CompositionPrincipe actif:
Perampanel
Excipients:
Comprimé pelliculé: lactosum, excip. pro compr. obduct. 10 mg et 12 mg: color. E132.
Suspension buvable: Sorbitol liquide cristallisant (contient 175 mg de sorbitol (E420)), cellulose mikrocrist., carmellose sodique (contient moins de 23 mg de sodium), poloxamerère 188, diméthicone, polysorbat 65, méthylcellulose, gel de silice, stéarate de polyéthylène glycol, acide sorbique (E 200), acide benzoïque (E210) 0,005 mg, acide sulfurique, acide citrique, benzoate de sodium (E211) 1,10 mg, Eau purifiée par 1 ml de suspension.
Indications/Possibilités d’emploiFycompa est indiqué:
en association dans le traitement des crises d'épilepsie focales avec ou sans généralisation secondaire chez des patients épileptiques âgés de 12 ans et plus.
en association dans le traitement des crises généralisées tonico-cloniques primaires chez des patients épileptiques âgés de 12 ans et plus.
Posologie/Mode d’emploiAdultes et adolescents âgés au moins de 12 ans
La dose de Fycompa doit être augmentée progressivement en fonction de la réponse de chaque patient, afin d'optimiser le rapport efficacité-tolérance.
Il convient de respecter un délai au moins d'une ou deux semaines entre chaque augmentation posologique en fonction du traitement initial et/ou des traitements concomitants (voir ci-dessous et rubrique «Interactions»), et du rapport bénéfice-risque individuel.
Crises d'épilepsie focales
Le pérampanel est efficace aux doses de 4 mg/jour à 12 mg/jour dans le traitement des crises d'épilepsie focales. Il convient cependant, dans un premier temps, d'administrer le traitement à une dose comprise entre 4 mg/jour et 8 mg/jour en raison des effets indésirables augmentant avec des doses croissantes.
Le traitement par Fycompa doit être instauré à la dose de 2 mg/jour (correspond à 4 ml de suspension par jour). La dose peut être augmentée par paliers de 2 mg (4 ml) (à intervalle d'une ou deux semaines selon les recommandations figurant ci-après) en fonction de la réponse clinique et de la tolérance jusqu'à une dose d'entretien de 4 mg/jour (8 ml/jour) à 8 mg/jour (16 ml/jour). Dans certains cas, si les crises épileptiques ne peuvent être suffisamment contrôlées après administration de cette dose, une augmentation posologique supplémentaire peut être effectuée par paliers de 2 mg (4 ml) jusqu'à atteindre une dose maximale de 12 mg/jour (24 ml/jour) en respectant les délais mentionnés ci-dessus et ci-après, en fonction de la tolérance de chaque patient et toujours après une évaluation soigneuse du rapport bénéfice-risque.
Crises généralisées tonico-cloniques primaires
Le pérampanel est efficace à une dose allant jusqu'à 8 mg/jour dans le traitement des crises d'épilepsie généralisées tonico-cloniques primaires.
Le traitement par Fycompa doit être instauré à la dose de 2 mg/jour (correspond à 4 ml de suspension par jour). La dose peut être augmentée par paliers de 2 mg (4 ml) en fonction de la réponse clinique et de la tolérance (à intervalle d'une ou deux semaines selon les recommandations figurant ci-après) jusqu'à une dose d'entretien de 8 mg/jour (16 mg/jour). En fonction de la réponse clinique et de la tolérance, une augmentation posologique supplémentaire peut être effectuée jusqu'à atteindre une dose maximale de 12 mg/jour (24 ml/jour) qui peut être efficace chez certains patients (voir«Propriétés/Effets»,rubrique «Efficacité clinique - Crises généralisées tonico-cloniques primaires»).
Intervalle entre chaque augmentation posologique
Chez les patients dont les traitements concomitants n'entraînent pas une réduction de la demi-vie du pérampanel (voir rubrique «Interactions»), il convient de respecter un délai d'au moins deux semaines entre chaque augmentation posologique.
Chez les patients dont les traitements concomitants diminuent la demi-vie du pérampanel (voir rubrique «Interactions»), un délai d'au moins une semaine devra être observé.
Recommandation posologique en cas de comédication avec un inducteur du CYP3A
Après administration complémentaire de pérampanel en doses fixes, les taux de réponse étaient plus faibles si les patients recevaient concomitamment des antiépileptiques inducteurs du CYP3A (carbamazépine, phénytoïne, oxcarbazépine) que chez des patients traités concomitamment avec des antiépileptiques ne présentant pas cet effet inducteur. Il convient de surveiller le taux de réponse des patients si ceux-ci passent d'un antiépileptique non inducteur enzymatique administré concomitamment à une substance inductrice enzymatique ou inversement. En fonction de la réponse clinique individuelle et de la tolérance, la dose peut être augmentée ou réduite par paliers de 2 mg.
Mode d'administration
Fycompa doit être pris par voie orale au coucher. Fycompa peut être pris au moment des repas ou en dehors des repas (voir «Pharmacocinétique»). Le relais entre les formulations en comprimé et en suspension doit être effectué avec précaution (voir «Pharmacocinétique»).
Comprimé pelliculés:
Le comprimé doit être avalé entier à l'aide d'un verre d'eau. Il ne doit pas être croqué, écrasé ou divisé. En l'absence de barre de cassure, les comprimés ne peuvent pas être coupés en deux.
Suspension:
Pour l'administration, veuillez utiliser la seringue pour administration orale. Les instructions d'utilisation de l'adaptateur et de la seringue sont présentées ci-dessous:
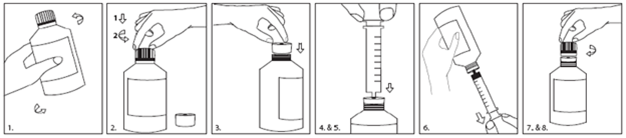
1.Agitez pendant au moins 5 secondes avant utilisation.
2.Appuyez sur le bouchon en tournant pour ouvrir le flacon.
3.Insérez l'adaptateur dans le goulot du flacon jusqu'à obtention d'un joint étanche.
4.Enfoncez complètement le piston de la seringue.
5.Insérez la seringue pour administration orale aussi loin que possible dans l'orifice de l'adaptateur.
6.Retournez l'ensemble et prélevez la quantité prescrite de Fycompa à partir du flacon.
7.Retournez le flacon et retirez la seringue pour administration orale.
8.Laissez l'adaptateur en place et refermez le flacon avec le bouchon. Rincez la seringue avec de l'eau propre et séchez-la soigneusement.
Arrêt du traitement de Fycompa
Il est recommandé d'arrêter progressivement le traitement afin de réduire le risque de crises d'épilepsie par effet rebond (voir rubrique Posologie/Mode d'emploi). Cependant, compte tenu de sa longue demi-vie et de la lente diminution des concentrations plasmatiques qui en résulte, le pérampanel peut être arrêté brutalement en cas d'absolue nécessité.
Arrêt de la co-médication
En cas d'arrêt d'inducteurs enzymatiques pris concomitamment, la concentration plasmatique de pérampanel peut augmenter et un ajustement de la posologie être nécessaire.
Prise oubliée:
En cas d'oubli d'une seule dose et compte tenu de la demi-vie longue du pérampanel, le patient doit attendre et prendre la dose suivante au moment habituel.
En cas d'oubli de plus d'une dose pendant une période continue de moins de cinq demi-vies (3 semaines pour les patients ne recevant pas de médicaments antiépileptiques induisant le métabolisme du pérampanel, 1 semaine pour les patients prenant des médicaments antiépileptiques induisant le métabolisme du pérampanel (voir «Interactions»), il convient d'envisager la reprise du traitement à la dernière dose administrée.
Si un patient a arrêté l'administration de pérampanel pendant une période continue de plus de 5 demi-vies, il est conseillé de suivre les recommandations d'instauration du traitement initial indiquées ci-dessus.
Adolescents
Chez les adolescents il faut surveiller les signes indiquant des modifications du comportement (voir rubrique «Mises en garde et précautions» concernant la suicidalité et l'agressivité)
Enfants âgés de moins de 12 ans
La sécurité et l'efficacité de Fycompa chez les enfants âgés de moins de 12 ans n'ont pas encore été établies. Aucune donnée n'est disponible. L'utilisation de ce médicament dans ce groupe d'âge n'est pas recommandée (voir «Mises en garde et précautions» concernant la suicidalité et l'agressivité)
Patients âgés (65 ans et plus)
Les études cliniques de Fycompa portant sur le traitement de l'épilepsie n'ont pas inclus suffisamment de patients âgés de 65 ans et plus pour déterminer si leur réponse au traitement est différente de celle des patients plus jeunes. Le rapport bénéfice-risque et/ou la nécessité de l'utilisation du pérampanel chez les patients âgés doivent être soigneusement évalués en particulier chez les patients âgés polymédiqués en raison des interactions médicamenteuses éventuelles. En outre, se référer également à la rubrique «Mises en garde et précautions», en particulier concernant les troubles de l'équilibre et de la coordination et le risque de chutes.
Patients insuffisants rénaux
Aucune adaptation posologique n'est requise chez les patients présentant une insuffisance rénale légère. L'utilisation de Fycompa n'est pas recommandée chez les patients présentant une insuffisance rénale modérée ou sévère ou chez les patients sous hémodialyse (voir également rubrique «Contre-indications» et «Données précliniques»).
Patients insuffisants hépatiques
Les augmentations posologiques chez les patients présentant une insuffisance hépatique légère ou modérée doivent reposer sur la réponse clinique et la tolérance et être effectuées en respectant un délai d'au moins deux semaines. Le traitement peut être instauré à la dose de 2 mg et la dose doit être augmentée par paliers de 2 mg à intervalles d'au moins 2 semaines jusqu'à une dose maximale de 8 mg, en fonction de l'efficacité et de la tolérance. Chez les patients présentant une insuffisance hépatique légère à modérée, la dose de pérampanel ne doit donc pas dépasser 8 mg. L'utilisation de Fycompa n'est pas recommandée chez les patients présentant une insuffisance hépatique sévère (voir également rubrique «Contre-indications» et «Données précliniques»).
Contre-indicationsHypersensibilité connue au pérampanel ou à l'un des excipients.
Insuffisance rénale modérée ou sévère et patients sous hémodialyse.
Insuffisance hépatique sévère.
Mises en garde et précautionsAggravation clinique, risque de suicide et modifications du comportement: Nécessité d'une surveillance étroite et, le cas échéant, modifications de la stratégie thérapeutique en temps utile
Il y a des indices laissant présumer que les patients épileptiques présentent un risque accru de comportement suicidaires. Une analyse de la FDA (USA), publiée en janvier 2008, sur les données de 199 études cliniques contrôlées contre placebo, réalisées avec au total 11 médicaments antiépileptiques, a révélé un risque 3,6 fois plus élevé de comportement suicidaires chez les patients épileptiques traités par ces produits que chez ceux sous placebo. Il n'y avait pas de différences importantes entre les diverses substances étudiées quant à leur risque de comportement suicidaires. Dans cette analyse, l'augmentation du risque était même plus prononcée chez les patients épileptiques que chez ceux atteints d'affections psychiatriques (telles que les troubles bipolaires), où une augmentation du risque d'un facteur 1,6 a été établie. Au total, des idées ou un comportement suicidaires ont été observés, toutes indications confondues, chez 0,43% des patients traités par les médicaments antiépileptiques et chez seulement 0,22% des patients sous placebo.
Par conséquent, les patients doivent être attentivement et étroitement surveillés afin de déceler des signes de modifications du comportement, par exemple irritabilité accrue, irascibilité, modifications de l'humeur, hostilité, réapparition ou aggravation d'un comportement agressif pouvant aller jusqu'à des menaces ou des actes agressifs à l'égard d'autrui et des signes indiquant des idées suicidaires.
Les données disponibles actuellement ne permettent pas d'exclure la possibilité d'un risque accru avec Fycompa.
Les patients traités par Fycompa pour une épilepsie doivent en conséquence être étroitement surveillés en cas d'aggravation clinique (y compris l'apparition de nouveaux symptômes) et de comportement suicidaires, en particulier au début d'un nouveau cycle de traitement ou en cas de modifications posologiques ou de modifications des concentrations plasmatiques (phase de titration, augmentations posologiques mais également diminution posologique et en cas d'éventuelles modifications des concentrations plasmatiques dues à des modifications des traitements concomitants).
Certains groupes de patients, tels que les patients présentant des antécédents de comportement ou d'idées suicidaires et les jeunes adultes semblent être sujets à un risque plus élevé d'idées suicidaires ou de tentatives de suicides et doivent faire l'objet d'une surveillance étroite lors du traitement. Ceci est particulièrement valable également pour les patients chez lesquels de tels symptômes ont été observés directement avant l'instauration du traitement.
Il convient d'envisager un changement de traitement chez les patients dont l'état clinique se dégrade (y compris le développement de nouveaux symptômes) et/ou chez lesquels des signes de comportement suicidaire apparaissent en particulier si ces symptômes sont prononcés, surviennent brutalement ou s'ils ne faisaient pas partie des symptômes initiaux des patients. Dans de tels cas, un arrêt du médicament peut s'avérer nécessaire.
Un traitement approprié doit être immédiatement envisagé dans tous les cas. Il convient d'indiquer aux patients (et à leurs aidants) de consulter immédiatement un médecin en cas d'apparition d'idées et de comportement suicidaires ou de comportement agressif envers autrui.
Ceci est particulièrement valable pour le groupe des patients adolescents. Chez les adolescents (âgés de 12 à 18 ans) une incidence élevé d'évènements neuropsychiatriques a été observée, en particulier la survenue répétée d'agressivité, de troubles psychotiques, d'irritabilité accrue et de comportement suicidaire.
Il convient de signaler aux patients (et à leurs aidants) la nécessité d'une surveillance en cas d'apparition d'idées suicidaires, de comportement suicidaire ou d'intentions auto-nuisibles ou nuisibles pour autrui. En cas de survenue de tels symptômes, un médecin doit immédiatement être consulté.
Syndrome d'hypersensibilité médicamenteuse (Drug Reaction with Eosinophilia and Systemic Symptoms, DRESS)/hypersensibilité multi-organique
Un syndrome d'hypersensibilité médicamenteuse a été décrit chez des patients qui prenaient des antiépileptiques, pérampanel inclus. Le DRESS peut être très dangereux, voire mortel. Le DRESS se manifeste par une éruption cutanée avec éosinophilie, conjointement à l'une ou plusieurs des caractéristiques suivantes: fièvre, lymphadénopathie, œdème du visage, atteinte des organes (foie, reins, poumons). Le DRESS peut se caractériser par un long temps de latence (de 2 à 8 semaines) entre l'exposition au médicament et l'apparition de la maladie. Si des signes ou des symptômes de DRESS apparaissent, le patient doit être examiné immédiatement et il convient d'interrompre le traitement par le pérampanel, pour autant qu'aucune autre cause des symptômes n'ait été identifiée.
Troubles du système nerveux
Le pérampanel peut provoquer des vertiges et une somnolence et peut donc avoir une forte influence sur l'aptitude à la conduite d'un véhicule ou à l'utilisation de machines (voir rubrique «Effet sur l'aptitude à la conduite et l'utilisation de machines»).
Agressivité
Des cas d'agressivité ont été décrits, lesquels étaient dose-dépendants puisqu'ils survenaient plus fréquemment à des doses élevées. Ces cas d'agressivité ont été observés plus fréquemment chez les adolescents que chez les adultes. Par conséquent, une augmentation progressive des doses doit être respectée et une diminution de la posologie et le cas échéant, l'arrêt du traitement, doivent être envisagés si les symptômes d'agressivité persistent. (voir rubrique «Posologie/Mode d'emploi»). Ces éventualités et les mesures à prendre doivent être au préalable discutées et planifiées avec un spécialiste.
La plupart de ces événements survenus dans le cadre d'études cliniques étaient d'intensité légère à modérée et cessaient spontanément ou avec un ajustement de la posologie. Dans certains cas, toutefois, il a été rapporté une agressivité marquée ayant conduit à un arrêt du traitement. Il est recommandé d'informer les patients (et leurs aidants) au début du traitement de l'éventualité de la survenue d'une irritabilité accrue, d'une agressivité et d'humeurs dépressives (voir la rubrique correspondante «Aggravation clinique, risque de suicide et modifications du comportement: Nécessité d'une surveillance étroite et, le cas échéant, modifications de la stratégie thérapeutique en temps utile»).
Risque de chutes
Il y a un risque accru de chutes, particulièrement chez les patients âgés. La raison sous-jacente n'est pas claire; d'éventuels effets secondaires, de type troubles de l'équilibre, vertiges et torpeur, pourraient jouer un rôle. Les patients et les aidants doivent être attentifs à ces phénomènes, en particulier en début de traitement et en cas de modification posologique et/ou des traitements concomitants (y compris les modifications posologiques des traitements concomitants).
Interactions avec des inducteurs ou inhibiteurs du cytochrome P450
La réponse clinique et la tolérance des patients doivent être suivies étroitement lors de l'ajout ou du retrait d'inducteurs ou d'inhibiteurs du cytochrome P450, car les concentrations plasmatiques du pérampanel peuvent être diminuées ou augmentées; il est possible que la dose de pérampanel doive être ajustée en conséquence (voir rubrique «Posologie/Mode d'emploi»).
Contraceptifs oraux
En raison de l'interaction prouvée avec le lévonorgestrel, Fycompa peut diminuer l'efficacité des contraceptifs hormonaux contenant des progestatifs (voir rubrique «Interactions»). Dans ce cas, il faut en tenir compte et il est recommandé d'utiliser une méthode de contraception non hormonale supplémentaire (dispositif intra-utérin (DIU), préservatif).
Potentiel d'utilisation abusive
Des précautions s'imposent chez les patients ayant des antécédents d'abus de substances addictives et les signes d'utilisation abusive du pérampanel doivent être surveillés chez ces patients.
Composants présentant un intérêt particulier
Fycompa comprimés pelliculés:
Lactose: Les patients atteints de la rare intolérance héréditaire au galactose, d'un déficit total en lactase ou d'une malabsorption du glucose-galactose ne doivent pas utiliser les comprimés pelliculés.
Fycompa suspension:
Sorbitol: Fycompa suspension contient 175 mg de sorbitol par ml, qui est une source de fructose. Les patients atteints d'une intolérance héréditaire au fructose (IHF) ne doivent pas prendre ce médicament. Le sorbitol peut provoquer des troubles gastro-intestinaux et avoir un léger effet laxatif. La prudence est de mise en cas d'utilisation concomitante de Fycompa suspension et d'autres antiépileptiques contenant du sorbitol, car la prise de plus de 1 gramme de sorbitol au total peut influer sur la résorption de certains médicaments.
Acide benzoïque/benzoate: Fycompa suspension contient de l'acide benzoïque et du benzoate de sodium.
Sodium: Fycompa suspension contient moins de 1 mmol (23 mg) de sodium par ml, c.-à-d. qu'il est quasiment «sans sodium».
InteractionsInteractions pharmacocinétiques
Données in vitro
Inhibition des enzymes du métabolisme des médicaments
Dans des microsomes hépatiques humains, le pérampanel (30 μmol/l) a présenté un faible effet inhibiteur sur le CYP2C8 et sur l'UGT1A9 parmi les principaux cytochromes et UDP-glucuronosyltransférases hépatiques.
Induction des enzymes du métabolisme des médicaments
Par rapport aux témoins positifs (dont le phénobarbital et la rifampicine), le pérampanel est un inducteur faible du CYP2B6 (30 μmol/l) et du CYP3A4/5 (≥3 μmol/l) parmi les principaux CYP et UGT hépatiques dans des hépatocytes humains en culture.
Données in vivo
Substrats du CYP3A
Chez des volontaires sains, Fycompa (6 mg/jour pendant 20 jours) a diminué de 13% l'ASC du midazolam. Une diminution plus importante de l'exposition au midazolam (ou à d'autres substrats sensibles du CYP3A) à des doses plus élevées de Fycompa ne peut être exclue.
Inducteurs du cytochrome P450
Les inducteurs puissants du cytochrome P450, tels que la rifampicine et le millepertuis, sont susceptibles de diminuer les concentrations du pérampanel et la possibilité de concentrations plasmatiques augmentées de ses métabolites réactifs n'est au contraire pas à exclure. Le felbamate diminue les concentrations de certains médicaments et pourrait également diminuer celles du pérampanel.
Inhibiteurs du cytochrome P450
Chez des volontaires sains, un inhibiteur du CYP3A4, le kétoconazole (400 mg/jour pendant 10 jours), a augmenté de 20% l'ASC du pérampanel et a allongé de 15% sa demi-vie (67,8 h contre 58,4 h). Des effets plus importants ne peuvent être exclus lorsque le pérampanel est associé à un inhibiteur du CYP3A ayant une demi-vie plus longue que le kétoconazole ou lorsque la durée du traitement par l'inhibiteur est plus longue. Des inhibiteurs puissants d'autres isoformes du cytochrome P450 pourraient également augmenter les concentrations du pérampanel.
Lévodopa
Chez des volontaires sains, Fycompa (4 mg/jour pendant 19 jours) n'a pas eu d'effet sur la Cmax ou l'ASC de la lévodopa.
Médicaments antiépileptiques
Les interactions éventuelles entre Fycompa (jusqu'à 12 mg/jour) et d'autres médicaments antiépileptiques ont été évaluées dans le cadre des études cliniques et dans l'analyse pharmacocinétique de population de 4 études de phase III combinées réalisées chez des patients présentant des crises d'épilepsie focales et chez des patients présentant des crises généralisées tonico-cloniques primaires. Le tableau ci-dessous présente une synthèse de l'effet de ces interactions sur la concentration moyenne à l'état d'équilibre. (Les valeurs indiquées dans le tableau se basent sur les modèles de cinétique de population).
|
Médicament antiépileptique co-administré
|
Effet du médicament antiépileptique sur la concentration de pérampanel
|
Effet de pérampanel sur la concentration du médicament antiépileptique
| |
Carbamazépine
|
Diminution d'un tiers
|
Diminution <10%
| |
Clobazam
|
Pas d'effet
|
Diminution <10%
| |
Clonazépam
|
Pas d'effet
|
Pas d'effet
| |
Lamotrigine
|
Pas d'effet
|
Diminution <10%
| |
Lévétiracétam
|
Pas d'effet
|
Pas d'effet
| |
Oxcarbazépine
|
Diminution de moitié
|
Augmentation de 35%1)
| |
Phénobarbital
|
Pas d'effet
|
Pas d'effet
| |
Phénytoïne
|
Diminution de moitié
|
Pas d'effet
| |
Topiramate
|
Diminution <20%
|
Pas d'effet
| |
Acide valproïque
|
Pas d'effet
|
Diminution <10%
| |
Zonisamide
|
Pas d'effet
|
Pas d'effet
|
1) Le métabolite actif monohydroxycarbazépine n'a pas été évalué.
Certains médicaments antiépileptiques connus pour être des inducteurs enzymatiques (carbamazépine, phénytoïne, oxcarbazépine) augmentent la clairance du pérampanel et, par conséquent, diminuent les concentrations plasmatiques du pérampanel. A l'inverse, l'arrêt d'inducteurs enzymatiques administrés concomitamment peut augmenter la concentration plasmatique du pérampanel et nécessiter une adaptation posologique. La carbamazépine, connue pour être un inducteur enzymatique puissant, a diminué de deux tiers les concentrations du pérampanel dans une étude menée chez des volontaires sains.
Un résultat similaire a été observé dans une analyse pharmacocinétique de population de patients présentant des crises d'épilepsie partielles et de patients présentant des crises généralisées tonico-cloniques primaires recevant le pérampanel à des doses allant jusqu'à 12 mg/jour dans des études cliniques contrôlés contre placebo. La clairance totale de Fycompa a augmenté lors de l'administration concomitante de carbamazépine (multiplication par 3), de phénytoïne (multiplication par 2) et d'oxcarbazépine (multiplication par 2), qui sont des inducteurs connus des enzymes du métabolisme (voir rubrique «Pharmacocinétique»). Il convient de tenir compte de cet effet dans le cadre du schéma thérapeutique et de prendre les mesures thérapeutiques appropriées lors de l'ajout ou du retrait de ces médicaments antiépileptiques.
Dans une analyse pharmacocinétique de population de patients présentant des crises d'épilepsie partielles et recevant du pérampanel à des doses allant jusqu'à 12 mg/jour et de patients présentant des crises généralisées tonico-cloniques primaires recevant des doses allant jusqu'à 8 mg par jour dans le cadre d'études cliniques contrôlées contre placebo, Fycompa n'a pas eu d'effet cliniquement pertinent sur la clairance du clonazépam, du lévétiracétam, du phénobarbital, de la phénytoïne, du topiramate, du zonisamide, de la carbamazépine, du clobazam, de la lamotrigine et de l'acide valproïque à la plus forte dose de pérampanel évaluée (12 mg/jour).
Dans l'analyse pharmacocinétique de population de patients épileptiques, le pérampanel a diminué de 26% la clairance de l'oxcarbazépine. L'oxcarbazépine est rapidement métabolisée en métabolite actif, la monohydroxycarbazépine, par la réductase cytosolique. L'effet du pérampanel sur les concentrations de la monohydroxycarbazépine n'est pas connu.
La dose du pérampanel est déterminée par l'effet clinique indépendamment des autres médicaments antiépileptiques.
Contraceptifs oraux
En raison de l'interaction prouvée avec le lévonorgestrel, Fycompa peut diminuer l'efficacité des contraceptifs hormonaux contenant des progestatifs. Il est recommandé d'utiliser une méthode de contraception non hormonale supplémentaire (dispositif intra-utérin (DIU), préservatif).
Chez des femmes en bonne santé, l'administration concomitante de Fycompa à la dose de 12 mg (mais pas de 4 ou 8 mg/jour) pendant 21 jours et d'un contraceptif oral combiné a diminué l'exposition au lévonorgestrel (les valeurs moyennes de la Cmax et de l'ASC ont été diminuées chacune de 40%). L'ASC de l'éthinylestradiol n'a pas été modifiée par Fycompa à la dose de 12 mg, tandis que sa Cmax a été diminuée de 18%. Il convient de prendre en compte de manière appropriée le risque de diminution de l'efficacité des contraceptifs oraux contenant des progestatifs chez les jeunes filles ou femmes en âge de procréer nécessitant un traitement par Fycompa (voir rubrique «Mises en garde et précautions»).
Population pédiatrique
Des études d'interaction n'ont été réalisées que chez l'adulte. Les données disponibles à ce sujet dans la population pédiatrique sont limitées: une analyse pharmacocinétique de population de patients adolescents inclus dans les études cliniques de phase III n'a mis en évidence aucune différence notable entre cette population et la population générale.
Interactions pharmacodynamiques
Alcool
Les effets du pérampanel et ceux de l'alcool étaient additifs ou synergiques sur les activités nécessitant attention et vigilance. L'administration répétée de pérampanel à raison de 12 mg/jour a potentialisé les effets de l'alcool également concernant la vigilance et l'attention et a augmenté les degrés de colère, de désorientation et de dépression (voir rubrique «Propriétés/Effets»). Ces effets peuvent également être observés lorsque Fycompa est utilisé en association avec d'autres médicaments dépresseurs du système nerveux central (SNC).
Grossesse/AllaitementFemmes en âge de procréer et contraception
L'utilisation de Fycompa n'est pas recommandée chez les femmes en âge de procréer n'utilisant pas de contraception. En raison de l'interaction prouvée avec le lévonorgestrel, Fycompa peut diminuer l'efficacité des contraceptifs hormonaux contenant des progestatifs (voir rubrique «Mises en gardes et précautions» et «Interactions»).
Fertilité
L'effet du pérampanel sur la fertilité humaine n'a pas été établi. Les effets observés dans les études animales n'affectaient pas la fertilité (voir «Données précliniques»).
Grossesse
On ne dispose jusqu'ici que d'une expérience limitée (moins de 300 grossesses) sur l'utilisation du pérampanel chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets tératogènes chez le rat ou le lapin, mais une embryotoxicité a été observée chez le rat à des doses entraînant une toxicité maternelle (voir «Données précliniques»).
Allaitement
Les études menées chez des rates allaitantes ont mis en évidence l'excrétion de pérampanel et/ou de ses métabolites dans le lait (pour plus d'informations, voir rubrique «Données précliniques»). On ne sait pas si le pérampanel est excrété dans le lait maternel chez la femme. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise entre interrompre l'allaitement ou arrêter le traitement par Fycompa en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Effet sur l’aptitude à la conduite et l’utilisation de machinesFycompa a une influence importante sur l'aptitude à la conduite et l'utilisation de machines.
Le pérampanel peut, entre autres, provoquer une torpeur, des vertiges, une somnolence, des nausées, une irascibilité et une irritabilité accrue et peut donc altérer fortement l'aptitude à la conduite et l'utilisation de machines. Il convient de conseiller aux patients de ne pas conduire de véhicule, de ne pas utiliser de machines et de ne pas entreprendre d'autres activités susceptibles d'être dangereuses jusqu'à ce qu'ils soient certains que le pérampanel n'altère par leur capacité à effectuer ces tâches.
Effets indésirablesDans l'ensemble des études contrôlées et non contrôlées menées chez des patients présentant des crises d'épilepsie partielles, 1'639 patients ont reçu le pérampanel, parmi lesquels 1'147 ont été traités pendant 6 mois et 703 pendant plus de 12 mois.
Dans les études contrôlées et non contrôlées menées chez des patients présentant des crises généralisées tonico-cloniques primaires, 114 patients ont reçu le pérampanel, parmi lesquels 68 ont été traités pendant 6 mois et 36 pendant plus de 12 mois.
Crises d'épilepsie partielles
Effets indésirables ayant entraîné l'arrêt du traitement: dans les études cliniques de phase III contrôlées, le taux d'arrêt de traitement en raison d'effets indésirables était de 2,9%, 7,7% et 19,2% respectivement chez les patients randomisés pour recevoir le pérampanel aux doses recommandées de 4 mg, 8 mg et 12 mg/jour et de 4,8% chez les patients du groupe placebo. Les effets indésirables les plus fréquents (>10% dans l'ensemble des groupes pérampanel et incidence supérieure chez les patients sous placebo) ayant entraîné l'arrêt du traitement étaient les vertiges et la somnolence.
Crises généralisées tonico-cloniques primaires
Dans les études cliniques de phase III contrôlées menées chez des patients présentant des crises généralisées tonico-cloniques primaires, le taux d'arrêt de traitement en raison d'effets indésirables a été de 4,9% chez les patients du groupe pérampanel (jusqu'à 8 mg/j) et de 1,2% chez les patients du groupe placebo. L'effet indésirable le plus fréquent (>2% dans l'ensemble des groupes pérampanel et incidence supérieure chez les patients sous placebo) ayant entraîné l'arrêt du traitement était les vertiges.
Dans la liste ci-dessous, les effets indésirables sont présentés par classe de systèmes d'organes et par fréquence. Les indications de fréquence utilisées pour la classification des effets indésirables sont les suivantes: très fréquents ≥1/10, fréquents ≥1/100, <1/10, occasionnels ≥1/1000, <1/100, rares ≥1/10'000, <1/1000, fréquence inconnue: fréquence non évaluable sur la base des données disponibles..
Troubles du métabolisme et de la nutrition
Fréquents: perte d'appétit, augmentation de l'appétit.
Troubles psychiatriques
Fréquents: anxiété, agressivité, colère, désorientation, symptômes dépressifs.
Occasionnels: suicidalité, risque de mise en danger de soi-même et d'autrui.
Système nerveux
Très fréquents: vertiges (32%), somnolence (15%).
Fréquents: ataxie, troubles de l'équilibre, dysarthrie, irritabilité.
Troubles oculaires
Fréquents: diplopie, vision trouble.
Troubles auditifs
Fréquents: vertige.
Troubles gastro-intestinaux
Fréquents: nausées.
Affections de la peau et des tissus sous-cutanés
Fréquence inconnue: Syndrome d'hypersensibilité médicamenteuse (Drug Reaction with Eosinophilia and Systemic Symptoms [DRESS]).
Troubles musculosquelettiques
Fréquents: dorsalgies.
Troubles généraux
Fréquents: fatigue, troubles de la marche, prise de poids.
Lésions, intoxications et complications liées aux procédures
Fréquents: chutes.
Investigations
Fréquents: prise de poids.
Population pédiatrique
Sur la base des données d'études cliniques de 196 adolescents (de 12 à 18 ans) sous pérampanel dans des études en double aveugle lors de crises focales et de crises généralisées tonico-cloniques primaires, le profil de sécurité chez les adolescents est comparable à celui des adultes, à l'exception de l'agressivité qui est apparue plus fréquemment que chez les adultes (voir également «Mises en garde et précautions»).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageL'expérience clinique concernant un surdosage du pérampanel chez l'homme est limitée. Dans un cas de surdosage volontaire rapporté lors duquel une dose ayant pu éventuellement atteindre au maximum 264 mg de Fycompa a été ingérée, le patient a présenté des épisodes d'altération de l'état mental, une excitabilité/agitation accrue et un comportement agressif; il s'est rétabli sans séquelles.
Il n'existe pas d'antidote spécifique pour les effets du pérampanel. Un traitement symptomatique et de maintien des fonctions vitales, comprenant la surveillance des signes vitaux et l'observation de l'état clinique du patient est indiqué. Du fait de sa demi-vie longue, les effets provoqués par le pérampanel peuvent être prolongés. En raison de la faible clairance rénale, les mesures spécifiques telles qu'une diurèse forcée, une dialyse ou une hémoperfusion sont peu susceptibles d'être efficaces.
Propriétés/EffetsCode ATC: N03AX22
Mécanisme d'action
Le pérampanel est le premier représentant de la classe des antagonistes sélectifs, non compétitifs des récepteurs ionotropiques au glutamate de type AMPA (acide α-amino-3-hydroxy-5-méthyl-4-isoxazole propionique) présents sur les neurones post-synaptiques. Le glutamate est le principal neurotransmetteur excitateur du système nerveux central. L'activation des récepteurs AMPA par le glutamate serait responsable de l'essentiel de la transmission synaptique excitatrice rapide dans le cerveau. Dans les études in vitro, le pérampanel a inhibé l'augmentation du calcium intracellulaire induite par l'activation des récepteurs AMPA.
Le mécanisme précis par lequel le pérampanel exerce ses effets antiépileptiques chez l'homme n'est pas complètement élucidé.
Effets pharmacodynamiques
Une analyse (d'efficacité) pharmacocinétique-pharmacodynamique a été réalisée à partir des données groupées issues de trois études d'efficacité portant sur les crises d'épilepsie partielles. Une analyse similaire a été également réalisée dans le cadre d'une étude portant sur les crises généralisées tonico-cloniques primaires. Dans les deux analyses, l'exposition au pérampanel est corrélée à la diminution de la fréquence des crises.
Fonctions psychomotrices
Dans une étude réalisée chez des volontaires sains, on a eu recours à des méthodes standard d'évaluation comprenant une simulation de la conduite d'un véhicule afin de déterminer les effets du pérampanel sur les fonctions psychomotrices. Des doses uniques et répétées de 4 mg de pérampanel n'ont pas altéré les activités psychomotrices simples, l'aptitude à la conduite ou la coordination sensorielle et motrice. Des doses uniques et répétées de 8 mg et 12 mg ont altéré les fonctions psychomotrices de façon dose-dépendante. L'aptitude à conduire un véhicule était diminuée après une dose de 12 mg de pérampanel mais la stabilité posturale n'était pas altérée de manière significative. Les fonctions psychomotrices étaient revenues au niveau initial dans les 2 semaines suivant l'arrêt de l'administration de pérampanel. Après l'administration d'alcool aux volontaires sains dans la même étude, afin d'atteindre une concentration sanguine de 80 à 100 mg/100 ml, le pérampanel a altéré la fonction psychomotrice simple généralement après absorption de doses uniques de 4 à 12 mg (n=35) et après 21 jours avec des doses répétées de 12 mg/jour (n=24). Les effets du pérampanel sur des activités complexes telles que la conduite de véhicules et les effets délétères de l'alcool étaient additifs ou synergiques.
Fonction cognitive
Dans une étude réalisée chez des volontaires sains, on a eu recours à des méthodes standard d'évaluation afin de déterminer les effets du pérampanel sur la vigilance et la mémoire. Après l'administration de doses uniques et répétées maximales de 12 mg de pérampanel par jour aucune altération n'a été observée.
Dans une étude contrôlée contre placebo chez de jeunes patients, des mesures au moyen du Global Cognition Scores du Cognitive Drug Research (CDR) System n'ont montré aucune modification significative de la cognition pour le pérampanel par rapport au placebo. Dans la phase de prolongation ouverte de cette étude, aucune modification significative du score global CDRSystem n'a été observée après 52 semaines de traitement par le pérampanel (voir paragraphe «Enfants et adolescents» ci-après).
Attention et humeur
Chez des volontaires sains ayant reçu de 4 à 12 mg/jour de pérampanel, le niveau de vigilance (réactivité) a diminué de façon dose-dépendante. L'humeur ne s'est détériorée chez les volontaires sains qu'après l'administration de 12 mg/jour; les changements d'humeur étaient minimes et traduisaient une baisse générale de la vigilance. L'administration répétée de pérampanel à raison de 12 mg/jour a potentialisé les effets de l'alcool également concernant la vigilance et l'attention et a augmenté l'intensité de la colère, de la désorientation et de la dépression.
Electrophysiologie cardiaque
Les effets du pérampanel sur l'électrophysiologie cardiaque ont été déterminés dans le cadre d'une étude pharmacologique clinique randomisée, en double aveugle et contrôlée contre placebo et versus moxifloxacine chez des volontaires sains. Le pérampanel a été administré à des doses journalières maximales de 12 mg/jour pendant 7 jours. Le pérampanel n'a pas induit d'allongement de l'intervalle QTc et n'a pas eu d'effet dose-dépendant ou cliniquement significatif sur la durée du complexe QRS.
Efficacité clinique – crises d'épilepsie focales
L'efficacité du pérampanel en association dans les crises d'épilepsie focales a été établie par trois études multicentriques, randomisées, en double aveugle, contrôlées contre placebo d'une durée de 19 semaines chez des patients adultes et adolescents.
Les patients présentaient des crises d'épilepsie focales avec ou sans généralisation secondaire et non correctement contrôlées par un à trois médicaments antiépileptiques concomitants.
Durant une période initiale de 6 semaines, les patients devaient présenter plus de cinq crises d'épilepsie et des périodes sans crise n'excédant pas 25 jours. Dans ces trois études cliniques, l'ancienneté moyenne de l'épilepsie était d'environ 21,06 ans. Entre 85,3% et 89,1% des patients prenaient deux à trois médicaments antiépileptiques concomitants avec ou sans stimulation vagale en parallèle.
Deux études ont été menées pour comparer des doses de 8 et 12 mg/jour de pérampanel à un placebo et la troisième étude pour comparer des doses de 2, 4 et 8 mg/jour à un placebo. Dans les trois études, après une phase initiale de 6 semaines avant la randomisation destinée à établir la fréquence initiale des crises d'épilepsie, les patients ont été randomisés et la posologie a été augmentée progressivement jusqu'à la dose attribuée par randomisation. Pendant la phase de titration des trois études, le traitement a été instauré à la dose de 2 mg/jour et la dose était augmentée chaque semaine par paliers de 2 mg/jour jusqu'à la dose cible. Les patients présentant des événements indésirables intolérables pouvaient poursuivre le traitement à la même dose ou diminuer la posologie à la dose antérieure tolérée. Dans les trois études, la phase de titration a été suivie d'une phase d'entretien d'une durée de 13 semaines au cours de laquelle les patients devaient recevoir une dose stable de pérampanel.
Les taux de répondeurs à 50% combinés ont été de 19% pour le placebo, 29% pour la dose de 4 mg, 35% pour la dose de 8 mg et 35% pour la dose de 12 mg. Un effet statistiquement significatif sur la diminution de la fréquence des crises au cours d'une période de 28 jours (entre la phase initiale et la phase de traitement) a été observé par rapport au groupe placebo avec le traitement par Fycompa aux doses de 4 mg/jour (étude 306), 8 mg/jour (études 304, 305 et 306) et 12 mg/jour (études 304 et 305). Ces études montrent que le pérampanel administré en association aux doses de 4 mg à 12 mg une fois par jour est significativement plus efficace que le placebo dans cette population.
Les données des études contrôlées contre placebo démontrent une amélioration du contrôle des crises d'épilepsie avec une dose de 4 mg de Fycompa une fois par jour et que ce bénéfice est majoré lorsque la dose est augmentée à 8 mg/jour. Dans la population globale, l'efficacité observée à la dose de 12 mg n'a pas été meilleure que celle observée à la dose de 8 mg. Un bénéfice de la dose de 12 mg a été observé chez certains patients supportant bien la dose de 8 mg et lorsque la réponse clinique à cette dose était insuffisante.
Enfants et adolescents:
Dans les trois études pivots de phase III en double aveugle, contrôlées contre placebo, au total N=143 adolescents âgés de 12 à 18 ans avaient été inclus en complément des patients adultes dans ces études. Les résultats chez ces adolescents étaient comparables à ceux observés dans la population adulte. On ne dispose actuellement d'aucun résultat d'études complémentaires réalisées de manière ciblée dans une ou plusieurs classes d'âge pédiatrique pour la même indication.
Une étude de 19 semaines randomisée, en double aveugle, contrôlée contre placebo et avec phase de prolongation ouverte (étude 235) a évalué les effets à court terme de Fycompa sur la cognition (dose cible: 8 à 12 mg une fois par jour) pendant son utilisation comme thérapie additionnelle chez 133 (Fycompa n=85, placebo n=48) patients adolescents âgés de 12 à 18 ans avec crises focales insuffisamment contrôlées. La fonction cognitive a été évaluée au moyen du Global Cognition t-Scores du Cognitive Drug Research (CDR) System. Il s'agit d'un score composite rassemblant les 5 domaines testés suivants: Power of Attention (capacité de concentration), Continuity of Attention (durée de concentration), Quality of Episodic Secondary Memory (qualité de la mémoire secondaire épisodique), Quality of Working Memory (qualité de la mémoire de travail) et Speed of Memory (vitesse de la mémoire). Par rapport à la valeur avant traitement, le changement moyen (SD) du CDR System Global Cognition t-Scores jusqu'à la fin de la phase de traitement en double aveugle (19 semaines) était de 1,1 (7,14) dans le groupe placebo et de (moins) –1,0 (8,86) dans le groupe pérampanel, la différence entre les groupes de traitement était de (moins) -2,2 (-5,2; 0,8) en termes de moyenne des moindres carrés (IC à 95%). Il n'y avait aucune différence statistiquement significative entre les groupes de traitement (p = 0,145). Les Global Cognition t-Scores du CDR System pour le placebo et le pérampanel étaient de 41,2 (10,7), respectivement 40,8 (13,0) lors du relevé des valeurs avant traitement. Chez les patients qui recevaient le pérampanel dans la phase de prolongation ouverte (n = 112), le changement moyen (SD) du Global Cognition t-Scores du CDR System a été de (moins) -1,0 (9,91) jusqu'à la fin du traitement ouvert par rapport à la valeur avant traitement. Ce résultat n'était pas statistiquement significatif (p = 0,96). Aucun effet sur le développement osseux n'a été observé durant 52 semaines de traitement par le pérampanel (n = 114). Aucun effet n'a été constaté sur le poids, la taille et le développement sexuel durant 104 semaines de traitement (n = 114).
Efficacité clinique - Crises généralisées tonico-cloniques primaires
L'efficacité du pérampanel chez les patients âgés de 12 ans et plus atteints d'épilepsie idiopathique généralisée présentant des crises généralisées tonico-cloniques primaires a été évaluée dans le cadre d'une étude multicentrique, randomisée, en double aveugle, contrôlée contre placebo.
Les patients qui ont été traités par un à trois médicaments antiépileptiques administrés simultanément et ayant présenté au moins trois crises généralisées tonico-cloniques primaires pendant la période de pré-inclusion de 8 semaines ont été inclus. La population de l'étude était composée de 164 patients (pérampanel n=82, placebo n=82). La posologie était augmentée progressivement sur quatre semaines jusqu'à une dose cible de 8 mg par jour ou jusqu'à la dose maximale tolérée et les patients ont été traités pendant 13 semaines supplémentaires au dernier palier de dose atteint à la fin de la période de titration. La durée totale de la période de traitement était de 17 semaines. Le médicament à l'étude était administré une fois par jour.
Le taux de répondeurs à 50% pour les crises généralisées tonico-cloniques primaires pendant la période d'entretien a été statistiquement significativement plus élevé dans le groupe pérampanel (58,0%) que dans le groupe placebo (35,8%), p=0,0059.
La variation médiane en pourcentage de la fréquence des crises généralisées tonico-cloniques primaires sur une période de 28 jours pendant les périodes de titration et d'entretien (combinées) par rapport à la période avant la randomisation a été plus importante avec le pérampanel (-76,5%) qu'avec le placebo (-38,4%), p <0,0001. Pendant la période d'entretien de 3 mois, 30,9% des patients (25/81) traités par le pérampanel ont été libres de crises généralisées tonico-cloniques primaires par rapport à 12,3% des patients (10/81) recevant le placebo. L'étude montre que le pérampanel administré en association à une dose de 8 mg une fois par jour est significativement plus efficace que le placebo.
Phase d'extension en ouvert: sur les 140 patients ayant terminé l'étude, 114 (81,4%) sont entrés dans la phase d'extension. Les patients recevant le placebo sont passés au pérampanel dans un intervalle de six semaines. Une période d'entretien (≥1 an) a suivi. Dans la phase d'extension, ≥1 an, 73,7% des patients ont reçu une dose de pérampanel supérieure à 4 à 8 mg par jour et 16,7% une dose supérieure à 8 à 12 mg par jour.
L'étude a inclus 22 adolescents âgés de 12 à 18 ans. Les résultats chez ces adolescents ont été comparables à ceux observés dans la population adulte.
Autres types d'épilepsie généralisée:
L'efficacité et la sécurité du pérampanel chez les patients présentant des crises myocloniques n'ont pas été établies. Les données disponibles sont insuffisantes pour tirer des conclusions. L'efficacité du pérampanel dans le traitement des absences n'a pas été démontrée.
Dans l'étude 332, multicentrique, randomisée, en double aveugle, contrôlée contre placebo chez les patients présentant des crises GTCP et des crises myocloniques concomitantes, 16,7% des patients (4/24) traités par le pérampanel ont été libres de crises par rapport à 13,0% des patients (3/23) recevant le placebo. Chez les patients présentant des crises d'absence concomitantes, 22,2% des patients (6/27) traités par le pérampanel ont été libres de crises par rapport à 12,1% des patients (4/33) recevant le placebo. 23,5% des patients (19/81) traités par le pérampanel ont été libres de crises (crises GTCP, crises myocloniques et absences) par rapport à 4,9% des patients (4/81) recevant le placebo.
Arrêt de la comédication antiépileptique
Dans une étude rétrospective de la pratique clinique, 51 patients atteints d'épilepsie qui recevaient le pérampanel en traitement en association ont changé pour le pérampanel en monothérapie. La majorité de ces patients avait des antécédents de crises d'épilepsie partielles. Parmi ces patients, 14 (27%) sont revenus au traitement en association au cours des mois suivants. Trente-quatre (34) patients ont été suivis pendant au moins 6 mois et sur ceux-ci, 24 patients (71%) sont restés sous pérampanel en monothérapie pendant au moins 6 mois. Dix (10) patients ont été suivis pendant au moins 18 mois et sur ceux-ci, 3 patients (30%) sont restés sous pérampanel en monothérapie pendant au moins 18 mois.
PharmacocinétiqueLa pharmacocinétique du pérampanel a été étudiée chez des volontaires sains adultes (âgés de 18 à 79 ans), des adultes et adolescents présentant des crises d'épilepsie partielles et des patients présentant des crises généralisées tonico-cloniques primaires, des adultes atteints de la maladie de Parkinson, des adultes présentant une neuropathie diabétique, des adultes atteints de sclérose en plaques et des patients présentant une insuffisance hépatique.
Absorption
Après administration orale, le pérampanel est rapidement et totalement absorbé et a un effet de premier passage hépatique négligeable (la biodisponibilité absolue est d'environ 100%). Les aliments ne modifient pas l'ampleur de l'absorption, mais ralentissent sa vitesse. En cas d'administration concomitante avec des aliments, les concentrations plasmatiques maximales sont diminuées et retardées de 2 heures par rapport à l'administration à jeun.
Chez des volontaires sains, l'augmentation des concentrations plasmatiques du pérampanel a été directement proportionnelle à celle des doses administrées dans la fourchette de doses comprises entre 2 mg et 12 mg. Dans une analyse pharmacocinétique de population de patients présentant des crises d'épilepsie partielles qui recevaient le pérampanel à des doses allant jusqu'à 12 mg/jour et de patients présentant des crises généralisées tonico-cloniques primaires qui recevaient le pérampanel à des doses allant jusqu'à 8 mg/jour dans des études cliniques contrôlées contre placebo, une relation linéaire entre la dose et la concentration plasmatique de pérampanel a été observée.
En cas d'administration à jeun, le pérampanel suspension buvable est bioéquivalent sur une base mg/mg au pérampanel comprimés. Après administration d'une dose unique de 12 mg des deux formulations avec un repas riche en graisses, l'ASC0-inf du pérampanel suspension buvable est équivalente, la Cmax est inférieure d'environ 23% et le temps jusqu'à l'exposition plasmatique maximale (tmax) est prolongé de 2 heures par rapport à la formulation en comprimés. Cependant, une analyse pharmacocinétique de population a démontré qu'en conditions d'exposition à l'état d'équilibre simulées, la Cmax et l'ASC0-24h du pérampanel suspension buvable étaient bioéquivalentes à celles de la formulation en comprimés, tant après administration à jeun qu'avec un repas.
Après administration avec un repas riche en graisses d’une dose unique de 12 mg de péampanel suspension buvable, la Cmax et ASC0-inf étaient inférieures d’environ 22 % et 13 % respectivement par rapport àl’administration à jeun.
Distribution
Les données des études in vitro indiquent que la liaison du pérampanel aux protéines plasmatiques est d'environ 95%.
Les études in vitro montrent que le pérampanel n'est pas un substrat ni un inhibiteur significatif des polypeptides transporteurs d'anions organiques (OATP) 1B1 et 1B3, des transporteurs d'anions organiques (OAT) 1, 2, 3 et 4, des transporteurs de cations organiques (OCT) 1, 2 et 3 et des pompes d'efflux que sont la glycoprotéine P et la protéine BCRP (Breast Cancer Resistance Protein).
Métabolisme
Le pérampanel est fortement métabolisé par oxydation primaire suivie d'une glucuroconjugaison. Le métabolisme du pérampanel est contrôlé en priorité par le CYP3A4 et/ou le CYP3A5, et pour une part plus faible par le CYP1A2 et le CYP2B6, sur la base de résultats d'études in-vitro, dans lesquelles du CYP humain recombinant et des microsomes hépatiques humains étaient utilisés. Après l'administration de pérampanel radiomarqué, les métabolites du pérampanel n'ont été retrouvés qu'à l'état de traces dans le plasma.
Élimination
Après l'administration d'une dose de pérampanel radiomarqué chez 8 volontaires sains âgés, 30% de la radioactivité ont été retrouvés dans les urines et 70% dans les fèces. Dans les urines et les fèces, la radioactivité retrouvée était essentiellement composée d'un mélange de métabolites oxydés et conjugués. Dans une analyse pharmacocinétique de population réalisée sur les données combinées de 19 études de phase I, la demi-vie t½ moyenne du pérampanel était de 105 heures. Lorsque le pérampanel a été administré en association avec la carbamazépine, un inducteur puissant du CYP3A, le t½ moyen a été de 25 heures.
Cinétique des populations particulières de patients
Patients insuffisants hépatiques
Les paramètres pharmacocinétiques du pérampanel après administration d'une dose unique de 1 mg ont été évalués chez 12 sujets présentant une insuffisance hépatique légère ou modérée (Child-Pugh A et B, respectivement) en comparaison avec 12 volontaires sains appariés pour les caractéristiques démographiques. La clairance apparente moyenne du pérampanel libre était de 188 ml/min chez les sujets présentant une insuffisance légère contre 338 ml/min dans le groupe témoin et de 120 ml/min chez les sujets présentant une insuffisance modérée contre 392 ml/min dans le groupe témoin. Le t½ a été plus long chez les sujets présentant une insuffisance légère (306 h contre 125 h) et chez les sujets présentant une insuffisance modérée (295 h contre 139 h) que chez les volontaires sains du groupe témoin.
Patients insuffisants rénaux
La pharmacocinétique du pérampanel n'a pas été évaluée de façon systématique chez les patients insuffisants rénaux. Le pérampanel est éliminé presque exclusivement par métabolisme suivi d'une excrétion rapide des métabolites; les métabolites du pérampanel ne sont retrouvés qu'à l'état de traces dans le plasma. Dans une analyse pharmacocinétique de population de patients présentant des crises d'épilepsie partielles ayant une clairance de la créatinine comprise entre 39 et 160 ml/min et qui recevaient le pérampanel à des doses allant jusqu'à 12 mg/jour dans le cadre d'études cliniques contrôlés contre placebo, la clairance du pérampanel n'a pas été influencée par la clairance de la créatinine.
Dans une analyse pharmacocinétique de population de patients présentant des crises généralisées tonico-cloniques primaires qui recevaient du pérampanel à des doses allant jusqu'à 8 mg/jour dans le cadre d'une étude clinique contrôlée contre placebo, la clairance du pérampanel n'a pas été influencée par la clairance de la créatinine initiale.
Sexe
Dans une analyse pharmacocinétique de population de patients présentant des crises d'épilepsie partielles et recevant le pérampanel à des doses allant jusqu'à 12 mg/jour et de patients présentant des crises généralisées tonico-cloniques primaires qui recevaient le pérampanel à des doses allant jusqu'à 8 mg/jour dans le cadre d'études cliniques contrôlés contre placebo, la clairance du pérampanel chez les femmes (0,54 l/h) était plus faible de 18% que chez les hommes (0,66 l/h).
Patients âgés (65 ans et plus)
Dans une analyse pharmacocinétique de population de patients (âgés de 12 à 74 ans), présentant des crises d'épilepsie partielles et recevant le pérampanel à des doses allant jusqu'à 8 mg/jour et de patients présentant des crises généralisées tonico-cloniques primaires (âgés de 12 à 58 ans) qui recevaient le pérampanel à des doses allant jusqu'à 12 mg/jour dans le cadre d'études cliniques contrôlés contre placebo, aucun effet significatif dû à l'âge sur la clairance du pérampanel n'a été observé.
Adolescents (âgés de 12 à 18 ans)
Une analyse pharmacocinétique de population des patients adolescents inclus dans les études de phase II et III n'a mis en évidence aucune différence notable entre cette population et la population générale.
Données précliniquesLes données précliniques pouvant être éventuellement pertinentes pour l'utilisation clinique sont résumées ci-dessous.
Dans une étude de pharmacologie de sécurité réalisé chez le rat, une augmentation dose-dépendante de la température corporelle (allant jusqu'à +0,6 °C pour 5 mg/kg) a été observée. Dans les études de toxicité une hypothermie a parfois été observée après administration répétée de doses élevées. Dans les études cliniques, il n'a pas été constaté d'augmentation ou de diminution sensible de la température corporelle.
Il a été établi que le foie est l'organe cible après administration répétée de doses élevées dans les études de toxicité (augmentation du poids, atrophie ou/et nécrose cellulaires). Basé sur la liaison covalente aux macromolécules, il y a accumulation de pérampanel qui atteint des concentrations tissulaires élevées dans cet organe et interagit avec le cytochrome P450, les enzymes UGT et la protéine de transport OAT2. Sur la base de l'exposition systémique (ASC) pour la dose sans effet toxique observable (no-observed-adverse-effect level: NOAEL) établie chez les animaux et pour les patients insuffisants hépatiques, l'écart de sécurité est de ≤1.
Le pérampanel n'a pas mis en évidence de toxicité génétique dans les études in vitro et in vivo réalisées.
Dans une étude de cancérogénicité d'une durée de 2 ans menée chez le rat, des kératoacanthomes (chez les animaux mâles) et des phéochromocytomes (chez les animaux femelles) ont été observés dans les groupes témoins et avec une incidence plus élevée dans les groupes traités par le pérampanel, sans qu'ils soient statistiquement significatifs. L'incidence se situait dans la marge d'une survenue spontanée chez les rats âgés.
Dans les études effectuées chez l'animal une incidence plus élevée de réactions cutanées après exposition à la lumière UV (formation de rides, apparition de zones blanches ou de zones erythémateuses/rouges) avec une marge de sécurité de ≤3 (basée sur l'exposition systémique, l'ASC et une dose clinique de 12 mg de pérampanel/jour) a été observée. Des études de distribution tissulaire avec le 14C-pérampanel ont mis en évidence de longs temps de rétention concernant la radioactivité (jusqu'à 110 semaines) dans le globe oculaire chez les rongeurs et les non rongeurs. Les études de toxicité générales n'ont, certes, pas mis en évidence de lésions ophtalmoscopiques ou histopathologiques oculaires liés au traitement après administration répétée. Cependant, dans l'étude de cancérogénicité d'une durée de 2 ans, une kératite dose-dépendante (≥3 mg/kg/jour, marge de sécurité <1 basée sur l'exposition systémique, l'ASC et une dose clinique de 12 mg de pérampanel/jour) et des infiltrations des cellules inflammatoires/des ulcères de la peau (≥3 mg/kg/jour, marge de sécurité <1 basée sur l'exposition systémique, l'ASC et une dose clinique de 12 mg de pérampanel/jour) ont été observées chez la souris. Les effets non néoplasiques sur le tractus uro-génital (dilatations et lésions mésenchymateuses) qui sont apparus dans le cadre de cette étude peuvent s'expliquer par une rétention urinaire/des obstructions rénales et vésiculaires.
Dans les études de fécondité chez le rat, des cycles prolongés et irréguliers ont été observés chez les femelles après administration de doses élevées (30 mg/kg; marge de sécurité <1 basée sur l'exposition systémique à la dose sans effet observable (no-observed-effect level, NOEL) et une dose clinique de 12 mg de pérampanel/jour); ces changements n'ont toutefois pas altéré la fécondité et les premiers stades du développement embryonnaire. Il n'y a pas eu d'effet sur la fécondité des mâles.
L'excrétion dans le lait a été mesurée chez les rates 10 jours après la mise à bas. Les concentrations étaient maximales après une heure et étaient 3,65 fois plus élevées que les concentrations plasmatiques. Le pérampanel traverse la barrière placentaire. Le passage à travers la barrière placentaire était cependant relativement faible; 0,09% ou moins de la dose administrée a été détecté chez les fœtus.
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets tératogènes chez le rat ou le lapin, mais une embryotoxicité a été observée chez le rat à des doses entraînant une toxicité maternelle (marge de sécurité <1 basée sur l'exposition systémique, l'ASC et une dose clinique de 12 mg de pérampanel/jour). Dans une étude de toxicité sur le développement péri- et post-natal chez le rat, des conditions anormales de parturition et d'allaitement à des doses toxiques et une augmentation de la mortinatalité dans la descendance (marge de sécurité <1 basée sur l'exposition systémique, l'ASC, à la dose sans effet observable (no-observed-effect level: NOEL) et une dose clinique de 12 mg de pérampanel/jour) ont été observées. De plus, les valeurs de l'indice de natalité et de l'indice de viabilité avaient tendance à diminuer 4 jours après la mise à bas et on a observé dans la descendance une baisse de la prise de poids et une différenciation morphologique retardée (retard à l'ouverture vaginale ou retard de la séparation balano-préputiale). Une altération du comportement et des fonctions de reproduction de la descendance n'a cependant pas été observée.
Dans des études de toxicité effectuée chez de jeunes rats et chiens une hypersensibilité au pérampanel a été observée en comparaison avec des animaux adultes à une exposition systémique similaire.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après ouverture
À utiliser dans les 90 jours après ouverture.
Remarques concernant le stockage
Ne pas conserver au-dessus de 30 °C. Conserver hors de portée des enfants.
Numéro d’autorisation62440, 67665 (Swissmedic).
PrésentationFycompa 2 mg: 7 comprimés pelliculés (B)
Fycompa 4 mg: 28 comprimés pelliculés (B)
Fycompa 6 mg: 28 comprimés pelliculés (B)
Fycompa 8 mg: 28 comprimés pelliculés (B)
Fycompa 10 mg: 28 comprimés pelliculés (B)
Fycompa 12 mg: 28 comprimés pelliculés (B)
Fycompa 0,5 mg/ml: 340 ml de suspension en flacon avec bouchon de sécurité enfant; chaque emballage contient un adaptateur à pression pour flacon et deux seringues de 20 ml avec une division de 0,5 ml.
Titulaire de l’autorisationEisai Pharma SA, Zurich.
Mise à jour de l’informationDécembre 2019.
|