Propriétés/EffetsCode ATC
L01XL07
Mécanisme d'action
Abecma est une thérapie par cellules T positives pour le récepteur d'antigène chimérique (thérapie par cellules CAR-T positives) qui cible l'antigène de maturation des cellules B exprimé à la surface des plasmocytes normaux et malins. La construction CAR contient un fragment scFv anti-BCMA pour la spécificité antigénique, un domaine transmembranaire, un domaine d'activation des lymphocytes T CD3 zeta et un domaine de costimulation 4-1BB. L'activation spécifique de l'antigène d'Abecma entraîne la prolifération de lymphocytes CAR-T positifs, la sécrétion de cytokines et la destruction cytolytique ultérieure des cellules exprimant BCMA.
Pharmacodynamique
Sans objet.
Efficacité clinique
KarMMa-3
KarMMa-3 est une étude clinique multicentrique, randomisée, contrôlée, ouverte, visant à évaluer l'efficacité et la sécurité d'Abecma par rapport aux schémas thérapeutiques standards chez des patients adultes atteints d'un myélome multiple récidivant et réfractaire, ayant reçu deux à quatre lignes de traitement antérieures contre le myélome, dont un immunomodulateur, un inhibiteur du protéasome et le daratumumab. Les patients étaient réfractaires à leur dernier traitement contre le myélome. Avant la randomisation, chaque patient a été assigné à un traitement standard en fonction de son dernier traitement contre le myélome. Les thérapies standards étaient composées de: daratumumab, pomalidomide, dexaméthasone (DPd); daratumumab, bortézomib, dexaméthasone (DVd); ixazomib, lénalidomide, dexaméthasone (IRd); carfilzomib, dexaméthasone (Kd) ou élotuzumab, pomalidomide, dexaméthasone (EPd). Lorsque cliniquement indiqué, les patients randomisés sur le bras Abecma ont utilisé la thérapie standard assignée comme thérapie de transition.
L'étude incluait des patients ayant répondu à au moins 1 traitement antérieur (faible réponse ou mieux) et présentant un statut de performance ECOG (Eastern Cooperative Oncology Group) de 0 ou 1. Les patients présentant une clairance de la créatinine sérique < 45 ml/min, un taux d'aspartate aminotransférase (AST) ou d'alanine aminotransférase (ALT) sériques correspondant à > 2,5 fois la valeur normale supérieure et une fraction d'éjection du ventricule gauche (FEVG) < 45 % ont été exclus de l'étude. Les patients avec un nombre absolu de neutrophiles < 1000/µl et une numération plaquettaire < 75000/μl, chez lesquels < 50 % des cellules nucléées de la moelle osseuse étaient des plasmocytes, ainsi que les patients avec une numération plaquettaire < 50000/μl, chez lesquels ≥50 % des cellules nucléées de la moelle osseuse étaient des plasmocytes, étaient également exclus.
L'âge moyen de la population de l'étude était de 63 ans (plage entre 30 et 83 ans); 40,9 % avaient 65 ans ou plus et 60,9 % étaient des hommes. Le statut de performance ECOG au début de l'étude était de 0 chez 48,2 %, de 1 chez 50,5 % et de 2 chez 0,8 % des patients.
Nonante pour cent des patients étaient réfractaires à un médicament immunomodulateur (IMiD), 74 % étaient réfractaires à un inhibiteur du protéasome (IP) et 95 % étaient réfractaires à un anticorps monoclonal anti-CD38. Soixante-six pour cent étaient triplement réfractaires aux classes (réfractaires à un IP, un IMiD et un anticorps monoclonal anti-CD38).
Les patients ont été randomisés avec un rapport de 2:1 sur un traitement par Abecma (n = 254) ou le traitement standard (n = 132) pour le myélome multiple récidivant et réfractaire. La randomisation était stratifiée selon l'âge, le nombre de traitements antérieurs du myélome et les anomalies cytogénétiques de haut risque. Les patients recevant le traitement standard pouvaient être traités par Abecma en cas de progression confirmée de la maladie.
Les patients randomisés sur Abecma devaient recevoir une chimiothérapie lymphodéplétive composée de cyclophosphamide (300 mg/m2 comme perfusion i. v. une fois par jour pendant 3 jours) et de fludarabine (30 mg/m2 comme perfusion i. v. une fois par jour pendant 3 jours), qui devait être instaurée 5 jours avant la date cible de la perfusion d'Abecma. Entre l'aphérèse et jusqu'à 14 jours avant le début de la chimiothérapie lymphodéplétive, un cycle de traitement anticancéreux par DPd, DVd, IRd, Kd ou EPd (thérapie de transition) était autorisé pour contrôler la maladie.
Parmi les 254 patients randomisés sur Abecma, 249 (98 %) ont reçu une leucaphérèse et 225 (88,6 %) patients ont reçu Abecma. Parmi les 225 patients, 192 (85,3 %) ont reçu une thérapie de transition. Abecma n'a pas été administré chez 29 patients en raison du décès (n = 4), d'événements indésirables (n = 5), de retrait (n = 2), de décision du médecin (n = 7), de la non-conformité aux critères de la chimiothérapie lymphodéplétive (n = 8) ou d'un défaut de fabrication (n = 3).
L'intervalle de doses autorisé était de 150 à 540 x 106 cellules CAR-T positives. La dose médiane effectivement administrée était de 445,3 x 106 cellules CAR-T positives (fourchette: 174,9 à 529,0 x 106 cellules CAR-T positives). Le délai médian entre la leucaphérèse et la disponibilité de la préparation était de 35 jours (fourchette: 24 à 102 jours) et le délai médian entre la leucaphérèse et la perfusion était de 49 jours (fourchette: 34 à 117 jours).
Parmi les 132 patients randomisés sur le traitement standard, 126 (95,5 %) ont reçu un traitement. Six patients ont arrêté l'étude à cause de la progression de la maladie (n = 1), d'un retrait (n = 3) ou d'une décision du médecin (n = 2) sans avoir reçu de traitement. Sur demande du médecin investigateur, les patients recevant un traitement standard ont pu recevoir Abecma lorsque le comité de contrôle indépendant (IRC) avait confirmé une progression de la maladie sur la base des critères de l'International Myeloma Working Group (IMWG) et l'aptitude à la participation à l'étude. Parmi les patients admissibles, 69 (54,8 %) ont reçu une leucaphérèse et 60 (47,6 %) ont reçu Abecma.
Le critère principal pour l'évaluation de l'efficacité était la survie sans progression (PFS), définie selon les critères IMWG Uniform Response Criteria for Multiple Myeloma et évaluée par l'IRC. Les autres critères d'efficacité étaient le taux de réponse global (ORR), la survie globale (OS) et le patient reported outcome (PRO). Dans la population cible (Intent-to-Treat, ITT), la durée de suivi médiane de la randomisation au jour de référence de la collecte des données était de 18,6 mois. Le tableau 3 présente un résumé de l'analyse intermédiaire des résultats d'efficacité.
Dans le bras Abecma, la durée médiane de réponse (DOR) chez les patients avec réponse partielle (partial response, PR) ou mieux était de 13,9 mois (IC à 95 %: 11,2; 17,8). Chez les patients ayant atteint une réponse complète (complete response, CR) ou mieux, la durée médiane de réponse (duration of response, DOR) était de 20 mois (IC à 95 %: 15,8; 24,3).
Tableau 3: Résumé des résultats d'efficacité sur la base de KarMMa-3 (Intent-to-Treat-Population)
|
|
Bras Abecma
(n = 254)
|
Bras de traitement standard (n = 132)
| |
Survie sans progression (PFS)
| |
Nombre d'événements, n (%)
|
149 (58,7)
|
93 (70,5)
| |
Médiane, mois [IC à 95 %] a
|
13,3 [11,8; 16,1]
|
4,4 [3,4; 5,9]
| |
Hazard ratio [IC à 95 %]b
|
0,49 [0,38; 0,65]
| |
Valeur de p unilatéralec
|
< 0,0001
| |
Taux de réponse global (ORR)
| |
n (%)
|
181 (71,3)
|
55 (41,7)
| |
IC à 95 (%)d
|
(65,7; 76,8)
|
(33,3; 50,1)
| |
Valeur de p unilatéralee
|
< 0,0001
| |
CR ou mieux (sCR+CR)
|
98 (38,5)
|
7 (5,3)
| |
sCR
|
90 (35,4)
|
6 (4,5)
| |
CR
|
8 (3,1)
|
1 (0,8)
| |
VGPR
|
55 (21,7)
|
13 (9,8)
| |
PR
|
28 (11,0)
|
35 (26,5)
| |
MRD négative par NGS et ≥ CR
| |
Taux de négativité de la MRD, n (%)f
|
51 (20,1)
|
1 (0,8)
| |
IC à 95 (%)d
|
(15,2; 25,0)
|
(0,0; 2,2)
|
IC = intervalle de confiance; CR = réponse complète; MRD = minimal residual disease; PR = réponse partielle; sCR = réponse complète stricte; VGPR = très bonne réponse partielle.
a Estimation de Kaplan-Meier.
b Basé sur le Cox proportional hazards model stratifié univarié.
c Valeur de p unilatérale basée sur le test du log-rank stratifié.
d Intervalle de confiance bilatéral de Forest.
e Valeur de p unilatérale basée sur le test de Cochran-Mantel-Haenszel (CMH) stratifié.
f La négativité de la MRD était définie comme le pourcentage de patients de la population en ITT atteignant une CR ou CR stricte et étant MRD négatifs à un moment quelconque dans les trois mois avant l'atteinte de la CR ou de la CR stricte jusqu'à la progression ou le décès. Basé sur une valeur seuil de 10-5 en utilisant ClonoSEQ, un test de séquençage nouvelle génération (NGS).
Au moment de l'analyse PFS finale (date de référence de la collecte des données: 28.04.2023) avec une durée médiane de suivi de 30,9 mois, la PFS médiane pour Abecma était de 13,8 mois (IC à 95 %: 11,8; 16,1) comparé à 4,4 mois pour le traitement standard (IC à 95 %: 3,4; 5,8); HR = 0,49 (IC à 95 %: 0,38; 0,63), ce qui concorde avec les résultats de l'analyse intermédiaire.
74 % des événements OS planifiés étaient atteints lors de l'analyse finale de la PFS. Les patients ayant reçu le traitement standard pouvaient recevoir Abecma en cas de progression confirmée de la maladie. Les données OS sont donc influencées par les 74 (56,1 %) patients du bras de traitement standard ayant reçu Abecma comme traitement de suivi. L'OS médiane pour Abecma était de 41,4 mois (IC à 95 %: 30,9; NA) contre 37,9 mois pour le traitement standard (IC à 95 %: 23,4; NA); HR = 1,01 (IC à 95 %: 0,73; 1,40).
Patient reported outcome (PRO)
Analyses descriptives
Trois mesures des PRO (EORTC QLQ-C30, EORTC QLQ-MY20, EQ-5D-5L) ont été réalisées au début de l'étude (sélection), tous les mois jusqu'au mois 24 et tous les trois mois ensuite. Parmi les patients ayant répondu au questionnaire (Abecma n = 211; traitement standard n = 108), des tendances à l'amélioration et des différences au niveau des variations de la valeur moyenne par rapport à la valeur de référence, incluant la fatigue, la douleur, la capacité fonctionnelle physique et le statut GHS/QoL, ont été observées chez les patients traités par Abecma comparé aux patients recevant le traitement standard (voir figure 1).
Figure 1: EORTC QLQ-C30 - variations de la valeur moyenne aux mois 3, 6, 9, 12 et 15 par rapport à la valeur de référence pour les domaines fatigue, douleur, capacité fonctionnelle physique et GHS/QoL dans l'étude KarMMa-3
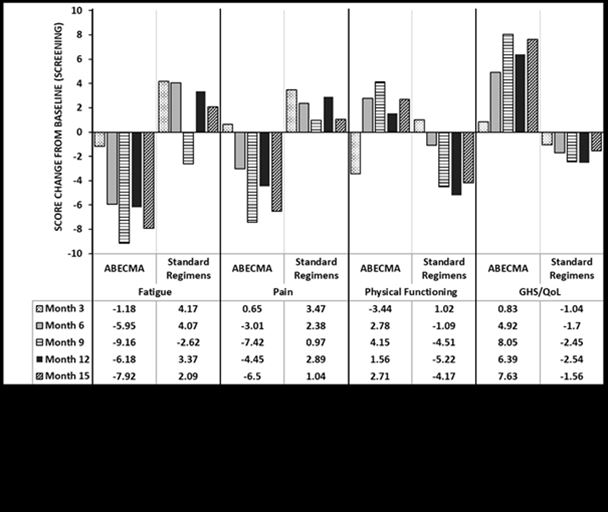
Analyse longitudinale de données avec contraintes (Constrained longitudinal data analysis (cLDA))
En comparant les variations moyennes des moindres carrés (Least Square - LS) entre la valeur de référence et le 25e mois à l'aide de la cLDA, les valeurs moyennes de variation des LS étaient en faveur des patients traités par Abecma pour la plupart des domaines des trois mesures PRO avec des tailles d'effet significatives (Hedge's g > 0,2) (voir figure 2).
Figure 2: Forest plot des différences entre les groupes pour l'ensemble de la cLDA concernant la variation moyenne des LS par rapport à la valeur de référence selon les groupes de traitement a, b, c dans l'étude KarMMa-3 (Abecma n = 211; traitement standard n = 108)
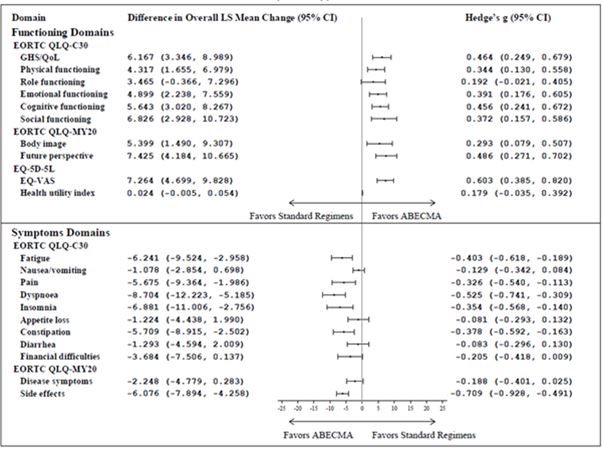
IC = intervalle de confiance; EORTC QLQ-C30 = European Organization for Research and Treatment of Cancer Quality of Life-of-Core 30 Questionnaire; EORTC QLQ-MY20 = European Organization for Research and Treatment of Cancer Quality of Life-of-Questionnaire Multiple Myeloma Module; EQ-VAS = Visual Analogue Scale; GHS = Global health status; LS = Least square; QoL = Quality of Life.
a Selon les directives de Cohen (1998, 1992) pour l'interprétation des valeurs g de Hedges, 0,20 indique de petits effets, 0,50 des effets moyens et 0,80 de grands effets.
b Domaines d'intérêt principaux: EORTC QLQ-C30 domaines de l'état de santé global/qualité de vie (QoL), fonction physique, fonction cognitive, fatigue et douleur; EORTC QLQ-MY20 domaines des symptômes de la maladie et effets indésirables du traitement; EQ-5D-5L indice de l'état de santé et EQ-VAS. Les autres domaines sont considérés comme secondaires.
c L'analyse ne comprenait pas d'ajustement en fonction des multiplicateurs.
Analyses PRO du temps écoulé avant l'événement (PRO-Time-to-Event)
Dans l'étude KarMMa-3, le temps écoulé avant l'événement (Time-to-Event) a été évalué à l'aide de l'estimation de Kaplan-Meier. L'analyse a montré le temps écoulé jusqu'à la confirmation d'une amélioration cliniquement significative ou une dégradation des domaines d'intérêt PRO chez les patients traités par Abecma versus ceux recevant le traitement standard. La survenue d'une dégradation ou d'une amélioration était définie comme une variation par rapport à la valeur de référence sur la base des valeurs seuils validées et confirmée par une évaluation consécutive ≥84 jours après l'apparition. Elle était indiquée sous forme de Hazard Ratio (HR); un HR<1,0 pour la dégradation et un HR>1,0 pour l'amélioration étaient en faveur d'Abecma. Chez les patients traités par Abecma, le temps écoulé jusqu'à une dégradation confirmée était significativement prolongé pour la plupart des domaines des trois PRO (HR>1,0). Chez les patients traités par Abecma, le temps écoulé jusqu'à une amélioration confirmée était également plus court pour la plupart des domaines des mesures PRO (HR>1,0).
KarMMa
KarMMa est une étude multicentrique ouverte, à un bras, visant à évaluer l'efficacité et la sécurité d'Abecma chez des patients adultes atteints d'un myélome multiple récidivant et réfractaire, qui ont reçu au moins trois traitements antérieurs contre le myélome, dont un immunomodulateur, un inhibiteur de protéasome et un anticorps anti-CD38.
L'étude comprenait un prétraitement (dépistage, leucaphérèse et thérapie de transition [si nécessaire]), un traitement (chimiothérapie lymphodéplétive [CLD] et perfusion d'Abecma) et un suivi (continu) pendant au moins 24 mois après la perfusion d'Abecma ou jusqu'à la progression documentée de la maladie, selon la période la plus longue. La période de CLD consistait en un cycle de 3 jours de cyclophosphamide (300 mg/m2 en perfusion i.v. quotidienne pendant 3 jours) et de fludarabine (30 mg/m2 en perfusion i.v. quotidienne pendant 3 jours) commençant 5 jours avant la date de perfusion prévue d'Abecma.
Les patients devaient rester à l'hôpital pendant 14 jours après la perfusion d'Abecma; la survenue potentielle d'un CRS et d'une neurotoxicité a été surveillée et les patients ont été traités en conséquence le cas échéant.
La population traitée par Abecma a été fortement réfractaire aux traitements précédents contre le myélome: 84,4 % des participants à l'étude étaient triplement réfractaires (c'est-à-dire réfractaires à un agent immunomodulateur, à un inhibiteur de protéase et à un anticorps anti-CD38).
Les doses cibles dans l'étude clinique étaient de 150, 300 ou 450 x 106 cellules CAR-T positives par perfusion. La plage posologique autorisée était de 150 à 540 x 106 cellules CAR-T positives. Le tableau 4 ci-dessous présente les doses cibles utilisées dans l'essai clinique, basées sur le nombre total de cellules CAR-T positives, et la plage correspondante de doses réellement administrées, définies comme cellules T viables CAR-positives.
Tableau 4: Dose totale de cellules CAR-T positives avec la plage posologique correspondante de cellules T viables CAR positives (x106) – étude KarMMa
|
Dose cible basée sur le nombre total de cellules CAR-T positives, incluant les cellules viables et non viables (x106)
|
Cellules T viables CAR-positives (x106)
(min, max)
| |
150
|
133 à 181
| |
300
|
254 à 299
| |
450
|
307 à 485
|
Sur 140 patients ayant subi une leucaphérèse, 128 ont reçu de l'Abecma. Un des 140 patients n'a pas reçu le produit en raison de défauts de fabrication. 11 autres patients n'ont pas été traités par Abecma sur décision du médecin (n = 3), en raison de l'abandon du patient (n = 4), d'événements indésirables (n = 1), de la progression de la maladie (n = 1) ou du décès (n = 2) avant de recevoir Abecma.
L'âge médian de la population étudiée était de 60,5 ans (plage: 33 à 78 ans); 35 % avaient au moins 65 ans et 59 % étaient des hommes. Le statut de performance de l'Eastern Cooperative Oncology Group (ECOG) au départ était de 0 chez 45 % des patients, de 1 chez 53 % des patients et de 2 chez 2 % des patients.
La plupart des patients (87,5 %) traités par Abecma ont reçu un traitement de transition pour contrôler leur myélome multiple pendant le processus de fabrication. Le délai médian entre la leucaphérèse et la disponibilité du produit a été de 32 jours (plage: entre 24 et 55 jours) et le temps médian entre la leucaphérèse et la perfusion de 40 jours (plage: entre 33 et 79 jours). La dose médiane réelle reçue dans tous les niveaux de dose cible a été de 315,3 x 106 cellules CAR-positives (plage: 150,5 à 518,4).
L'efficacité a été déterminée sur la base du taux de réponse global (overall response rate, ORR), du taux de réponse complète (complete response, CR) et de la durée de la réponse (duration of response, DOR) par un comité de contrôle indépendant (Independent Review Committee, IRC).
Un autre critère d'évaluation était la maladie résiduelle minimale (minimal residual disease, MRD), évaluée au moyen d'un séquençage de nouvelle génération (next generation sequencing, NGS)
Les résultats d'efficacité pour les niveaux de dose cibles de 150 à 450 x 106 cellules CAR-T positives sont présentés dans le tableau 4.
Dans l'analyse principale, portant sur la population traitée, l'ORR a été de 73,4 % (IC à 95 %: 65,8; 81,1) et le taux de réponse complète (CR) de 32,87 % (IC à 95 %: 24,7; 40,9). Pour les patients présentant une réponse partielle (partial response, PR) ou mieux, la durée médiane de la DOR a été de 10,6 mois (IC à 95 %: 8,0, 11,4). Chez les patients présentant une CR ou mieux, la durée médiane de la DOR a été de 23,3 mois (IC à 95 %: 11,4; 23,3). La durée de suivi médiane a été de 15,4 mois pour tous les patients traités (plage: entre 0,2 et 24,2).
Sur 140 patients de la population incluse, l'ORR a été de 67,1 % et la CR de 30 %. D'autres résultats d'efficacité pour la population incluse étaient conformes à ceux de la population traitée.
Tableau 5: Résumé des résultats d'efficacité sur la base de l'étude KarMMa
|
|
Population incluse
(n = 140)
|
Population traitée
Dose cible d'Abecma (cellules CAR-T positives)
| |
[150 x 106]
(n = 4)
|
[300 x 106]
(n = 70)
|
[450 x 106]
(n = 54)
|
[150 bis 450 x 106]
(n = 128)
| |
Taux de réponse global (SCR + CR + VGPR + PR), n (%)
|
94 (67,1)
|
2 (50,0)
|
48 (68,6)
|
44 (81,5)
|
94 (73,4)
| |
IC à 95 %a
|
59,4; 74,9
|
6,8; 93,2
|
56,4; 79,1
|
68,6; 90,7
|
65,8; 81,1
| |
CR ou mieux, n (%)
|
42 (30,0)
|
1 (25,0)
|
20 (28,6)
|
21 (38,9)
|
42 (32,8)
| |
IC à 95 %a
|
22,4; 37,6
|
0,6; 80,6
|
18,4; 40,6
|
25,9; 53,1
|
24,7; 40,9
| |
VGPR ou mieux, n (%)
|
68 (48,6)
|
2 (50,0)
|
31 (44,3)
|
35 (64,8)
|
68 (53,1)
| |
IC à 95 %a
|
40,3; 56,9
|
6,8; 93,2
|
32,4; 56,7
|
50,6; 77,3
|
44,5; 61,8
| |
Patients avec statut MRD négatifb et ≥ CR, n
|
|
1
|
17
|
15
|
33
| |
Sur la base de la population traitée, %
|
─
|
25,0
|
24,3
|
27,8
|
25,8
| |
IC à 95 %a
|
|
0,6; 80,6
|
14,8; 36,0
|
16,5; 41,6
|
18,5; 34,3
| |
Sur la base des participants à l'étude avec ≥ CR, %
|
|
100
|
85,0
|
71,4
|
78,6
| |
IC à 95 %a
|
|
2,5; 100,0
|
62,1; 96,8
|
47,8; 88,7
|
63,2; 89,7
| |
Temps jusqu'à la réponsec, n
|
94
|
2
|
48
|
44
|
94
| |
Médiane (mois)
|
1
|
1
|
1
|
1
|
1
| |
min., max.
|
0,5; 8,8
|
1,0; 1,0
|
0,5; 8,8
|
0,9; 2,0
|
0,5; 8,8
| |
Durée de la réponsec (PR ou mieux), n
|
94
|
2
|
48
|
44
|
94
| |
Médianed (mois)
|
10,6
|
13,0
|
8,5
|
11,3
|
10,6
| |
IC à 95 %a
|
8,0; 11,4
|
2,8; 23,3
|
5,4; 10,9
|
10,3; NE
|
8,0; 11,4
| |
Durée de la réponse (CR ou mieux), n
|
42
|
1
|
20
|
21
|
42
| |
Médianed (mois)
|
23,3
|
23,3
|
16,2
|
NE
|
23,3
| |
IC à 95 %a
|
11,4; 23,3
|
NE; NE
|
8,0; NE
|
11,4; NE
|
11,4; 23,3
| |
Survie globalee (OS), mois, n
|
140
|
4
|
70
|
54
|
128
| |
Médiane (mois)
|
21,4
|
18,2
|
NE
|
NE
|
NE
| |
IC à 95 %a
|
19,3; NE
|
9,4; NE
|
18,0; NE
|
NE; NE
|
18,9; NE
| |
Taux sans événement à 6 mois, %
|
87,4
|
100
|
89,6
|
86,9
|
88,8
| |
Taux sans événement à 12 mois, %
|
75,8
|
75,0
|
78,5
|
77,3
|
77,9
|
CAR = récepteur antigénique chimérique; IC = intervalle de confiance; CR = complete response (réponse complète); Max = maximum; Min = minimum; MRD = Minimal Residual Disease (maladie résiduelle minimale); NE = non évaluable; PR = partial response (réponse partielle); sCR = stringent complete response (réponse complète stricte); VGPR = very good partial response (très bonne réponse partielle).
a Pour le total («population traitée» et «population enregistrée»): IC de Forest; pour des niveaux de dose cibles individuels: IC exact selon Clopper-Pearson.
b Sur la base d'une valeur seuil de 10-5 en utilisant un test de séquençage de nouvelle génération.
c La réponse est définie comme l'atteinte d'une sCR, d'une CR, d'une VGPR ou d'une PR selon les critères IMWG.
d La médiane est basée sur l'estimation Kaplan-Meier.
e L'OS était définie comme la période allant de la date de la leucaphérèse (population enregistrée) ou de la perfusion d'Abecma (population traitée) jusqu'au décès, toutes causes confondues.
Remarque: La dose cible est de 450 × 106 cellules CAR-T positives dans une plage de 150 à 540 × 106 cellules CAR-T positives. La dose de 150 × 106 cellules CAR-T positives ne fait pas partie de la fourchette de doses approuvées.
Qualité de vie liée à la santé (health-related Quality of life, HRQoL)
La HRQoL a été évaluée à l'aide du questionnaire C30 sur la qualité de vie (EORTC-QLQ-C30) et du module sur le myélome multiple (EORTC-QLQ-MY20) de l'Association européenne pour la recherche et le traitement des cancers (European Organisation for Research and Treatment of Cancer EORTC) en se concentrant principalement sur la fatigue, les douleurs, la fonction physique, la fonction cognitive et l'état de santé général/la qualité de vie, les effets secondaires et les symptômes de la maladie comme sous-échelles. Selon les résultats des données obtenues 10 mois après la perfusion d'Abecma, les patients traités par Abecma ont connu des améliorations cliniquement significatives des scores de fatigue, des douleurs, du fonctionnement physique et de l'état de santé général peu après la perfusion, qui sont devenues statistiquement significatives (p < 0,05) à plusieurs moments du mois 3 au mois 9 après le traitement, sans détérioration du fonctionnement cognitif ni aggravation des symptômes de la maladie ou des effets secondaires. Pour la plupart des critères d'évaluation et des points d'observation, un pourcentage plus important de patients a rapporté une amélioration cliniquement significative qu'une détérioration.
Étude de preuves en conditions réelles (real world, RW)
L'étude de preuves RW (étude NDS-MM-003) était une étude d'observation rétrospective, qui a recueilli en conditions réelles des données sur des patients atteints de myélome multiple récidivant et réfractaire (RRMM) qui avaient reçu au moins trois traitements antérieurs, dont un agent immunomodulateur, un IP et un anticorps anti-CD38. Dans ce groupe, on a sélectionné les patients qui répondaient aux critères de sélection les plus proches possibles de ceux de l'étude KarMMa (c'est-à-dire l'absence de comorbidités et l'initiation d'un nouveau traitement après être devenu réfractaire au dernier traitement anti-myélome). L'ORR et la survie globale (OS) ont été évalués pour les deux groupes en utilisant la méthode du Propensity Score pour évaluer l'efficacité comparative des patients traités avec les thérapies disponibles par rapport à Abecma dans l'essai KarMMa. Le risque relatif d'ORR a été de 2,4 (IC à 95 %: 1,7; 3,3) p < 0,0001. Le hazard ratio de l'OS a été de 0,41 (IC 95 % 0,26, 0,65), favorisant significativement la cohorte traitée par Abecma par rapport à la cohorte RRMM correspondante traitée par les thérapies disponibles (p = 0,0002).
Sécurité et efficacité chez les patients âgés
Dans les études cliniques sur Abecma, 163 patients (39,9 %) étaient âgés d'au moins 65 ans et 17 d'au moins 75 ans (4,2 %). Aucune différence cliniquement significative n'a été observée concernant la sécurité ou l'efficacité d'Abecma entre ces patients et les patients âgés de moins de 65 ans.
|