CompositionPrincipe actif: guselkumab, produit par des cellules CHO (Chinese Hamster Ovary) génétiquement modifiées.
Excipients: L-histidine, monochlorhydrate de L-histidine monohydraté, polysorbate 80, saccharose, eau pour préparations injectables q.s. ad solutionem pour 1 ml.
Forme galénique et quantité de principe actif par unitéSolution injectable en seringue préremplie pour administration sous-cutanée:
Chaque seringue préremplie contient 100 mg de guselkumab dans 1 ml de solution.
Solution injectable en stylo prérempli pour administration sous-cutanée:
Chaque stylo prérempli contient 100 mg de guselkumab dans 1 ml de solution.
Indications/Possibilités d’emploiTREMFYA est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les patients adultes ayant présenté une réponse insuffisante à d'autres traitements systémiques tels que la ciclosporine, le méthotrexate (MTX) ou la PUVA (psoralène et UV-A) ou présentant une contre-indication ou une intolérance à de tels traitements.
Posologie/Mode d’emploiTREMFYA doit être utilisé sous la conduite et la surveillance d'un médecin expérimenté dans le diagnostic et le traitement du psoriasis en plaques.
Avant d'instaurer le traitement, le médecin doit s'assurer que le patient a compris que TREMFYA est un nouveau traitement dont l'expérience est limitée et les risques à long terme inconnus. L'efficacité et la sécurité de TREMFYA sont démontrées sur une période allant jusqu'à 1 an.
Posologie: La dose recommandée de TREMFYA est de 100 mg en injection sous-cutanée aux semaines 0 et 4, suivie d'une dose d'entretien toutes les 8 semaines.
L'arrêt du traitement doit être envisagé chez les patients ne présentant pas de réponse au bout de 16 semaines de traitement.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Mode d'administration:
TREMFYA doit être administré par voie sous-cutanée, dans l'abdomen ou la cuisse. Dans la mesure du possible, les zones cutanées présentant des signes de psoriasis ne doivent pas être utilisées comme sites d'injection.
Après une formation adaptée à la technique d'injection sous-cutanée, les patients peuvent s'injecter eux-mêmes TREMFYA si le médecin estime cela approprié. Cependant, le médecin doit assurer un suivi médical adéquat des patients. Les patients doivent être informés de la nécessité d'injecter la dose complète de TREMFYA conformément aux «Instructions d'utilisation» fournies séparément dans l'emballage. Pour des informations plus précises concernant la préparation et les précautions particulières de manipulation, voir «Remarques particulières: Remarques concernant la manipulation».
Instructions spéciales pour la posologie
Patients âgés (≥65 ans)
Aucun ajustement de la dose n'est nécessaire (voir «Pharmacocinétique»).
Patients atteints d'insuffisance rénale ou hépatique
TREMFYA n'a pas été étudié chez ces populations de patients. Aucune recommandation posologique ne peut être faite.
Enfants et adolescents (<18 ans)
La sécurité et l'efficacité de TREMFYA pour les enfants et les adolescents de moins de 18 ans n'ont pas été étudiées. Aucune donnée n'est disponible.
Contre-indicationsHypersensibilité sévère au principe actif ou à l'un des excipients selon la composition.
Infections actives cliniquement importantes (p.ex. tuberculose active).
Mises en garde et précautionsInfections
TREMFYA peut augmenter le risque d'infections. Dans les études cliniques, des infections sont survenues au cours d'un traitement de 16 semaines chez 23% des patients du groupe TREMFYA versus 21% des patients du groupe placebo. Le taux d'infections sévères était ≤0,2% dans le groupe TREMFYA et dans le groupe placebo. Chez les patients présentant une infection active cliniquement importante, le traitement par TREMFYA ne doit pas être instauré tant que l'infection n'est pas guérie ou convenablement traitée. Les patients séropositifs au VHC ou au VIH, les patients pour lesquels le test de dépistage de l'hépatite B est positif et les patients ayant des antécédents d'infections chroniques ou récidivantes ont été exclus des études cliniques.
Les patients traités par TREMFYA doivent être informés de la nécessité de consulter un médecin en cas de survenue de signes ou de symptômes d'une infection aiguë ou chronique cliniquement importante. Si un patient développe une infection sévère ou cliniquement importante, ou ne répond pas à un traitement standard, il devra être étroitement surveillé et le traitement par TREMFYA devra être interrompu jusqu'à guérison de l'infection.
Dépistage de la tuberculose avant le traitement
Dans les études cliniques, les patients présentant une tuberculose (TB) latente et recevant simultanément un traitement par TREMFYA et une prophylaxie anti-TB appropriée n'ont pas développé de TB. Avant d'instaurer le traitement par TREMFYA, les patients doivent être examinés pour dépister une infection TB. Le traitement d'une TB latente doit être instauré avant d'administrer TREMFYA. Les patients recevant TREMFYA doivent être surveillés pendant et après le traitement afin de dépister des signes et des symptômes d'une TB active. TREMFYA ne doit pas être utilisé chez les patients présentant une TB active. Chez les patients présentant des antécédents de TB latente ou active pour lesquels le suivi d'un traitement adapté ne peut être confirmé, un traitement anti-TB devra être envisagé avant l'instauration d'un traitement par TREMFYA.
Tumeurs malignes
Dans les études cliniques, le traitement par TREMFYA n'a pas été associé à un risque accru d'affections malignes.
Les patients atteints de psoriasis ayant reçu auparavant un traitement UV devront faire l'objet d'un examen minutieux avant et pendant le traitement par TREMFYA afin de dépister la présence de tumeurs cutanées.
Traitement concomitant par d'autres immunosuppresseurs systémiques ou une photothérapie
La sécurité et l'efficacité de TREMFYA en association avec des immunosuppresseurs, y compris des agents biologiques, ou la photothérapie n'ont pas été évaluées.
Hypersensibilité
De graves réactions d'hypersensibilité ont été rapportées après la commercialisation. Certains cas, dont des cas d'urticaire et de dyspnée, sont survenus plusieurs jours à plusieurs semaines après le traitement par le guselkumab. En cas de survenue d'une réaction d'hypersensibilité grave, l'administration de TREMFYA doit être immédiatement interrompue et un traitement approprié doit être instauré.
Immunisations
Avant d'instaurer le traitement par TREMFYA, la réalisation de toutes les vaccinations nécessaires doit être achevée conformément aux recommandations vaccinales en vigueur. Aucune donnée n'est disponible concernant la réponse aux vaccins inactivés. L'administration concomitante de TREMFYA et de vaccins vivants doit être évitée. Un délai suffisant entre des vaccinations avec des vaccins vivants et le début du traitement devra être respecté conformément aux recommandations vaccinales actuelles sur les principes actifs immunosuppresseurs.
Des informations pertinentes sur l'utilisation de principes actifs immunosuppresseurs avec des vaccins spécifiques sont également disponibles dans les informations professionnelles correspondantes.
InteractionsDans une étude de phase I menée chez des sujets atteints de psoriasis en plaques modéré à sévère, les modifications de l'exposition systémique (Cmax et AUCinf) au midazolam, à la S-warfarine, à l'oméprazole, au dextrométhorphane et à la caféine, observées après une dose unique de guselkumab, n'étaient pas cliniquement significatives. Ceci indique que des interactions médicamenteuses entre le guselkumab et les substrats de différentes enzymes du CYP (CYP3A4, CYP2C9, CYP2C19, CYP2D6 et CYP1A2) sont peu probables. Aucun ajustement posologique n'est nécessaire en cas d'administration concomitante de guselkumab et de substrats du CYP450.
Comme aucune donnée n'est disponible concernant la réponse aux vaccins sous traitement par TREMFYA, l'administration concomitante de TREMFYA et de vaccins vivants doit être évitée (voir «Mises en garde et précautions»).
Grossesse/AllaitementFemmes en âge de procréer
Les femmes en âge de procréer doivent utiliser des méthodes de contraception efficaces pendant le traitement et au moins 12 semaines après la fin de celui-ci.
Grossesse
Il n'existe pas de données concernant l'emploi du guselkumab chez la femme enceinte. Les expérimentations animales n'ont révélé aucune toxicité directe ou indirecte ayant une incidence sur la grossesse, le développement embryonnaire, le développement fœtal, l'accouchement ou le développement postnatal (voir «Données précliniques»). Par mesure de précaution, l'utilisation de TREMFYA doit être évitée pendant la grossesse.
Allaitement
On ignore si le guselkumab est excrété dans le lait maternel humain. Comme de nombreux médicaments et immunoglobulines sont excrétés dans le lait maternel et que le guselkumab est associé à un risque potentiel d'effets indésirables chez l'enfant allaité, la décision d'arrêter l'allaitement ou le traitement par TREMFYA doit être prise en tenant compte du bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement par TREMFYA pour la femme.
Fertilité
Les effets du guselkumab sur la fertilité humaine n'ont pas été évalués. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesRésumé du profil de sécurité
Le profil de sécurité de TREMFYA chez les patients atteints de psoriasis en plaques modéré à sévère repose sur les données d'une étude de phase II et de quatre études de phase III. Parmi les 1823 patients traités par TREMFYA, 1393 patients y ont été exposés pendant au moins 6 mois (24 semaines) et 728 patients pendant au moins 1 an (c.-à-d. traités jusqu'à la semaine 48). La plupart des patients (n=1658) ont reçu un schéma posologique de 100 mg de TREMFYA en injection sous-cutanée toutes les 8 semaines.
L'effet indésirable (EI) le plus fréquent (>1%) était l'infection des voies respiratoires supérieures.
Liste des effets indésirables
Les fréquences des effets indésirables mentionnés ont été déterminées d'après l'analyse des données regroupées de 823 patients atteints de psoriasis en plaques modéré à sévère ayant reçu TREMFYA pendant les périodes contrôlées contre placebo de deux études de phase III.
Les effets indésirables provenant d'études cliniques portant sur le psoriasis et de l'expérience post-commercialisation sont présentés par classes de systèmes d'organes MedDRA et par fréquence, selon la convention suivante: très fréquents (≥1/10), fréquents (≥1/100 et <1/10), occasionnels (≥1/1000 et <1/100), rares (≥1/10'000 et <1/1000), très rares (≥1/10'000) et fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Infections et infestations
Très fréquents: infection des voies respiratoires supérieures (13,9%).
Fréquents: gastro-entérite, infections à Herpes simplex, dermatophytoses.
Affections du système immunitaire
Occasionnels: hypersensibilité.
Affections du système nerveux
Fréquents: céphalées.
Affections gastro-intestinales
Fréquents: diarrhée.
Affections de la peau et du tissu sous-cutané
Fréquents: urticaire.
Occasionnels: éruption cutanée.
Affections musculo-squelettiques et du tissu conjonctif
Fréquents: arthralgie.
Troubles généraux et anomalies au site d'administration
Fréquents: érythème au niveau du site d'injection.
Occasionnels: douleurs au niveau du site d'injection.
Description d'effets indésirables sélectionnés
Gastroentérite
Pendant la période contrôlée contre placebo de deux études cliniques de phase III, l'incidence des gastro-entérites a été plus élevée dans le groupe traité par TREMFYA (1,1%) que dans le groupe placebo (0,7%). Ces effets indésirables à type de gastro-entérite étaient non graves et n'ont pas conduit à l'arrêt du traitement par TREMFYA jusqu'à la semaine 48.
Réactions au site d'injection
Lors de deux études cliniques de phase III, des réactions au site d'injection sont survenues lors de 0,7% des injections de TREMFYA et de 0,3% des injections de placebo jusqu'à la semaine 48. Les effets indésirables à type d'érythème et de douleurs au site d'injection étaient tous d'intensité légère à modérée, aucun n'était grave et aucun n'a conduit à l'arrêt du traitement par TREMFYA.
Immunogénicité
L'immunogénicité de TREMFYA a été évaluée à l'aide d'une méthode sensible de dosage immunologique, tolérante au médicament. D'après l'analyse des données regroupées des études de phase II et de phase III, moins de 6% des patients traités par TREMFYA ont développé des anticorps anti-médicament pendant une durée de traitement allant jusqu'à 52 semaines. Parmi les patients ayant développé des anticorps anti-médicament, environ 7% présentaient des anticorps classés comme neutralisants, soit 0,4% de l'ensemble des patients traités par TREMFYA.
Vu la faible fréquence de l'immunogénicité, aucune déclaration pertinente ne peut être faite concernant les éventuels effets des anticorps anti-médicament sur l'efficacité et la sécurité.
SurdosageAu cours des études cliniques, des doses uniques de guselkumab allant jusqu'à 987 mg (10 mg/kg) ont été administrées par voie intraveineuse à des volontaires sains, et des doses uniques de guselkumab allant jusqu'à 300 mg ont été administrées par voie sous-cutanée à des patients atteints de psoriasis en plaques, sans qu'une toxicité dose-limitante n'ait été observée. En cas de surdosage, le patient doit être surveillé afin de détecter tout signe ou symptôme d'effets indésirables et un traitement symptomatique approprié doit immédiatement être instauré.
Propriétés/EffetsCode ATC: L04AC16
Mécanisme d'action
Le guselkumab est un anticorps monoclonal (AcM) IgG1λ humain qui se lie de façon sélective à l'interleukine 23 (IL-23) avec une spécificité et une affinité élevées. L'IL-23 agit notamment sur la différenciation, l'expansion et la survie de certaines sous-populations de lymphocytes T (lymphocytes Th17 p. ex.) et de certaines sous-populations de cellules de l'immunité innée, ainsi que sur la libération des cytokines proinflammatoires IL-17A, IL-17F et IL-22. Chez l'homme, il a été montré que le blocage sélectif de l'IL-23 permet de normaliser la production de ces cytokines.
Les taux d'IL-23 sont élevés dans la peau des patients atteints de psoriasis en plaques. Dans les modèles in vitro, il a été montré que le guselkumab inhibe la bioactivité de l'IL-23 en bloquant son interaction avec le récepteur de surface cellulaire de l'IL-23, perturbant ainsi la signalisation, l'activation et la cascade cytokinique médiées par l'IL-23. Le guselkumab exerce ses effets cliniques sur le psoriasis en plaques en inhibant la voie cytokinique de l'IL-23 par modulation de profils d'expression de gènes dans les zones cutanées concernées. Ces effets locaux entraînent une réduction de l'épaisseur de l'épiderme et de la concentration des lymphocytes T. Par ailleurs, lors des études de phase II et de phase III, une réduction des taux sériques d'IL-17A, d'IL-17F et d'IL-22 a été observée chez les patients traités par le guselkumab comparés au groupe placebo.
Efficacité clinique
L'efficacité et la sécurité du guselkumab ont été étudiées au cours de quatre études de phase III randomisées, en double aveugle, contrôlées versus placebo et/ou comparateur actif, menées chez des patients adultes atteints de psoriasis en plaques modéré à sévère, candidats à une photothérapie ou à un traitement systémique. Deux études (VOYAGE 1 et VOYAGE 2) ont évalué l'efficacité et la sécurité du guselkumab versus placebo et adalimumab chez 1829 patients adultes. Dans l'étude VOYAGE 2, l'arrêt du guselkumab et la reprise du traitement ont été en outre étudiés à la semaine 28 chez les patients répondeurs, par rapport à la poursuite du traitement en continu. Les patients qui avaient déjà été traités par le guselkumab ou l'adalimumab, ainsi que les patients présentant un psoriasis érythrodermique, un psoriasis en gouttes ou un psoriasis pustuleux ont été exclus des études VOYAGE 1 et VOYAGE 2. Une autre étude (NAVIGATE) a évalué l'efficacité et la sécurité du guselkumab versus ustékinumab chez 268 patients adultes ayant présenté une réponse insuffisante à l'ustékinumab.
L'étude clinique (ORION) visait à évaluer l'efficacité, la sécurité, la PK, l'immunogénicité, les possibilités d'utilisation et l'acceptation du guselkumab administré avec le stylo prérempli.
VOYAGE 1 et VOYAGE 2
Les patients randomisés dans le groupe guselkumab ont reçu 100 mg aux semaines 0 et 4, puis toutes les 8 semaines jusqu'à la semaine 48 (VOYAGE 1) ou jusqu'à la semaine 20 (VOYAGE 2). Les patients randomisés dans le groupe adalimumab ont reçu 80 mg à la semaine 0 et 40 mg à la semaine 1, puis 40 mg toutes les deux semaines jusqu'à la semaine 48 (VOYAGE 1) ou jusqu'à la semaine 23 (VOYAGE 2). Dans les deux études, les patients randomisés dans le groupe placebo ont reçu 100 mg de guselkumab aux semaines 16 et 20, puis toutes les 8 semaines. Dans l'étude VOYAGE 2, les patients qui avaient été randomisés dans le groupe guselkumab à la semaine 0 et présentaient une amélioration du PASI (Psoriasis Area and Severity Index) d'au moins 90% (réponse PASI 90) à la semaine 28 ont été re-randomisés soit pour poursuivre le traitement par le guselkumab toutes les 8 semaines (traitement d'entretien), soit pour recevoir le placebo (arrêt du traitement). Les patients appartenant au dernier groupe (re-randomisation après placebo) ont à nouveau été traités par le guselkumab en cas de perte de 50% de l'amélioration de leur PASI à la semaine 28. Les patients du groupe guselkumab sans réponse PASI 90 ont poursuivi le traitement par le guselkumab. Chez les patients qui avaient été randomisés dans le groupe adalimumab et qui présentaient une réponse PASI 90 à la semaine 28, le traitement a été arrêté et un traitement par le guselkumab a été instauré en cas de perte de 50% de l'amélioration du PASI à la semaine 28. Les patients du groupe adalimumab qui n'ont pas obtenu de réponse PASI 90 ont reçu le guselkumab pour la première fois aux semaines 28 et 32, puis toutes les 8 semaines. Tous les patients ont été suivis pendant jusqu'à 48 semaines après la première administration du traitement de l'étude.
Dans les études VOYAGE 1 et 2, les caractéristiques de la maladie à l'inclusion étaient homogènes au sein des populations des études, avec respectivement une moyenne de surface corporelle atteinte (SCA) de 22% et 24%, une médiane de score PASI à l'inclusion de 19 dans les deux études, un score IGA à l'inclusion (Investigator's Global Assessment, évaluation globale par l'investigateur) respectivement jugé «modéré» ou «sévère» chez 74,6% et 75,5% ou 25,1% et 24,5% des patients. 19% et 18% des patients avaient des antécédents d'arthrite psoriasique.
Parmi l'ensemble des patients inclus dans VOYAGE 1 et 2, respectivement 32% et 29% étaient naïfs à la fois de traitement systémique conventionnel et de traitement biologique, 54% et 57% avaient déjà reçu précédemment une photothérapie et 62% et 64% avaient déjà reçu un traitement systémique conventionnel. Dans les deux études, 21% des patients avaient déjà reçu précédemment un traitement biologique, parmi lesquels 11% avaient reçu au moins un anti-TNFα (facteur de nécrose tumorale alpha) et environ 10% un anti-IL-12/IL-23.
Un traitement topique ou systémique associé ou une photothérapie concomitante contre le psoriasis n'étaient pas autorisés dans l'étude.
L'efficacité du guselkumab a été évaluée sur la base de l'atteinte cutanée globale, de l'atteinte localisée du cuir chevelu, des mains, des pieds et des ongles, ainsi que de la qualité de vie. Les co-critères principaux d'évaluation dans les études VOYAGE 1 et 2 étaient le pourcentage de patients ayant obtenu un score IGA de type «blanchi» ou «lésion minime» (IGA 0/1) et une réponse PASI 90 à la semaine 16 versus placebo (voir le tableau 1).
Effets sur les symptômes cutanés
Le traitement par le guselkumab a entraîné des améliorations significatives des paramètres de l'activité de la maladie par rapport au placebo à la semaine 16 et par rapport à l'adalimumab aux semaines 16 et 48. Les principaux résultats d'efficacité sont présentés dans le tableau 1 ci-dessous.
Tableau 1: résumé des réponses cliniques observées lors des études VOYAGE 1 et VOYAGE 2
|
|
Nombre de patients (%)
| |
Placebo (n = 174)
|
VOYAGE 1 Guselkumab (n = 329)
|
Adalimumab (n = 334)
|
Placebo (n = 248)
|
VOYAGE 2 Guselkumab (n = 496)
|
Adalimumab (n = 248)
| |
Semaine 16
| |
PASI 75
|
10 (5,7)
|
300 (91,2)a
|
244 (73,1)b
|
20 (8,1)
|
428 (86,3)a
|
170 (68,5)b
| |
PASI 90
|
5 (2,9)
|
241 (73,3)c
|
166 (49,7)b
|
6 (2,4)
|
347 (70,0)c
|
116 (46,8)b
| |
IGA 0/1
|
12 (6,9)
|
280 (85,1)c
|
220 (65,9)b
|
21 (8,5)
|
417 (84,1)c
|
168 (67,7)b
| |
IGA 0
|
2 (1,1)
|
157 (47,7)a
|
88 (26,3)d
|
2 (0,8)
|
215 (43,3)a
|
71 (28,6)d
| |
Semaine 48
| |
PASI 75
|
-
|
289 (87,8)
|
209 (62,6)e
|
-
|
-
|
-
| |
PASI 90
|
-
|
251 (76,3)
|
160 (47,9)b
|
-
|
-
|
-
| |
IGA 0/1
|
-
|
265 (80,5)
|
185 (55,4)b
|
-
|
-
|
-
| |
IGA 0
|
-
|
166 (50,5)
|
86 (25,7)b
|
-
|
-
|
-
|
a p <0,001 pour la comparaison entre le guselkumab et le placebo.
b p <0,001 pour la comparaison entre le guselkumab et l'adalimumab sur les critères secondaires majeurs d'évaluation.
c p <0,001 pour la comparaison entre le guselkumab et le placebo sur les co-critères principaux d'évaluation.
d Des comparaisons entre le guselkumab et l'adalimumab n'ont pas été effectuées.
e p <0,001 pour la comparaison entre le guselkumab et l'adalimumab.
Des améliorations statistiquement significatives du psoriasis des ongles, de l'atteinte du cuir chevelu et de l'atteinte palmoplantaire ont également été observées à la semaine 16 par rapport au placebo.
Réponse au cours du temps
Le guselkumab a montré une efficacité d'apparition rapide, avec un pourcentage d'amélioration du score PASI significativement plus élevé comparé au placebo dès la semaine 2 (p <0,001), avec une différence maximale atteinte autour de la semaine 20 (VOYAGE 1 et 2) et se maintenant jusqu'à la semaine 48 (VOYAGE 1).
Figure 1: pourcentage de patients ayant obtenu une réponse PASI 90 lors des différentes visites jusqu'à la semaine 48 (patients randomisés à la semaine 0) dans l'étude VOYAGE 1
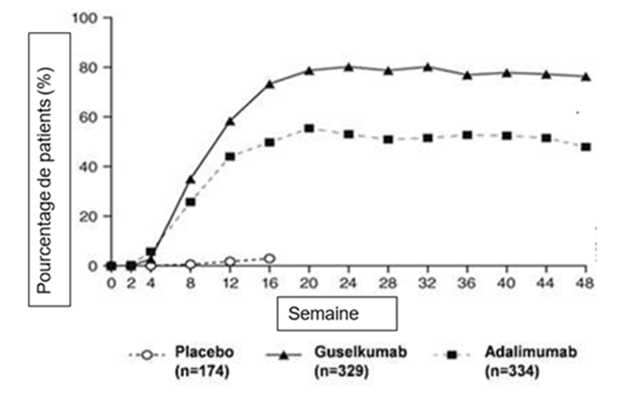
L'efficacité et la sécurité du guselkumab ont été démontrées indépendamment de l'âge, du sexe, de l'appartenance ethnique, du poids corporel, de la localisation des plaques, du score de sévérité PASI à l'inclusion, de la présence concomitante d'une arthrite psoriasique et de la prise d'un traitement antérieur biologique. Le guselkumab s'est avéré efficace chez les patients naïfs de traitement systémique conventionnel, chez les patients naïfs de traitement biologique et chez les patients précédemment exposés à un traitement biologique.
Arrêt, puis reprise du traitement
Dans l'étude VOYAGE 2, à la semaine 48, 88,6% des patients ayant reçu un traitement d'entretien par le guselkumab présentaient une réponse PASI 90 versus 36,8% des patients ayant arrêté le traitement à la semaine 28 (p <0,001). Une perte de la réponse PASI 90 a été observée dès 4 semaines après l'arrêt du traitement par le guselkumab avec un délai médian de perte de la réponse PASI 90 d'environ 15 semaines. 84,1% des patients ayant arrêté le traitement par le guselkumab et l'ayant repris ultérieurement ont obtenu une réponse PASI 90 dans les 16 à 20 semaines après la reprise du traitement par le guselkumab.
Résultats du traitement concernant différentes régions atteintes
Dans les études VOYAGE 1 et 2, à la semaine 16, des améliorations significativement plus importantes de l'atteinte du cuir chevelu (ss-IGA), des mains et des pieds (hf-PGA) et des ongles (NAPSI, f-PGA) ont été observées chez les patients traités par le guselkumab par rapport aux patients du groupe placebo.
Qualité de vie liée à la santé/Résultats rapportés par les patients
Dans les études VOYAGE 1 et 2, à la semaine 16, des améliorations significativement plus importantes de la qualité de vie liée à la santé, mesurée à l'aide du Dermatology Life Quality Index (DLQI), et des symptômes (démangeaisons, douleurs, brûlures, picotements et tiraillements cutanés) et signes (sécheresse cutanée, fissures, desquamation, exfoliation, rougeurs et saignements) du psoriasis, rapportés par les patients dans le carnet de suivi Psoriasis Symptoms and Signs Diary (PSSD) ont été observées chez les patients traités par le guselkumab par rapport aux patients ayant reçu le placebo.
NAVIGATE
L'étude NAVIGATE a évalué l'efficacité du guselkumab chez des patients ayant présenté une réponse insuffisante à l'ustékinumab à la semaine 16 (c.-à-d. n'ayant pas de réponse de type «blanchi» ou «lésion minime», définie par un score IGA ≥2). Les patients ne devaient pas avoir reçu un traitement antérieur par le guselkumab et/ou l'ustékinumab. Tous les patients ont reçu un traitement par l'ustékinumab en ouvert aux semaines 0 et 4. À la semaine 16, 268 patients présentant un score IGA ≥2 ont été randomisés pour poursuivre le traitement par l'ustékinumab toutes les 12 semaines ou pour débuter un traitement par le guselkumab aux semaines 16 et 20, puis toutes les 8 semaines. Les caractéristiques à l'inclusion des patients randomisés étaient similaires à celles des patients des études VOYAGE 1 et 2.
12 semaines après la randomisation, la proportion de patients ayant obtenu un score IGA 0/1 et une amélioration ≥2 points a été plus élevée dans le groupe guselkumab que dans le groupe ustékinumab (31,1% vs 14,3%; p = 0,001), de même que la proportion de patients ayant obtenu une réponse PASI 90 (48% vs 23%; p <0,001). Aucune donnée n'est disponible sur le passage inverse du guselkumab à l'ustékinumab.
ORION
L'étude ORION visait à évaluer l'efficacité, la sécurité, la PK, l'immunogénicité, les possibilités d'utilisation et l'acceptation du guselkumab administré avec le stylo prérempli. Dans cette étude, 78 patients atteints de psoriasis en plaques modéré à sévère ont été randomisés pour recevoir soit TREMFYA (100 mg aux semaines 0 et 4, puis toutes les 8 semaines) soit un placebo. La population de l'étude ORION était comparable à celle des études VOYAGE 1 et 2. L'efficacité, mesurée par le score IGA (0,1) et le PASI 90 à la semaine 16, était comparable dans les trois études ORION, VOYAGE 1 et 2. L'acceptation par les patients et la sécurité d'emploi du stylo ont été établies.
PharmacocinétiqueAbsorption
Après administration d'une dose unique de 100 mg par voie sous-cutanée chez des sujets sains, le guselkumab a atteint une concentration sérique maximale (Cmax) moyenne (± ET) de 8,09 ± 3,68 μg/ml environ 5,5 jours après l'injection.
Après administration de 100 mg de guselkumab par voie sous-cutanée aux semaines 0 et 4, puis toutes les 8 semaines, les concentrations sériques de guselkumab ont atteint l'état d'équilibre autour de la semaine 20. Dans deux études de phase III, les concentrations sériques résiduelles moyennes (± ET) du guselkumab à l'état d'équilibre ont été de 1,15 ± 0,73 μg/ml et de 1,23 ± 0,84 μg/ml.
La biodisponibilité absolue du guselkumab après injection sous-cutanée d'une dose unique de 100 mg dans la cuisse a été estimée à environ 49% chez les sujets sains.
Distribution
Le volume de distribution moyen pendant la phase terminale (Vz) après administration unique par voie intraveineuse chez le sujet sain était dans toutes les études compris entre 7 et 10 l environ.
Métabolisme
La voie exacte de métabolisation du guselkumab n'a pas encore été caractérisée. Le guselkumab étant un AcM IgG humain, il devrait être dégradé en petits peptides et en acides aminés par les voies cataboliques, de la même manière que les IgG endogènes.
Élimination
Dans toutes les études, la clairance systémique (Cl) moyenne après administration unique par voie intraveineuse chez les sujets sains est comprise entre 0,288 et 0,479 l/jour. La demi-vie (t½) moyenne du guselkumab était d'environ 17 jours chez les sujets sains et d'environ 15 à 18 jours chez les patients atteints de psoriasis en plaques dans les études.
Linéarité/Non linéarité
Après injection sous-cutanée unique de doses comprises entre 10 mg et 300 mg chez des sujets sains ou chez des patients atteints de psoriasis en plaques, l'exposition systémique au guselkumab (Cmax et AUC) a augmenté approximativement de façon dose-proportionnelle.
Cinétique pour certains groupes de patients
Patients âgés
Aucune étude spécifique n'a été réalisée chez des patients âgés. Sur les 1384 patients atteints de psoriasis en plaques exposés au guselkumab et inclus dans l'analyse pharmacocinétique de population, 70 patients étaient âgés de 65 ans ou plus, dont 4 patients âgés de 75 ans ou plus. Les analyses pharmacocinétiques de population n'ont révélé aucune modification apparente de la Cl/F estimée chez les patients âgés de 65 ans ou plus par rapport aux patients âgés de moins de 65 ans. Ceci suggère qu'aucun ajustement posologique n'est nécessaire chez les patients âgés.
Patients atteints d'insuffisance rénale ou hépatique
Aucune étude spécifique n'a été réalisée pour déterminer les effets de l'insuffisance rénale ou hépatique sur la pharmacocinétique du guselkumab. On s'attend à ce que l'élimination rénale du guselkumab intact, un AcM IgG, soit faible et d'importance mineure; de même, l'insuffisance hépatique ne devrait pas influer sur la clairance du guselkumab, car les AcM IgG sont principalement éliminés par catabolisme intracellulaire.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicité après administration répétée, et de toxicité de la reproduction n'ont pas révélé de risque particulier pour l'homme.
Lors des études de toxicité après administration répétée chez des macaques crabiers, le guselkumab administré par voie intraveineuse et sous-cutanée a été bien toléré à des doses allant jusqu'à 50 mg/kg/semaine pendant jusqu'à 5 semaines par voie i.v. et 24 semaines par voie s.c. Par ailleurs, aucun effet indésirable pharmacologique à type d'immunotoxicité ou de toxicité cardiovasculaire n'a été observé au cours des études de toxicité après administration répétée ou lors d'une étude spécifique de pharmacologie de sécurité cardiovasculaire chez des macaques crabiers.
Dans une étude sur l'évaluation combinée de la toxicité pendant le développement embryofœtal et le développement prénatal et postnatal, des macaques crabiers gravides (respectivement 19, 20 et 20 animaux dans les groupes ayant reçu 0, 10 ou 50 mg/kg) ont reçu des doses sous-cutanées hebdomadaires de guselkumab du début de l'organogenèse jusqu'à la mise bas. Chez 1 des 16 guenons du groupe contrôle et chez 3 des 14 guenons de tous les groupes ayant reçu le guselkumab, des morts néonatales sont survenues chez les jeunes (les taux de Cmax et d'AUClast étaient 31 et 8 fois supérieurs aux concentrations chez l'homme). Ces morts néonatales ont été attribuées à un manque de soins de la part de la mère, à un traumatisme ou à une naissance prématurée ou tardive, même si un effet dû au principe actif n'était pas exclu. À tous les paliers de doses, des pertes fœtales (avortements spontanés, y compris mort-nés) ont en outre été observées, toutes situées dans la fourchette du contrôle historique du laboratoire, mais pour lesquelles un effet dû au principe actif n'était pas exclu. L'importance clinique de ces observations n'est pas connue. Aucun effet du guselkumab sur le développement fonctionnel ou immunologique des jeunes animaux n'a été constaté entre la naissance et l'âge de 6 mois.
Aucune modification prénéoplasique n'a été observée à l'examen histopathologique d'animaux traités pendant jusqu'à 24 semaines, ou après la phase de rétablissement de 12 semaines pendant laquelle le médicament était décelable dans le sérum. Les niveaux de dose dans les études chez l'animal étaient environ 45 fois supérieurs à la dose de 100 mg prévue pour l'administration à des patients atteints de psoriasis en plaques (sur la base d'un patient d'un poids corporel de 90 kg) et ont entraîné chez les singes des concentrations sériques maximales plus de 100 fois supérieures à celles observées chez l'homme.
Aucune étude de mutagénicité ou de carcinogénicité n'a été réalisée avec le guselkumab.
Remarques particulièresIncompatibilités
Aucune étude de compatibilité n'ayant été effectuée, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques concernant le stockage
Conserver hors de la portée des enfants.
Conserver au réfrigérateur (2-8 °C). Ne pas congeler.
Conserver la seringue préremplie et le stylo prérempli dans son carton pour les protéger de la lumière.
Remarques concernant la manipulation
Après avoir sorti Tremfya du réfrigérateur, laisser la seringue préremplie ou le stylo prérempli dans le carton et attendre 30 minutes avant d'injecter TREMFYA, afin que la solution puisse atteindre la température ambiante. Ne pas secouer la seringue préremplie ou le stylo prérempli.
Un contrôle visuel de la seringue préremplie ou du stylo prérempli est recommandé avant l'utilisation. La solution doit être limpide, incolore à jaune clair et peut contenir quelques petites particules protéiques blanches ou translucides. TREMFYA ne doit pas être utilisé si la solution est trouble ou présente un changement de coloration, ou si elle contient de grosses particules.
Chaque emballage de TREMFYA contient une brochure séparée intitulée «Instructions d'utilisation», décrivant en détail la préparation et l'utilisation de la seringue préremplie ou du stylo prérempli.
TREMFYA ne contenant pas de conservateur, tout médicament non utilisé restant dans la seringue ou dans le stylo doit être jeté.
Le médicament non utilisé et/ou les déchets doivent être éliminés conformément aux exigences nationales.
TREMFYA est proposé sous forme de solution stérile à usage unique dans
·une seringue préremplie en verre de 1 ml avec une aiguille fixe et un protège aiguille sans latex, intégrée dans un dispositif de protection passive de l'aiguille.
·une seringue préremplie en verre de 1 ml, intégrée dans un stylo prérempli avec un protège-aiguille.
Numéro d’autorisation66583, 67490 (Swissmedic).
PrésentationTREMFYA, solution injectable en seringue préremplie:
boîte contenant 1 seringue préremplie de 100 mg/ml [B]
TREMFYA solution injectable en stylo prérempli:
boîte contenant 1 stylo prérempli de 100 mg/ml [B]
Titulaire de l’autorisationJanssen-Cilag AG, Zoug, ZG.
Mise à jour de l’informationSeptembre 2019.
|