiatique) et l'origine ethnique (hispano-américain ou latino) n'ont eu aucun effet sur la pharmacocinétique du sémaglutide.
Poids corporel
Le poids corporel a influencé l'exposition au sémaglutide. Un poids corporel plus élevé a été associé à une exposition plus faible. Dans les essais cliniques, Rybelsus a assuré une exposition systémique adéquate à un poids corporel compris entre 40 et 188 kg.
Maladie du tractus gastro-intestinal supérieur
Une maladie du tractus gastro-intestinal supérieur (gastrite chronique et/ou reflux gastro-œsophagien) n'a pas eu d'impact cliniquement significatif sur la pharmacocinétique du sémaglutide. La pharmacocinétique a été évaluée chez des patients diabétiques de type 2 avec ou sans maladie du tractus gastro-intestinal supérieur recevant des doses de sémaglutide une fois par jour pendant 10 jours consécutifs.
Ces résultats ont également été observés chez des patients diabétiques de type 2 souffrant d'une maladie des voies gastro-intestinales supérieures dans des études cliniques de phase 3a.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée ou génotoxicité n'ont pas révélé de risque particulier pour l'être humain.
Carcinogénicité
Les tumeurs non létales des cellules C de la thyroïde observées chez les rongeurs constituent un effet spécifique à la classe des agonistes des récepteurs du GLP-1. Lors d'études de carcinogénicité sur 2 ans chez le rat et la souris, le sémaglutide a provoqué des tumeurs des cellules C de la thyroïde à des expositions cliniquement significatives. Les tumeurs des cellules C chez les rongeurs sont dues à un mécanisme non génotoxique, spécifique, médié par les récepteurs du GLP 1, auquel les rongeurs sont particulièrement sensibles. La pertinence de ces résultats pour l'être humain est considérée comme faible mais ne peut pas être complètement exclue.
Toxicité sur la reproduction
Lors d'études de fertilité chez le rat, le sémaglutide n'a pas affecté les performances d'accouplement ni la fertilité des mâles. Chez le rat femelle, une prolongation du cycle œstrien et une faible baisse du nombre d'ovulations ont été observées à des doses associées à une réduction du poids maternel.
Lors d'études du développement embryo-fœtal chez le rat, le sémaglutide a entraîné une embryotoxicité à des expositions inférieures aux niveaux cliniquement significatifs. Le sémaglutide a entraîné une nette réduction du poids maternel et une diminution de la croissance et de la survie embryonnaires. Chez les fœtus, des malformations viscérales et squelettiques majeures ont été observées, notamment des effets sur les os longs, les côtes, les vertèbres, la queue, les vaisseaux sanguins et les ventricules cérébraux. Des évaluations mécanistes ont indiqué que l'embryotoxicité impliquait une anomalie médiée par les récepteurs du GLP-1 au niveau de l'apport de nutriments à l'embryon via le sac vitellin du rat. En raison des différences d'anatomie et de fonction du sac vitellin entre les espèces, et en raison de l'absence d'expression des récepteurs du GLP-1 dans le sac vitellin des primates non humains, ce mécanisme médié par les récepteurs du GLP 1 observé chez le rat n'est probablement pas pertinent chez l'être humain. Cependant, un effet direct du sémaglutide sur le fœtus ne peut être exclu.
Lors d'études de toxicité pour le développement chez le lapin et le singe cynomolgus, une augmentation des fausses couches et une légère hausse de l'incidence des anomalies fœtales ont été observées à des expositions cliniquement significatives. Ces résultats coïncidaient avec une nette réduction du poids maternel allant jusqu'à 16 %. Il n'est pas établi si ces effets sont liés à la réduction de consommation d'aliments par la mère en tant qu'effet direct du GLP-1.
La croissance et le développement postnataux ont été évalués chez le singe cynomolgus. Les nourrissons étaient légèrement plus petits à la naissance, mais ont récupéré pendant l'allaitement.
Analyses de toxicité chez des animaux juvéniles
Chez les jeunes rats mâles et femelles, le sémaglutide a retardé la maturation sexuelle. Ces retards n'ont eu aucun impact sur la fertilité et la capacité de reproduction des deux sexes, ni sur la capacité des femelles à maintenir une grossesse.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C.
Conserver le contenu dans le blister d'origine pour le protéger de la lumière et de l'humidité.
Tenir hors de la portée et de la vue des enfants.
Numéro d’autorisation67446 (Swissmedic)
PrésentationComprimés de 3 mg, 7 mg ou 14 mg sous blister.
3 mg: emballages de 30 comprimés [B]
7 mg: emballages de 30 et 90 comprimés [B]
14 mg: emballages de 30 et 90 comprimés [B]
Titulaire de l’autorisationNovo Nordisk Pharma AG, Kloten
Domicile: Zürich
Mise à jour de l’informationNovembre 2024
|