CompositionPrincipes actifs
Dupilumab (produit à partir de cellules de hamster chinois génétiquement modifiées).
Excipients
L-Histidinum, Histidini hydrochloridum monohydricum, Arginini hydochloridum, Natrii Acetas Trihydricus, Acidum Aceticum Glaciale, Polysorbatum 80, Saccharum, Aqua ad iniectabilia q.s. ad solutionem 2 ml ou 1,14 ml.
Indications/Possibilités d’emploiDermatite atopique:
Pour le traitement des enfants de moins de 12 ans, seule la seringue préremplie est indiquée (voir «Posologie et mode d'emploi» => Mode d'administration).
Dupixent est indiqué pour le traitement des patients âgés de 6 ans et plus atteints de dermatite atopique modérée à sévère, dont la maladie n'est pas contrôlée de manière adéquate par des thérapies topiques sur ordonnance ou lorsque ces thérapies ne sont pas recommandées.
Dupixent peut être utilisé avec ou sans corticostéroïdes topiques.
Asthme:
Dupixent est indiqué chez l'adulte et l'adolescent âgé de 12 ans ou plus en traitement d'entretien d'appoint pour l'asthme sévère répondant aux critères suivants:
·nombre de cellules éosinophiles dans le sang ≥0,15 g/litre (soit ≥150 cellules/µl), aucun contrôle complet de l'asthme et au moins 1 exacerbation sévère au cours des 12 mois précédents, malgré un traitement associant des corticostéroïdes par inhalation et des bronchodilatateurs à action prolongée;
·ou nécessité d'un traitement permanent avec des corticostéroïdes systémiques.
Pour une information détaillée concernant les populations de patients investiguées, voir section «Efficacité clinique».
Polypose naso-sinusienne (PNS)
Dupixent est indiqué en traitement additionnel aux corticostéroïdes par voie nasale chez les adultes présentant une polypose naso-sinusienne sévère insuffisamment contrôlés par des corticostéroïdes systémiques et/ou la chirurgie.
Posologie/Mode d’emploiLe traitement doit être initié par des professionnels de santé expérimentés dans le diagnostic et le traitement des pathologies pour lesquelles le dupilumab est indiqué (voir section «indication»).
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Dermatite atopique:
Adultes:
La posologie de dupilumab recommandée chez l'adulte est une dose initiale de 600 mg administrée par voie sous-cutanée (deux injections à 300 mg), suivi d'une dose de 300 mg en injection sous-cutanée toutes les deux semaines.
Enfants et adolescents (6-17 ans):
Le schéma posologique recommandé du dupilumab pour les enfants et adolescents âgés de 6 à 17 ans est détaillé dans le tableau ci-dessous.
Schéma posologique du dupilumab pour administration sous-cutanée chez les enfants et adolescents âgés de 6 à 17 ans atteints de dermatite atopique
|
Poids corporel du patient
|
Dose initiale
|
Doses suivantes
| |
15 kg à moins de 30 kg
|
300 mg (une injection de 300 mg) au jour 1,
suivie de 300 mg au jour 15
|
300 mg
(toutes les quatre semaines)
commençant 4 semaines après la dose du jour 15
| |
30 kg à moins de 60 kg
|
400 mg
(deux injections de 200 mg)
|
200 mg
(toutes les 2 semaines)
| |
60 kg et plus
|
600 mg
(deux injections de 300 mg)
|
300 mg
(toutes les 2 semaines)
|
Dupilumab peut être utilisé avec ou sans corticostéroïdes topiques. L'utilisation d'inhibiteurs topiques de la calcineurine est possible, mais doit être réservée à des zones problématiques telles que le visage, le cou, les régions intertrigineuses et les zones génitales. L'arrêt du traitement devra être envisagé chez les patients qui ne présentent aucune réponse après 16 semaines de traitement.
Asthme
La dose recommandée de dupilumab chez l'adultes et l'adolescent (âgé de 12 ans et plus) est:
·Pour les patients avec l'asthme sévère traité par des corticostéroïdes par inhalation et des bronchodilatateurs à action prolongée: une dose initiale de 400 mg (soit 2 injections de 200 mg), suivie d'une dose de 200 mg administrée toutes les deux semaines en injection sous-cutanée.
·Dans le cas de l'asthme sévère traité par des corticostéroïdes oraux ou asthme sévère associé à une dermatite atopique modérée à sévère selon l'indication approuvée, une dose initiale de 600 mg (soit 2 injections de 300 mg), suivie d'une dose de 300 mg administrée toutes les deux semaines, en injection sous-cutanée.
En cas de corticothérapie orale associée, la dose de corticostéroïdes pourra être diminuée lorsqu'une amélioration clinique avec le dupilumab est observée. Les corticostéroïdes doivent être réduits progressivement.
Le dupilumab est destiné à un traitement au long cours. La décision de poursuivre ou non le traitement doit être réévaluée au moins une fois par an, par le médecin, en fonction du niveau de contrôle de l'asthme.
Polypose naso-sinusienne
La dose recommandée de dupilumab chez l'adulte est une dose initiale de 300 mg suivie d'une dose de 300 mg administrée toutes les deux semaines.
Dupilumab est destiné à un traitement au long cours. L'interruption du traitement doit être envisagée en cas d'absence de réponse après 24 semaines de traitement. Certains patients présentant initialement une réponse partielle peuvent bénéficier d'une amélioration en poursuivant le traitement après 24 semaines.
Dose oubliée (pour toutes les indications):
En cas d'oubli d'une dose, demandez au patient d'administrer l'injection dans les 7 jours qui suivent la dose manquée, puis reprendre les injections selon son calendrier d'administration habituel. Si la dose oubliée n'est pas administrée dans les 7 jours, demandez au patient d'attendre la dose suivante prévue dans son calendrier d'administration.
Ajustement de dosage:
Patients présentant des troubles de la fonction hépatique
Aucune donnée n'est disponible pour les patients souffrant d'une insuffisance hépatique (voir section «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée. Les données disponibles concernant les patients atteints d'insuffisance rénale sévère sont très limitées (voir section «Pharmacocinétique»).
Patients âgés (≥65 ans)
Des données très limitées sont disponibles chez les patients de 65 ans et plus. Bien qu'aucune différence en termes de sécurité ou d'efficacité n'ait été observée entre les patients jeunes et âgés, le nombre de patients âgés de ≥65 ans n'est pas suffisant pour déterminer s'ils répondent différemment des sujets plus jeunes.
Enfants et adolescents
La sécurité d'emploi et l'efficacité de dupilumab chez l'enfant de moins de 6 ans atteint de dermatite atopique n'ont pas été établies. La sécurité d'emploi et l'efficacité de dupilumab chez l'enfant de poids corporel < 15 kg n'ont pas été établies. Aucune donnée n'est disponible.
La sécurité et l'efficacité du dupilumab n'ont pas été établies chez les enfants de moins de 12 ans présentant un asthme sévère. Aucune donnée n'est disponible.
La polypose naso-sinusienne n'apparait habituellement pas chez les enfants.
La sécurité et l'efficacité n'ont pas été établies dans le traitement de la polypose naso-sinusienne chez les enfants âgés de moins de 18 ans (voir section «Propriétés/Effets»). Aucune donnée n'est disponible.
Poids corporel
Aucun ajustement posologique n'est recommandé en fonction du poids corporel pour les patients adultes atteints de dermatite atopique.
Pour les patients âgés de 6 à 17 ans atteints de dermatite atopique, la dose recommandée est de 300 mg toutes les 4 semaines (15 kg à <30 kg), 200 mg toutes les 2 semaines (30 kg à <60 kg) ou 300 mg toutes les 2 semaines (≥60 kg).
Mode d'administration
Voie sous-cutanée.
Concernant l'utilisation du stylo prérempli: les patients doivent être avisés de lire et de suivre les instructions d'utilisation et de veiller à injecter tout le contenu du stylo prérempli. L'injection peut prendre jusqu'à 20 secondes.
Le stylo prérempli de dupilumab n'est pas destiné à être utilisé chez les enfants de moins de 12 ans. Pour les enfants de 6 à 11 ans atteints de dermatite atopique, la seringue préremplie de dupilumab est la présentation appropriée pour l'administration à cette population.
Le dupilumab doit être administré par injection sous-cutanée dans la cuisse ou l'abdomen, en excluant une zone de 5 cm autour du nombril. Si l'injection est réalisée par un tiers, on peut également opter pour le haut du bras.
Pour la dose initiale (400 mg ou 600 mg selon l'indication concernée), deux injections consécutives de dupilumab à 200 mg ou 300 mg (selon l'indication), doivent être pratiquées en deux endroits différents.
Il faut changer de site d'injection à chaque nouvelle injection. Il convient de ne pas injecter dupilumab dans une peau sensible, abîmée ou présentant des ecchymoses ou des cicatrices.
Le patient peut s'injecter dupilumab seul ou l'injection peut être réalisée par un soignant si le professionnel de santé juge que cela est approprié. Avant utilisation, le patient et/ou les soignants doivent être correctement formés à la préparation et à l'administration de dupilumab conformément à la notice d'utilisation disponible dans la notice d'emballage.
Chez les adolescents de 12 ans et plus, il est recommandé que dupilumab soit administré par ou sous la surveillance d'un adulte.
Comme pour tout agent thérapeutique auto-administré, les médecins doivent surveiller de près l'observance du traitement par le patient. Dans les études cliniques de phase 3, les patients qui se sont auto-injectés ont oublié environ un sixième des doses; 17,5% des doses de dupilumab ont été manquées et 26,2% des doses placebo.
Pour de plus amples informations sur l'administration du médicament, consulter la section «Remarques concernant la manipulation».
Contre-indicationsHypersensibilité à la substance active ou à l'un des excipients répertoriés à la section «Composition».
Mises en garde et précautionsLe dupilumab ne doit pas être utilisé pour traiter des symptômes aigus d'asthme ou les exacerbations. Le dupilumab ne doit pas être utilisé pour traiter un bronchospasme aigu ou un état de mal asthmatique.
Les corticostéroïdes systémiques, topiques ou inhalés ne doivent pas être interrompus brutalement après l'instauration du traitement par le dupilumab. Les réductions de la dose de corticostéroïdes, le cas échéant, doivent être progressives et effectuées sous le contrôle direct d'un médecin. Une réduction de la dose de corticostéroïdes peut être associée à des symptômes systémiques de sevrage et/ou révéler des affections précédemment disparues avec la corticothérapie systémique.
L'expression des biomarqueurs de l'inflammation de type 2 peut être inhibée par l'utilisation de corticostéroïdes systémiques. Cela doit être pris en compte lors de l'évaluation du phénotype de type 2 chez les patients sous corticostéroïdes oraux (voir section «Propriétés/Effets»).
Hypersensibilité:
En cas de réaction d'hypersensibilité systémique générale (immédiate ou retardée), l'administration de dupilumab doit être stoppée immédiatement et un traitement approprié doit être instauré.
Des réactions d'hypersensibilité incluant l'anaphylaxie, la maladie sérique ou la pseudo maladie sérique et l'angio-œdème ont été rapportées (Voir section «Effets Indésirables»).
Une réaction anaphylactique a été signalée très rarement dans le cadre du programme de développement de l'asthme après l'administration du dupilumab (voir section «Effets indésirables»).
Hyperéosinophilie:
Des cas de pneumopathie à éosinophiles et des cas de vascularite en rapport avec une granulomatose éosinophilique avec polyangéite ont été rapportés chez des patients ayant participé au programme de développement dans l'asthme. L'apparition de lésions cutanées de vascularite, une aggravation des symptômes pulmonaires, des complications cardiaques et/ou une neuropathie survenant chez des patients présentant une hyperéosinophilie doivent mettre le médecin en alerte. Les patients traités pour un asthme peuvent présenter une hyperéosinophilie systémique grave parfois révélée par les symptômes cliniques d'une pneumopathie à éosinophiles ou d'une vascularite en rapport avec une granulomatose éosinophilique avec polyangéite; ces affections étant souvent traitées par une corticothérapie orale. Ces cas, en général, mais pas toujours, peuvent être liés à la réduction de la corticothérapie orale.
Helminthiase:
Toute participation à une étude clinique de patients présentant une helminthiase connue a été exclue. Dupilumab peut influer sur la réponse immunitaire à l'helminthiase en inhibant la voie de signalisation de l'IL-4/IL-13. Les patients avec une helminthiase préexistante doivent être traités avant le début du traitement par dupilumab. En cas d'infection pendant le traitement par dupilumab et en l'absence de réponse au traitement contre les helminthes, le traitement par dupilumab doit être arrêté jusqu'à la disparition de l'infection.
Evènements liés à la conjonctivite et kératite:
Des cas de conjonctivite et de kératite ont été signalés avec Dupixent chez des patients atteints de dermatite atopique.
Certains patients ont signalé des troubles visuels (par exemple, une vision floue) associés à une conjonctivite ou à une kératite (voir section «Effets indésirables»).
Les patients doivent signaler toute nouvelle apparition ou l'aggravation des symptômes oculaires à leur médecin.
Les patients traités par dupilumab qui développent une conjonctivite qui n'est pas résolue après un traitement standard doivent suivre un examen ophtalmologique le cas échéant (voir section «Effets indésirables»).
Patients atteints de dermatite atopique ou de polypose naso-sinusienne présentant un asthme associé:
Les patients traités par le dupilumab pour une dermatite atopique modérée à sévère ou pour une polypose naso-sinusienne sévère qui ont également un asthme associé ne doivent pas modifier ou arrêter leur traitement anti-asthmatique sans avoir consulté leur médecin.
Les patients présentant un asthme associé doivent faire l'objet d'un suivi attentif après l'arrêt du traitement par dupilumab.
Vaccinations:
Les vaccins vivants et les vaccins vivants atténués ne doivent pas être administrés pendant le traitement par le dupilumab, la sécurité et l'efficacité n'ayant pas été établies. Les réponses immunitaires au vaccin dcaT et au vaccin polysaccharidique méningococcique ont été évaluées. Il est recommandé de vérifier que les patients sont à jour dans leur vaccination par les vaccins vivants et atténués selon le calendrier vaccinal en vigueur avant d'initier un traitement par le dupilumab (voir section «Interactions»).
Excipients:
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose de 300 mg de même que pour la dose de 200 mg.
InteractionsInteractions pharmacocinétiques
Données in vivo
Interactions avec les substrats du CYP:
Dermatite atopique: Les effets du dupilumab sur la pharmacocinétique des substrats des CYP ont été évalués au cours d'une étude clinique menée chez des patients atteints de dermatite atopique. Les données recueillies grâce à cette étude ne mettent en évidence aucun effet cliniquement significatif du dupilumab sur l'activité du CYP1A2, CYP3A4, CYP2C19, CYP2D6 ou CYP2C9.
Asthme: Il n'est pas attendu d'interaction pharmacocinétique avec dupilumab. Les analyses de population n'ont pas mis en évidence d'effet sur la pharmacocinétique du dupilumab chez les patients présentant un asthme modéré à sévère.
Autres interactions
Vaccination:
La sécurité et l'efficacité de l'utilisation simultanée de dupilumab avec des vaccins vivants n'ont pas été étudiées. Éviter l'utilisation de vaccins vivants chez les patients traités par dupilumab. Il est recommandé de vérifier que les patients sont à jour dans leur vaccination par les vaccins vivants et atténués selon le calendrier vaccinal en vigueur avant d'initier un traitement par le dupilumab (voir section «Mises en garde et précautions».
Vaccins inactivés: Les réponses immunitaires à la vaccination ont été évaluées dans une étude dans laquelle les sujets atteints de dermatite atopique ont été traités une fois par semaine pendant 16 semaines avec 300 mg de dupilumab (deux fois la fréquence de dosage recommandée). Après 12 semaines d'administration de dupilumab, les sujets ont été vaccinés avec un vaccin Tdap (Adacel®) et un vaccin contre le polysaccharide méningococcique (Menomune®). Les réponses des anticorps à l'anatoxine tétanique et au polysaccharide méningococcique du sérogroupe C ont été évaluées 4 semaines plus tard. Les réponses humorales au vaccin contre le tétanos et au vaccin polysaccharidique contre le méningocoque étaient semblables chez les sujets traités par le dupilumab et ceux recevant le placebo. Les réponses immunitaires aux autres composants actifs des vaccins Adacel et Menomune n'ont pas été évaluées.
Grossesse, allaitementGrossesse
Il existe peu de données concernant l'utilisation de dupilumab chez les femmes enceintes.
Les études menées chez l'animal n'ont pas montré d'effets nocifs directs ou indirects en ce qui concerne la toxicité sur la reproduction (voir «Données précliniques»). Dupilumab ne doit pas être utilisé pendant la grossesse, sauf si le bénéfice potentiel justifie le risque potentiel encouru par le fœtus.
Allaitement
On ne sait pas si le dupilumab est excrété dans le lait maternel ou absorbé par voie systémique après ingestion. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement par dupilumab en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Les études menées chez l'animal n'ont montré aucune altération de la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'effet de dupilumab sur l'aptitude à conduire des véhicules ou à utiliser des machines n'a pas été investigué directement.
Effets indésirables1. Adultes atteints dermatite atopique
Résumé du profil de sécurité
Les réactions indésirables les plus fréquentes ont été: les réactions au site d'injection, la conjonctivite, la blépharite et l'herpès labial. De rares cas de maladie sérique ou de réaction de type maladie sérique ont été rapportés au cours des études conduites dans le cadre du programme de développement dans la dermatite atopique (voir section «Mises en garde et précautions»).
Au cours des études sur l'emploi de dupilumab chez les adultes en monothérapie, la part des patients qui ont arrêté le traitement en raison d'effets indésirables était de 1,9 % dans le groupe sous placebo, de 1,9 % dans le groupe sous dupilumab à raison de 300 mg toutes les deux semaines (Q2W) et de 1,5 % dans le groupe sous dupilumab à raison de 300 mg toutes les semaines (QW; cette posologie n'est pas approuvée en Suisse).
Au cours de l'étude avec emploi concomitant de corticostéroïdes topiques (CST) chez les adultes, la part des patients qui ont arrêté le traitement en raison d'effets indésirables était de 7,6 % dans le groupe sous placebo + CST, de 1,8 % dans le groupe traité par l'association dupilumab 300 mg Q2W + CST et de 2,9 % dans le groupe recevant l'association dupilumab 300 mg QW + CST (cette posologie n'est pas approuvée en Suisse).
Dans une étude d'extension de phase 3, multicentrique, ouverte (OLE, AD-1225), l'innocuité à long terme des doses répétées de dupilumab a été évaluée chez des adultes atteints de dermatite atopique modérée à sévère ayant déjà participés à des études contrôlées avec dupilumab ou avaient été sélectionnés pour une étude de phase 3 (SOLO1 ou SOLO2). Les données de sécurité de l'AD-1225 reflètent l'exposition au dupilumab chez 2677 patients adultes atteints de dermatite atopique, dont 2254 qui ont complété au moins 52 semaines de traitement, 1192 qui ont complété au moins 100 semaines et 357 patients qui ont complété au moins 148 semaines dans l'étude. La majorité des patients de l'essai 5 (99,7%) ont été exposés à une dose hebdomadaire de 300 mg de dupilumab (QW). Le profil de sécurité à long terme observé dans cette étude jusqu'à 3 ans était généralement cohérent avec le profil de sécurité de dupilumab observé dans les études contrôlées.
Liste des réactions indésirables
La sécurité d'emploi de dupilumab a été évaluée dans le cadre de quatre études randomisées, contrôlées contre placebo et menées en double aveugle et d'une étude de recherche de dose chez des patients atteints de dermatite atopique modérée à sévère. Au cours de ces cinq études, 1689 sujets ont été traités par des injections sous-cutanées de dupilumab, en association ou non avec un traitement par des corticostéroïdes topiques (CST). Au total, 305 patients ont été traités par dupilumab pendant au moins un an.
Les effets indésirables portant sur les études cliniques portant sur la dermatite atopique sont présentés ci-après par système organe classe et par fréquence, selon les catégories suivantes: très fréquent (≥1/10); fréquent (≥1/100, < 1/10); peu fréquent (≥1/1 000, < 1/100), rare (≥1/10 000, < 1/1 000) ou très rare (< 1/10 000). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Infections et Infestations:
Fréquent: Conjonctivite, herpès labial.
Peu fréquent: Autres infections du virus herpès simplex (excluant l'eczéma herpétique).
Affections hématologiques et du système lymphatique:
Fréquent: Eosinophilie.
Affections du système immunitaire:
Rare: Maladie sérique/réaction de type maladie sérique.
Affections du système nerveux:
Fréquent: Céphalées.
Troubles des yeux:
Fréquent: Conjonctivite, prurit oculaire, blépharite.
Peu fréquent: Kératites.
Troubles généraux et anomalies au site d'administration:
Très fréquent: Réactions au site d'injection (9.6%).
2. Adolescents atteints de dermatite atopique
La sécurité du dupilumab a été évaluée dans une étude réalisée chez 250 patients âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère, dont 166 ont été traités par dupilumab (AD-1526). Le profil de sécurité du dupilumab chez ces patients suivis jusqu'à la semaine 16 était similaire au profil de sécurité issu des études réalisées chez des adultes atteints de dermatite atopique.
La sécurité à long terme du dupilumab a été évaluée dans une étude d'extension en ouvert réalisée chez des patients âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère (AD-1434). Le profil de sécurité du dupilumab chez ces patients suivis jusqu'à la semaine 52 était similaire au profil de sécurité observé à la semaine 16 dans l'étude AD-1526. Le profil de sécurité à long terme du dupilumab observé chez les adolescents était en accord avec celui observé chez les adultes atteints de dermatite atopique.
3. Enfants (6 à 11 ans) atteints de dermatite atopique
La sécurité du dupilumab en association avec le corticostéroïdes topiques (CST) a été évaluée dans un essai portant sur 367 sujets âgés de 6 à 11 ans atteints de dermatite atopique sévère (AD-1652). Le profil de sécurité du dupilumab + CST chez ces patients était similaire jusqu'à la semaine 16 à celui des essais menés chez les adultes et les adolescents atteints de dermatite atopique.
La sécurité à long terme du dupilumab + CST a été évaluée dans une étude ouverte étendue portant sur 368 sujets âgés de 6 à 11 ans atteints de dermatite atopique (AD-1434). Parmi les patients qui ont participé à cette étude, 110 (29,9%) souffraient de dermatite atopique modérée et 72 (19,6%) de dermatite atopique grave au moment de l'inclusion dans l'étude AD-1434. Le profil de sécurité du dupilumab + CST chez les sujets suivis jusqu'à la semaine 52 était similaire au profil de sécurité observé jusqu'à la semaine 16 dans l'AD-1652. Le profil de sécurité à long terme du dupilumab + CST observé chez les sujets pédiatriques était similaire à celui observé chez les adultes et les adolescents atteints de dermatite atopique.
4. Asthme
Résumé du profil de sécurité
L'effet indésirable le plus fréquent était un érythème au site d'injection. Une réaction anaphylactique a été signalée dans de rares cas au cours des études conduites dans le cadre du programme de développement dans l'asthme (voir section «Mises en garde et précautions»).
Dans les études DRI12544 et QUEST, le pourcentage de patients ayant interrompu le traitement en raison d'effets indésirables était de 4,3 % dans le groupe placebo, de 3,2 % dans le groupe dupilumab 200 mg 1x/2 semaines et de 6,1 % dans le groupe dupilumab 300 mg 1x/2 semaines.
Effets indésirables
Un total de 2 888 patients adultes et adolescents présentant un asthme modéré à sévère ont été évalués dans 3 essais multicentriques randomisés, contrôlée contre placebo, d'une durée de 24 à 52 semaines (DRI12544, QUEST et VENTURE). Parmi eux, 2 678 avaient des antécédents d'une ou de plusieurs exacerbations sévères dans l'année précédant l'inclusion malgré l'utilisation régulière d'une dose moyenne à élevée de corticostéroïdes inhalés associée à un ou plusieurs autres traitements de fond (DRI12544 et QUEST). Au total, 210 patients présentant un asthme cortico-dépendant et recevant une dose élevée de corticostéroïdes inhalés associées à deux autres traitements au maximum, ont été inclus (VENTURE).
La sécurité à long terme du dupilumab a été évaluée dans une étude de d'extension en ouverte réalisée chez 2282 patients âgés de 12 ans et plus souffrant d'asthme modéré à sévère (TRAVERSE). Dans cette étude, les patients ont été suivis pendant 96 semaines, ce qui représente une exposition cumulative de 3169 patients-années au dupilumab. Le profil de sécurité du dupilumab dans l'étude TRAVERSE était conforme au profil de sécurité observé dans les études pivotales sur l'asthme chez les patients suivis jusqu'à la semaine 52.
Voici la liste des effets indésirables observés pendant les essais cliniques dans l'asthme, par classe de systèmes d'organes et fréquence, en utilisant les catégories suivantes: très fréquent (≥1/10); fréquent (≥1/100 à < 1/10); peu fréquent (≥1/1 000 à < 1/100); rare (≥1/10 000 à < 1/1 000); très rare (< 1/10 000). Pour chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de sévérité:
Affections du système immunitaire:
Rare: Réaction anaphylactique
Troubles généraux et anomalies au site d'administration:
Très fréquent: Érythème au site d'injection (14,6%)
Fréquent: Œdème au site d'injection (4,8%), douleur au site d'injection (3,8%), prurit au site d'injection (4,7%).
5. Polypose naso-sinusienne
Résumé du profil de sécurité
Les évènements indésirables les plus fréquents étaient les réactions et les œdèmes au site d'injection.
Sur l'ensemble des données de sécurité, la proportion de patients ayant interrompu le traitement en raison d'événements indésirables était de 2,0% dans le groupe dupilumab 300 mg 1 fois toutes les 2 semaines et de 4,6% dans le groupe placebo.
Liste des effets indésirables:
Au total, 722 patients adultes présentant une polypose naso-sinusienne ont été évalués dans 2 études cliniques randomisées, contrôlées contre placebo, multicentriques d'une durée de 24 et de 52 semaines (SINUS-24 et SINUS-52).
Les données de sécurité présentées sont issues des 24 premières semaines de traitement.
La liste des effets indésirables observés pendant les études cliniques portant sur la polypose naso-sinusienne par classe de systèmes d'organes et fréquence sont présentés ci-dessous, en utilisant les catégories suivantes: très fréquent (≥1/10); fréquent (≥1/100 à < 1/10); peu fréquent (≥1/1 000 à < 1/100); rare (≥1/10 000 à < 1/1 000); très rare (< 1/10 000). Pour chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de sévérité.
Infections et infestations:
Fréquent: conjonctivite
Affections hématologiques et du système lymphatique:
Fréquent: Hyperéosinophilie.
Troubles généraux et anomalies au site d'administration:
Fréquent: Réaction au site d'injection; Œdème au site d'injection.
Affections gastro-intestinales:
Fréquent: gastrite, maux de dents.
Affections musculosquelettiques, du tissu conjonctif et des os
Fréquent: arthalgie.
Affections psychiatriques:
Fréquent: insomnie.
Description de certains effets indésirables
Description de réactions indésirables sélectionnées dans les indications dermatite atopique, de l'asthme et la polypose naso-sinusienne:
Réactions d'hypersensibilité:
Des réactions d'hypersensibilité incluant l'anaphylaxie et la maladie sérique ou la pseudo maladie sérique ont été rapportées (voir sections «Contre-Indications» et «Mises en garde et précautions»).
Événements liés à la conjonctivite et kératite:
Les événements liés à la conjonctivite et à la kératite sont survenus plus fréquemment chez les patients atteints de dermatite atopique ayant reçu le dupilumab dans les études contrôlées contre placebo. La conjonctivite ou la kératite était guérie ou en cours de guérison pendant la période de traitement.
Les taux respectifs de conjonctivite et de kératite sont restés similaires à 3 ans dans l'étude à long terme OLE (AD-1225).
Parmi les patients asthmatiques, la fréquence de survenue des conjonctivites et des kératites était faible et similaire entre le dupilumab et le placebo.
Chez les patients asthmatiques, la fréquence des conjonctivites et des kératites était faible et similaire à celle de Dupixent et du placebo.
Chez les patients atteints de polypose naso-sinusiennes (PNS), la fréquence de la conjonctivite était faible, bien que la fréquence dans le groupe traité par Dupixent ait été plus élevée que dans le groupe placebo.
Aucun cas de kératite n'a été signalé pendant le programme de développement du traitement de la PNS (voir section «Mises en garde et précautions»).
Eczéma herpétiforme:
Au cours des études de 16 semaines portant sur l'emploi de dupilumab en monothérapie dans la dermatite atopique, un eczéma herpétiforme a été rapporté chez <1 % des patients des groupes sous dupilumab et chez <1 % des patients du groupe sous placebo.
Dans l'étude sur l'association dupilumab + CST d'une durée de 52 semaines, un eczéma herpétiforme a été signalé chez 0,2% des patients du groupe sous dupilumab + CST et chez 1,9 % du groupe sous placebo + CST.
Hyperéosinophilie:
Les patients traités par le dupilumab ont initialement présenté une augmentation moyenne de leur taux d'éosinophiles par rapport à leur taux à l'inclusion dans l'étude, supérieure à celle du groupe placebo. Les taux d'éosinophiles ont ensuite diminué au cours de l'étude, revenant quasiment à leurs valeurs à l'inclusion dans l'étude. Les taux d'éosinophiles a continué à diminuer en dessous du niveau de référence pendant l'étude d'extension en ouverte chez les patients asthmatiques.
Une hyperéosinophilie apparue sous traitement (≥5 000 cellules/µl) a été rapportée chez < 2 % des patients traités par dupilumab et < 0,5 % des patients traités par placebo.
Infections:
Au cours des études cliniques de 16 semaines portant sur le traitement en monothérapie de la dermatite atopique, des infections graves ont été signalées chez 1,0 % des patients sous placebo et chez 0,5 % des patients traités par dupilumab. Dans l'étude CHRONOS de 52 semaines, des infections graves ont été rapportées chez 0,6 % des patients sous placebo et 0,2 % des patients traités par dupilumab. Les taux d'infections graves sont restés stables à 3 ans dans l'étude à long terme OLE (AD-1225).
Zona:
Le zona a été rapporté chez <0,1% des patients des groupes dupilumab et chez <1% des patients du groupe placebo dans les études de monothérapie de 16 semaines. Dans l'étude dupilumab + TCS de 52 semaines, le zona a été signalé chez 1% des patients du groupe dupilumab + TCS et chez 2% des patients du groupe placebo + TCS.
Immunogénicité:
Comme toutes les protéines thérapeutiques, dupilumab présente un risque d'immunogénicité.
Environ 6 % des patients atteints de dermatite atopique, présentant un asthme ou une polypose naso-sinusienne et ayant reçu le dupilumab 300 mg 1x/2 semaines pendant 52 semaines ont développé des ADA contre le dupilumab. Environ 2 % d'entre eux présentaient des réponses en ADA persistantes et environ 2 % présentaient des anticorps neutralisants. Des résultats similaires ont été observés chez des sujets pédiatriques (6 à 11 ans) atteints de dermatite atopique qui ont reçu du dupilumab 200 mg toutes les 2 semaines ou 300 mg toutes les 4 semaines pendant 16 semaines.
Environ 16% des sujets adolescents atteints de dermatite atopique qui ont reçu dupilumab 300 mg ou 200 mg Q2S pendant 16 semaines ont développé des anticorps dirigés contre le dupilumab; environ 3% présentaient des réponses ADA persistantes et environ 5% avaient des anticorps neutralisants.
Environ 4 % des patients présentant une dermatite atopique ou une polypose naso-sinusienne dans les groupes placebo des études de 52 semaines dont les schémas posologiques étaient de 200 mg et de 300 mg administrés toutes les 2 semaines étaient également positifs pour les anticorps dirigés contre le dupilumab. Environ 2 % d'entre eux ont présenté une réponse en ADA persistante et environ 1 % présentaient des anticorps neutralisants.
Environ 9 % des patients asthmatiques qui ont reçu le dupilumab à raison de 200 mg 1x/2 semaines pendant 52 semaines ont développé des anticorps contre le dupilumab. Environ 4 % d'entre eux présentaient des réponses en ADA persistantes et environ 4 % présentaient des anticorps neutralisants.
Deux sujets adultes ayant présenté des réponses en ADA à titre élevé ont développé une maladie sérique ou des réactions ressemblant à une maladie sérique et pendant le traitement par dupilumab. (voir section «Mises en garde et précautions»).
Quel que soit l'âge ou la population, environ 2 à 4% des sujets adolescents atteints de dermatite atopique dans le groupe placebo étaient positifs pour les anticorps anti- dupilumab; environ 1% présentaient des réponses ADA persistantes et environ 1% avaient des anticorps neutralisants.
Chez les sujets ayant reçu Dupixent, le développement d'anticorps dirigés contre le dupilumab a été associé à des concentrations sériques plus faibles de dupilumab. Quelques sujets qui avaient des titres élevés d'anticorps n'avaient également aucune concentration détectable de dupilumab sérique.
Population pédiatrique
Le profil de sécurité observé chez les enfants et adolescents âgés de 6 à 17 ans dans les études cliniques pour la dermatite atopique était similaire à celui observé chez les adultes.
Effets indésirables après commercialisation
Les effets indésirables supplémentaires suivants ont été signalés lors de l'utilisation post-approbation de dupilumab. Les effets indésirables sont issus de rapports spontanés et leur fréquence est donc inconnue (ne peut être estimée à partir des données disponibles).
Troubles du système immunitaire:
·Angio-œdème
Troubles de la peau et des tissus sous-cutanés:
·Eruption cutanée au visage
Troubles musculo-squelettiques et du tissu conjonctif:
·Arthralgie
Troubles oculaires:
·Kératite et kératite ulcéreuse
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéficerisque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe aucun traitement spécifique pour un surdosage à dupilumab. En cas de surdosage, il convient de surveiller le patient pour détecter tout signe ou symptôme de réaction indésirable et d'instaurer immédiatement un traitement symptomatique approprié.
Propriétés/EffetsCode ATC
D11AH05
Mécanisme d'action
Le dupilumab est un anticorps monoclonal humain recombinant de type IgG4 qui inhibe la voie de signalisation de l'interleukine 4 (IL-4) et de l'interleukine 13 (IL-13). Le dupilumab inhibe la voie de signalisation de l'IL-4 via le récepteur de type 1 (IL-4Rα/γc) et les voies de signalisation de l'IL-4 et de l'IL-13 via le récepteur de type II (IL-4Rα/IL-13Rα).
L'IL-4 et l'IL-13 sont des cytokines des pathologies inflammatoires de type 2 telles que la dermatite atopique et l'asthme. Le blocage de la voie de l'IL-4/IL-13 avec le dupilumab diminue plusieurs médiateurs de l'inflammation de type 2.
Pharmacodynamique
Au cours des études cliniques conduites dans la dermatite atopique, le traitement par le dupilumab a été associé à une baisse, par rapport aux valeurs initiales, des concentrations en biomarqueurs de l'immunité de type 2 telles que le taux de Chimiokine thymique régulée par activation (TARC/CCL17), les IgE sériques totales et la concentration sérique en IgE spécifiques à certains allergènes. Une réduction du taux de lactate déshydrogénase (LDH) a été observée sous traitement par dupilumab chez les adultes et les adolescents atteints de dermatite atopique.
Le traitement s'accompagne d'une augmentation des taux d'IgG4, due à la présence de dupilumab. Les effets d'une exposition à un anticorps monoclonal IgG4 sur le long terme n'ont pas été investigués de façon conclusive.
Au cours des essais cliniques portant sur l'asthme, le traitement par le dupilumab a nettement réduit la fraction de monoxyde d'azote expirée (FeNO) et les concentrations circulantes d'éotaxine-3, d'IgE totale, d'IgE spécifique d'un allergène, de TARC et de périostine chez les sujets asthmatiques comparativement au placebo. Ces réductions de biomarqueurs inflammatoires de type 2 étaient comparables pour les schémas posologiques de 200 mg 1x/2 semaines et de 300 mg 1x/2 semaines. Ces marqueurs étaient quasiment non détectables après 2 semaines de traitement, sauf l'IgE, qui a diminué plus lentement. Ces effets étaient maintenus pendant toute la durée du traitement.
Efficacité clinique
1. Efficacité et sécurité clinique dans la dermatite atopique chez l'adulte:
L'efficacité et la sécurité de l'emploi de dupilumab en monothérapie et en association avec un traitement par des corticostéroïdes topiques ont été évaluées lors de trois études pivots randomisées, contrôlées contre placebo et menées en double aveugle (SOLO 1, SOLO 2 et CHRONOS) chez 2119 patients de 18 ans et plus qui étaient atteints de dermatite atopique (DA) modérée à sévère définie par un score ≥3 selon l'évaluation globale de l'investigateur (Investigator's Global Assessment, IGA), un score EASI (Eczema Area and Severity Index) ≥16 et 10 % ou plus de surface corporelle atteinte. Les patients éligibles inclus dans ces trois études avaient précédemment répondu de manière inappropriée à un traitement topique.
Dans les trois études, les patients ont reçu 1) une dose initiale de 600 mg de dupilumab (deux injections à 300 mg) le jour 1, suivie d'une dose de 300 mg toutes les deux semaines (Q2W); 2) une dose initiale de 600 mg de dupilumab le jour 1, suivie d'une dose de 300 mg une fois par semaine (QW, cette posologie n'est pas approuvée en Suisse); ou 3) un placebo correspondant. dupilumab a été administré par injection sous-cutanée (SC) dans toutes les études. En cas de besoin de contrôle de symptômes intolérables de la dermatite atopique, les patients ont été autorisés à recevoir un traitement de secours (comprenant des stéroïdes topiques plus puissants ou des immunosuppresseurs administrés par voie générale) à la discrétion du médecin-investigateur. Les patients qui ont reçu un traitement de secours ont été considérés comme ne répondant pas au traitement.
L'étude SOLO 1 comprenait 671 patients (224 sous placebo, 224 sous dupilumab 300 mg Q2W et 223 sous dupilumab 300 mg QW) qui ont été traités sur une durée de 16 semaines.
L'étude SOLO 2 comprenait 708 patients (236 sous placebo, 233 sous dupilumab 300 mg Q2W et 239 sous dupilumab 300 mg QW (cette posologie n'est pas approuvée en Suisse) qui ont été traités sur une durée de 16 semaines.
L'étude CHRONOS incluait 740 patients (315 sous placebo + CST, 106 sous dupilumab 300 mg Q2W + CST et 319 sous dupilumab 300 mg QW + CST, cette dernière posologie n'est pas approuvée en Suisse) qui ont été traités sur une durée de 52 semaines. Les patients ont reçu dupilumab ou un placebo en association avec un traitement concomitant par des CST selon un schéma thérapeutique standard dès le début de l'étude. Les patients ont également été autorisés à recevoir des inhibiteurs topiques de la calcineurine.
Critères primaires:
Dans les trois études pivots, les co-critères d'évaluation principaux étaient la part des patients qui présentaient un score IGA de 0 ou 1 («disparition» ou «quasi-disparition») avec une réduction ≥2 points sur une échelle IGA de 0 à 4 ainsi que la part des patients dont l'EASI s'est amélioré d'au moins 75 % (EASI-75) entre le début de l'étude et la semaine 16. Les autres résultats évalués comprenaient la part des patients dont l'EASI s'est amélioré d'au moins 50 % (EASI-50) ou 90 % (EASI-90), la diminution des démangeaisons évaluée sur une échelle numérique (EN) pour l'évaluation du prurit (peak pruritus Numerical Rating Scale [NRS]) et le pourcentage de variation du score SCORAD (SCORing Atopic Dermatitis) entre le début de l'étude et la semaine 16. Les critères d'évaluation secondaires supplémentaires incluaient la variation moyenne des scores POEM (Patient Oriented Eczema Measure), DLQI (Dermatology Life Quality Index) et HADS (Hospital Anxiety and Depression Scale) entre le début de l'étude et la semaine 16. Dans l'étude CHRONOS, l'efficacité a également été évaluée à la semaine 52.
Caractéristiques de base:
Sur l'ensemble des groupes de traitement des études sur l'utilisation en monothérapie (SOLO 1 et SOLO 2), l'âge moyen était de 38,3 ans et le poids moyen de 76,9 kg, 42,1 % des participants étaient de sexe féminin, 68,1 % étaient de race blanche, 21,8 % étaient asiatiques et 6,8 % étaient noirs. Dans ces études, 51,6 % des patients présentaient un score IGA initial de 3 (DA modérée) et 48,3 % des patients un score IGA initial de 4 (DA sévère), et 32,4 % des patients avaient reçu précédemment des immunosuppresseurs par voie générale. En moyenne, le score EASI initial était de 33,0, le score de prurit hebdomadaire initial sur l'EN de 7,4, le SCORAD initial de 67,8, le score POEM initial de 20,5, le DLQI initial de 15,0, et le score total HADS initial de 13,3.
Sur l'ensemble des groupes de traitement de l'étude avec emploi concomitant de CST (CHRONOS), l'âge moyen était de 37,1 ans et le poids moyen de 74,5 kg, 39,7 % des participants étaient de sexe féminin, 66,2 % étaient de race blanche, 27,2 % étaient asiatiques et 4,6 % étaient noirs. Dans cette étude, 53,1 % des patients présentaient un score IGA initial de 3 et 46,9 % des patients un score IGA initial de 4, et 33,6 % des patients avaient reçu précédemment des immunosuppresseurs par voie générale. En moyenne, le score EASI initial était de 32,5, le score de prurit hebdomadaire initial sur l'EN de 7,3, le SCORAD initial de 66,4, le score POEM initial de 20,1, le DLQI initial de 14,5 et le score total HADS initial de 12,7.
Réponse clinique: études de 16 semaines sur l'emploi en monothérapie (SOLO 1 et SOLO 2)
Dans les études SOLO 1 et SOLO 2, la part des patients chez lesquels on a obtenu, entre le début de l'étude et la semaine 16, un score IGA 0 ou 1, un score EASI-75 et/ou une amélioration >4 points du score de prurit sur l'EN était supérieure chez les patients ayant reçu dupilumab après randomisation que chez les patients sous placebo (voir Tableau 1).
Une amélioration rapide du score de prurit sur l'EN (définie comme une amélioration ≥4 points dès la semaine 2; p <0,01) a été constatée chez une part significativement plus élevée de patients sous dupilumab après randomisation que sous placebo, et la part des patients répondant au traitement selon cette échelle a continué à augmenter pendant toute la période sous traitement. L'amélioration du score de prurit sur l'EN s'est accompagnée d'une amélioration des signes objectifs de la dermatite atopique.
La Figure 1 et la Figure 2 montrent respectivement le pourcentage moyen de variation du score EASI initial et du score de prurit initial sur l'EN jusqu'à la semaine 16.
Tableau 1: Résultats relatifs à l'efficacité de dupilumab en monothérapie à la semaine 16
|
|
SOLO 1 (EAI)a
|
SOLO 2 (EAI)a
| |
|
Placebo
|
dupilumab 300 mg Q2S
|
Placebo
|
dupilumab 300 mg Q2S
| |
Patients randomisés
|
224
|
224
|
236
|
233
| |
IGA 0 ou 1b,
% de répondeursc
|
10,3 %
|
37,9 %e
|
8,5 %
|
36,1 %e
| |
EASI-50,
% de répondeursc
|
24,6 %
|
68,8 %e
|
22,0 %
|
65,2 %e
| |
EASI-75,
% de répondeursc
|
14,7 %
|
51,3 %e
|
11,9 %
|
44,2 %e
| |
EASI-90,
% de répondeursc
|
7,6 %
|
35,7 %e
|
7,2 %
|
30,0 %e
| |
Nombre de patients avec un score de prurit initial sur l'EN ≥4
|
212
|
213
|
221
|
225
| |
Prurit sur l'EN (amélioration ≥4 points),
% de répondeursc, d
|
12,3 %
|
40,8 %e
|
9,5 %
|
36,0 %e
|
MC = moindres carrés; ET = erreur-type
a L'ensemble d'analyse intégral (EAI) comprend tous les patients randomisés.
b A été considéré comme répondant au traitement («répondeur») tout patient présentant un score IGA 0 ou 1 («disparition» ou «quasi-disparition») avec une réduction >2 points sur l'échelle IGA de 0 à 4.
c Les patients qui ont reçu un traitement de secours ou pour lesquels il manquait des données ont été considérés comme ne répondant pas au traitement.
d A la semaine 2, la part des patients chez lesquels on a enregistré une amélioration du score de prurit sur l'EN ≥4 points était significativement plus élevée sous dupilumab que sous placebo (p <0,01).
e p <0,0001
Figure 1: Variation moyenne (en %) du score EASI initial au cours des études SOLO 1a et SOLO 2a (EAI)b

Figure 2: Variation moyenne (en %) du score de prurit initial sur l'EN au cours des études SOLO 1a et SOLO 2a (EAI)b

MC = moindres carrés
a Dans les analyses primaires des critères d'évaluation de l'efficacité, les patients qui ont reçu un traitement de secours ou pour lesquels il manquait des données ont été considérés comme ne répondant pas au traitement.
b L'ensemble d'analyse intégral (EAI) comprend tous les patients randomisés.
Les effets du traitement dans les sous-groupes (poids, âge, sexe, origine ethnique et traitement de fond, y compris immunosuppresseurs) des études SOLO 1 et SOLO 2 concordaient avec les résultats obtenus dans la population globale des études.
Réponse clinique: étude de 52 semaines avec emploi concomitant de CST (CHRONOS)
Dans l'étude CHRONOS, la part des patients chez lesquels on a obtenu un score IGA 0 ou 1, un score EASI-75 et/ou une amélioration >4 points du score de prurit sur l'EN entre le début de l'étude et les semaines 16 et 52 était significativement plus élevée chez les patients qui ont reçu l'association dupilumab 300 mg Q2W + CST après randomisation que chez les patients sous placebo + CST (voir Tableau 2).
Une amélioration rapide du score de prurit sur l'EN (définie comme une amélioration de >4 points dès la semaine 2; p <0,05) a été constatée chez une part significativement plus élevée de patients ayant reçu l'association dupilumab + CST après randomisation que sous placebo + CST et la part des patients répondant au traitement selon cette échelle a continué à augmenter pendant toute la période sous traitement. L'amélioration du score de prurit sur l'EN s'est accompagnée d'une amélioration des signes objectifs de la dermatite atopique.
La Figure 3 et la Figure 4 montrent respectivement le pourcentage moyen de variation du score EASI initial et du score initial sur l'EN jusqu'à la semaine 52 dans l'étude CHRONOS.
Tableau 2: Résultats relatifs à l'efficacité de dupilumab utilisé en association avec un traitement concomitant par des CSTa aux semaines 16 et 52 dans l'étude CHRONOS
|
|
Semaine 16 (EAI)b
|
Semaine 52 (EAI Semaine 52)b
| |
|
Placebo
+
CST
|
Dupilumab 300 mg
Q2S + CST
|
Placebo
+
CST
|
Dupilumab 300 mg
Q2S + CST
| |
Patients
randomisés
|
315
|
106
|
264
|
89
| |
IGA 0 ou 1c,
% de répondeursd
|
12,4 %
|
38,7 %f
|
12,5 %
|
36,0 %f
| |
EASI-50,
% de répondeursd
|
37,5 %
|
80,2 %f
|
29,9 %
|
78,7 %f
| |
EASI-75,
% de répondeursd
|
23,2 %
|
68,9 %f
|
21,6 %
|
65,2 %f
| |
EASI-90,
% de répondeursd
|
11,1 %
|
39,6 %f
|
15,5 %
|
50,6 %f
| |
Nombre de patients avec un score de prurit initial sur l'EN ≥4
|
299
|
102
|
249
|
86
| |
Prurit sur l'EN
(amélioration ≥4 points)
% de répondeursd, e
|
19,7 %
|
58,8 %f
|
12,9 %
|
51,2 %f
|
MC = moindres carrés; ET = erreur-type
a Tous les patients recevaient un traitement de fond par des corticostéroïdes topiques. Les patients étaient autorisés à prendre des inhibiteurs topiques de la calcineurine.
b L'ensemble d'analyse intégral (EAI) comprend tous les patients randomisés. L'EAI à la semaine 52 comprend tous les patients randomisés au moins un an avant la date limite de l'analyse primaire.
c A été considéré comme répondant au traitement («répondeur») tout patient présentant un score IGA 0 ou 1 («disparition» ou «quasi-disparition») avec une réduction ≥2 points sur l'échelle IGA de 0 à 4.
d Les patients qui ont reçu un traitement de secours ou pour lesquels il manquait des données ont été considérés comme ne répondant pas au traitement.
e A la semaine 2, la part des patients chez lesquels on a enregistré une amélioration du score de prurit sur l'EN ≥4 points était significativement plus élevée sous dupilumab
que sous placebo (p <0,05).
f p < 0,0001
g p = 0,0015
h p = 0,0003
i p = 0,0005
Figure 3: Variation moyenne (en %) du score EASI initial au cours de l'étude CHRONOSa (EAI à la semaine 52)b
CHRONOS
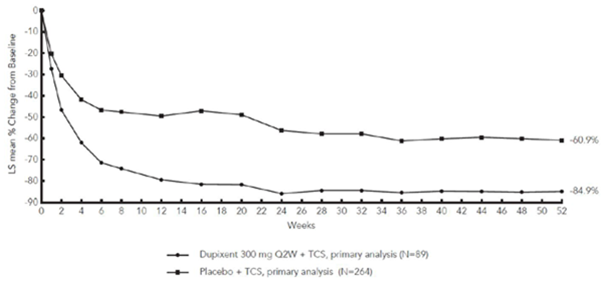
Figure 4: Variation moyenne (en %) du score de prurit initial sur l'EN au cours de l'étude CHRONOSa (EAI à la semaine 52)b
CHRONOS
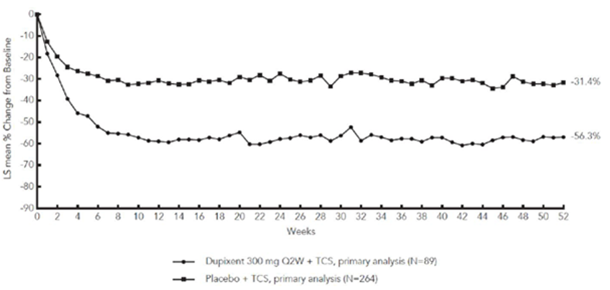
MC = moindres carrés
a Dans les analyses primaires des critères d'évaluation de l'efficacité, les patients qui ont reçu un traitement de secours ou pour lesquels il manquait des données ont été considérés comme ne répondant pas au traitement.
b L'EAI à la semaine 52 comprend tous les patients randomisés au moins un an avant la date limite de l'analyse primaire.
Les effets du traitement dans les sous-groupes (poids, âge, sexe, origine ethnique et traitement de fond, y compris immunosuppresseurs) de l'étude CHRONOS correspondaient aux résultats obtenus dans la population globale des études.
Réponse Clinique: patients chez qui un traitement par des cyclosporines ne permet pas de contrôler efficacement la maladie, n'est pas toléré ou n'est pas recommandé (étude CAFE)
L'étude CAFE visait à évaluer l'efficacité de dupilumab associé à un traitement concomitant par des CST face à un placebo sur une période de traitement de 16 semaines chez des patients adultes atteints de dermatite atopique sévère chez qui un traitement oral par une cyclosporine ne permet pas de contrôler efficacement la maladie, n'est pas toléré, est actuellement contre-indiqué ou n'est pas recommandé d'un point de vue médical.
Au total, l'étude incluait 325 patients, dont 210 avaient été exposés précédemment à une cyclosporine et 115 n'avaient jamais reçu auparavant de cyclosporines ou chez qui un traitement par une cyclosporine n'était pas recommandé d'un point de vue médical. L'âge moyen des patients était de 38,4 ans et 38,8 % des patients étaient de sexe féminin. En moyenne, l'EASI initial était de 33,1, le BSA de 55,7, le score de prurit hebdomadaire initial sur l'EN était de 6,4, le SCORAD initial de 67,2 et le DLQI initial de 13,8.
Le critère d'évaluation principal était la part des patients avec un score EASI-75 à la semaine 16.
Les critères d'évaluation principaux et secondaires de l'étude CAFE de 16 semaines sont résumés dans le Tableau 3.
Tableau 3: Résultats correspondant aux critères d'évaluation principaux et secondaires de l'étude CAFE
|
|
Placebo + CST
|
Dupilumab 300 mg Q2W + CST
| |
Patients randomisés
|
108
|
107
| |
EASI-75, répondeurs (en %)
|
29,6 %
|
62,6 %
| |
EASI, pourcentage de variation moyen (MC) par rapport aux valeurs initiales (+/- ET)
|
-46,6
(2,76)
|
-79,8
(2,59)
| |
Score de prurit sur l'EN, pourcentage de variation moyen (MC) par rapport aux valeurs initiales (+/- ET)
|
-25,4 % (3,39)
|
-53,9 % (3,14)
| |
SCORAD, pourcentage de variation moyen (MC) par rapport aux valeurs initiales (+/- ET)
|
-29,5 % (2,55)
|
-62,4 % (2,48)
| |
DLQI, variation moyenne (MC) par rapport aux valeurs initiales (ET)
|
-4,5
(0,49)
|
-9,5
(0,46)
|
Dans le sous-groupe de patients de l'étude CHRONOS de 52 semaines considéré comme similaire à la population de l'étude CAFE, un score EASI-75 a été obtenu chez 69,6 % des patients traités par dupilumab 300 mg Q2W contre 18,0 % des patients sous placebo à la semaine 16, et chez 52,4 % des patients traités par dupilumab 300 mg Q2W contre 18,6 % des patients sous placebo à la semaine 52. Dans ce sous-groupe, le pourcentage moyen de variation du score de prurit initial sur l'EN atteignait, à la semaine 16, -51,4 % dans le groupe sous dupilumab 300 mg Q2W contre -30,2 % sous placebo et, à la semaine 52, -54,8 % dans le groupe sous dupilumab 300 mg Q2W contre -30,9 % sous placebo.
Entretien et durabilité de la réponse (étude SOLO CONTINUE)
Pour évaluer l'entretien et la durabilité de la réponse, les sujets traités par dupilumab pendant 16 semaines dans le cadre des études SOLO 1 et SOLO 2 chez qui on avait obtenu un score IGA 0 ou 1 ou un score EASI-75 ont été soumis à une nouvelle procédure de randomisation dans le cadre de l'étude SOLO CONTINUE afin de recevoir soit un traitement supplémentaire par dupilumab, soit un placebo pendant 36 semaines, pour atteindre une durée cumulée de traitement de 52 semaines. Les critères d'évaluation ont été évalués aux semaines 51 ou 52.
Les co-critères d'évaluation principaux étaient l'écart entre le début (semaine 0) et la semaine 36 du pourcentage de variation du score EASI par rapport aux valeurs initiales des études SOLO 1 et SOLO 2, et le pourcentage de patients avec un score EASI-75 à la semaine 36 parmi les patients ayant un score EASI-75 initialement.
Les patients qui ont poursuivi le même schéma posologique que dans les études SOLO 1 et SOLO 2 (300 mg Q2S ou 300 mg QS; cette dernière posologie n'est pas approuvée en Suisse) ont montré l'effet optimal du maintien de la réponse clinique, alors que l'efficacité pour d'autres schémas posologiques a baissé de manière dose-dépendante.
Les critères d'évaluation principaux et secondaires de l'étude SOLO CONTINUE de 52 semaines sont résumés dans le tableau 4.
Tableau 4: Résultats correspondant aux critères d'évaluation principaux et secondaires de l'étude SOLO CONTINUE
|
|
Placebo
|
Dupilumab 300 mg
| |
|
N = 83
|
Q8S
N = 84
|
Q2S/QS
N = 169
| |
Co-critères d'évaluation principaux
| |
Écart moyen (MC) (+/- ET) entre le début et la semaine 36 du pourcentage de variation du score EASI par rapport aux valeurs initiales des études parents
|
21,7
(3,13)
|
6,8***
(2,43)
|
0,1***
(1,74)
| |
Pourcentage de patients avec un score EASI-75 à la semaine 36 parmi les patients ayant un score EASI-75 initialement, n (%)
|
24/79
(30,4 %)
|
45/82*
(54,9 %)
|
116/162***
(71,6 %)
| |
Critères d'évaluation secondaires clés
| |
Pourcentage de patients dont la réponse IGA à la semaine 36 ne s'écartait pas de plus d'1 point de la valeur initiale dans le sous-ensemble de patients ayant un score IGA (0,1) initialement, n (%)
|
18/63
(28,6)
|
32/64†
(50,0)
|
89/126***
(70,6)
| |
Pourcentage de patients avec un score IGA (0,1) à la semaine 36 dans le sous-ensemble de patients ayant un score IGA (0,1) initialement, n (%)
|
9/63
(14,3)
|
21/64†
(32,8)
|
68/126***
(54,0)
| |
Pourcentage de patients dont la valeur maximale du prurit sur l'EN augmentait de ≥3 points entre le début et la semaine 35 dans le sous-ensemble de patients ayant une valeur maximale du prurit sur l'EN ≤7 initialement, n (%)
|
56/80
(70,0)
|
45/81
(55,6)
|
57/168***
(33,9)
|
†p < 0,05; *p < 0,01; **p < 0,001; ***p ≤0,0001
Dans l'étude SOLO CONTINUE, une tendance à une production plus forte d'anticorps anti-médicament (ADA) émergeant en cours de traitement a été observée avec l'allongement des intervalles d'administration.
ADA émergeant en cours de traitement: QS: 1,2 %; Q2S: 4,3 %; Q4S: 6,0 %; Q8S: 11,7 %. Réponses de type AAM subsistant plus de 12 semaines: QS: 0,0 %; Q2S: 1,4 %; Q4S: 0,0 %; Q8S: 2,6 %.
Qualité de vie / résultats rapportés par les patients
Dans les études SOLO1 et SOLO2, dupilumab a significativement amélioré les symptômes liés à l'AD, la qualité de vie liée à la santé et les symptômes d'anxiété et de dépression, tels que mesurés par les scores totaux POEM, DLQI et HADS, respectivement, en comparaison au placebo.
Dans l'étude CHRONOS, dupilumab a significativement amélioré les symptômes liés à l'AD et la qualité de vie liée à la santé, tels que mesurés par les scores totaux POEM et DLQI, respectivement, à 16 semaines, comparé au placebo.
Dans SOLO1 et SOLO2, les variations moyennes (± SE) des moindres carrés des scores totaux POEM, DLQI et HADS entre le début et la 16ème semaine étaient significativement plus élevés chez les patients recevant dupilumab que chez ceux recevant un placebo (p <0,0001 pour toutes les comparaisons excepté HADS dans SOLO2, p <0,001), voir le tableau 5.
Dans l'étude CHRONOS, les variations moyennes (± SE) des moindres carrés des scores totaux POEM et DLQI entre le début et la 16ème semaine étaient significativement plus importants chez les patients dupilumab que chez ceux recevant le placebo (p <0,0001 pour toutes les comparaisons), voir Tableau 5.
Tableau 5. Résultats additionnels du critère d'évaluation secondaire de dupilumab avec ou sans TCS à la semaine 16.
|
|
Monotherapie à la semaine 16
|
Traitement concomitant par des TCS à la semaine 16
| |
|
SOLO1
|
SOLO2
|
CHRONOS
| |
|
Placebo
|
Dupilumab 300 mg Q2S
|
Placebo
|
Dupilumab 300 mg Q2S
|
Placebo
|
Dupilumab 300 mg Q2S
| |
Patients
randomisés
|
224
|
224
|
236
|
233
|
315
|
106
| |
POEM, variation moyenne (MC) par rapport aux valeurs initiales (ET)
|
-5,1
(0,67)
|
-11,6a
(0,49)
|
-3,3
(0,55)
|
-10,2a
(0,49)
|
-5,3
(0,41)
|
-12,7a
(0,64)
| |
DLQI, variation moyenne (MC) par rapport aux valeurs initiales (ET)
|
-5,3
(0,50)
|
-9,3a
(0,40)
|
-3,6
(0,50)
|
-9,3a
(0,38)
|
-5,8
(0,34)
|
-10,0a
(0,50)
|
MC = moindres carrés; ET = écart-type
a p < 0,0001
2. Efficacité et securité clinique chez les adolescents atteints de dermatite atopique:
L'efficacité et la sécurité du dupilumab en monothérapie chez les patients adolescents ont été évaluées dans une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo (AD-1526) chez 251 patients adolescents âgés de 12 à 17 ans atteints de dermatite atopique (DA) modérée à sévère définie par un score IGA ≥3 dans l'évaluation globale des lésions de la DA sur une échelle de sévérité de 0 à 4, un score EASI ≥16 sur une échelle de 0 à 72, et une surface corporelle atteinte minimale (BSA) ≥10 %. Les patients éligibles inclus dans cette étude présentaient auparavant une réponse insuffisante aux traitements topiques.
Les patients ont reçu:
1.une dose initiale de 400 mg du dupilumab (deux injections de 200 mg) le jour 1, suivie par 200 mg une fois toutes les deux semaines (1x/2 semaines) pour les patients ayant un poids corporel initial < 60 kg, ou une dose initiale de 600 mg de dupilumab (deux injections de 300 mg) le jour 1, suivie par 300 mg 1x/2 semaines pour les patients ayant un poids corporel initial ≥60 kg;
2.une dose initiale de 600 mg de dupilumab (deux injections de 300 mg) le jour 1, suivie par 300 mg une fois toutes les quatre semaines (1x/4 semaines) quel que soit le poids corporel initial ou
3.un placebo.
Le dupilumab a été administré par injection sous-cutanée (SC). En cas de symptômes intolérables, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l'investigateur. Les patients ayant reçu un traitement de secours ont été considérés comme non-répondeurs.
Dans cette étude, la moyenne d'âge était de 14,5 ans, le poids médian était de 59,4 kg, 41,0 % des sujets étaient de sexe féminin, 62,5 % étaient de race blanche, 15,1 % étaient asiatiques, et 12,0 % étaient de race noire. À l'inclusion dans l'étude, 46,2 % des patients avaient un score IGA de 3 (DA modérée), 53,8 % des patients avaient un score IGA à l'inclusion dans l'étude de 4 (DA sévère), l'atteinte moyenne de la BSA était de 56,5 %, et 42,4 % des patients avaient reçu un traitement antérieur par des immunosuppresseurs systémiques. Le score EASI (Eczema Area and Severity Index) moyen à l'inclusion dans l'étude était de 35,5, le score NRS de prurit moyen hebdomadaire à l'inclusion dans l'étude était de 7,6, le SCORAD (SCORing Atopic Dermatitis) moyen à l'inclusion dans l'étude était de 70,3, le score POEM (Patient Oriented Eczema Measure) moyen à l'inclusion dans l'étude était de 21,0, et le score CDLQI (Children Dermatology Life Quality Index [index de qualité de vie des enfants en dermatologie]) moyen à l'inclusion dans l'étude était de 13,6. Globalement, 92,0 % des patients présentaient au moins un état allergique coexistant; 65,6 % présentaient une rhinite allergique, 53,6 % de l'asthme, et 60,8 % des allergies alimentaires.
Les co-critères d'évaluation principaux étaient la proportion de patients avec un score IGA égal à 0 ou 1 , avec une amélioration d'au moins 2 points, et la proportion de patients avec un EASI-75 (amélioration d'au moins 75 % du score EASI) entre l'inclusion dans l'étude et la semaine 16. Les autres critères d'évaluation incluaient la proportion de sujets avec un EASI-50 ou EASI-90 (amélioration du score EASI d'au moins 50 % et 90 % respectivement par rapport à l'inclusion dans l'étude), la réduction du prurit mesuré par le score de prurit maximal NRS, et le pourcentage de variation du score de l'échelle SCORAD entre l'inclusion dans l'étude et la semaine 16. Les autres critères d'évaluation secondaires incluaient la variation moyenne entre l'inclusion dans l'étude et la semaine 16 des scores POEM et CDLQI.
Réponse clinique:
Les résultats de l'efficacité à la semaine 16 pour l'étude réalisée chez les adolescents atteints de dermatite atopique sont présentés dans le Tableau ci-dessous.
Tableau 6: Résultats de l'efficacité du dupilumab dans l'étude réalisée chez les adolescents atteints de dermatite atopique à la semaine 16 (population FAS)
|
|
AD-1526 (FAS)a
| |
|
Placebo
|
Dupilumab 200 mg (< 60 kg) et 300 mg (≥60 kg) 1x/2 sem.
| |
Patients randomisés
|
85a
|
82a
| |
IGA 0 ou 1b, % de répondeursc
|
2,4 %
|
24,4 %
| |
EASI-50, % de répondeursc
|
12,9 %
|
61,0 %
| |
EASI-75, % de répondeursc
|
8,2 %
|
41,5 %
| |
EASI-90, % de répondeursc
|
2,4 %
|
23,2 %
| |
EASI, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-23,6 %
(5,49)
|
-65,9 %
(3,99)
| |
SCORAD, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude
(+/- ET)
|
-17,6 %
(3,76)
|
-51,6 %
(3,23)
| |
Score NRS de prurit, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-19,0 %
(4,09)
|
-47,9%
(3,43)
| |
Score NRS de prurit (amélioration > 4 points), % de répondeursc
|
4,8 %
|
36,6 %
| |
BSA, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-11,7 %
(2,72)
|
-30,1 %
(2,34)
| |
CDLQI, variation moyenne (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-5,1
(0,62)
|
-8,5
(0,50)
| |
CDLQI (amélioration ≥6 points), % de répondeurs
|
19,7 %
|
60,6 %
| |
POEM, variation moyenne (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-3,8
(0,96)
|
-10,1
(0,76)
| |
POEM (amélioration ≥ 6 points), % de répondeurs
|
9,5 %
|
63,4 %
|
a La population FAS (Full Analysis Set) comprend tous les patients randomisés.
b Un répondeur est défini comme un sujet avec un score IGA de 0 ou 1 («blanchi» ou «presque blanchi») avec une réduction ≥2 points sur l'échelle IGA 0 - 4.
c Les patients ayant reçu un traitement de secours ou avec des données manquantes ont été considérés comme non-répondeurs (58,8 % et 20,7 % respectivement dans les bras placebo et Dupixent).
Toutes les valeurs p < 0,0001.
Une proportion plus importante de patients randomisés dans le groupe placebo a eu besoin d'un traitement de secours (dermocorticoïdes topiques, corticostéroïdes systémiques ou immunosuppresseurs non-stéroïdiens systémiques) par comparaison au groupe dupilumab (58,8 % et 20,7 % respectivement).
Une proportion significativement plus importante de patients randomisés dans le groupe dupilumab a présenté une amélioration rapide du score NRS de prurit par comparaison au placebo (définie comme une amélioration > 4 points dès la semaine 4; valeurs nominales de p < 0,001) et la proportion de patients ayant une amélioration du score NRS de prurit a continué à augmenter pendant toute la période de traitement (voir figure ci-dessous). L'amélioration du score NRS de prurit a été accompagnée d'une amélioration des signes objectifs de dermatite atopique.
Proportion de patients adolescents avec une amélioration ≥4 points du score NRS de prurit dans l'étude AD-1526a (FAS)b
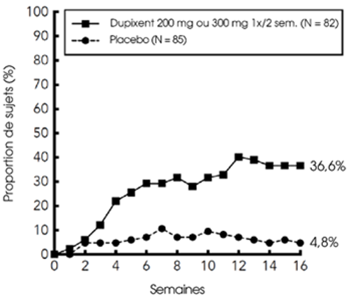
a Dans les analyses principales des critères d'évaluation de l'efficacité, les sujets ayant reçu un traitement de secours ou ayant des données manquantes ont été considérés comme non-répondeurs.
b La population FAS (Full Analysis Set) inclut tous les sujets randomisés.
Dans le groupe dupilumab, une amélioration significative a été observée en termes de symptômes rapportés par le patient et d'impact de la DA sur le sommeil et la qualité de vie liée à la santé, tel que mesuré par les scores POEM, SCORAD et CDLQI à 16 semaines par comparaison au placebo.
L'efficacité à long terme du dupilumab chez les patients adolescents atteints de DA modérée à sévère qui avaient participé à des essais cliniques antérieurs portant sur le dupilumab a été évaluée dans une étude d'extension en ouvert (AD-1434). Les données d'efficacité issues de cette étude suggèrent que le bénéfice clinique obtenu à la semaine 16 a été maintenu jusqu'à la semaine 52.
3. Efficacité et sécurité cliniques chez les enfants (6 à 11 ans) souffrant de dermatite atopique
L'efficacité et la sécurité de l'utilisation du dupilumab en même temps que les CST chez des sujets pédiatriques ont été évaluées dans un essai multicentrique, randomisé, en double aveugle et contrôlé par placebo (AD-1652) chez 367 sujets âgés de 6 à 11 ans, la dermatite atopique étant définie par un score IGA de 4 (échelle de 0 à 4), un score EASI ≥21 (échelle de 0 à 72) et une BSA minimale de ≥15%. Les sujets admissibles inscrits à cet essai avaient auparavant une réponse inadéquate au médicament topique. L'inclusion a été stratifiée en fonction du poids de base (<30 kg; ≥30 kg).
Les sujets du groupe dupilumab Q4W + CST ont reçu une dose initiale de 600 mg le jour 1, suivie de 300 mg Q4W de la semaine 4 à la semaine 12, quel que soit leur poids. Les sujets du groupe dupilumab Q2W + CST ayant un poids de base <30 kg ont reçu une dose initiale de 200 mg le jour 1, suivie de 100 mg Q2W de la semaine 2 à la semaine 14, et les sujets ayant un poids de base de ≥30 kg ont reçu une dose initiale de 400 mg le jour 1, suivie de 200 mg Q2W de la semaine 2 à la semaine 14. Les sujets ont été autorisés à recevoir un traitement de secours à la discrétion de l'enquêteur. Les sujets qui ont reçu un traitement de secours ont été considérés comme non-répondants.
Dans l'étude AD-1652, l'âge moyen était de 8,5 ans, le poids médian était de 29,8 kg, 50 % des sujets étaient des femmes, 69,2 % étaient des Blancs, 16,7 % des Noirs et 7,6 % des Asiatiques. Au départ, l'implication moyenne de la BSA était de 57,6 %, et 16,9 % avaient déjà reçu des immunosuppresseurs systémiques non stéroïdiens. En outre, au départ, le score moyen de l'EASI était de 37,9 et la moyenne hebdomadaire du score quotidien de la pire démangeaison était de 7,8 sur une échelle de 0 à 10, le score SCORAD moyen à l'inclusion dans l'étude était de 73,6, le score POEM à l'inclusion dans l'étude était de 20,9 et le CDLQI moyen à l'inclusion dans l'étude était de 15,1. Dans l'ensemble, 91,7% des sujets avaient au moins une affection comorbide allergique; 64,4% avaient des allergies alimentaires, 62,7% avaient d'autres allergies, 60,2% avaient une rhinite allergique et 46,7% étaient asthmatiques.
Le principal critère d'évaluation était la proportion de sujet présentant un IGA 0 (clair) ou 1 (presque clair) à la semaine 16. Les autres résultats évalués comprenaient la proportion de sujets présentant un EASI-75 ou un EASI-90 (amélioration d'au moins 75 % ou 90 % de l'EASI par rapport au niveau de référence, respectivement), le pourcentage de changement du score EASI entre l'inclusion dans l'étude et la semaine 16 et la réduction des démangeaisons telle que mesurée par le score NRS de prurit (amélioration ≥4 points). Les autres critères d'évaluation secondaires incluaient la variation moyenne entre l'inclusion dans l'étude et la semaine 16 des scores POEM et CDLQI.
Le tableau 7 présente les résultats par tranche de poids de base pour les schémas posologiques approuvés.
Tableau 7: Résultats d'efficacité de DUPIXENT avec CST concomitant dans AD-1652 à la semaine 16 (population FAS)a
|
|
DUPIXENT
300 mg Q4Wd
+ CST
(N=61)
|
Placebo
+ CST
(N=61)
|
DUPIXENT
200 mg Q2We
+ CST
(N=59)
|
Placebo
+ CST
(N=62)
| |
<30 kg
|
<30 kg
|
≥30 kg
|
≥30 kg
| |
IGA 0 ou 1 b, % de répondeurs c
|
29,5%
|
13,1%
|
39,0%
|
9,7%
| |
EASI-50, % de répondeurs c
|
95.1%
|
42,6%
|
86,4%
|
43,5%
| |
EASI-75, % de répondeurs c
|
75,4%
|
27,9%
|
74,6%
|
25,8%
| |
EASI-90, % de répondeurs c
|
45,9%
|
6,6%
|
35,6%
|
8,1%
| |
EASI, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-84,3%
(3,08)
|
-49,1%
(3,30)
|
-80,4%
(3,61)
|
-48,3%
(3,63)
| |
SCORAD, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude
(+/- ET)
|
-65,3%
(2,87)
|
-28,9%
(3,05)
|
-62,7%
(3,14)
|
-30,7%
(3,28)
| |
Score NRS de prurit, variation moyenne en % (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-55,1%
(3,94)
|
-27,0%
(4,24)
|
-58,2%
(4,01)
|
-25,0%
(3,95)
| |
Score NRS de prurit (amélioration ≥4 points), % de répondeurs c
|
54,1%
|
11,7%
|
61,4%
|
12,9%
| |
CDLQI, variation moyenne (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-11,5
(0,69)
|
-7,2
(0,76)
|
-9,8
(0,63)
|
-5,6
(0,66)
| |
CDLQI (amélioration ≥6 points), % de répondeurs
|
81,8%
|
48,3%
|
80,8%
|
35,8%
| |
POEM, variation moyenne (MC) par rapport à l'inclusion dans l'étude (+/- ET)
|
-14,0
(0,95)
|
-5,9
(1,04)
|
-13,6
(0,90)
|
-4,7
(0,91)
|
a La population FAS (Full Analysis Set) comprend tous les sujets randomisés.
b Le sujet répondant a été défini comme un sujet ayant un IGA 0 ou 1 («clair» ou «presque clair»).
c Les sujets ayant reçu un traitement de secours ou pour lesquels des données manquaient ont été considérés comme des non-répondants.
d Au jour 1, les sujets ont reçu 600 mg de dupilumab.
e Au jour 1, les sujets ont reçu 200 mg (poids de référence <30 kg) ou 400 mg (poids de référence ≥30 kg) de DUPIXENT.
Une plus grande proportion de sujets randomisés avec dupilumab + CST ont obtenu une amélioration du score NRS de prurit par rapport au placebo + CST (défini comme une amélioration de ≥4 points à la semaine 16). Voir la figure 5.
Figure 5: Proportion de sujets pédiatriques avec une amélioration du score NRS de prurit ≥4 points à la semaine 16 en AD-1652a (FAS)b
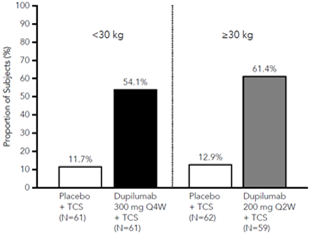
a Dans les analyses primaires des paramètres d'efficacité, les sujets qui ont reçu un traitement de secours ou pour lesquels il manquait des données ont été considérés comme des non-répondants.
b La population FAS (Full Analysis Set) comprend tous les sujets randomisés.
Dans le groupe dupilumab, une amélioration significative a été observée en termes de symptômes rapportés par le patient et d'impact de la DA sur le sommeil et la qualité de vie liée à la santé, tel que mesuré par les scores POEM, SCORAD et CDLQI à 16 semaines par comparaison au placebo.
L'efficacité à long terme du dupilumab + TCS chez les patients pédiatriques atteints de dermatite atopique qui avaient a participé aux précédents essais cliniques du dupilumab + TCS a été évalué dans le cadre d'une extension ouverte étude (AD-1434). Dans cet essai, les données non contrôlées disponibles à la semaine 52 pour 45 patients ont démontré que la proportion de patients présentant des améliorations cliniquement pertinentes pour les scores IGA ou EASI se maintenait au fil du temps.
4. Efficacité et sécurité clinique chez les adultes et adolescents atteints d'asthme:
Le programme de développement de l'asthme incluait trois études multicentriques, randomisées, en double aveugle, contrôlées contre placebo, en groupes parallèles (DRI12544, QUEST et VENTURE) d'une durée de traitement de 24 à 52 semaines, qui incluaient un total de 2 888 patients (âgés de 12 ans ou plus).
Les patients ont été inclus, quel que soit leur taux d'éosinophiles dans le sang ou des niveaux de FeNO ou d'IgE.
Dans les études DRI12544 et QUEST, les analyses de sous-groupes prédéterminées incluaient des éosinophiles sanguins ≥150 et ≥300 cellules/µl.
L'étude DRI12544 était une étude de recherche de dose de 24 semaines, qui incluait 776 patients (âgés de 18 ans et plus). Le dupilumab a été évalué comparativement au placebo chez des patients adultes présentant un asthme modéré à sévère et recevant une dose moyenne à élevée de corticoïdes inhalés et un bêta-agoniste de longue durée d'action. Le critère d'évaluation principal était la variation du VEMS (L) entre l'inclusion dans l'étude et la semaine 12 chez les sujets avec des niveaux de base d'eosinophiles ≥300 cells/mcL. Le taux annualisé d'exacerbations sévères d'asthme a également été évalué au cours de la période contrôlée contre placebo de 24 semaines. Les résultats ont été évalués dans la population globale (sans restriction sur le taux minimal d'éosinophiles) et dans les sous-groupes en fonction de la numération d'éosinophiles sanguins à l'inclusion dans l'étude.
L'étude QUEST était une étude de confirmation de 52 semaines, qui comprenait 1 902 patients (âgés de 12 ans ou plus). Le dupilumab a été évalué comparativement au placebo chez 107 adolescents et 1 795 patients adultes présentant un asthme persistant et recevant une corticothérapie inhalée à dose moyenne ou élevée et un second traitement de fond. Les patients nécessitant un troisième traitement de fond pouvaient également être inclus dans cet essai. Les patients ont été randomisés pour recevoir soit 200 mg de dupilumab toutes les deux semaines (N=631), soit 300 mg de dupilumab toutes les deux semaines (N=633) (ou dans le groupe placebo correspondant pour le 200 mg (N=317) ou le 300 mg (N=321)) après administration d'une dose initiale de 400 mg, 600 mg ou de placebo, respectivement. Les critères d'évaluation principaux étaient le taux annualisé d'exacerbations sévères survenant au cours de la période contrôlée contre placebo de 52 semaines et la variation du VEMS mesuré avant administration du bronchodilatateur entre l'inclusion dans l'étude et la semaine 12 dans la population globale (sans restriction sur le nombre minimal d'éosinophiles).
Des critères seconfaire supplémentires incluaient le taux d'exacerbation sévère annualisés et FEV1 dans les sous-groupes en fonction du taux d'éosinophiles sanguins.
L'étude VENTURE était une étude de réduction des corticostéroïdes oraux de 24 semaines conduites chez 210 patients présentant un asthme, sans restriction sur les taux des biomarqueurs de l'inflammation de type 2 à l'inclusion, et qui nécessitaient une corticothérapie orale quotidienne en complément de l'utilisation régulière d'une dose élevée de corticostéroïdes inhalés associés à un autre traitement de fond. Après avoir optimisé la dose de corticostéroïdes oraux (CSO) pendant la période de sélection, les patients ont reçu 300 mg de dupilumab (n = 103) ou un placebo (n = 107) une fois toutes les deux semaines pendant 24 semaines après une dose initiale de 600 mg ou un placebo. Le traitement de fond initial était maintenu pendant l'étude. La dose de CSO était réduite toutes les 4 semaines pendant la phase de réduction des CSO (semaines 4 à 20), tant que l'asthme restait contrôlé. Le critère d'évaluation principal était le pourcentage de réduction de la dose de corticostéroïdes oraux évaluée dans la population globale, correspondant à la différence entre la dose de corticostéroïdes oraux lors des semaines 20 à 24, tout en maintenant le contrôle de l'asthme avec une dose de corticostéroïdes oraux préalablement optimisée optimisée dans la population générale (sans restriction du nombre minimum d'éosinophiles de base dans le sang) Des critères secondaires additionnels ont inclu le taux annualisé d'événements d'exacerbation sévère pendant la période de traitement et le taux de répondeurs dans les scores ACQ-5 et AQLQ (S).
Les données démographiques et les caractéristiques de l'asthme à l'inclusion de ces 3 études sont fournies dans le Tableau 8 ci-dessous.
Tableau 8: Données démographiques et caractéristiques de l'asthme à l'inclusion dans les études
|
Paramètres
|
DRI12544
(n = 776)
|
QUEST
(n = 1 902)
|
VENTURE
(n = 210)
| |
Moyenne d'âge (ans) (ET)
|
48,6 (13,0)
|
47,9 (15,3)
|
51,3 (12,6)
| |
% de femmes
|
63,1
|
62,9
|
60,5
| |
% de race blanche
|
78,2
|
82,9
|
93,8
| |
Durée de l'asthme (ans), moyenne ± ET
|
22,03 (15,42)
|
20,94 (15,36)
|
19,95 (13,90)
| |
Patients n'ayant jamais fumé, (%)
|
77,4
|
80,7
|
80,5
| |
Moyenne des exacerbations l'année précédente ± ET
|
2,17 (2,14)
|
2,09 (2,15)
|
2,09 (2,16)
| |
Utilisation de CSI à dose élevée (%)a
|
49,5
|
51,5
|
88,6
| |
VEMS (L) avant une dose lors de la visite à l'inclusion ± ET
|
1,84 (0,54)
|
1,78 (0,60)
|
1,58 (0,57)
| |
Pourcentage de variation moyenne du VEMS à l'inclusion dans l'étude (%) (± ET)
|
60,77 (10,72)
|
58,43 (13,52)
|
52,18 (15,18)
| |
% de réversibilité (± ET)
|
26,85 (15,43)
|
26,29 (21,73)
|
19,47 (23,25)
| |
Score ACQ-5 moyen (± ET)
|
2,74 (0,81)
|
2,76 (0,77)
|
2,50 (1,16)
| |
Score AQLQ moyen (± ET)
|
4,02 (1,09)
|
4,29 (1,05)
|
4,35 (1,17)
| |
Antécédents médicaux de maladie atopique % global
(DA %, PN %, RA %)
|
72,9
(8,0, 10,6, 61,7)
|
77,7
(10,3, 12,7, 68,6)
|
72,4
(7,6, 21,0, 55,7)
| |
FeNO moyen en ppb (± ET)
|
39,10 (35,09)
|
34,97 (32,85)
|
37,61 (31,38)
| |
% de patients avec FeNO ppb
| |
≥25
≥50
|
49,9
21,6
|
49,6
20,5
|
54,3
25,2
| |
IgE totale moyenne en UI/ml (± ET)
|
435,05
(753,88)
|
432,40
(746,66)
|
430,58
(775,96)
| |
Numération moyenne d'éosinophiles à l'inclusion dans l'étude (± ET) en cellules/µl
|
350 (430)
|
360 (370)
|
350 (310)
| |
% de patients avec EOS
| |
≥150 cellules/µl
|
77,8
|
71,4
|
71,4
| |
≥300 cellules/µl
|
41,9
|
43,7
|
42,4
|
CSI = corticostéroïdes inhalés; VEMS = volume expiratoire maximal par seconde; ACQ-5 = Asthma Control Questionnaire-5 (questionnaire sur le contrôle de l'asthme à 5 items); AQLQ = Asthma Quality of Life Questionnaire (questionnaire sur la qualité de vie liée à l'asthme); DA = dermatite atopique; PN = polypose nasale; RA = rhinite allergique; FeNO = fraction de monoxyde d'azote expiré; EOS= éosinophiles sanguins
a La population des études du dupilumab dans l'asthme incluait des patients recevant des doses moyenne ou élevée de CSI. La dose moyenne de CSI était définie en terme d'équivalent à 500 mg de fluticasone ou équivalent par jour.
Exacerbations
Les études DRI12544 et QUEST ont évalué la fréquence des exacerbations sévères de l'asthme définies comme une détérioration de l'asthme nécessitant l'utilisation de corticostéroïdes systémiques pendant au moins 3 jours ou une hospitalisation ou une visite aux urgences en raison d'un asthme nécessitant des corticostéroïdes systémiques.
Dans la population d'analyse primaire (sujets avec un taux d'éosinophiles sanguins de base ≥300 cellules / ml dans l'essai DRI12544 et la population globale dans l'essai QUEST), les sujets recevant DUPIXENT 200 mg ou 300 mg Q2W avaient des réductions significatives du taux d'exacerbations asthmatique par rapport au placebo. Dans la population globale de l'étude QUEST, le taux d'exacerbations sévères était de 0,46 et 0,52 pour DUPIXENT 200 mg Q2W et 300 mg Q2W, respectivement, par rapport aux taux de placebo de 0,87 et 0,97.
Le rapport des taux d'exacerbations sévères par rapport au placebo était de 0,52 (IC à 95%: 0,41, 0,66) et 0,54 (IC à 95%: 0,43, 0,68) pour DUPIXENT 200 mg Q2W et 300 mg Q2W, respectivement. Les résultats chez les sujets avec une numération des éosinophiles sanguins au départ ≥150 cellules / mcL et ≥300 cellules / mcL dans les essais DRI12544 et QUEST sont présentés dans le tableau 9.
Les taux de réponse par les éosinophiles sanguins de base pour l'étude QUEST sont illustrés à la figure 6. Les analyses de sous-groupes prédéfinies des essais DRI12544 et QUEST ont montré qu'il y avait des réductions plus importantes des exacerbations sévères chez les sujets ayant des niveaux d'éosinophiles sanguins de base plus élevés. Dans l'essai QUEST, les réductions des exacerbations étaient significatives dans le sous-groupe de sujets avec des éosinophiles sanguins initiaux ≥150 cellules / mcL. Chez les sujets dont la numération éosinophile dans le sang était < à 150 cellules / ml, des taux d'exacerbation sévère similaires ont été observés entre DUPIXENT et le placebo.
Dans l'étude QUEST, le rapport des taux d'exacerbations entraînant des hospitalisations et / ou des visites aux urgences par rapport au placebo était de 0,53 (IC à 95%: 0,28, 1,03) et de 0,74 (IC à 95%: 0,32, 1,70) avec DUPIXENT 200 mg ou 300 mg Q2W, respectivement.
Tableau 9: Taux d'exacerbation sévère dans DRI12544 et QUEST
|
Traitement
|
Niveau sanguin EOS
| |
|
≥150 cellules/mcL
|
≥300 cellules/mcL
| |
Exacerbations par An
|
% Reduction
|
Exacerbations par An
|
%
Reduction
| |
N
|
Rate
(95% CI)
|
Rate
Ratio (95%CI)
|
N
|
Rate
(95% CI)
|
Rate
Ratio (95%CI)
| |
Toutes les exacerbations sévères
| |
DRI12544 study
| |
Dupilumab 200 mg Q2W
|
120
|
0.29
(0.16, 0.53)
|
0.28a
(0.14, 0.55)
|
72%
|
65
|
0.30
(0.13, 0.68)
|
0.29c
(0.11, 0.76)
|
71%
| |
Dupilumab 300 mg
Q2W
|
129
|
0.28
(0.16, 0.50)
|
0.27b
(0.14, 0.52)
|
73%
|
64
|
0.20
(0.08, 0.52)
|
0.19d
(0.07, 0.56)
|
81%
| |
Placebo
|
127
|
1.05
(0.69, 1.60)
|
|
|
68
|
1.04
(0.57, 1.90)
|
|
| |
QUEST study
| |
Dupilumab 200 mg
Q2W
|
437
|
0.45
(0.37, 0.54)
|
0.44e
(0.34,0.58)
|
56%
|
264
|
0.37
(0.29, 0.48)
|
0.34e
(0.24,0.48)
|
66%
| |
Placebo
|
232
|
1.01
(0.81, 1.25)
|
|
|
148
|
1.08
(0.85, 1.38)
|
|
| |
Dupilumab 300 mg
Q2W
|
452
|
0.43
(0.36, 0.53)
|
0.40 e
(0.31,0.53)
|
60%
|
277
|
0.40
(0.32, 0.51)
|
0.33e
(0.23,0.45)
|
67%
| |
Placebo
|
237
|
1.08
(0.88, 1.33)
|
|
|
142
|
1.24
(0.97, 1.57)
|
|
|
Figure 6: Risque relatif dans le taux d'événements annualisé d'exacerbations sévères à travers la numération des éosinophiles sanguins de base (cellules / mcL) dans QUEST.
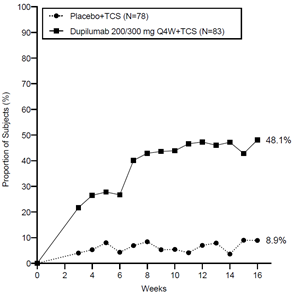
Le délai avant la première exacerbation était plus long pour les sujets recevant DUPIXENT que pour le placebo dans l'étude QUEST (Figure 7).
Figure 7: Courbe d'incidence de Kaplan Meier pour le délai avant la première exacerbation sévère chez les sujets avec des éosinophiles sanguins de base ≥300 cellules / mcL (QUEST)a
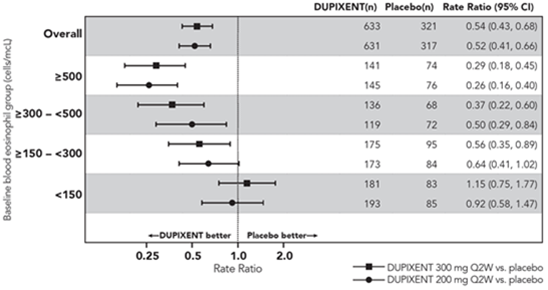
a Au moment de la fermeture de la base de données, tous les patients n'avaient pas terminé la semaine 52.
Fonction pulmonaire
Des augmentations significatives du VEMS pré-bronchodilatateur ont été observées à la semaine 12 pour les essais DRI12544 et QUEST dans les populations analysées primaires (sujets avec un taux éosinophile sanguin de base ≥300 cellules / mcL dans l'essai DRI12544 et la population globale dans l'essai QUEST). Dans la population globale de l'étude QUEST, la variation moyenne du FEV1 LS par rapport à la valeur initiale était de 0,32 L (21%) et 0,34 L (23%) pour DUPIXENT 200 mg Q2W et 300 mg Q2W, respectivement, par rapport à la moyenne placebo de 0,18 L (12%) et 0,21 L (14%).
La différence moyenne de traitement par rapport au placebo était de 0,14 L (IC à 95%: 0,08, 0,19) et de 0,13 L (IC à 95%: 0,08, 0,18) pour DUPIXENT 200 mg Q2W et 300 mg Q2W, respectivement. Les résultats chez les sujets ayant un taux d'éosinophiles sanguins au départ ≥150 cellules / mcL et ≥300 cellules / mcL dans les essais DRI12544 et QUEST sont présentés dans le tableau 10. Les améliorations du VEMS par les éosinophiles sanguins au départ pour l'essai QUEST sont illustrées à la figure 8. L' analyse en sous-groupe des essais DRI12544 et QUEST a démontré une plus grande amélioration chez les sujets ayant des éosinophiles sanguins de base plus élevés.
Tableau 10 - Changement moyen par rapport à la valeur initiale du VEMS pré-bronchodilatateur à la semaine 12 dans DRI12544 et QUEST.
|
Traitement
|
Niveau sanguin de base EOS
| |
|
≥150 cellules /mcL
|
≥300 cellules /mcL
| |
N
|
LS Mean Δ From baseline
L (%)
|
LS Mean
Difference vs. placebo (95% CI)
|
N
|
LS mean Δ From baseline
L (%)
|
LS Mean
Difference vs. placebo (95% CI)
| |
Etude DRI12544
| |
Dupilumab200 mg Q2W
|
120
|
0.32 (18.25)
|
0.23a
(0.13, 0.33)
|
65
|
0.43 (25.9)
|
0.26c
(0.11, 0.40)
| |
Dupilumab300 mg Q2W
|
129
|
0.26 (17.1)
|
0.18b
(0.08, 0.27)
|
64
|
0.39 (25.8)
|
0.21d
(0.06, 0.36)
| |
Placebo
|
127
|
0.09 (4.36)
|
|
68
|
0.18 (10.2)
|
| |
Etude QUEST
| |
Dupilumab200 mg Q2W
|
437
|
0.36 (23.6)
|
0.17e
(0.11, 0.23)
|
264
|
0.43 (29.0)
|
0.21e
(0.13, 0.29)
| |
Placebo
|
232
|
0.18 (12.4)
|
|
148
|
0.21 (15.6)
|
| |
Dupilumab300 mg Q2W
|
452
|
0.37 (25.3)
|
0.15e
(0.09, 0.21)
|
277
|
0.47 (32.5)
|
0.24e
(0.16, 0.32)
| |
Placebo
|
237
|
0.22 (14.2)
|
|
142
|
0.22 (14.4)
|
|
a p-value <0.0001, bp-value = 0.0004, cp-value = 0.0008, dp-value = 0.0063, ep-value <0.0001
Figure 8: Différence moyenne de changement LS par rapport à la valeur initiale par rapport au placebo à la semaine 12 dans le pré-bronchodilatateur FEV1 selon le nombre d'éosinophiles sanguins de base (cellules / mcL) dans QUEST.

Les variations moyennes du VEMS au fil du temps dans QUEST sont illustrées à la Figure 9.
Figure 9: Variation moyenne par rapport à la valeur de base dans le pré-bronchodilatateur FEV1 (L) au fil du temps chez les sujets présentant des éosinophiles sanguins initiaux ≥300 cellules / mcL (QUEST).
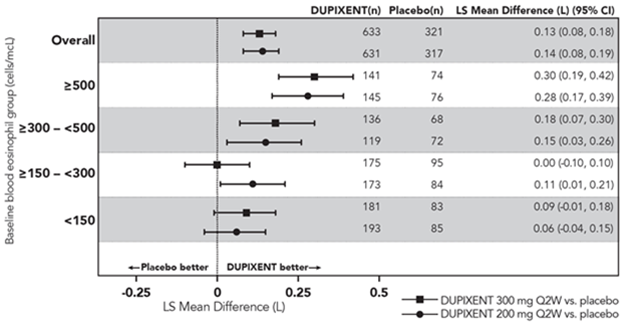
Critères secondaires additionels:
Qualité de vie/Résultats déclarés par le patient dans l'asthme
ACQ-5 et AQLQ (S) ont été évalués dans QUEST à 52 semaines. Le taux de répondeurs a été défini comme une amélioration du score de 0,5 ou plus (échelle de 0-6 pour ACQ-5 et 1-7 pour AQLQ (S)).
·Le taux de répondeurs ACQ-5 pour Dupilumab 200 mg et 300 mg Q2W dans la population globale était de 69% vs 62% placebo (odds ratio 1,37; IC 95%: 1,01, 1,86) et 69% vs 63% placebo (odds ratio 1,28 IC 95%: 0,94, 1,73), respectivement; et les taux de répondeurs AQLQ (S) étaient respectivement de 62% vs 54% placebo (odds ratio 1,61; IC 95%: 1,17, 2,21) et 62% vs 57% placebo (odds ratio 1,33; IC 95%: 0,98, 1,81).
·Le taux de répondeurs ACQ-5 pour Dupilumab 200 mg et 300 mg Q2W chez les sujets avec des éosinophiles sanguins à la base ≥300 cellules / ml était de 75% vs 67% placebo (odds ratio: 1,46; IC 95%: 0,90, 2,35) et 71% vs placebo à 64% (rapport de cotes: 1,39; IC à 95%: 0,88, 2,19), respectivement; et les taux de répondeurs AQLQ (S) étaient de 71% vs 55% placebo (odds ratio: 2,02; 95% IC: 1,24, 3,32) et 65% vs 55% placebo (odds ratio: 1,79; 95% IC: 1,13, 2,85) , respectivement.
Étude de réduction des corticostéroïdes oraux (VENTURE)
VENTURE a évalué l'effet de dupilumab sur la réduction de l'utilisation des corticostéroïdes oraux d'entretien. La dose moyenne initiale de corticostéroïdes oraux était de 12 mg dans le groupe placebo et de 11 mg dans le groupe recevant dupilumab. Le critère d'évaluation principal était le pourcentage de réduction par rapport à la valeur initiale de la dose finale de corticostéroïdes oraux à la semaine 24 tout en maintenant le contrôle de l'asthme.
Par rapport au placebo, les sujets recevant dupilumab ont obtenu des réductions plus importantes de la dose quotidienne de corticostéroïdes par voie orale, tout en maintenant un contrôle de l'asthme. Le pourcentage moyen de réduction de la dose quotidienne d'OCS par rapport à l'inclusion était de 70% (médiane 100%) chez les sujets recevant dupilumab (IC 95%: 60%, 80%) par rapport à 42% (médiane 50%) chez les sujets recevant le placebo (IC 95%): 33%, 51%). Des réductions de 50% ou plus de la dose d'OCS ont été observées chez 82 (80%) sujets recevant dupilumab contre 57 (53%) chez ceux recevant le placebo. La proportion de sujets avec une dose finale moyenne inférieure à 5 mg aux semaines 24 était de 72% pour dupilumab et de 37% pour le placebo (rapport de cotes 4,48 IC 95%: 2,39, 8,39). Un total de 54 (52%) sujets recevant dupilumab versus 31 (29%) sujets dans le groupe placebo ont eu une réduction de 100% de leur dose d'OCS.
Dans cette étude de 24 semaines, les exacerbations de l'asthme (définies comme une augmentation temporaire de la dose de corticostéroïdes oraux pendant au moins 3 jours) étaient plus faibles chez les sujets recevant dupilumab par rapport à ceux recevant le placebo (taux annualisé 0,65 et 1,60 pour le groupe dupilumab et le placebo, respectivement; le rapport de taux de 0,41 [IC à 95% 0,26, 0,63]) et l'amélioration du VEMS pré-bronchodilatateur de la ligne de base à la semaine 24 étaient plus importants chez les sujets recevant dupilumab que chez ceux recevant le placebo (différence moyenne LS pour dupilumab versus placebo de 0,22 L [95% IC: 0,09 à 0,34 L]). Les effets sur la fonction pulmonaire et sur la réduction des stéroïdes et des exacerbations par voie orale étaient similaires, indépendamment des taux d'éosinophiles sanguins de base. L'ACQ-5 et l'AQLQ (S) ont également été évalués dans VENTURE et ont montré des améliorations similaires à celles de QUEST.
Etude d'extension à long terme (TRAVERSE)
La sécurité à long terme du dupilumab a été évaluée dans l'étude d'extension ouverte (TRAVERSE) chez 2 193 adultes et 89 adolescents souffrant d'asthme modéré à sévère, incluant 185 adultes souffrant d'asthme dépendant aux corticostéroïdes oraux, qui avaient participé aux précédents essais cliniques du dupilumab (DRI12544, QUEST et VENTURE) (voir section «Effets indésirables»). L'efficacité, mesurée comme critère d'évaluation secondaire, était similaire aux résultats observés dans les études pivots et s'est maintenue jusqu'à 96 semaines. Chez les adultes souffrant d'asthme dépendant aux corticostéroïdes oraux, on a observé une réduction maintenue des exacerbations et une amélioration de la fonction pulmonaire jusqu'à 96 semaines, malgré la diminution ou l'arrêt de la dose de corticostéroïdes oraux.
Informations complémentaires
Population pédiatrique
Dermatite atopique:
La sécurité et l'efficacité du dupilumab ont été établies chez des patients pédiatriques de 6 ans et plus souffrant de dermatite atopique modérée à sévère.
L'utilisation du dupilumab dans ce groupe d'âge est soutenue par l'étude AD 1526 qui a inclus 251 adolescents âgés de 12 à 17 ans atteints de dermatite atopique modérée à sévère et l'étude AD-1652 qui a inclus 367 enfants âgés de 6 à 11 ans atteints de dermatite atopique sévère. La sécurité et l'efficacité étaient généralement cohérentes entre les patients pédiatriques et adultes.
L'utilisation du dupilumab est également supportée par l'étude AD-1434, une étude d'extension en ouverte qui a recruté des sujets ayant terminé l'AD 1526 et l'AD-1652. L'étude AD-1434 a inclus 136 adolescents de l'étude AD 1526 et 110 enfants de l'étude AD-1652 souffrant de dermatite atopique modérée au moment de l'inclusion dans l'étude d'extension. L'étude AD-1434 comprenait 64 adolescents de l'étude AD-1526 et 72 enfants de l'étude AD-1652 atteints de dermatite atopique sévère au moment de l'inclusion. Aucun nouveau signal de sécurité n'a été identifié dans l'étude AD-1434 (voir section «Effets indésirables»).
La sécurité et l'efficacité chez les patients pédiatriques de moins de 6 ans atteints de dermatite atopique n'ont pas été établies.
Asthme:
Au total, 107 adolescents âgés de 12 à 17 ans présentant un asthme modéré à sévère ont été inclus dans l'étude QUEST et ont reçu 200 mg (n = 21) ou 300 mg (n = 18) de dupilumab (ou 200 mg [n = 34] ou 300 mg [n = 34] du placebo correspondant) toutes les deux semaines. L'efficacité a été observée chez les adolescents et les adultes en termes d'exacerbations d'asthme sévère et de fonction pulmonaire. Les deux dosages à 200 mg et à 300 mg administrés toutes les deux semaines ont montré des améliorations significatives du VEMS (différence moyenne des MC par rapport à l'inclusion à la semaine 12) de 0,36 L et 0,27 L, respectivement. Une réduction du taux d'exacerbations sévères était observée à la dose de 200 mg administrée toutes les 2 semaines, comme pour l'adulte. La sécurité et l'efficacité n'ont pas été établies chez les patients pédiatriques (âgés de < 12 ans) asthmatiques sévères. Le profil d'effets indésirables chez les adolescents était généralement similaire à celui des adultes.
5. Efficacité clinique dans la polypose naso-sinusienne
Le programme de développement dans la polypose naso-sinusienne comprenait deux études randomisées, en double aveugle, en groupes parallèles, multicentriques et contrôlées contre placebo (SINUS-24 et SINUS 52) chez 724 patients âgés de 18 ans et plus avec un traitement de fond par des corticostéroïdes administré par voie nasale. Ces études ont inclus des patients présentant une polypose naso-sinusienne sévère malgré une chirurgie naso-sinusienne ou un traitement préalable, ou des patients non éligibles à un traitement par des corticostéroïdes systémiques au cours des 2 dernières années. Un traitement de secours par des corticostéroïdes systémiques ou par chirurgie au cours des études était autorisé sur décision de l'investigateur.
Dans l'étude SINUS-24, un total de 276 patients a été randomisé pour recevoir 300 mg de dupilumab (n = 143) ou un placebo (n = 133) toutes les deux semaines pendant 24 semaines. Dans l'étude SINUS-52, 448 patients ont été randomisés pour recevoir 300 mg de dupilumab (n = 150) toutes les deux semaines pendant 52 semaines, 300 mg de dupilumab (n = 145) toutes les deux semaines jusqu'à la semaine 24 suivi de 300 mg toutes les 4 semaines jusqu'à la semaine 52, ou un placebo (N = 153). Tous les patients présentaient des signes d'opacification des sinus sur le score de tomodensitométrie (TDM) des sinus de Lund MacKay et 73 % à 90 % des patients présentaient une opacification de l'ensemble des sinus. Les patients ont été stratifiés selon leurs antécédents chirurgicaux et la présence concomitante d'un asthme/maladie respiratoire exacerbée par les anti-inflammatoires non stéroïdiens.
Les critères principaux associés d'évaluation de l'efficacité étaient la variation du score endoscopique bilatéral des polypes nasaux entre l'entrée dans l'étude et la semaine 24 évalué par des lecteurs centralisés en aveugle et la différence moyenne du score d'obstruction/congestion nasale sur 28 jours, reporté quotidiennement par les patients dans leur cahier de suivi. Pour le score endoscopique les polypes de chaque côté du nez étaient classés selon une échelle catégorielle (0 = aucun polype; 1 = petits polypes dans le méat médian n'atteignant pas le bord inférieur du cornet moyen; 2 = polypes atteignant le bord inférieur du cornet moyen; 3 = gros polypes atteignant le bord inférieur du cornet inférieur ou polypes au milieu du cornet moyen; 4 = gros polypes entraînant une obstruction complète de la cavité nasale inférieure). Le score total correspondait à la somme des scores du côté droit et du côté gauche. La congestion nasale a été évaluée quotidiennement par les patients selon une échelle de gravité catégorielle de 0 à 3 (0 = aucun symptôme; 1 = symptômes légers; 2 = symptômes modérés; 3 = symptômes sévères).
Dans les deux études, les principaux critères d'évaluation secondaires à la semaine 24 étaient les variations par rapport aux valeurs à l'inclusion dans l'étude: score sinusal TDM de Lund MacKay score total des symptômes, test d'identification des odeurs de l'Université de Pennsylvanie (UPSIT), perte quotidienne de l'odorat et questionnaire à 22 items d'évaluation des symptômes naso-sinusiens (SNOT-22). Dans les deux études regroupées, la réduction de la proportion de patients ayant eu recours à un traitement de secours par corticothérapie systémique et/ou chirurgie naso-sinusienne ainsi que l'amélioration du VEMS dans le sous-groupe asthme ont été évaluées. Les autres critères d'évaluation secondaires comprenaient le questionnaire de contrôle de l'asthme à 6 items (ACQ-6) dans le sous-groupe de patients présentant un asthme concomitant.
Les données démographiques et les caractéristiques initiales dans ces 2 études sont présentées dans le Tableau 11 ci-dessous.
Tableau 11: Données démographiques et caractéristiques initiales des études portant sur la PNS:
|
Paramètres
|
SINUS-24
(N=276)
|
SINUS-52
(N=448)
| |
Age moyen (années) (ET)
|
50,49 (13,39)
|
51,95 (12,45)
| |
% patients de sexe masculin
|
57,2
|
62,3
| |
Durée moyenne de la polypose naso-sinusienne(années) (ET)
|
11,11 (9,16)
|
10,94 (9,63)
| |
Patients avec ≥1 chirurgie (%)
|
71,7
|
58,3
| |
Patients traités par corticothérapie systémique au cours des 2 dernières années (%)
|
64,9
|
80,1
| |
SPN endoscopique moyen bilatéral (ET), score 0 à 8
|
5,75 (1,28)
|
6,10 (1,21)
| |
Score de congestion nasale moyena (ET), score 0 à 3
|
2,35 (0,57)
|
2,43 (0,59)
| |
Score sinusal TDM de Lund MacKay total, moyennea , (ET), score 0 à 24
|
19,03 (4,44)
|
17,96 (3,76)
| |
Score moyen du test de l'odorat (UPSIT)a (ET), score 0 à 40
|
14,56 (2,71)
|
13,61 (8.02)
| |
Score de la perte de l'odorat (matin) moyena , (ET), score 0 à 3
|
2,71 (0,54)
|
2,75 (0,52)
| |
Score SNOT-22 total moyena (ET), score 0 à 110
|
49,40 (20,20)
|
51,86 (20,90)
| |
Score de sévérité de la rhinosinusite a , (ET), 0 à 10 cm
|
7,68 (2,05)
|
8,00 (2,08)
| |
Taux moyen d'éosinophiles dans le sang (cellules/µl) (ET)
|
437 (333)
|
431 (353)
| |
Taux total moyen (IgE) IU/mL (ET)
|
211,97 (275,73)
|
239,84
(341,53)
| |
% global d'atopie (maladies inflammatoires de type 2) dans les antécédents médicaux
|
75,4%
|
82,4%
| |
Asthme (%)
|
58,3
|
59,6
| |
VEMS moyen (L)(ET)
|
2,69
(0,96)
|
2,57
(0,83)
| |
Pourcentage de la valeur prédite du VEMS (%) (ET)
|
85,30
(20,23)
|
83,39
(17,72)
| |
Score ACQ-6 moyena (ET)
|
1,62
(1,14)
|
1,58
(1,09)
| |
Maladie respiratoire exacerbée par les anti-inflammatoires non stéroïdiens (%)
|
30,4
|
26,8
|
a Les scores les plus élevés indiquent une sévérité plus importante de la maladie, à l'exception du test UPSIT, où les scores les plus élevés indiquent une maladie moins sévère, ET= écart type; SPN = score des polypes nasaux; UPSIT = score d'identification des odeurs de l'Université de Pennsylvanie; SNOT-22 = questionnaire d'évaluation des symptômes naso-sinusiens à 22 items; EVA = échelle visuelle analogique; VEMS = volume expiratoire maximal par seconde; ACQ-6 = questionnaire du contrôle de l'asthme à 6 items.
Réponse clinique (SINUS-24 et SINUS-52)
Les résultats des critères d'évaluation principaux et secondaires pour les études portant sur la polypose naso-sinusienne sont présentés dans le Tableau 12 ci-dessous:
Tableau 12: Résultats des critères d'évaluation principaux et secondaires dans les études cliniques portant sur la polypose naso-sinusienne:
|
|
SINUS -24
|
SINUS -52
| |
|
Placebo
(n=133)
|
Dupilumab
300mg 1x/2sem
(n=143)
|
Variation moyenne des MC par rapport au placebo
(IC à 95%)
|
Placebo
(n=153)
|
Dupilumab
300mg 1x/2sem
(n=295)
|
Variation moyenne des MC par rapport au placebo
(IC à 95%)
| |
Critères d'évaluation Principaux à la Semaine 24
| |
Scores
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
| |
SPN
|
5,86
|
0,17
|
5,64
|
-1,89
|
-2,06
(-2,43; -1,69)
|
5,96
|
0,10
|
6,18
|
-1,71
|
-1,80
(-2,10; -1,51)
| |
CN
|
2,45
|
-0,45
|
2,26
|
-1,34
|
-0,89
(-1,07; -0,71)
|
2,38
|
-0,38
|
2,46
|
-1,25
|
-0,87
(-1,03; -0,71)
| |
Critères d'évaluation secondaires clés à la Semaine 24
| |
Scores
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
| |
Score TDM de LMK des sinus
|
19,55
|
-0,74
|
18,55
|
-8,18
|
-7,44
(-8,35; -6,53)
|
17,65
|
-0,09
|
18,12
|
-5,21
|
-5,13
(-5,80; -4,46)
| |
Score total des symptômes
|
7,28
|
-1,17
|
6,82
|
-3,77
|
-2,61
(-3,04; -2,17)
|
7,08
|
-1,00
|
7,30
|
-3,45
|
-2,44
(-2,87; -2,02)
| |
UPSIT
|
14,44
|
0,70
|
14,68
|
11,26
|
10,56
(8,79; 12,34)
|
13,78
|
-0,81
|
13,53
|
9,71
|
10,52
(8,98; 12,07)
| |
Perte de l'odorat
|
2,73
|
-0,29
|
2,70
|
-1,41
|
-1,12
(-1,31; -0,93)
|
2,72
|
-0,23
|
2,77
|
-1,21
|
-0,98
(-1,15; -0,81)
| |
SNOT-22
|
50,87
|
-9,31
|
48,0
|
-30,43
|
-21,12
(-25,17; -17,06)
|
53,48
|
-10,40
|
51,02
|
-27,77
|
-17,36
(-20,87; -13,85)
| |
EVA
|
7,96
|
-1,34
|
7,42
|
-4,54
|
-3,20
(-3,79; -2,60)
|
7,98
|
-1,39
|
8,01
|
-4,32
|
-2,93
(-3,45, -2,40)
|
Une réduction du score représente une amélioration, à l'exception du test UPSIT où une augmentation du score représente une amélioration.
Le score total des symptômes est un score composite de la sévérité consistant en la somme des symptômes quotidiens de la congestion nasale, perte de l'odorat et de la rhinorrhée antérieure/postérieure.
CN = congestion nasale, SPN = score de polypose nasale; LMK = score TDM de Lund-Mackay total; UPSIT = score d'identification des odeurs de l'Université de Pennsylvanie; SNOT-22 = questionnaire d'évaluation des symptômes naso-sinusiens à 22 items; STS = score total des symptômes; EVA = échelle visuelle analogique pour la rhinosinusite (toutes valeurs p <0,0001, nominales pour l'EVA)
Les résultats de l'étude SINUS-52 à la semaine 52 sont présentés dans le Tableau 13 ci-dessous.
Tableau 13: Résultats d'efficacité de l'étude SINUS-52 à la semaine 52:
|
|
Placebo
(n=153)
|
Dupilumab
300mg 1x/2sem
(n=150)
|
Variation moyenne des MC par rapport au placebo (IC à 95%)
|
Dupilumab
300mg 1x/2sem-1x/4sem
(n=145)
|
Variation moyenne des MC par rapport au placebo (IC à 95%)
| |
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
|
Moyenne à l'inclusion dans l'étude
|
Variation moyenne des MC
| |
SPN
|
5,96
|
0,15
|
6,07
|
-2,24
|
-2,40
(-2,77; -2,02)
|
6,29
|
-2,06
|
-2,21
(-2,59; -1,83)
| |
CN
|
2,38
|
-0,37
|
2,48
|
-1,35
|
-0,98
(-1,17; -0,79)
|
2,44
|
-1,48
|
-1,10
(-1,29; -0,91)
| |
Score TDM de LMK des sinus
|
17,65
|
0,11
|
18,42
|
-6,83
|
-6,94
(-7,87; -6,01)
|
17,81
|
-5,60
|
-5,71
(-6,64; -4,77)
| |
Score total des symptômes
|
7,08
|
-0,94
|
7,31
|
-3,79
|
-2,85
(-3,35; -2,35)
|
7,28
|
-4,16
|
-3,22
(-3,73; -2,72)
| |
UPSIT
|
13,78
|
-0,77
|
13,46
|
9,53
|
10,30
(8,50; 12,10)
|
13,60
|
9,99
|
10,76
(8,95; 12,57)
| |
Perte de l'odorat
|
2,72
|
-0,19
|
2,81
|
-1,29
|
-1,10
(-1.31, -0.89)
|
2,73
|
-1,49
|
-1,30
(-1,51; -1,09)
| |
SNOT-22
|
53,48
|
-8,88
|
50,16
|
-29,84
|
-20,96
(-25,03; -16,89)
|
51,89
|
-30,52
|
-21,65
(-25,71; -17,58)
| |
EVA
|
7,98
|
-0,93
|
8,24
|
-4,74
|
-3,81
(-4,46; -3,17)
|
7,78
|
-4,39
|
-3,46
(-4,10, -2,81)
|
Une réduction du score représente une amélioration, sauf dans le cas du test UPSIT où c'est une augmentation du score qui représente une amélioration.
Le score total des symptômes est un score composite de sévérité consistant en la somme des symptômes quotidiens de la congestion nasale, la perte de l'odorat et la rhinorrhée antérieure/postérieure.
CN = congestion nasale, SPN = score des polypes nasaux; LMK = score TDM de Lund-Mackay total; UPSIT = score d'identification des odeurs de l'Université de Pennsylvanie; SNOT-22 = questionnaire d'évaluation des symptômes naso-sinusiens à 22 items; STS = score total des symptômes; EVA = échelle visuelle analogique pour la rhinosinusite (toutes les valeurs p <0,0001)
Une efficacité statistiquement et cliniquement significative a été observée dans l'étude SINUS-24 en ce qui concerne l'amélioration du SPN endoscopique bilatéral à la semaine 24. Au cours de la période post-traitement durant laquelle l'administration du dupilumab avait été interrompue, l'effet du traitement a diminué au cours du temps (voir Figure 10a). Des résultats similaires ont également été observés dans l'étude SINUS-52 à la semaine 24 et à la semaine 52 avec une amélioration progressive au cours du temps (voir Figure 10b).
Figure 10. Variation moyenne des moindres carrés (MC) du score des polypes nasaux bilatéraux (SPN) entre l'inclusion dans l'étude dans les études SINUS-24 et SINUS-52 – Population en ITT
|
Figure 10a. SINUS-24
|
Figure 10b. SINUS-52
| |
|

|
|
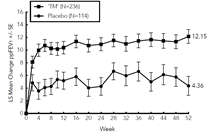
|
Dans les deux études, des améliorations significatives de la congestion nasale et de la sévérité de la perte quotidienne de l'odorat ont été observées dès la première évaluation à la semaine 4. La variation moyenne des moindre carrés (MC) pour la congestion nasale (CN) à la semaine 4 dans le groupe dupilumab par rapport au groupe placebo était de -0,41 (IC à 95%: -0,52; -0,30) dans l'étude SINUS-24 et de -0,37 (IC à 95%: -0,46; -0,27) dans l'étude SINUS-52. La variation moyenne des moindres carrés (MC) pour la perte d'odorat à la semaine 4 dans le groupe dupilumab par rapport au groupe placebo était de -0,34 (IC à 95%: -0,44; -0,25) dans l'étude SINUS-24 et de -0,31 (IC à 95%: -0,41; -0,22) dans l'étude SINUS-52.
Une réduction de la proportion de patients présentant une anosmie a été observée dans les études SINUS-24 et SINUS-52. A l'inclusion 74% à 79% des patients présentaient une anosmie, qui a été réduite à 24% dans SINUS-24 et à 30% dans SINUS-52 à la semaine 24 comparé à l'absence de variation dans les bras placebo. Une amélioration du débit nasal inspiratoire de pointe (DNIP) a été observée à la semaine 24 dans les études SINUS-24 et SINUS-52. La variation moyenne des MC dans le groupe dupilumab par rapport au groupe placebo était de 40,4 L/min (IC à 95%: 30,4; 50,4) et de 36,6 L/min (IC à 95%: 28,0; 45,3), respectivement.
Parmi les patients présentant un score EVA de rhinosinusite >7 à l'inclusion, un pourcentage plus élevé de patients a atteint un score EVA ≤7 dans le bras dupilumab comparé au bras placebo (83,3% versus 39,4% dans l'étude SINUS-24 et 75,0% versus 39,3% dans l'étude SINUS-52) à la semaine 24.
Dans une analyse prédéfinie des données regroupées des deux études ajustée en fonction de la multiplicité, le traitement par le dupilumab a entraîné une réduction significative de l'utilisation d'une corticothérapie systémique et du besoin de chirurgie naso-sinusienne par rapport au placebo (HR de 0,24; IC à 95%: 0,17; 0,35) (voir Figure 11). La proportion de patients nécessitant une corticothérapie systémique a été réduite de 74% (HR de 0,26; IC à 95%: 0,18; 0,38). Le nombre total de cures de corticothérapie systémique par an a été réduit de 75% (RR de 0,25; IC à 95%: 0,17; 0,37). La dose totale annualisée individuelle moyenne de corticostéroïdes systémiques prescrits (en mg) au cours de la période de traitement était de 71% plus faible dans le groupe dupilumab que dans le groupe placebo (60,5 [531,3] mg versus 209,5 [497,2] mg, respectivement).La proportion de patients nécessitant une intervention chirurgicale a été réduite de 83% (HR de 0,17; IC à 95%: 0,07; 0,46).
Figure 11. Courbe de Kaplan Meier du temps écoulé avant la première corticothérapie systémique et/ou intervention chirurgicale naso-sinusienne pendant la période de traitement – Population en ITT [SINUS-24 et SINUS-52 poolées]
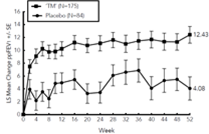
Les effets de dupilumab sur les critères d'évaluation principaux que sont le score des polypes nasaux et la congestion nasale ainsi que les critères d'évaluation secondaires clés sur le score TDM de LMK des sinus étaient similaires chez les patients avant chirurgie et ceux sans chirurgie.
Chez les patients présentant un asthme associé, des améliorations significatives du VEMS et de l'ACQ-6 ont été observées à semaine 24, indépendamment des taux d'éosinophiles sanguins à l'inclusion dans l'étude. La variation moyenne des MC des données regroupées du VEMS entre l'inclusion dans l'étude et la semaine 24 pour le groupe dupilumab 300 1x/2 semaines était de 0,14 contre -0,07 L pour le groupe placebo, avec une différence de 0,21 L (IC à 95%: 0,13; 0,29). De plus, des améliorations du VEMS étaient notées dès la première évaluation post inclusion, à la semaine 8 dans l'étude SINUS-24 et à la semaine 4 dans l'étude SINUS-52. Des améliorations de l'ACQ-6 chez les patients présentant un asthme associé ont été observées dans les deux études. La réponse était définie comme une amélioration du score de 0,5 ou plus. La variation moyenne des MC dans le groupe dupilumab et le placebo était de -0,76 (IC à 95%: -1,00 à -0,51) à la semaine 24 dans l'étude SINUS-24 et de -0,94 (IC à 95%: -1,19; -0,69) dans l'étude SINUS-52.
Le taux de réponse ACQ-6 dans le groupe dupilumab 300 mg 1x/2 semaines, à la semaine 24 dans l'étude SINUS-24 était de 56% versus 28% dans le groupe placebo (OR 3,17; IC à 95%: 1,65; 6,09). Le taux de réponse ACQ-6 dans le groupe dupilumab 300 mg 1x/2 semaines, à la semaine 52 dans l'étude SINUS-52 était de 46% versus 14% dans le groupe placebo (OR 7,02; IC à 95%: 3,10; 15,90).
Chez les patients présentant une maladie respiratoire exacerbée par les anti-inflammatoires non stéroïdiens, les effets du dupilumab sur les critères d'évaluation principaux du SPN et de la CN ainsi que les critères d'évaluation secondaires clés sur le score TDM de LMK des sinus étaient similaires avec ceux observés dans la population totale de la PNS.
PharmacocinétiqueLa pharmacocinétique du dupilumab est similaire chez les patients atteints de dermatite atopique et d'asthme.
Absorption
Après une dose initiale de 600 mg administrée par voie sous-cutanée (SC), le dupilumab a atteint un pic de concentration ± SD (Cmax) de 70,1 ± 24,1 μg / mL environ une semaine après l'administration de la dose.
Selon une analyse pharmacocinétique de population, la biodisponibilité absolue du dupilumab après administration par voie sous-cutanée est estimée à 64 %.
Après l'administration d'une dose initiale de 600 mg suivie d'une dose de 300 mg toutes les deux semaines ou d'une dose de 300 mg administrée toutes les deux semaines sans dose de charge, les concentrations à l'état d'équilibre ont été obtenues à la semaine 16.
Dans l'ensemble des essais cliniques, les concentrations sériques minimales moyennes à l'état d'équilibre (±ET) étaient comprises entre 60,3 ± 35,1 µg/ml et 79,9 ± 41,4 mcg/ml pour une dose de 300 mg et de 29,2 ± 18,7 à 36,5 ± 22,2 µg/ml pour une dose de 200 mg administrée une semaine sur deux.
Distribution
Selon l'analyse pharmacocinétique de population, le volume de distribution du dupilumab est estimé à environ 4,6 l.
Métabolisme
Aucune étude spécifique sur le métabolisme n'a été menée, car le dupilumab est une protéine. Le dupilumab devrait être dégradé en petits peptides et en acides aminés individuels.
Élimination
Le dupilumab est éliminé par des voies parallèles linéaires et non linéaires. A des concentrations élevées, le dupilumab est principalement éliminé par une voie protéolytique non saturable, alors qu'à des concentrations faibles, c'est la voie saturable non linéaire médiée par la cible IL-4R α qui prédomine.
Selon une analyse pharmacocinétique de population, le délai médian pour que les concentrations en dupilumab baissent en dessous du seuil de détection après la dernière dose à l'état d'équilibre est estimé à 9 semaines pour le schéma thérapeutique 200 mg 1x/2 semaines, de 10 à 11 semaines pour le schéma thérapeutique 300 mg 1x/2 semaines et de 13 semaines pour le schéma thérapeutique 300 mg 1x/semaine.
Linéarité/non-linéarité
En raison de la clairance non linéaire, l'exposition au dupilumab, mesurée à partir de l'aire sous la courbe, augmente de manière plus que proportionnelle à la dose après l'administration de doses uniques de 75-600 mg par voie sous-cutanée.
Cinétique pour certains groupes de patients
Sexe, origine ethnique et poids:
Une analyse pharmacocinétique de population n'a pas permis d'établir que le sexe et l'origine ethnique avaient un quelconque impact cliniquement significatif sur l'exposition systémique au dupilumab.
Les concentrations minimales de dupilumab étaient inférieures chez les sujets avec un poids corporel plus élevé, sans que cela ait un impact significatif sur l'efficacité.
Troubles de la fonction hépatique
Le dupilumab étant un anticorps monoclonal, l'élimination hépatique ne devrait pas être significative. Aucune étude clinique n'a été réalisée pour évaluer l'impact de l'insuffisance hépatique sur les paramètres pharmacocinétiques du dupilumab.
Troubles de la fonction rénale
Le dupilumab étant un anticorps monoclonal, l'élimination rénale ne devrait pas être significative. Aucune étude clinique n'a été réalisée pour évaluer l'impact de l'insuffisance rénale sur les paramètres pharmacocinétiques du dupilumab. L'analyse pharmacocinétique de population n'a pas montré qu'une insuffisance rénale légère à modérée avait un impact cliniquement significatif sur l'exposition systémique au dupilumab. Les données disponibles sont très limitées pour les patients atteints d'insuffisance rénale sévère.
Patients âgés
Parmi les 1472 patients atteints de dermatite atopique qui ont été exposés à dupilumab au cours d'une étude de recherche de dose de phase 2 ou lors d'études de phase 3 contrôlées contre placebo, 67 patients, au total, étaient âgés de 65 ans ou plus. Bien qu'aucune différence en termes de sécurité ou d'efficacité n'ait été constatée entre les patients plus jeunes et ceux plus âgés, le nombre de patients de 65 ans et plus n'est pas suffisant pour déterminer si ces derniers répondent différemment par rapport aux patients plus jeunes.
Une analyse pharmacocinétique de population n'a pas permis d'établir que l'âge avait un quelconque impact cliniquement significatif sur l'exposition systémique au dupilumab. Seuls 61 patients de plus de 65 ans étaient inclus dans cette étude.
Seulement 79 patients âgés de plus de 65 ans ont été exposés à dupilumab dans les études portant sur la polypose naso-sinusienne dont 11 étaient âgés de plus de 75 ans.
Sur les 1 977 patients asthmatiques exposés au dupilumab, 240 patients au total avaient 65 ans ou plus et 39 patients étaient âgés de 75 ans ou plus. L'efficacité et la sécurité dans ce groupe d'âge étaient similaires à celles observées de la population globale de l'étude.
Enfants et adolescents
Dermatite atopique:
La pharmacocinétique du dupilumab chez les patients pédiatriques (âgés < 6 ans) ou de poids corporel < 15 kg atteints de dermatite atopique n'a pas été étudiée.
Pour les adolescents âgés de 12 à 17 ans atteints de dermatite atopique et recevant une fois toutes les 2 semaines soit 200 mg (< 60 kg) ou 300 mg (≥60 kg), la moyenne ± ET des concentrations résiduelles du dupilumab à l'état d'équilibre était de 54,5 ± 27,0 µg/ml.
Chez les enfants de 6 à 11 ans atteints de dermatite atopique recevant une dose de 200 mg toutes les deux semaines (Q2W) (≥30 kg) ou une dose de 300 mg toutes les quatre semaines (Q4W) (<30 kg), la moyenne ± ET des concentrations résiduelles du dupilumab à l'état d'équilibre étaient respectivement de 86,0 ± 34,6 µg/ml et 98,7 ± 33,2 µg/ml.
Asthme:
Au total, 107 adolescents âgés de 12 à 17 ans présentant un asthme modéré à sévère ont été inclus dans l'étude QUEST. La moyenne ± ET des concentrations résiduelles à l'état d'équilibre de dupilumab étaient respectivement de 107 µg/ml ± 51,6 µg/ml et 46,7 µg/ml ± 26,9 µg/ml pour une dose de 300 mg ou de 200 mg administrés une semaine sur deux. Aucune différence pharmacocinétique liée à l'âge n'a été observée chez les patients adolescents après correction en fonction du poids corporel.
Polypose naso-sinusienne:
La polypose naso-sinusienne ne survient habituellement pas chez les enfants. La pharmacocinétique de dupilumab chez les patients pédiatriques (< 18 ans) présentant une polypose naso-sinusienne n'a pas été étudiée.
Données précliniquesLes données non cliniques ne révèlent aucun risque particulier pour l'être humain d'après les études conventionnelles de toxicité des doses répétées (y compris les critères d'évaluation d'innocuité pharmacologique) et de toxicité pour la reproduction et le développement.
Mutagénicité
Le potentiel mutagène du dupilumab n'a pas été évalué; toutefois, les anticorps monoclonaux ne sont pas censés altérer l'ADN ou les chromosomes
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée avec le dupilumab. Une évaluation des éléments probants disponibles concernant l'inhibition de l'IL-4Rα et les données toxicologiques chez l'animal avec des anticorps de substitution ne suggère aucune majoration du risque cancérigène avec le dupilumab.
Toxicité sur la reproduction
Au cours d'une étude de toxicologie pour la reproduction réalisée chez le singe avec un anticorps de substitution spécifique à l'IL-4Rα du singe, aucune anomalie fœtale n'a été observée à des posologies entraînant une saturation de l'IL-4Rα.
Une étude plus approfondie sur le développement prénatal et postnatal n'a révélé aucun effet indésirable chez les femelles ou leur progéniture jusqu'à 6 mois après la mise-bas/la naissance.
Les études de la fertilité menées chez la souris mâle et la souris femelle avec un anticorps de substitution dirigé contre l'IL-4Rα n'ont révélé aucun effet défavorable sur la fertilité (voir rubrique «Grossesse/Allaitement»).
Remarques particulièresLes remarques suivantes concernent l'ensemble des présentations disponibles.
Incompatibilités
Aucune donnée de compatibilité n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Tenir hors de la vue et de la portée des enfants.
Conserver la seringue pré-remplie ou le stylo dans son emballage d'origine à l'abri de la lumière.
Conserver au réfrigérateur (2-8°C). Ne pas congeler ni chauffer le produit.
Une fois sorti du réfrigérateur, si besoin, Dupixent peut être conservé à température ambiante ne dépassant pas 25°C pendant 14 jours au maximum. Passé ce délai, la seringue ou le stylo doit impérativement être jeté(e).
Remarques concernant la manipulation
Dupixent se présente dans une seringue pré-remplie avec système de sécurité ou encore sous forme de stylo pré-rempli. Les instructions d'utilisations sont présentées à la suite de l'information destinée aux patients.
Solution pour injection sous-cutanée, limpide à légèrement opalescente et incolore à jaune clair qui ne contient aucune particule visible.
Sortir la seringue ou le stylo du réfrigérateur, et laisser atteindre la température ambiante (environ 45 min) avant de procéder à l'injection de Dupixent.
Ne pas exposer la seringue ou le stylo à la chaleur ou à la lumière directe du soleil et ne pas secouer.
Examiner la seringue pré-remplie ou le stylo avant de l'utiliser. La solution ne doit pas être utilisée si elle est trouble, colorée ou si elle contient des particules, ou si une partie du dispositif semble endommagée.
Tout médicament non utilisé ou déchet doit être éliminé conformément aux exigences locales. Après utilisation, placer la seringue pré-remplie ou le stylo dans un conteneur résistant aux perforations et l'éliminer selon les réglementations locales. Ne pas recycler le conteneur.
Numéro d’autorisation66649, 67661 (Swissmedic)
PrésentationDupixent 300 mg/2 ml, solution injectable en seringue pré-remplie avec système de sécurité: boîtes de 2 seringues (B).
Dupixent 300 mg/2 ml, solution injectable en stylo pré-rempli: boîte de 2 stylos (B).
Dupixent 200 mg/1.14 ml, solution injectable en seringue pré-remplie avec système de sécurité: boîtes de 2 seringues (B).
Dupixent 200 mg/1,14 ml, solution injectable en stylo pré-rempli: boîte de 2 stylos (B).
Titulaire de l’autorisationsanofi-aventis (suisse) sa, 1214 Vernier/GE
Mise à jour de l’informationAvril 2022
|