CompositionPrincipes actifs
Rilpivirine (sous forme de base libre de rilpivirine).
Excipients
Acide citrique monohydraté, glucose monohydraté, poloxamère 338, phosphate monosodique monohydraté, hydroxyde de sodium pour ajuster le pH et garantir l'isotonicité, eau pour préparations injectables.
Teneur totale en sodium pour 600 mg de principe actif: 1,66 mg.
Teneur totale en sodium pour 900 mg de principe actif: 2,49 mg.
Indications/Possibilités d’emploiREKAMBYS en association avec le cabotégravir injectable est indiqué dans le traitement de l'infection par le virus de l'immunodéficience humaine de type 1 (VIH-1) chez l'adulte et l'adolescents âgés d'au moins 12 ans et pesant au moins 35 kg, virologiquement contrôlés (ARN du VIH-1< 50 copies/ml) sous traitement antirétroviral stable depuis au moins 6 mois avant le passage à l'association rilpivirine-cabotégravir, sans résistance connue ou suspectée et sans antécédents d'échec virologique à des principes actifs de la classe des INNTI et des INI (voir «Efficacité clinique»).
Posologie/Mode d’emploiLe traitement doit être effectué par un médecin expérimenté dans la prise en charge des infections par le VIH.
Les injections de REKAMBYS doivent être administrée par un professionnel de la santé.
Avant d'instaurer le traitement par REKAMBYS, le médecin traitant doit soigneusement sélectionner les patients qui acceptent le schéma d'injection requis et informer les patients de l'importance de respecter les rendez-vous programmés pour l'administration du médicament, afin de maintenir le contrôle virologique et de réduire le risque de rebond viral et de développement éventuel de résistances associées à l'oubli de doses.
Après l'arrêt de REKAMBYS en association avec une injection de carbotégravir, il est indispensable d'initier un régime antirétroviral alternatif, complètement suppresseur, dans le mois qui suit la dernière injection de REKAMBYS dans le cas d'un schéma posologique mensuel et dans les deux mois qui suivent la dernière injection de REKAMBYS dans le cas d'un schéma posologique bimestriel.
REKAMBYS doit toujours être administré avec une injection de cabotégravir. Il convient donc de toujours prendre en compte également l'information professionnelle correspondante pour l'injection du cabotégravir.
Posologie usuelle
Adultes et adolescents âgés d'au moins 12 ans et pesant au moins 35 kg
Le traitement par REKAMBYS peut commencer par un traitement d'induction par voie orale ou directement par les injections. Le médecin et le patient peuvent décider d'utiliser les comprimés de rilpivirine en tant que traitement d'induction par voie orale avant de commencer les injections de REKAMBYS afin d'évaluer la tolérance (voir tableau 1) ou de commencer directement avec le traitement par REKAMBYS. Les injections de REKAMBYS peuvent être administrée mensuellement ou tous les 2 mois (voir tableau 2 pour les recommandations posologiques mensuelles et le tableau 3 pour les recommandations posologiques bimestrielles). Le médecin et le patient doivent discuter des deux options posologiques avant de commencer les injections de REKAMBYS et choisir la fréquence la mieux adaptée au patient.
En cas de suspicion d'un échec virologique, un régime alternatif doit être initié dès que possible.
Traitement d'induction (Lead-in-Phase) par voie orale
Lors du traitement d'induction par voie orale, il est recommandé que les patients virologiquement contrôlés prennent des comprimés de rilpivirine par voie orale pendant environ un mois (au moins 28 jours, au maximum 2 mois) avant de commencer le traitement avec les injections de REKAMBYS en vue d'évaluer la comptabilité avec le cabotégravir. Un comprimé de 25 mg de rilpivirine une fois par jour au moment des repas et un comprimé de 30 mg de cabotégravir (une fois par jour) doivent être pris (voir l'information professionnelle des comprimés de rilpivirine).
|
Tableau 1: Schéma posologique du traitement d'induction par voie orale
| |
|
Traitement d'induction par voie orale
| |
Médicament
|
1 mois (au moins 28 jours, au maximum 2 mois), suivi de la première injectiona
| |
Rilpivirine
|
25 mg une fois par jour pendant un repas
| |
Cabotégravir
|
30 mg une fois par jour
| |
a
voir le tableau 2 pour le schéma posologique des injections mensuelles et le tableau 3 pour le schéma posologique des injections tous les 2 mois
|
Administration mensuelle de la dose
Première injection (900 mg correspondant à une dose de 3 ml)
La dose initiale recommandée de REKAMBYS est d'une injection intramusculaire unique de 3 ml (900 mg) le dernier jour du traitement antirétroviral actuel ou du traitement d'induction par voie orale. (Voir «Mode d'administration»).
Injections suivantes (600 mg correspondant à une dose de 2 ml)
Après la première injection, la dose recommandée de REKAMBYS pour les injections ultérieures est d'une injection intramusculaire mensuelle de 2 ml (600 mg). (Voir «Mode d'administration»). Les injections peuvent être administrées aux patients jusqu'à 7 jours avant ou après la date prévue de l'injection mensuelle de 2 ml.
|
Tableau 2: Schéma posologique pour l'injection intramusculaire mensuelle
| |
Médicament
|
Première injection
Première injection au mois 1 (le dernier jour du TAR en cours ou de la phase d'induction par voie orale, si elle a lieu)
|
Injections suivantes
Un mois après la première injection et mensuellement par la suite
| |
Rilpivirine
|
3 ml (900 mg)
|
2 ml (600 mg)
| |
Cabotégravir
|
3 ml (600 mg)
|
2 ml (400 mg)
|
Administration de la dose tous les 2 mois
Injections d'induction (900 mg correspondant à une dose de 3 ml)
La première injection intramusculaire d'induction de REKAMBYS de 3 ml (900 mg) doit être effectuée le dernier jour du traitement antirétroviral actuel ou du traitement d'induction par voie orale. Un mois plus tard, une deuxième injection intramusculaire d'induction de 3 ml (900 mg) doit être administrée.
(Voir «Mode d'administration»). La deuxième injection d'induction de 3 ml (900 mg) peut être administrée aux patients jusqu'à 7 jours avant ou après l'administration de la dose.
Injections suivantes (900 mg correspondant à une dose de 3 ml)
Après la deuxième injection d'induction, la dose recommandée de REKAMBYS pour les injections ultérieures est d'une injection intramusculaire unique de 3 ml (900 mg) tous les 2 mois. (Voir «Mode d'administration»). Les injections peuvent être administrées aux patients jusqu'à 7 jours avant ou après l'administration de l'injection de 3 ml tous les 2 mois.
|
Tableau 3: Schéma posologique pour l'injection intramusculaire tous les 2 mois
| |
Médicament
|
Première injection
|
Injections suivantes
| |
Injections d'induction au mois 1 (le dernier jour du TAR en cours ou de la phase d'induction par voie orale, si elle a lieu) et au mois 2
|
Deux mois après la dernière injection d'induction et tous les 2 mois par la suite
| |
Rilpivirine
|
3 ml (900 mg)
|
3 ml (900 mg)
| |
Cabotégravir
|
3 ml (600 mg)
|
3 ml (600 mg)
|
Ajustement de la fréquence d'administration
Recommandations posologiques lors du passage d'injections mensuelles à des injections tous les deux mois
Les patients passant d'injections mensuelles en continu à des injections en continu tous les deux mois doivent recevoir une injection intramusculaire de 3 ml (900 mg) de REKAMBYS un mois après la dernière injection continue de 2 ml (600 mg). Les injections suivantes de 3 ml (900 mg) sont ensuite administrées tous les 2 mois.
Recommandations posologiques lors du passage d'injections tous les deux mois à des injections mensuelles
Les patients passant d'injections en continu tous les deux mois à des injections mensuelles en continu dans le cadre de la phase d'entretien doivent recevoir une injection intramusculaire unique de 2 ml (600 mg) de REKAMBYS deux mois après la dernière injection de 3 ml (900 mg) de REKAMBYS, puis une injection de 2 ml (600 mg) tous les mois.
Omission d'une injection
Il est fortement recommandé de respecter le schéma d'administration des injections. Les patients qui manquent un rendez-vous pour une injection doivent faire l'objet d'une réévaluation clinique afin de s'assurer que la reprise du traitement est appropriée. Voir les tableaux 4 et 5 pour les recommandations posologiques après l'omission d'une injection.
Omission d'une dose injectable mensuelle
Traitement oral de transition et reprise des injections mensuelles:
Si un retard de plus de 7 jours par rapport à la date prévue de l'injection est inévitable, un traitement oral (25 mg de rilpivirine et 30 mg de cabotégravir une fois par jour sous forme de comprimés) peut être utilisé pendant 2 mois consécutifs. En variante, un autre traitement antirétroviral oral totalement suppressif peut être utilisé jusqu'à la reprise des injections. Le choix du traitement doit tenir compte des lignes directrices actuelles de traitement du VIH. Pour les données sur le traitement oral de transition avec d'autres traitements antirétroviraux totalement suppressifs, voir section «Efficacité clinique».
La première dose du traitement oral doit être prise 1 mois (± 7 jours) après la dernière dose injectable de REKAMBYS. L'administration par injection doit être reprise le dernier jour de l'administration orale (voir les recommandations dans le tableau 4). Si plus de deux injections mensuelles consécutives ont été omises et doivent être remplacées, un autre traitement oral doit être instauré un mois (± 7 jours) après la dernière injection de REKAMBYS.
|
Tableau 4: Recommandations pour la reprise des injections mensuelles de REKAMBYS après l'omission d'injections ou après un traitement oral de transition
| |
Temps écoulé depuis la dernière injection
|
Recommandation
| |
≤2 mois:
|
Poursuivre le plus tôt possible les injections mensuelles de 2 ml (600 mg).
| |
> 2 mois:
|
Reprendre la dose de 3 ml (900 mg), puis poursuivre les injections mensuelles de 2 ml (600 mg).
|
Omission d'une dose injectable tous les deux mois
Traitement oral de transition et reprise des injections tous les deux mois:
Si un retard de plus de 7 jours par rapport à la date prévue de l'injection est inévitable, un traitement oral quotidien (25 mg de rilpivirine et 30 mg de cabotégravir sous forme de comprimés) peut être utilisé pendant 2 mois consécutifs. En variante, un autre traitement antirétroviral oral totalement suppressif peut être utilisé jusqu'à la reprise des injections. Le choix du traitement doit tenir compte des lignes directrices actuelles de traitement du VIH. Pour les données sur le traitement oral de transition avec d'autres traitements antirétroviraux totalement suppressifs, voir section «Efficacité clinique».
La première dose du traitement oral doit être prise deux mois (± 7 jours) après la dernière injection de REKAMBYS et de cabotégravir. L'administration par injection doit être reprise le dernier jour de l'administration orale (voir les recommandations dans le tableau 5). S'il faut remplacer un écart de plus de deux mois, c.-à.-d. si plus d'une injection tous les deux mois a été omise, un autre traitement oral doit être instauré deux mois (± 7 jours) après la dernière injection de REKAMBYS.
|
Tableau 5: Recommandations pour la reprise des injections tous les deux mois de REKAMBYS après l'omission d'injections ou après le traitement oral de transition
| |
Visite manquée pour une injection
|
Temps écoulé depuis la dernière injection
|
Recommandation (toutes les injections sont de 3 ml)
| |
Injection 2
|
≤2 mois
|
Réaliser l'injection de 3 ml (900 mg) dès que possible et poursuivre le schéma d'administration des injections tous les 2 mois.
| |
> 2 mois
|
Reprendre la dose de 3 ml (900 mg) chez le patient, puis réaliser une deuxième injection de 3 ml (900 mg) en dose initiale un mois plus tard. Suivre ensuite le schéma d'administration des injections tous les 2 mois.
| |
Injection 3 ou suivantes
|
≤3 mois
|
Réaliser l'injection de 3 ml (900 mg) dès que possible et poursuivre le schéma d'administration des injections tous les 2 mois.
| |
> 3 mois
|
Reprendre la dose de 3 ml (900 mg) chez le patient, puis réaliser une deuxième injection de 3 ml (900 mg) en dose initiale un mois plus tard. Suivre ensuite le schéma d'administration des injections tous les 2 mois.
|
Mode d'administration
Administration uniquement sous forme d'injection par voie intramusculaire (IM) dans le muscle fessier (glutéal). Injecter lentement (voir «Mises en garde et précautions»). Ne pas injecter par voie intraveineuse.
Lors de l'administration de REKAMBYS, le professionnel de santé doit tenir compte de l'indice de masse corporelle (IMC) du patient afin de s'assurer que la longueur de l'aiguille est suffisante pour atteindre le muscle fessier.
REKAMBYS et le cabotégravir doivent être injectés dans le muscle fessier lors de la même visite, dans des sites d'injection séparés (controlatéralement ou si cela n'est pas possible, à une distance de 2 cm). Il convient également de respecter une distance de 2 cm par rapport aux sites d'injection précédents ou à d'éventuelles réactions au niveau des sites d'injection précédents.
Pour réaliser l'injection, tenir compte des instructions détaillées étape par étape qui figurent dans le mode d'emploi joint à l'emballage. Ces instructions doivent être attentivement suivies lors de la préparation de la suspension injectable afin d'éviter les fuites.
REKAMBYS doit être administré par un professionnel de la santé.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
On ne dispose que d'informations limitées sur l'utilisation de la rilpivirine chez les patients présentant des troubles légers ou modérés de la fonction hépatique (score de Child-Pugh A ou B). Aucun ajustement de la dose de REKAMBYS n'est nécessaire chez les patients présentant des troubles légers à modérés de la fonction hépatique (score de Child-Pugh A ou B). REKAMBYS n'a pas été évalué chez les patients présentant des troubles sévères de la fonction hépatique (score de Child-Pugh C) (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose de REKAMBYS n'est nécessaire chez les patients présentant des troubles légers ou modérés de la fonction rénale. Les patients présentant une clairance de la créatinine estimée < 50 ml/min/1,73 m2 n'ont pas été inclus dans les études de phase III. REKAMBYS doit être utilisé avec prudence chez les patients présentant des troubles sévères de la fonction rénale (clairance de la créatinine de 15 à ≤30 ml/min) ou une insuffisance rénale terminale (clairance de la créatinine ≤15 ml/min) (surveillance accrue des effets indésirables). Chez les patients présentant des troubles sévères de la fonction rénale ou une insuffisance rénale terminale, l'association de REKAMBYS et d'un inhibiteur puissant du CYP3A ne doit être utilisée que si le bénéfice est supérieur au risque (voir «Pharmacocinétique»).
Patients âgés (à partir de 65 ans)
Aucun ajustement de la dose de REKAMBYS n'est nécessaire chez les patients âgés (voir «Pharmacocinétique»).
Enfants âgés de moins de 12 ans et adolescents pesant moins de 35 kg
La sécurité et l'efficacité de REKAMBYS n'ont pas été établies chez les enfants âgés de moins de 12 ans et les adolescents pesant moins de 35 kg. On ne dispose d'aucune donnée.
Contre-indicationsHypersensibilité à la rilpivirine ou à l'un des excipients.
L'administration concomitante de REKAMBYS et de médicaments qui sont des inducteurs modérés à puissants des enzymes du CYP3A est contre-indiquée, car des diminutions importantes des concentrations plasmatiques de rilpivirine peuvent survenir, ce qui peut entraîner une perte de l'effet thérapeutique de REKAMBYS (voir «Interactions»).
Ces médicaments sont par exemple:
·les anticonvulsivants carbamazépine, oxcarbazépine, phénobarbital, phénytoïne;
·les principes actifs antimycobactériens rifabutine, rifampicine, rifapentine;
·le glucocorticoïde systémique dexaméthasone, sauf en cas de traitement par une dose unique;
·le millepertuis (Hypericum perforatum).
Les inhibiteurs de la pompe à protons sont également contre-indiqués lors de l'administration de la rilpivirine orale.
Il convient par ailleurs de tenir compte de l'information professionnelle du cabotégravir.
Mises en garde et précautionsAction prolongée d'une injection de rilpivirine
Des concentrations résiduelles de rilpivirine peuvent rester dans la circulation systémique des patients pendant une longue durée (jusqu'à 4 ans) allant au-delà de la période d'administration active. Il convient de tenir compte de la longue durée de libération de la rilpivirine lors de l'utilisation de REKAMBYS, lors de l'évaluation du rapport bénéfice-risque individuel avant de choisir le traitement, pendant le traitement et après le traitement par REKAMBYS (voir «Interactions», «Effets indésirables», «Grossesse», «Allaitement», «Fertilité», «Pharmacocinétique» et «Surdosage»).
En raison de la forte liaison aux protéines, il n'existe aucun mécanisme permettant d'arrêter ou de neutraliser la libération de la rilpivirine ou de retirer celle-ci du muscle (p.ex. par aspiration) ou du sang (p.ex. par hémodialyse).
Transmission du VIH
Les résultats des études d'observation ont montré qu'il n'y a pas de risque de transmission sexuelle du VIH, si la suppression virologique a été atteinte et maintenue. Toutefois, le risque de transmission sexuelle du VIH ne peut pas être exclu si le TAR prescrit n'est pas pris régulièrement et/ou si la suppression virologique n'a pas été atteinte et maintenue.
Patients co-infectés par le virus de l'hépatite B et de l'hépatite C
Les patients co-infectés par le virus de l'hépatite B ont été exclus des études menées avec REKAMBYS. Il n'est pas recommandé d'instaurer REKAMBYS chez les patients co-infectés par le virus de l'hépatite B. L'incidence d'une élévation des enzymes hépatiques était plus élevée chez les patients co-infectés par le virus de l'hépatite B et traités par la rilpivirine orale que chez les patients non co-infectés par le virus de l'hépatite B et traités par la rilpivirine orale. Il convient de suivre les directives thérapeutiques actuelles relatives au traitement de l'infection par le VIH chez les patients co-infectés par le virus de l'hépatite B et de tenir compte de la rubrique «Interactions avec d'autres médicaments» (voir ci-dessous).
Les données sont limitées chez les patients co-infectés par le virus de l'hépatite C. L'incidence d'une élévation des enzymes hépatiques était plus élevée chez les patients co-infectés par le virus de l'hépatite C et traités par la rilpivirine orale que chez les patients non co-infectés par le virus de l'hépatite C et traités par la rilpivirine orale. L'exposition pharmacocinétique à la rilpivirine orale et injectable chez les patients co-infectés par le virus de l'hépatite C était comparable à celle chez observée les patients non co-infectés par le virus de l'hépatite C. Une surveillance de la fonction hépatique est recommandée chez les patients co-infectés par le virus de l'hépatite C. En cas de survenue d'une infection par le virus de l'hépatite C au cours du traitement par REKAMBYS, il convient de suivre les directives thérapeutiques actuelles relatives au traitement de l'infection par le VIH chez les patients co-infectés par le virus de l'hépatite C et de tenir compte de la rubrique «Interactions avec d'autres médicaments» (voir ci-dessous).
Interactions avec d'autres médicaments
La prudence est recommandée lors de la prescription concomitante de rilpivirine et de médicaments pouvant diminuer l'exposition à la rilpivirine.
La prudence est recommandée lors de l'administration concomitante de REKAMBYS et d'un médicament présentant un risque connu de torsades de pointes (voir «Interactions»).
Pour les indications sur les interactions médicamenteuses, voir «Interactions».
Réactions cutanées et d'hypersensibilité
En association avec un traitement par la rilpivirine, des réactions cutanées et d'hypersensibilité graves ont été signalées, par exemple des cas de DRESS (Drug Rash with Eosinophilia and Systemic Symptoms). Certaines réactions cutanées ont été associées à des symptômes constitutionnels tels que la fièvre, d'autres à un dysfonctionnement des organes, y compris des valeurs hépatiques sériques élevées. REKAMBYS doit être arrêté immédiatement si des signes ou des symptômes de réactions cutanées ou d'hypersensibilité sévères se manifestent, notamment des exanthèmes sévères ou des exanthèmes accompagnés de fièvre, de vésicules, d'atteinte des muqueuses, de conjonctivite, d'œdème facial, d'angio-œdème, d'hépatite ou d'éosinophilie. L'état clinique et les valeurs de laboratoire doivent être surveillés et un traitement approprié doit être initié.
Infections opportunistes
Des infections opportunistes et d'autres complications de l'infection par le VIH peuvent en outre survenir chez les patients recevant de la rilpivirine ou un autre traitement antirétroviral. Les patients doivent donc faire l'objet d'une surveillance clinique étroite par des médecins expérimentés dans le traitement de telles affections associées au VIH.
Syndrome inflammatoire de restauration immunitaire
Un syndrome inflammatoire de restauration immunitaire a été rapporté chez des patients ayant reçu un traitement antirétroviral combiné, par exemple par la rilpivirine orale. Au cours de la phase initiale d'un traitement antirétroviral combiné, les patients dont le système immunitaire répond au traitement peuvent développer une réaction inflammatoire à des infections opportunistes asymptomatiques ou résiduelles (p.ex. au complexe Mycobacterium avium, à cytomégalovirus, pneumonie à Pneumocystis jiroveci et tuberculose), pouvant nécessiter des investigations et un traitement supplémentaires. Des maladies auto-immunes (telles qu'une maladie de Basedow ou une hépatite auto-immune) ont également été rapportées dans le cadre d'un syndrome inflammatoire de restauration immunitaire. Cependant, le délai d'apparition est plus variable et ces événements peuvent survenir plusieurs mois après l'instauration du traitement (voir «Effets indésirables»).
Hépatotoxicité
Une hépatotoxicité a été observée sous rilpivirine et cabotégravir chez un nombre limité de patients avec ou sans maladie hépatique préexistante connue (voir «Effets indésirables»).
La surveillance des paramètres hépatiques est recommandée. L'administration de la rilpivirine doit être arrêtée en cas de suspicion d'hépatotoxicité (voir «Action prolongée d'une injection de rilpivirine»).
Risque de développement de résistances après l'arrêt du traitement
Afin de réduire le risque de résistance au virus, il convient d'utiliser un autre schéma antirétroviral, pleinement actif, au plus tard un mois après la dernière injection mensuelle de REKAMBYS ou au plus tard 2 mois après la dernière injection de REKAMBYS en cas d'administration tous les 2 mois. En cas de suspicion d'échec virologique, un autre schéma thérapeutique devra être instauré dès que possible.
Administration intraveineuse accidentelle/réactions post-injection
Au cours des études cliniques, des réactions graves, dont les symptômes peuvent être dyspnée, bronchospasmes, agitation, crampes abdominales, éruption cutanée/urticaire, vertiges, bouffées vasomotrices, transpiration, sensation d'engourdissement buccal, variations de la tension artérielle et du pouls et douleurs (par exemple, dos et poitrine) ont été rapportées occasionnellement dans les minutes suivant l'injection de rilpivirine. Ces symptômes ont été observés chez <1% des patients et ont commencé à s'atténuer en quelques minutes après l'injection, mais leur disparition complète n'a parfois été observée que le jour suivant. Certains patients ont reçu un traitement de soutien. Ils peuvent être en rapport avec une administration intraveineuse accidentelle pendant l'injection intramusculaire, provoquant des concentrations plasmatiques de rilpivirine transitoirement élevées.
Suivre attentivement les instructions d'emploi lors de la préparation et de l'administration de REKAMBYS. Avant l'administration, le flacon de REKAMBYS doit être amené à température ambiante. La suspension est injectée lentement en veillant à éviter une administration intraveineuse accidentelle. Les patients doivent être maintenus sous surveillance pendant une courte période (env. 10 minutes) après l'injection. En cas de survenue d'une réaction post-injection, il convient de surveiller et de traiter le patient selon l'indication clinique (voir «Surdosage»).
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsAucune étude n'a été réalisée pour évaluer les interactions médicamenteuses après l'administration de REKAMBYS. Les données et recommandations suivantes reposent sur des études avec administration orale de rilpivirine ou sur des réflexions théoriques.
Effet d'autres médicaments sur la rilpivirine
Inducteurs et inhibiteurs du CYP3A4
La rilpivirine est principalement métabolisée par le cytochrome P450-3A (CYP3A). Les médicaments induisant ou inhibant le CYP3A peuvent donc influencer la clairance de la rilpivirine (voir «Pharmacocinétique»). Une réduction des concentrations plasmatiques de rilpivirine a été observée lors de l'administration concomitante de REKAMBYS et de médicaments induisant le CYP3A, ce qui peut éventuellement diminuer l'effet thérapeutique de REKAMBYS. C'est pourquoi l'administration concomitante de REKAMBYS et d'inducteurs modérés ou puissants du CYP3A est contre-indiquée.
Une augmentation des concentrations plasmatiques de rilpivirine a été observée lors de l'administration concomitante de REKAMBYS et de médicaments inhibant le CYP3A.
Médicaments allongeant l'intervalle QT
On ne dispose que d'informations limitées sur la possibilité d'interaction pharmacodynamique entre la rilpivirine et les médicaments allongeant l'intervalle QTc à l'électrocardiogramme. Dans le cadre d'une étude menée chez des volontaires sains, des doses suprathérapeutiques de rilpivirine orale (75 mg une fois par jour et 300 mg une fois par jour) ont entraîné un allongement de l'intervalle QTc à l'électrocardiogramme. Les concentrations plasmatiques de rilpivirine après des injections de REKAMBYS sont comparables à celles observées après la prise d'Edurant à une posologie de 25 mg une fois par jour (voir «Pharmacodynamique»). La prudence est recommandée lors de l'administration concomitante de REKAMBYS et d'un médicament présentant un risque connu de torsades de pointes.
Effet de la rilpivirine sur d'autres médicaments
Substrats du CYP
La rilpivirine ne devrait pas avoir d'effets cliniquement significatifs sur l'exposition aux médicaments métabolisés par les enzymes du CYP.
Autres médicaments antirétroviraux
REKAMBYS en association avec le cabotégravir injectable est un schéma complet indiqué pour le traitement des infections par le VIH-1 et ne doit pas être utilisé avec d'autres médicaments antirétroviraux destinés au traitement du VIH-1.
Le tableau 7 présente des informations sur les interactions avec d'autres médicaments antirétroviraux contre le VIH afin de faciliter le choix d'un autre schéma antirétroviral après l'arrêt de REKAMBYS (voir «Risque de développement de résistances après l'arrêt du traitement»). En ce qui concerne les interactions, tous les médicaments antirétroviraux contre le VIH énumérés dans le tableau 7 peuvent être utilisés après l'arrêt de REKAMBYS. En raison de la longue durée de libération de la rilpivirine après l'arrêt des injections de REKAMBYS, certaines interactions sont possibles avec d'autres médicaments antirétroviraux contre le VIH. La rilpivirine n'a probablement aucun effet cliniquement significatif sur la pharmacocinétique d'autres médicaments antirétroviraux contre le VIH. Les inhibiteurs de l'intégrase boostés et les inhibiteurs de la protéase boostés ou non boostés peuvent entraîner une augmentation des quantités résiduelles de rilpivirine dans le plasma. Les inhibiteurs non nucléosidiques de la transcriptase inverse éfavirenz, étravirine et névirapine peuvent en revanche entraîner une diminution des quantités résiduelles de rilpivirine dans le plasma. Ces interactions médicamenteuses survenant après l'arrêt de REKAMBYS sont considérées comme cliniquement non significatives et ne modifient pas la longue durée de libération de la rilpivirine à partir du site d'injection.
Le tableau 6 et le tableau 7 présentent des interactions sélectionnées, connues et théoriques, entre la rilpivirine et des médicaments co-administrés ainsi que des médicaments antirétroviraux contre le VIH devant être utilisés après l'arrêt de REKAMBYS. Les données des études d'interactions médicamenteuses présentent les rapports des moyennes géométriques (geometric mean ratios, GMR) des paramètres pharmacocinétiques en cas d'administration avec/sans traitement concomitant, avec des intervalles de confiance (IC) à 90%.
|
Tableau 6: Interactions et recommandations posologiques de REKAMBYS avec d'autres médicaments
| |
Principe actif par classe thérapeutique (schéma posologique)
|
Effets sur la concentration de principe actif
GMR (IC à 90%) Ω
(éventuel mécanisme d'interaction)
|
Recommandations concernant la co-administration
| |
AGENTS ANTIVIRAUX CONTRE LE VIH (pour l'utilisation avec REKAMBYS)
| |
Cabotégravir* Ω (30 mg q.d. pendant 12 jours)
Rilpivirine (25 mg q.d. pendant 24 jours)
|
Cabotégravir:
AUC: 1,12 (1,05 – 1,19)
Cmin: 1,14 (1,04 – 1,24)
Cmax: 1,05 (0,96 – 1,15)
Rilpivirine:
AUC: 0,99 (0,89 – 1,09)
Cmin: 0,92 (0,79 – 1,07)
Cmax: 0,96 (0,85 – 1,09)
|
Aucun ajustement posologique recommandé.
| |
AUTRES AGENTS ANTIVIRAUX
| |
Ribavirine
|
Non évaluée. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Telbivudine, entécavir, lamivudine, ténofovir alafénamide
|
Non évalués. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
Chez les patients co-infectés par le virus de l'hépatite B, l'instauration d'un traitement par REKAMBYS n'est pas recommandée. Les médecins doivent suivre les directives thérapeutiques actuelles relatives au traitement des infections par le VIH chez les patients co-infectés par le virus de l'hépatite B.
| |
Sofosbuvir/velpatasvir, sofosbuvir/velpatasvir/voxilaprévir, lédipasvir, pibrentasvir/glécaprivir, elbasvir/grazoprévir
|
Non évalués.
Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
ANTICONVULSIVANTS
| |
Carbamazépine
Oxcarbazépine
Phénobarbital
Phénytoïne
|
Non évalués. Des diminutions importantes des concentrations plasmatiques de rilpivirine sont attendues.
(Induction du CYP3A)
|
L'administration concomitante de REKAMBYS est contre-indiquée.
L'administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine (voir «Contre-indications»).
| |
ANTIFONGIQUES AZOLÉS
| |
Kétoconazole*#Ω (400 mg q.d. pendant 22 jours)
Rilpivirine (150 mg q.d. pendant 11 jours)
|
Kétoconazole:
AUC: 0,76 (0,70 – 0,82)
Cmin: 0,34 (0,25 – 0,46)
Cmax: 0,85 (0,80 – 0,90)
(Induction du CYP3A en raison d'une dose élevée de rilpivirine dans l'étude)
Rilpivirine:
AUC: 1,49 (1,31 – 1,70)
Cmin: 1,76 (1,57 – 1,97)
Cmax: 1,30 (1,13 – 1,48)
(Inhibition du CYP3A)
|
Aucun ajustement posologique recommandé.
| |
Fluconazole
Itraconazole
Posaconazole
Voriconazole
|
Non évalués. L'administration concomitante de REKAMBYS et d'antifongiques azolés peut entraîner une augmentation des concentrations plasmatiques de rilpivirine.
(Inhibition des enzymes du CYP3A)
|
Aucun ajustement posologique recommandé.
| |
ANTIMYCOBACTÉRIENS
| |
Rifabutine*#Ω
(300 mg q.d. pendant 11 jours)
Rilpivirine (150 mg q.d. pendant 11 jours)
|
Rifabutine:
AUC: 1,03 (0,97 – 1,09)
Cmin: 1,01 (0,94 – 1,09)
Cmax: 1,03 (0,93 – 1,14)
25-Odésacétyl-rifabutine:
AUC: 1,07 (1,02 – 1,11)
Cmin: 1,12 (1,03 – 1,22)
Cmax: 1,07 (0,98 – 1,17)
|
L'administration concomitante est contre-indiquée. Une administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine (voir «Contre-indications»).
| |
Rifabutine*#Ω
(300 mg q.d. pendant 17 jours)
Rilpivirine (25 mg q.d. pendant 11 jours)
|
Rilpivirine:
AUC: 0,58 (0,52 – 0,65)
Cmin: 0,52 (0,46 – 0,59)
Cmax: 0,69 (0,62 – 0,76)
| |
Rifabutine*#Ω
(300 mg q.d. pendant 17 jours)
Rilpivirine
(50 mg q.d. pendant 11 jours)
|
Rilpivirine:
AUC: 1,15 (1,06 – 1,26) †
Cmin: 0,93 (0,85 – 1,01) †
Cmax: 1,43 (1,30 – 1,56) †
† En comparaison avec l'administration isolée de 25 mg de rilpivirine q.d.
(Induction des enzymes du CYP3A)
| |
Rifampicine*#Ω
(600 mg q.d. pendant 7 jours)
Rilpivirine (150 mg q.d. pendant 7 jours)
|
Rifampicine:
AUC: 0,99 (0,92 – 1,07)
Cmin :NA
Cmax: 1,02 (0,93 – 1,12)
25désacétylrifampicine:
AUC: 0,91 (0,77 – 1,07)
Cmin: NA
Cmax: 1,00 (0,87 – 1,15)
Rilpivirine:
AUC: 0,20 (0,18 – 0,23)
Cmin: 0,11 (0,10 – 0,13)
Cmax: 0,31 (0,27 – 0,36)
(Induction des enzymes du CYP3A)
|
L'administration concomitante est contre-indiquée. L'administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine (voir «Contre-indications»).
| |
Rifapentine
|
Non évaluée. Des diminutions importantes des concentrations plasmatiques de rilpivirine sont attendues.
(Induction des enzymes du CYP3A)
|
L'administration concomitante est contre-indiquée. L'administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine
(voir «Contre-indications»).
| |
ANTIBIOTIQUES MACROLIDES
| |
Clarithromycine
Érythromycine
|
Non évaluées. Une augmentation de l'exposition à la rilpivirine est attendue.
(Inhibition des enzymes du CYP3A)
|
Si possible, d'autres traitements, tels que l'azithromycine, doivent être envisagés.
| |
GLUCOCORTICOÏDES ou corticostéroïdes
| |
Dexaméthasone (systémique, sauf en cas d'utilisation d'une dose unique)
|
Non évaluée. Des diminutions significatives des concentrations plasmatiques de rilpivirine sont attendues.
(Induction des enzymes du CYP3A)
|
L'administration concomitante est contre-indiquée (sauf en dose unique). L'administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine (voir «Contre-indications»). D'autres traitements doivent être envisagés.
| |
ANALGÉSIQUES NARCOTIQUES
| |
Méthadone*Ω
(60 à 100 mg q.d., dose individualisée)
Rilpivirine (25 mg q.d. pendant 11 jours)
|
R(-)-méthadone:
AUC: 0,84 (0,74 – 0,95)
Cmin: 0,78 (0,67 – 0,91)
Cmax: 0,86 (0,78 – 0,95)
S(+)-méthadone:
AUC: 0,84 (0,74 – 0,96)
Cmin: 0,79 (0,67 – 0,92)
Cmax: 0,87 (0,78 – 0,97)
Rilpivirine:
AUC: aucune interaction†
Cmin: aucune interaction†
Cmax: aucune interaction†
† Sur la base de contrôles historiques
|
Aucun ajustement posologique n'est nécessaire lors de l'instauration de l'administration concomitante de méthadone. Toutefois, une surveillance clinique est recommandée, car le traitement d'entretien par la méthadone doit éventuellement être ajusté chez certains patients.
| |
ANTIARYTHMIQUES
| |
Digoxine*Ω (0,5 mg en dose unique)
Rilpivirine (25 mg q.d. pendant 16 jours)
|
Digoxine:
AUC: 0,98 (0,93 – 1,04)
Cmin: NA
Cmax: 1,06 (0,97 – 1,17)
|
Aucun ajustement posologique recommandé.
| |
ANTIDIABÉTIQUES
| |
Metformine*Ω (850 mg en dose unique)
Rilpivirine (25 mg q.d. pendant 13 jours)
|
Metformine:
AUC: 0,97 (0,90 – 1,06)
Cmin: NA
Cmax: 1,02 (0,95 – 1,10)
|
Aucun ajustement posologique recommandé.
| |
PRÉPARATIONS PHYTOTHÉRAPEUTIQUES
| |
Millepertuis (Hypericum perforatum)
|
Non évalué. Des diminutions significatives des concentrations plasmatiques de rilpivirine sont attendues.
(Induction des enzymes du CYP3A)
|
L'administration concomitante est contre-indiquée. L'administration concomitante peut entraîner une perte de l'effet thérapeutique de la rilpivirine (voir «Contre-indications»).
| |
ANALGÉSIQUES
| |
Paracétamol*#Ω
(500 mg en dose unique)
Rilpivirine (150 mg q.d. pendant 11 jours)
|
Paracétamol:
AUC: 0,91 (0,86 – 0,97)
Cmin: NA
Cmax: 0,97 (0,86 – 1,10)
Rilpivirine:
AUC: 1,16 (1,10 – 1,22)
Cmin: 1,26 (1,16 – 1,38)
Cmax: 1,09 (1,01 – 1,18)
|
Aucun ajustement posologique recommandé.
| |
CONTRACEPTIFS ORAUX
| |
Éthinylestradiol*Ω
(0,035 mg q.d. pendant 21 jours)
Noréthistérone
(1 mg q.d. pendant 21 jours)
Rilpivirine (25 mg q.d. pendant 15 jours)
|
Éthinylestradiol:
AUC: 1,14 (1,10 – 1,19)
Cmin: 1,09 (1,03 – 1,16)
Cmax: 1,17 (1,06 – 1,30)
Noréthistérone:
AUC: 0,89 (0,84 – 0,94)
Cmin: 0,99 (0,90 – 1,08)
Cmax: 0,94 (0,83 – 1,06)
Rilpivirine:
AUC: aucune interaction †
Cmin: aucune interaction †
Cmax: aucune interaction †
† Sur la base de contrôles historiques
|
Aucun ajustement posologique recommandé.
| |
INHIBITEURS DE L'HMG CO-A RÉDUCTASE
| |
Atorvastatine*#Ω
(40 mg q.d. pendant 4 jours)
Rilpivirine (150 mg q.d. pendant 15 jours)
|
Atorvastatine:
AUC: 1,04 (0,97 – 1,12)
Cmin: 0,85 (0,69 – 1,03)
Cmax: 1,35 (1,08 – 1,68)
Rilpivirine:
AUC: 0,90 (0,81 – 0,99)
Cmin: 0,90 (0,84 – 0,96)
Cmax: 0,91 (0,79 – 1,06)
|
Aucun ajustement posologique recommandé.
| |
Fluvastatine
Lovastatine
Pitavastatine
Pravastatine
Rosuvastatine
Simvastatine
|
Non évaluées.
|
Aucun ajustement posologique recommandé.
| |
INHIBITEURS DE LA PHOSPHODIESTÉRASE DE TYPE 5 (PDE-5)
| |
Sildénafil*#Ω
(50 mg en dose unique)
Rilpivirine (75 mg q.d. pendant 12 jours)
|
Sildénafil:
AUC: 0,97 (0,87 – 1,08)
Cmin: NA
Cmax: 0,93 (0,80 – 1,08)
Rilpivirine:
AUC: 0,98 (0,92 – 1,05)
Cmin: 1,04 (0,98 – 1,09)
Cmax: 0,92 (0,85 – 0,99)
|
Aucun ajustement posologique recommandé.
| |
Vardénafil
Tadalafil
|
Non évalués.
|
Aucun ajustement posologique recommandé.
| |
Ω
GMR et IC à 90% sur la base d'études d'interactions médicamenteuses avec la rilpivirine orale.
* L'interaction entre la rilpivirine et le médicament a été évaluée au cours d'une étude clinique. Toutes les autres interactions médicamenteuses reposent sur des prédictions.
# Cette étude d'interaction a été réalisée à une dose supérieure à la dose recommandée de rilpivirine orale et l'effet maximal sur le médicament co-administré a été évalué. La recommandation posologique se rapporte à la dose recommandée de 25 mg de rilpivirine q.d. ainsi qu'au schéma posologique de REKAMBYS.
|
|
Tableau 7: Interactions et recommandations posologiques pour les MÉDICAMENTS ANTIVIRAUX CONTRE LE VIH (destinés à l'administration lors de l'arrêt de REKAMBYS)
| |
Principe actif par classe thérapeutique (schéma posologique)
|
Effets sur la concentration de principe actif
GMR (IC à 90%) Ω
(éventuel mécanisme d'interaction)
|
Recommandation concernant l'administration après l'arrêt de REKAMBYS
| |
Dolutégravir*Ω (50 mg q.d. pendant 5 jours)
Rilpivirine (25 mg q.d. pendant 16 jours)
|
Dolutégravir:
AUC: 1,12 (1,05-1,19)
Cmin: 1,22 (1,15-1,30)
Cmax: 1,13 (1,06-1,21)
Rilpivirine:
AUC: 1,06 (0,98-1,16)
Cmin: 1,21 (1,07-1,38)
Cmax: 1,10 (0,99-1,22)
|
Aucun ajustement posologique recommandé.
| |
Raltégravir*Ω (400 mg b.i.d. pendant 15 jours)
Rilpivirine (25 mg q.d. pendant 11 jours)
|
Raltégravir:
AUC: 1,09 (0,81-1,47)
Cmin: 1,27 (1,01-1,60)
Cmax: 1,10 (0,77-1,58)
Rilpivirine:
AUC: 1,12 (1,05-1,19)
Cmin: 1,03 (0,96-1,12)
Cmax: 1,12 (1,04-1,20)
|
Aucun ajustement posologique recommandé.
| |
Bictégravir
Elvitégravir/cobicistat
|
Non évalués. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Darunavir/ritonavir*Ω (800 mg q.d. pendant 22 jours)
Rilpivirine (150 mg q.d. pendant 11 jours)
|
Darunavir:
AUC: 0,89 (0,81-0,99)
Cmin: 0,89 (0,68-1,16)
Cmax: 0,90 (0,81-1,00)
Ritonavir:
AUC: 0,85 (0,78-0,91)
Cmin: 0,78 (0,68-0,90)
Cmax: 0,83 (0,72-0,95)
Rilpivirine:
AUC: 2,30 (1,98-2,67)
Cmin: 2,78 (2,39-3,24)
Cmax: 1,79 (1,56-2,06)
|
Aucun ajustement posologique recommandé.
| |
Lopinavir/ritonavir* Ω (400 mg b.i.d. pendant 20 jours)
Rilpivirine (150 mg q.d. pendant 10 jours)
|
Lopinavir:
AUC: 0,99 (0,89-1,10)
Cmin: 0,89 (0,73-1,08)
Cmax: 0,96 (0,88-1,05)
Ritonavir:
AUC: 0,96 (0,84-1,11)
Cmin: 1,07 (0,89-1,28)
Cmax: 0,89 (0,73-1,08)
Rilpivirine:
AUC: 1,52 (1,36-1,70)
Cmin: 1,74 (1,46-2,08)
Cmax: 1,29 (1,18-1,40)
|
Aucun ajustement posologique recommandé.
| |
Darunavir/cobicistat
Autres inhibiteurs de la protéase boostés (atazanavir, fosamprénavir, saquinavir, tipranavir)
Inhibiteurs de la protéase non boostés (atazanavir, fosamprénavir, indinavir, nelfinavir)
|
Non évalués. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Ténofovir disoproxil fumarate*Ω (300 mg q.d. pendant 16 jours)
Rilpivirine (150 mg q.d. pendant 8 jours)
|
Ténofovir (plasma):
AUC: 1,23 (1,16-1,31)
Cmin: 1,24 (1,10-1,38)
Cmax: 1,19 (1,06-1,34)
Rilpivirine:
AUC: 1,01 (0,87-1,18)
Cmin: 0,99 (0,83-1,16)
Cmax: 0,96 (0,81-1,13)
|
Aucun ajustement posologique recommandé.
| |
Didanosine*Ω (400 mg q.d. pendant 14 jours)
Rilpivirine (150 mg q.d. pendant 7 jours)
|
Didanosine:
AUC: 1,12 (0,99-1,27)
Cmin: NA
Cmax: 0,96 (0,80-1,14)
Rilpivirine:
AUC: 1,00 (0,95-1,06)
Cmin: 1,00 (0,92-1,09)
Cmax: 1,00 (0,90-1,10)
|
Aucun ajustement posologique recommandé.
| |
Abacavir, emtricitabine, lamivudine, stavudine, ténofovir alafénamide, zidovudine
|
Non évalués. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Éfavirenz, doravirine, étravirine, névirapine
Délavirdine
|
Non évalués. Aucune interaction médicamenteuse cliniquement significative n'est attendue lors de l'administration après l'arrêt de REKAMBYS.
|
Aucun ajustement posologique recommandé.
| |
Maraviroc
|
Non évalué. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Enfuvirtide
|
Non évalué. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
Fostemsavir
|
Non évalué. Aucune interaction médicamenteuse cliniquement significative n'est attendue.
|
Aucun ajustement posologique recommandé.
| |
* L'interaction entre la rilpivirine et le principe actif a été évaluée au cours d'une étude clinique. Toutes les autres interactions médicamenteuses présentées sont des prédictions.
Ω Les GMR et l'IC à 90% reposent sur des études d'interactions médicamenteuses avec la rilpivirine orale.
|
Grossesse, allaitementGrossesse
L'effet de REKAMBYS sur la grossesse chez la femme n'est pas connu. Il n'existe pas d'études cliniques concernant l'utilisation de REKAMBYS chez la femme enceinte.
Les expérimentations animales n'ont révélé aucune toxicité directe ou indirecte de la rilpivirine ni aucune incidence sur la grossesse, le développement embryonnaire, le développement fœtal ou le développement post-natal (voir «Données précliniques»).
REKAMBYS ne doit pas être utilisé chez les femmes en âge de procréer qui prévoient une grossesse ou n'utilisent pas de méthode contraceptive fiable, sauf si le bénéfice attendu justifie les risques éventuels pour l'enfant à naître.
Il convient de délivrer aux patientes des conseils sur l'utilisation de contraceptifs efficaces. REKAMBYS et les contraceptifs à base d'œstrogènes et/ou de progestérone peuvent être utilisés simultanément sans ajustement de la dose (voir «Interactions»).
REKAMBYS ne doit pas être utilisé pendant la grossesse sauf si le bénéfice attendu justifie le risque éventuel pour l'enfant à naître. Une diminution de la disponibilité de la rilpivirine orale ayant été observée pendant la grossesse, la charge virale doit être surveillée étroitement.
Après l'arrêt de REKAMBYS, la rilpivirine pourrait être présente pendant jusqu'à 4 ans dans la circulation systémique chez certains patients. Il convient donc de tenir compte du risque d'exposition du fœtus pendant la grossesse.
Pour surveiller l'issue des grossesses pour la mère et le fœtus, une étude de registre de grossesses a été démarrée chez des patientes enceintes traitées par des antirétroviraux (Antiretroviral Pregnancy Registry) (http://www.apregistry.com). Il s'agit d'une étude observationnelle prospective de l'exposition, sur la base du volontariat, visant à recueillir et évaluer les données sur l'issue de grossesses pendant lesquelles les mères ont été traitées par des antirétroviraux. Les données sur l'exposition à la rilpivirine pendant le premier trimestre de la grossesse sont suffisantes pour permettre d'identifier un risque de malformations congénitales multiplié au moins par deux. À ce jour, de telles élévations du risque n'ont pas été observées.
Allaitement
On ignore si la rilpivirine passe dans le lait maternel chez la femme. Dans les expérimentations animales, la rilpivirine a été excrétée dans le lait. En considérant que la rilpivirine a pu être détectée dans la circulation systémique de certaines patientes pendant jusqu'à 4 ans après l'arrêt de REKAMBYS, elle pourrait être présente dans le lait maternel pendant cette même période.
Étant donné la possibilité d'une transmission du VIH, mais aussi d'effets indésirables chez le nourrisson allaité, il convient de recommander aux mères de ne pas allaiter.
Fertilité
Aucune donnée n'est disponible concernant les effets de la rilpivirine sur la fertilité chez l'être humain. Aucun effet sur la fertilité n'a été observé dans les expérimentations animales lors de l'administration orale de la rilpivirine (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'influence de REKAMBYS sur l'aptitude à la conduite et l'utilisation de machines n'a pas été évaluée.
Effets indésirablesEffets indésirables rapportés dans les études cliniques chez l'adulte
Les effets indésirables (EI) survenus sous la rilpivirine seule ou sous un traitement associant la rilpivirine et le cabotégravir (administration mensuelle ou tous les deux moins), y compris les EI imputables aux formulations orales et injectables de rilpivirine plus cabotégravir, sont répertoriés ci-dessous. En cas de divergence des fréquences entre les études de phase III, la catégorie de fréquence la plus élevée est indiquée.
Les EI les plus fréquemment rapportés dans les études avec administration mensuelle de la dose étaient: réactions au site d'injection (jusqu'à 84% des patients), céphalées (jusqu'à 12% des patients) et pyrexie (10% des patients).
Les EI les plus fréquemment rapportés dans l'étude ATLAS-2M avec administration de la dose tous les deux mois étaient: réactions au site d'injection (76% des patients), céphalées (7% des patients) et pyrexie (7% des patients).
Les EI sont présentés par classe de systèmes d'organes (SOC) et par fréquence1. Les catégories de fréquence sont définies comme suit: «très fréquents» (≥1/10), «fréquents» (≥1/100 à < 1/10), «occasionnels» (≥1/1000 à < 1/100), «rares» (≥1/10 000 à < 1/1000) et «très rares» (< 1/10 000).
Troubles du métabolisme et de la nutrition
Fréquents: prise de poids.
Affections psychiatriques
Fréquents: dépression, états anxieux, rêves anormaux, insomnie.
Affections du système nerveux
Très fréquents: céphalées (12%).
Fréquents: vertiges.
Occasionnels: somnolence, réactions vasovagales (aux injections).
Affections gastro-intestinales
Fréquents: nausées, vomissements, douleurs abdominales2, flatulences, diarrhée, augmentation de la lipase (grade 3-4).
Affections hépatobiliaires
Occasionnels: augmentation des transaminases (ASAT/ALAT), hépatotoxicité (ASAT/ALAT).
Affections de la peau et du tissu sous-cutané
Fréquents: éruption cutanée3.
Affections musculosquelettiques et du tissu conjonctif
Fréquents: myalgie, augmentation de la créatine phosphokinase (grade 3–4).
Troubles généraux
Très fréquents: pyrexie4 (10%).
Fréquents: fatigue, asthénie, sensation de malaise.
Occasionnels: syndrome post-injection (voir «Mises en garde et précautions»).
Anomalies au site d'administration
Réactions au site d'injection5 (84%).
Très fréquents: douleurs (79%), nodules (17%), induration (12%).
Fréquents: troubles, gonflement, érythème, prurit, ecchymoses, sensation de chaleur, hématome.
Occasionnels: cellulite, abcès, anesthésie, hémorragie, changement de couleur.
1 La fréquence des effets indésirables identifiés est basée sur tous les événements rapportés et n'est pas limitée à ceux qui ont été considérés au moins comme ayant un lien possible par le médecin.
2 Douleurs abdominales inclut le groupe de termes préférentiels MedDRA suivant: douleurs abdominales, douleurs de la partie supérieure de l'abdomen (épigastriques)
3 Éruption cutanée inclut le groupe de termes préférentiels MedDRA suivant: éruption cutanée, éruption érythémateuse, éruption généralisée, éruption maculeuse, éruption maculo-papuleuse, éruption morbilliforme, éruption papuleuse, éruption prurigineuse.
4 Pyrexie inclut le groupe de termes préférentiels MedDRA suivant: sensation de chaleur, élévation de la température corporelle, pyrexie. La plupart des événements de pyrexie ont été rapportés dans la semaine suivant l'injection.
5 Les réactions au site d'injection mentionnées ont été rapportées chez au moins 2 participants.
Le profil de sécurité général établi dans l'étude FLAIR entre la semaine 96 et la semaine 124 correspondait au profil observé à la semaine 48, aucun nouvel élément relatif à la sécurité n'ayant pu être identifié. Dans la phase d'extension de l'étude FLAIR, à la semaine 124, lors de l'initiation directe d'injections de rilpivirine plus cabotégravir sans traitement d'induction par voie orale, aucune nouvelle information relative à la sécurité n'a été mise en évidence en lien avec l'abandon du traitement d'induction par voie orale.
Le profil de sécurité général établi dans l'étude ATLAS-2M à la semaine 152 correspondait au profil observé à la semaine 48 et à la semaine 96, aucun nouvel élément relatif à la sécurité n'ayant pu être identifié.
Effets indésirables rapportés dans les études cliniques chez les adolescents (âgés de 12 à < 18 ans)
Sur la base des données issues des analyses de l'étude MOCHA sur 16 semaines (cohorte 1) et 24 semaines (cohorte 2), aucun nouveau problème de sécurité n'a été observé chez les adolescents (âgés d'au moins 12 ans et ayant un poids corporel de 35 kg ou plus) par rapport au profil de sécurité établi pour l'adulte (voir «Études cliniques»).
Pour les effets indésirables rapportés lors de l'administration de la rilpivirine orale (Edurant), voir l'information professionnelle de rilpivirine en comprimés.
Description d'effets indésirables sélectionnés et informations complémentaires
Effets indésirables fréquents sous Vocabria/Rekambys une fois par mois par rapport au traitement oral standard quotidien (TAC)
|
Tableau 8: Effets indésirables systémiques chez ≥1% des participants infectés par le VIH-1 et virologiquement contrôlés dans les études regroupées FLAIR et ATLAS (semaine 48)
| |
Effet indésirable
|
CAB+RPV
(n=591)
|
TAC
(n=591)
| |
Céphalées
|
12%
|
6%
| |
Pyrexie3
|
10%
|
2%
| |
Diarrhée
|
9%
|
7%
| |
Augmentation de la créatine phosphokinase (grade 3-4)
|
8%
|
4%
| |
Augmentation de la lipase (grade 3-4)
|
6%
|
3%
| |
Nausées
|
5%
|
3%
| |
Fatigue
|
5%
|
2%
| |
Éruption cutanée2
|
5%
|
3%
| |
Sensation de vertige
|
4%
|
1%
| |
Myalgie
|
4%
|
1%
| |
Douleurs abdominales1
|
4%
|
2%
| |
Insomnie
|
4%
|
1%
| |
États anxieux
|
4%
|
2%
| |
Asthénie
|
3%
|
< 1%
| |
Vomissements
|
2%
|
1%
| |
Dépression
|
2%
|
2%
| |
Malaise
|
2%
|
< 1%
| |
Rêves anormaux
|
1%
|
< 1%
| |
Flatulences
|
1%
|
< 1%
|
1 Douleurs abdominales inclut le groupe de termes préférentiels MedDRA suivant: douleurs abdominales, douleurs de la partie supérieure de l'abdomen (épigastriques).
2 Éruption cutanée inclut le groupe de termes préférentiels MedDRA suivant: éruption cutanée, éruption érythémateuse, éruption généralisée, éruption maculeuse, éruption maculo-papuleuse, éruption morbilliforme, éruption papuleuse, éruption prurigineuse.
3 Pyrexie inclut le groupe de termes préférentiels MedDRA suivant: pyrexie, sensation de chaleur, élévation de la température corporelle.
TAC = traitement antirétroviral en cours (Current Antiretroviral Regimen, CAR)
Il convient de noter que les études FLAIR et ATLAS étaient des études ouvertes comportant un changement de traitement (voir les détails à la rubrique «Efficacité clinique»). Dans le bras traité par le cabotégravir et la rilpivirine, une incidence accrue d'effets indésirables a été rapportée, ce qui est imputable soit au schéma thérapeutique soit à un biais lié à la conception des études.
Réactions locales au site d'injection (RSI)
En cas d'administration mensuelle de la dose
Dans le cadre des études de phase III et de l'étude de phase IIIB (ATLAS, FLAIR et ATLAS-2M), un total de 1% des patients a arrêté le traitement par injections de REKAMBYS et de cabotégravir en raison de RSI.
Sur 30 393 injections, 6815 RSI ont été rapportées et les réactions au site d'injection étaient généralement d'intensité légère (grade 1, 75% des patients) ou modérée (grade 2, 36% des patients). 4% des patients ont développé des RSI sévères (grade 3) et aucun participant aux études n'a développé de RSI de grade 4.
La durée médiane des RSI a été au total de 3 jours (de 1 jour à 341 jours) et 11% des participants aux études ont rapporté la présence de RSI persistantes au moment de l'injection suivante.
Le pourcentage de patients ayant rapporté des RSI a diminué au fil du temps, passant de 70% dans la semaine 4 à 19% dans la semaine 48.
En cas d'administration de la dose tous les 2 mois
Dans le cadre de l'étude ATLAS-2M, moins de 1% des participants à l'étude a arrêté le traitement par REKAMBYS plus Vocabria en raison de RSI. Sur 8470 injections, 2507 RSI ont été rapportées et les réactions au site d'injection étaient généralement d'intensité légère (grade 1, 71% des patients) ou modérée (grade 2, 27% des patients). Des RSI sévères (grade 3) sont survenues chez 3% des patients et aucun patient n'a développé de RSI de grade 4.
La durée médiane des RSI a été au total de 3 jours (de 1 jour à 424 jours) et 5% des patients ont rapporté la présence de RSI persistantes au moment de l'injection suivante.
Le pourcentage de patients ayant rapporté des RSI a diminué au fil du temps, passant de 70% dans la semaine 4 à 20% dans la semaine 48.
Prise de poids
Au moment de l'analyse à la semaine 48, les patients ayant reçu la rilpivirine IM plus le cabotégravir IM au cours des études de phase III FLAIR et ATLAS présentaient une prise de poids médiane de 1,5 kg; les patients du groupe comparatif, chez lesquels le traitement antirétroviral standard (TAC) respectif avait été poursuivi, présentaient une prise de poids médiane de 1,0 kg (analyse combinée). Lors des évaluations individuelles des études FLAIR et ATLAS de phase III, les patients inclus dans les bras traités par la rilpivirine IM plus le cabotégravir IM présentaient une prise de poids médiane de respectivement 1,3 kg et 1,8 kg, contre 1,5 kg et 0,3 kg dans les bras sous TAC. Après 48 semaines de traitement, la prise de poids médiane dans le cadre de l'étude ATLAS-2M était de 1,0 kg aussi bien dans le bras avec administration mensuelle de la rilpivirine IM plus le cabotégravir IM que dans le bras avec administration tous les 2 mois.
Modification des paramètres biologiques
Dans les études cliniques portant sur REKAMBYS plus le cabotégravir, des taux élevés de lipase ont été observés, des élévations de la lipase de grades 3 et 4 se produisant plus souvent sous REKAMBYS plus cabotégravir que dans le groupe TAC. Ces augmentations étaient généralement asymptomatiques et n'ont pas conduit à une interruption du traitement.
Par ailleurs, au cours du traitement par la rilpivirine IM plus le cabotégravir IM, des augmentations asymptomatiques de la créatine phosphokinase (CPK) ont été rapportées, principalement en rapport avec une activité physique.
Hépatotoxicité
Une élévation des transaminases (ALAT/ASAT) a été observée au cours des études cliniques chez des patients ayant reçu l'association rilpivirine plus cabotégravir. Ces élévations étaient principalement imputables à une hépatite virale aiguë (hépatite A, B, C). Quelques participants traités par rilpivirine orale plus cabotégravir oral ont présenté des élévations des transaminases imputables à une suspicion d'hépatotoxicité médicamenteuse. Ces modifications ont été réversibles à l'arrêt du traitement (voir «Mises en garde et précautions»).
Une faible augmentation, non progressive, de la bilirubine totale (sans ictère clinique) a été observée au cours du traitement par rilpivirine plus cabotégravir. Ces modifications ne sont pas considérées comme cliniquement pertinentes, car elles reflètent probablement une compétition entre le cabotégravir et la bilirubine non conjuguée pour une voie de clairance commune (UGT1A1).
Informations complémentaires sur des groupes de patients particuliers
Utilisation chez les enfants et les adolescents
La sécurité et l'efficacité de REKAMBYS chez les enfants de moins de 18 ans n'ont pas encore été établies.
En ce qui concerne les effets indésirables liés au cabotégravir, tenir compte de l'information sur le médicament correspondante.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe pas d'antidote spécifique en cas de surdosage de REKAMBYS. L'expérience de surdosages de rilpivirine chez l'être humain est limitée. Des surdosages peuvent notamment survenir en cas d'administration intraveineuse accidentelle (voir «Mises en garde et précautions»). Le traitement en cas de surdosage de REKAMBYS comporte des mesures générales de soutien, comme la surveillance des signes vitaux et de l'ECG (intervalle QT, voir «Interactions»/«Médicaments allongeant l'intervalle QT») ainsi que l'observation de l'état clinique du patient. Il est conseillé de contacter un centre antipoison afin d'obtenir les dernières recommandations relatives au traitement d'un surdosage. La rilpivirine étant fortement liée aux protéines plasmatiques, il est peu probable qu'une dialyse entraîne une élimination significative du principe actif.
Propriétés/EffetsCode ATC
J05AG05
Mécanisme d'action
La rilpivirine est un INNTI de type diarylpyrimidine du VIH-1. L'activité de la rilpivirine est médiée par une inhibition non compétitive de la transcriptase inverse (TI) du VIH-1. La rilpivirine n'inhibe pas les ADN polymérases cellulaires humaines α, β et γ.
Pharmacodynamique
Microbiologie
Activité antivirale in vitro
La rilpivirine s'est avérée active contre les souches de laboratoire du VIH-1 de type sauvage dans des modèles d'infection aiguë de lignées lymphocytaires T avec une valeur médiane de la CE50 pour le VIH-1/IIIB de 0,73 nM (0,27 ng/ml). Bien que la rilpivirine ait présenté une activité in vitro limitée contre le VIH-2 avec des valeurs de CE50 comprises entre 2510 et 10 830 nM (de 920 à 3970 ng/ml), le traitement d'une infection à VIH-2 par REKAMBYS n'est pas recommandé étant donné l'absence de données cliniques.
De plus, une activité antivirale de la rilpivirine a été observée contre un large éventail d'isolats primaires du VIH-1 du groupe M (soustypes A, B, C, D, F, G, H) avec des valeurs de CE50 comprises entre 0,07 et 1,01 nM (de 0,03 à 0,37 ng/ml) et du groupe O avec des valeurs de CE50 comprises entre 2,88 et 8,45 nM (de 1,06 à 3,10 ng/ml).
Résistance
En prenant en compte l'ensemble des données in vitro et in vivo disponibles, obtenues avec la rilpivirine orale chez des patients non préalablement traités, les substitutions d'acides aminés suivantes peuvent affecter l'activité de la rilpivirine lorsqu'elles sont présentes à l'instauration du traitement: K101E, K101P, E138A, E138G, E138K, E138Q, E138R, V179L, Y181C, Y181I, Y181V, Y188L, H221Y, F227C, M230I et M230L ainsi que l'association de L100I et K103N.
Patients virologiquement contrôlés
Le nombre de patients satisfaisant aux critères d'échec virologique confirmé (EVC) (deux taux plasmatiques successifs d'ARN du VIH-1 ≥200 copies/ml après obtention d'un contrôle à < 200 copies/ml) était faible dans les études de phase III combinées ATLAS et FLAIR (voir «Efficacité clinique»). Dans l'analyse combinée, 7 cas d'EVC sous rilpivirine plus cabotégravir (7/591, 1,2%) et 7 cas d'EVC sous TAC (7/591, 1,2%) ont été observés jusqu'à la semaine 48. Un patient présentant un EVC (FLAIR) n'avait jamais reçu de dose injectable et n'avait pas développé de résistance. Dans le groupe traité par la rilpivirine plus le cabotégravir de l'analyse combinée, 5/591 (0,8%) participants ont développé une résistance: respectivement 5/591 (0,8%) et 4/591 (0,7%) participants avec des mutations associées à une résistance à la rilpivirine et/ou au cabotégravir (tableau 9).
|
Tableau 9: Mutations associées à une résistance avant l'instauration du traitement de l'étude et lors d'une suspicion d'échec virologique dans les cas présentant un échec virologique confirmé dans le cadre des études ATLAS et FLAIR
| |
Étude
|
Sous-type de VIH-1
|
Inclusion*
|
Suspicion d'échec virologique**
| |
|
|
Transcriptase inverse
|
Intégrase
|
Transcriptase inverse
|
Intégrase
| |
ATLAS
|
A
|
E138A/E
|
Aucune
|
E138A
|
Aucune
| |
AG
|
V108I/V+E138K
|
Aucune
|
V108I+E138K
|
Aucune
| |
A
|
Aucune
|
Aucune
|
E138E/K
|
N155H
| |
FLAIR
|
A1
|
Aucune
|
Aucune
|
E138A/E/K/T
|
Q148R
| |
A1
|
Aucune
|
Aucune
|
K101E
|
G140R
| |
A1
|
Aucune
|
Aucune
|
E138K
|
Q148R
| |
*FLAIR: tests post-hoc de la résistance aux INI et tests de détection de la résistance aux INNTI avec du plasma prélevé à l'inclusion, ATLAS: des tests post-hoc de la résistance aux INI et aux INNTI ont été réalisés avec des cellules mononucléaires du sang périphérique, prélevées dans le cadre des deux études à l'inclusion avant l'instauration du traitement de l'étude.
**Moment de la suspicion d'échec virologique: la première de deux visites consécutives avec une charge virale ≥200 copies/ml après obtention d'un contrôle à < 200 copies/ml.
|
Lors de l'étude de phase IIIB ATLAS-2M (voir «Efficacité clinique»), 10/1045 (1,0%) participants ont satisfait aux critères d'EVC jusqu'à la semaine 48: 8/522 (1,5%) dans le bras q8w (administration de la dose tous les 2 mois) et 2/523 (0,4%) dans le bras q4w (administration mensuelle). Dans le groupe q8w, le développement d'une résistance a été observé chez 5/522 (1,0%) patients: respectivement 4/522 (0,8%) et 5/522 (1,0%) patients avec des mutations associées à une résistance à la rilpivirine et/ou au cabotégravir. Dans le groupe q4w, le développement d'une résistance a été observé chez 2/523 (0,4%) patients: respectivement 1/523 (0,2%) et 2/523 (0,4%) patients avec des mutations associées à une résistance à la rilpivirine et/ou au cabotégravir (tableau 10).
|
Tableau 10: Mutations associées à une résistance avant l'instauration du traitement de l'étude et lors d'une suspicion d'échec virologique dans les cas présentant un échec virologique confirmé dans le cadre de l'étude ATLAS-2M
| |
Étude
|
Sous-type de VIH-1
|
Inclusion*
|
Suspicion d'échec virologique**
| |
|
|
Transcriptase inverse
|
Intégrase
|
Transcriptase inverse
|
Intégrase
| |
ATLAS-2M bras q8w
|
A
|
E138A/E
|
Aucune
|
K101E+E138A
|
N155H
| |
|
A1
|
Aucune
|
Aucune
|
E138E/K
|
Q148Q/R+N155N/H
| |
|
A1
|
Y188L+P225H
|
Aucune
|
Y188L+P225H
|
NA
| |
|
B
|
Aucune
|
Aucune
|
Aucune
|
Aucune
| |
|
B
|
K103N+V108I/V+E138A
|
Aucune
|
K103N+E138A
|
N155H
| |
|
C
|
V108I/V+H221H/Y+Y181C/Y
|
Aucune
|
K103N
|
Aucune
| |
|
C
|
Y188F/H/L/Y
|
G140G/R
|
Y188L
|
Q148Q/R+N155H/N
| |
|
Complexe
|
Aucune
|
Aucune
|
K101E
|
Q148R
| |
ATLAS-2M bras q4w
|
B
|
Aucune
|
Aucune
|
Aucune
|
N155N/H
| |
|
B
|
Aucune
|
Aucune
|
K101E+M230L
|
E138E/K+Q148R
| |
* Des tests post-hoc de la résistance ont été réalisés avec des cellules mononucléaires du sang périphérique, prélevées à l'inclusion avant l'instauration du traitement de l'étude. n.d.: non disponible.
** Moment de la suspicion d'échec virologique: la première de deux visites consécutives avec une charge virale ≥200 copies/ml après obtention d'un contrôle à < 200 copies/ml.
|
Jusqu'à la semaine 152, 13 patients remplissaient les critères d'EVC pendant la phase d'entretien et d'extension; 2 patients (bras Q8W) remplissaient les critères d'EVC depuis l'analyse à la semaine 96 (voir tableau 11). Dix patients ont présenté un EVC avant la semaine 48 (8 patients dans le bras Q8W et 2 patients dans le bras Q4W) et 1 patient (bras Q8W) a rempli les critères d'EVC entre les semaines 48 et 96.
|
Tableau 11: Proportion cumulée de patients satisfaisant à l'EVC après la visite jusqu'à la semaine 152 Phase d'entretien + Phase d'extension (population ITT-E): Analyse à la semaine 152
| |
Moment de l'EVSa
|
Q8W
(N = 522)
n (%)
|
Q4W
(N = 523)
n (%)
| |
Semaine 8
|
1 (0,2)
|
0
| |
Semaine 16
|
4 (0,8)
|
1 (0,2)
| |
Semaine 24
|
7 (1,3)
|
1 (0,2)
| |
Semaine 32
|
7 (1,3)
|
2 (0,4)
| |
Semaine 48
|
8 (1,5)
|
2 (0,4)
| |
Semaine 88
|
9 (1,7)
|
2 (0,4)
| |
Semaine 112
|
10 (1,9)
|
2 (0,4)
| |
Semaine 120
|
11 (2,1)
|
2 (0,4)
| |
a Première des 2 valeurs consécutives d'ARN de VIH-1 ≥200 copies/ml.
Note: ce résumé représente la proportion cumulée d'EVC jusqu'à la visite d'examen.
Remarque: Seules les visites pour lesquelles au moins un nouvel EVC est apparu sont mentionées.
|
En plus des 9 patients présentant un EVC dans le groupe Q8W, 2 patients ont atteint l'EVC entre les semaines 96 et 152 (voir tableau 11). Un patient ayant quitté l'étude ATLAS après avoir reçu 1 à 24 semaines de CAB + RPV LA, a satisfait aux critères d'EVC à la semaine 112. Au début de l'étude, ce patient présentait le polymorphisme IN L74I. Au moment de la suspicion d'échec virologique (EVS), 3 mutations INNTI ont été observées, K103N et des mutations associées à la résistance au RPV, E138A et Y181Y/C. La mutation Q148R associée à la résistance à l'INI a été détectée en même temps que le polymorphisme L74I. Une sensibilité phénotypique réduite au RPV (FC = 3,4) et au CAB (FC = 9,5) a été observée. Le sous-type du virus VIH-1 était A au moment de l'EVS. L'autre patient a atteint les critères d'EVC à la semaine 120 et ne présentait pas de mutations associées à une résistance à la référence. Au moment de l'EVS, les mutations de résistance au RPV E138A et M230M/L ainsi que la mutation de résistance INI Q148R ont été détectées. L'analyse phénotypique a montré une sensibilité réduite au RPV (FC = 16) et au CAB (FC = 3,3). Le patient était porteur de virus VIH-1 de sous-type B/C au moment de l'EVS. Il n'y a pas eu d'autres patients atteignant l'EVC dans le groupe Q4W.
Résistance croisée
Virus présentant des mutations ciblées au niveau des sites de liaison aux INNTI
Dans un groupe de 67 souches de laboratoire de VIH-1 recombinantes, présentant une substitution d'acides aminés aux positions de la TI associées à une résistance aux INNTI, incluant les substitutions les plus fréquemment observées K103N et Y181C, la rilpivirine a présenté une activité antivirale contre 64 de ces souches (96%). Les substitutions uniques d'acides aminés, associées à une perte de la sensibilité à la rilpivirine, étaient les suivantes: K101P, Y181I et Y181V. Tandis que la substitution K103N n'a pas entraîné de diminution de la sensibilité à la rilpivirine, l'association de K103N et L100I a réduit d'un facteur 7 la sensibilité à la rilpivirine.
Isolats cliniques recombinants
La rilpivirine a conservé sa sensibilité (FC ≤ BCO) contre 62% des 4786 isolats cliniques recombinants du VIH-1 présentant une résistance à l'éfavirenz et/ou la névirapine.
Patients virologiquement contrôlés
Dans le cadre de l'analyse à la semaine 48 des études de phase III ATLAS et FLAIR, 5 patients sur 7 avec un EVC présentaient un phénotype de résistance à la rilpivirine au moment de l'échec. Chez ces 5 patients, un phénotype de résistance croisée à l'éfavirenz (n = 4), l'étravirine (n = 3) et la névirapine (n = 4) a été observé.
Effets sur l'intervalle QT/QTc et l'électrophysiologie cardiaque
Les concentrations plasmatiques de rilpivirine après des injections de REKAMBYS sont comparables à celles observées après la prise de rilpivirine à une posologie de 25 mg une fois par jour. REKAMBYS à la dose recommandée de 600 mg une fois par mois ou de 900 mg tous les 2 mois n'est pas associée à un effet cliniquement significatif sur l'intervalle QTc. L'effet de la rilpivirine orale à la posologie recommandée de 25 mg une fois par jour sur l'intervalle QTcF a été évalué au cours d'une étude croisée, randomisée, contrôlée contre placebo et contre traitement actif (400 mg de moxifloxacine une fois par jour) chez 60 adultes sains, sur la base de 13 mesures effectuées pendant 24 heures à l'état d'équilibre. La rilpivirine orale à la posologie recommandée de 25 mg une fois par jour n'est pas associée à des effets cliniquement significatifs sur l'intervalle QTc.
Lors de l'examen de doses suprathérapeutiques de rilpivirine orale (75 mg une fois par jour et 300 mg une fois par jour) chez des adultes sains, les différences moyennes maximales en termes d'intervalle QTcF, appariées en fonction du temps (limite supérieure de l'intervalle de confiance à 95%), par rapport au placebo et après correction par rapport aux valeurs initiales, étaient respectivement de 10,7 (15,3) et 23,3 (28,4) ms. La prise de rilpivirine à des posologies de 75 mg une fois par jour et de 300 mg une fois par jour a entraîné une Cmax moyenne environ 4,4 et 11,6 fois supérieure à la Cmax moyenne à l'état d'équilibre à la posologie recommandée de 600 mg de REKAMBYS une fois par mois. L'administration orale de 75 mg ou 300 mg de rilpivirine une fois par jour à l'état d'équilibre a entraîné une Cmax moyenne environ 4,1 et 10,7 fois supérieure à la Cmax moyenne à l'état d'équilibre à la posologie recommandée de 900 mg de REKAMBYS tous les 2 mois.
Efficacité clinique
Études cliniques
En cas d'administration mensuelle de la dose
Patients adultes infectés par le VIH-1 virologiquement contrôlés
L'efficacité d'injections de REKAMBYS et de cabotégravir a été évaluée dans le cadre de deux études de non-infériorité de phase III FLAIR et ATLAS, randomisées, multicentriques, contrôlées contre traitement actif, à bras parallèles et en ouvert. L'analyse principale a été réalisée une fois que tous les patients avaient terminé leur visite de la semaine 48 ou étaient sortis prématurément de l'étude.
Lors de l'étude FLAIR, 629 patients infectés par le VIH-1, non préalablement traités par des antirétroviraux (naïfs d'ARV) ont reçu pendant 20 semaines un traitement par le dolutégravir, un inhibiteur du transfert de brin de l'intégrase (INI) (soit dolutégravir/abacavir/lamivudine soit dolutégravir + 2 autres inhibiteurs nucléosidiques de la transcriptase inverse (INTI) si les patients étaient HLA-B*5701 positifs). Les patients virologiquement contrôlés (ARN du VIH-1< 50 copies/ml, n = 566) ont ensuite été randomisés (1:1) pour recevoir un traitement par rilpivirine plus cabotégravir ou pour poursuivre leur traitement antirétroviral en cours (TAC). Chez les patients randomisés pour recevoir le traitement par rilpivirine plus cabotégravir, la thérapie a été commencée par un traitement d'induction oral d'environ 1 mois (au minimum 28 jours et au maximum 2 mois) comprenant un comprimé de cabotégravir de 30 mg et un comprimé de rilpivirine de 25 mg. Le traitement a ensuite été poursuivi par le cabotégravir par voie injectable (mois 1: injection de 600 mg, à partir du mois 2: injection de 400 mg) plus rilpivirine par voie injectable (mois 1: injection de 900 mg, à partir du mois 2: injection de 600 mg) une fois par mois pendant jusqu'à 96 semaines.
Lors de l'étude ATLAS, 616 patients infectés par le VIH-1, préalablement traités par un ARV et virologiquement contrôlés depuis au moins 6 mois (ARN du VIH-1< 50 copies par ml), ont été randomisés (1:1) pour recevoir un traitement par rilpivirine plus cabotégravir ou pour poursuivre leur traitement antirétroviral en cours (TAC). Chez les patients randomisés pour recevoir le traitement par rilpivirine plus cabotégravir, la thérapie a été commencée par un traitement d'induction oral d'environ 1 mois (au minimum 28 jours et au maximum 2 mois) comprenant un comprimé de cabotégravir de 30 mg et un comprimé de rilpivirine de 25 mg une fois par jour. Le traitement a ensuite été poursuivi par le cabotégravir par voie injectable (mois 1: injection de 600 mg, à partir du mois 2: injection de 400 mg) plus rilpivirine par voie injectable (mois 1: injection de 900 mg, à partir du mois 2: injection de 600 mg) une fois par mois pendant 44 semaines supplémentaires. Au début de l'étude ATLAS, respectivement 50%, 17% et 33% des patients avaient été traités avant la randomisation par un INNTI, un IP ou un INI comme troisième classe de principe actif. Cette répartition reste similaire après la randomisation dans le bras témoin (TAC).
L'analyse combinée des études FLAIR et ATLAS a montré, dans le bras traité par rilpivirine plus cabotégravir au début des études, que l'âge médian des patients était de 38 ans, 27% étaient des femmes, 27% étaient non blancs et 7% avaient un taux de cellules CD4+ inférieur à 350 cellules par mm3; les bras de traitement ne présentaient pas de différences significatives pour ce qui est de ces caractéristiques.
Le critère d'évaluation principal des deux études était la proportion de patients présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml à la semaine 48 (algorithme snapshot pour la population en ITT-E).
Dans le cadre d'une analyse combinée des deux études de phase III FLAIR et ATLAS, l'association rilpivirine plus cabotégravir était non inférieure au TAC en termes de proportion de patients présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml (respectivement de 1,9% et 1,7%) à la semaine 48. La différence ajustée entre le traitement par rilpivirine plus cabotégravir et le TAC (0,2; IC à 95%: –1,4; 1,7) a satisfait le critère de non-infériorité (limite supérieure de l'IC à 95% inférieure à 4%).
Les résultats de non-infériorité issus des études FLAIR et ATLAS ont montré que la durée du contrôle virologique de l'ARN du VIH-1 avant l'instauration du traitement par rilpivirine plus cabotégravir (c.-à-d. 5 mois ou ≥6 mois) n'a pas eu d'effet sur le taux de réponse globale.
Le critère d'évaluation principal et d'autres résultats à la semaine 48, incluant les résultats selon les caractéristiques initiales principales des patients (facteurs à l'inclusion) pour les études FLAIR et ATLAS et les données combinées de celles-ci, sont présentés dans le tableau 12 et le tableau 13.
|
Tableau 12: Résultats virologiques du traitement randomisé dans les études FLAIR et ATLAS à la semaine 48 (analyse snapshot)
| |
|
FLAIR
|
ATLAS
|
Données combinées
| |
|
RPV + CAB
n = 283
|
TAC
n = 283
|
RPV + CAB
n = 308
|
TAC
n = 308
|
RPV + CAB
n = 591
|
TAC
n = 591
| |
ARN du VIH-1 ≥50 copies/ml, n (%)†
|
6 (2,1)
|
7 (2,5)
|
5 (1,6)
|
3 (1,0)
|
11 (1,9)
|
10 (1,7)
| |
Différence entre les traitements en % (IC à 95%)*
|
-0,4 (-2,8, 2,1)
|
0,7 (-1,2, 2,5)
|
0,2 (-1,4, 1,7)
| |
ARN du VIH-1< 50 copies/ml, n (%)
|
265
(93,6)
|
264 (93,3)
|
285
(92,5)
|
294 (95,5)
|
550
(93,1)
|
558 (94,4)
| |
Aucune donnée virologique pendant la fenêtre de la semaine 48, n (%)
|
12
(4,2)
|
12 (4,2)
|
18
(5,8)
|
11 (3,6)
|
30
(5,1)
|
23 (3,9)
| |
Raisons
| |
Sortie de l'étude/arrêt du médicament à l'étude en raison d'EI ou du décès,
n (%)
|
8
(2,8)
|
2 (0,7)
|
11
(3,6)
|
5 (1,6)
|
19
(3,2)
|
7
(1,2)
| |
Sortie de l'étude/arrêt du médicament à l'étude pour d'autres raisons, n (%)
|
4 (1,4)
|
10 (3,5)
|
7 (2,3)
|
6 (1,9)
|
11 (1,9)
|
16 (2,7)
| |
Données manquantes durant cette fenêtre, mais dans l'étude
|
0
|
0
|
0
|
0
|
0
|
0
| |
* Ajustée en fonction des facteurs de stratification à l'inclusion.
† Inclut les patients ayant arrêté le traitement en raison d'un manque d'efficacité ou ayant arrêté alors qu'ils n'étaient pas contrôlés.
RPV = rilpivirine, CAB = cabotégravir, n = nombre de patients dans chaque groupe de traitement, IC = intervalle de confiance, TAC = traitement antiviral en cours.
|
|
Tableau 13: Proportion de patients avec un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml dans l'analyse des sous-groupes à la semaine 48 selon les caractéristiques initiales principales des patients (facteurs à l'inclusion) (snapshot des résultats)
| |
Caractéristiques initiales des patients
(facteurs à l'inclusion)
|
Données combinées de FLAIR et ATLAS
| |
RPV + CAB
n = 591
n/N (%)
|
TAC
n = 591
n/N (%)
| |
CD4+ à l'inclusion (cellules/mm3)
|
< 350
|
0/42
|
2/54 (3,7)
| |
≥350 à < 500
|
5/120 (4,2)
|
0/117
| |
≥500
|
6/429 (1,4)
|
8/420 (1,9)
| |
Sexe
|
Masculin
|
6/429 (1,4)
|
9/423 (2,1)
| |
Féminin
|
5/162 (3,1)
|
1/168 (0,6)
| |
Groupe ethnique
|
Blancs
|
9/430 (2,1)
|
7/408 (1,7)
| |
Noirs/Afro-Américains
|
2/109 (1,8)
|
3/133 (2,3)
| |
Asiatiques/autres
|
0/52
|
0/48
| |
IMC
|
< 30 kg/m2
|
6/491 (1,2)
|
8/488 (1,6)
| |
≥30 kg/m2
|
5/100 (5,0)
|
2/103 (1,9)
| |
Âge (années)
|
< 50
|
9/492 (1,8)
|
8/466 (1,7)
| |
≥50
|
2/99 (2,0)
|
2/125 (1,6)
| |
Traitement antiviral à l'inclusion lors de la randomisation
(3e classe de principe actif)
|
IP
|
1/51 (2,0)
|
0/54
| |
INI
|
6/385 (1,6)
|
9/382 (2,4)
| |
INNTI
|
4/155 (2,6)
|
1/155 (0,6)
| |
IMC = indice de masse corporelle, IP = inhibiteur de protéase, INI = inhibiteur de l'intégrase, INNTI = inhibiteur non nucléosidique de la transcriptase inverse
|
Lors des études FLAIR et ATLAS, les différences entre les traitements selon les caractéristiques initiales des patients (facteurs à l'inclusion) (numération des CD4+, sexe, âge, groupe ethnique, IMC, troisième classe de principe actif thérapeutique à l'inclusion) n'étaient pas cliniquement significatives.
Avant le jour 1 ou avant la randomisation, les patients étaient virologiquement contrôlés dans le cadre des études FLAIR et ATLAS et aucune modification cliniquement pertinente du nombre de cellules CD4+ par rapport au taux initial n'a été observée.
Les patients ayant reçu la rilpivirine IM plus le cabotégravir IM ont rapporté une amélioration importante en termes de satisfaction à l'égard du traitement par rapport à l'utilisation d'un traitement antirétroviral oral quotidien. Lors de la mesure à l'aide du questionnaire de satisfaction du traitement anti-VIH (HIV-Treatment Satisfaction Questionnaire, HIV-TSQ), la différence en termes de satisfaction vis-à-vis du traitement au HIV-TSQc (variation) était de + 4,1 à la semaine 48 dans l'étude FLAIR (IC à 95%: 2,8; 5,5; p < 0,001) et celle au HIV-TSQs (état) était de + 5,7 à la semaine 44 dans l'étude ATLAS (IC à 95%: 4,37; 7,0; p < 0,001).
Semaine 96 FLAIR
Dans le cadre de l'étude FLAIR, les résultats d'efficacité à 96 semaines sont restés cohérents avec ceux observés à 48 semaines. La proportion de patients présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml était de 3,2% pour la rilpivirine IM plus le cabotégravir IM (n = 283) et de 3,2% pour le TAC (n = 283) (différence ajustée entre le traitement par la rilpivirine IM plus le cabotégravir IM et le TAC [0,0; IC à 95%: -2,9; 2,9]).
Semaine 124 FLAIR – injection directe versus traitement d'induction par voie orale
Dans le cadre de l'étude FLAIR, une évaluation de la sécurité et de l'efficacité a été réalisée à la semaine 124 auprès des patients, qui ont décidé à la semaine 100, de passer d'abacavir/dolutégravir/lamivudine à rilpivirine plus cabotégravir dans la phase d'extension avec ou sans traitement d'induction par voie orale. Deux groupes ont donc été formés, l'un avec traitement d'induction par voie orale (groupe «Oral Lead-in» (OLI)) (n = 121) et l'autre avec injection directe (groupe «Direct to injection» (DTI)) (n = 111).
À la semaine 124, la proportion de patients avec un taux d'ARN du VIH-1 ≥50 copies/ml s'élevait à respectivement (1/111 [0,9%]) et (1/121 [0,8%]) dans le groupe avec injection directe et le groupe avec traitement d'induction par voie orale. Les taux de suppression virologique (ARN du VIH-1< 50 copies/ml) étaient similaires dans le groupe avec injection directe (110/111 [99,1%]) et dans le groupe avec traitement d'induction par voie orale (113/121 [93,4%]).
En cas d'administration tous les 2 mois
Patients adultes infectés par le VIH-1 virologiquement contrôlés
L'efficacité d'injections de rilpivirine administrées tous les 2 mois ont été évaluées dans l'étude de non-infériorité de phase IIIB ATLAS-2M, randomisée, multicentrique, à bras parallèles et en ouvert. L'analyse principale a été réalisée une fois que tous les participants avaient terminé leur visite de la semaine 48 ou étaient sortis prématurément de l'étude.
Lors de l'étude ATLAS-2M, 1045 participants infectés par le VIH-1, préalablement traités par des ARV et virologiquement contrôlés, ont été randomisés (1:1) pour recevoir des injections de rilpivirine plus cabotégravir tous les 2 mois ou tous les mois. Les patients qui n'avaient pas été traités par l'association cabotégravir/rilpivirine avant le début de l'étude ont reçu un traitement d'induction oral, comprenant un comprimé de 25 mg de rilpivirine et un comprimé de 30 mg de cabotégravir, une fois par jour pendant environ 1 mois (au minimum 28 jours et au maximum 2 mois). Les patients qui recevaient après la randomisation des injections mensuelles de rilpivirine (mois 1: injection de 900 mg, à partir du mois 2: injection de 600 mg) et des injections de cabotégravir (mois 1: injection de 600 mg, à partir du mois 2: injection de 400 mg) ont été traités pendant 44 semaines supplémentaires. Les patients qui, après la randomisation, recevaient des injections de rilpivirine tous les 2 mois (injection de 900 mg aux mois 1, 2 et 4, puis tous les 2 mois) et des injections de cabotégravir (injection de 600 mg aux mois 1, 2 et 4, puis tous les 2 mois) ont été traités pendant 44 semaines supplémentaires.
Avant la randomisation, 63%, 13% et 24% des participants avaient reçu rilpivirine + cabotégravir pendant respectivement 0 semaine, 1 à 24 semaines ou > 24 semaines.
Au début de l'étude, l'âge médian des participants était de 42 ans, 27% étaient des femmes, 27% étaient non blancs et 6% avaient un taux de cellules CD4+ inférieur à 350 cellules par mm3. Ces caractéristiques étaient similaires entre les différents bras de traitement.
Le critère d'évaluation principal de l'étude ATLAS-2M était la proportion de patients présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml à la semaine 48 (algorithme snapshot pour la population en ITT-E). Dans le tableau 14 et le tableau 15, le critère d'évaluation principal et d'autres résultats de la semaine 48 pour l'étude ATLAS-2M sont présentés, notamment les résultats concernant les caractérisitques initiales centrales des patients (caractéristiques «à l'inclusion»).
Lors de l'étude ATLAS-2M, le traitement tous les 2 mois par le cabotégravir IM plus la rilpivirine IM n'était pas inférieur à l'administration mensuelle du cabotégravir IM et de la rilpivirine IM en termes de proportion de patients présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml (respectivement 1,7% et 1,0%) à la semaine 48. La différence ajustée entre l'administration tous les 2 mois et l'administration tous les mois de cabotégravir IM + rilpivirine IM (0,8; IC à 95%: –0,6; 2,2) a satisfait au critère de non-infériorité (limite supérieure de l'IC à 95% inférieure à 4%). Les résultats d'efficacité à la semaine 96 et à la semaine 152 sont cohérents avec les résultats du critère d'évaluation principal à la semaine 48 (voir tableau 14).
Tableau 14: Résultats virologiques du traitement randomisé dans le cadre de l'étude ATLAS-2M à 48, 96 et 152 semaines (analyse snapshot)
|
|
Administration tous les 2 mois (q8w)
|
Administration tous les mois (q4w)
| |
|
n=522 (%)
|
n=523 (%)
| |
Semaine 48
|
|
| |
ARN du VIH-1 ≥ 50 copies/ml, n (%)†
|
9 (1.7)
|
5 (1.0)
| |
Différence entre les traitements en % (IC à 95%)*
|
0.8 (-0.6, 2.2)
| |
ARN du VIH-1 < 50 copies/ml, n (%)
|
492 (94.3)
|
489 (93.5)
| |
Aucune donnée virologique pendant la fenêtre de la semaine 48, n (%)
|
21 (4.0)
|
29 (5.5)
| |
Raisons:
| |
Sortie de l’étude en raison d’un EI ou du décès, n (%)
|
9 (1.7)
|
13 (2.5)
| |
Sortie de l’étude pour d’autres raisons, n (%) a
|
12 (2.3)
|
16 (3.1)
| |
Dans l’étude, mais données manquantes pendant cette fenêtre
|
0
|
0
| |
Semaine 96
|
|
| |
ARN du VIH-1 ≥ 50 copies/ml, n (%)†
|
11 (2.1)
|
6 (1.1)
| |
ARN du VIH-1 < 50 copies/ml, n (%)
|
475 (91.0)
|
472 (90.2)
| |
Aucune donnée virologique pendant la fenêtre de la semaine 48, n (%)
|
36 (6.9)
|
45 (8.6)
| |
Raisons:
|
|
| |
Sortie de l’étude en raison d’un EI ou du décès, n (%)
|
17 (3.3)
|
17 (3.3)
| |
Sortie de l’étude pour d’autres raisons, n (%)a
|
16 (3.1)
|
27 (5.2)
| |
Dans l’étude, mais données manquantes pendant cette fenêtre
|
3 (0.6)
|
1 (0.2)
| |
Woche 152
|
|
| |
ARN du VIH-1 ≥ 50 copies/ml, n (%)†
|
14 (2.7)
|
5 (1.0)
| |
ARN du VIH-1 < 50 copies/ml, n (%)
|
456 (87.4)
|
449 (85.9)
| |
Aucune donnée virologique pendant la fenêtre de la semaine 48, n (%)
|
52 (10.0)
|
69 (3.2)
| |
Raisons:
|
|
| |
Sortie de l’étude en raison d’un EI ou du décès, n (%)
|
23 (4.4)
|
24 (4.6)
| |
Sortie de l’étude pour d’autres raisons, n (%) a
|
28 (5.4)
|
44 (8.4)
| |
Dans l’étude, mais données manquantes pendant cette fenêtre
|
1 (0.2)
|
1 (0.2)
| |
* Ajustée en fonction de facteurs de stratification à l’inclusion.
† Inclut les participants ayant arrêté le traitement en raison d’un manque d’efficacité ou ayant arrêté alors qu’ils n’étaient pas virologiquement contrôlés.
n = nombre de participants dans chaque groupe de traitement, IC = intervalle de confiance, TAC = traitement antirétroviral en cours.
|
Tableau 15: Proportion de participants présentant un taux plasmatique d'ARN du VIH-1 ≥50 copies/ml dans l'analyse des sous-groupes à la semaine 48 dans le cadre de l'étude ATLAS-2M selon les caractéristiques initiales principales des patients (facteurs à l'inclusion) (analyse snapshot des résultats).
|
Nombre de patients présentant un taux d'ARN du VIH-1 ≥50 copies/ml/nombre total évalué (%)
| |
Caractéristiques initiales des patients
|
|
Administration tous les 2 mois (q8w)
|
Administration tous les mois (q4w)
| |
|
n/N (%)
|
n/N (%)
| |
Nombre de cellules CD4+ à l'inclusion (cellules/mm3)
|
< 350
|
1/35 (2,9)
|
1/27 (3,7)
| |
350 à < 500
|
1/96 (1,0)
|
0/89
| |
≥500
|
7/391 (1,8)
|
4/407 (1,0)
| |
Sexe
|
Masculin
|
4/385 (1,0)
|
5/380 (1,3)
| |
Féminin
|
5/137 (3,5)
|
0/143
| |
Groupe ethnique
|
Blancs
|
5/370 (1,4)
|
5/393 (1,3)
| |
Non blancs
|
4/152 (2,6)
|
0/130
| |
Noirs/Afro-Américains
|
4/101 (4,0)
|
0/90
| |
Non noirs/Afro-Américains
|
5/421 (1,2)
|
5/421 (1,2)
| |
IMC
|
< 30 kg/m2
|
3/409 (0,7)
|
3/425 (0,7)
| |
≥30 kg/m2
|
6/113 (5,3)
|
2/98 (2,0)
| |
Âge (années)
|
< 35
|
4/137 (2,9)
|
1/145 (0,7)
| |
35 à < 50
|
3/242 (1,2)
|
2/239 (0,8)
| |
> 50
|
2/143 (1,4)
|
2/139 (1,4)
| |
Traitement antérieur par CAB/RPV
|
Aucun
|
5/327 (1,5)
|
5/327 (1,5)
| |
1 à 24 semaines
|
3/69 (4,3)
|
0/68
| |
> 24 semaines
|
1/126 (0,8)
|
0/128
| |
IMC = indice de masse corporelle; CAB = cabotégravir, RPV = rilpivirine
|
Dans l'étude ATLAS-2M, aucune différence cliniquement significative entre les traitements en ce qui concerne le critère d'évaluation principal n'a été observée en corrélation avec les caractéristiques initiales des patients (caractéristiques «à l'inclusion») (nombre de lymphocytes CD4+, sexe, groupe ethnique, IMC, âge et traitement antérieur par cabotégravir/rilpivirine).
Analyses post-hoc
Des analyses multivariées (AMV) des études de phase III regroupées (ATLAS sur 96 semaines, FLAIR sur 124 semaines, ATLAS-2M sur 152 semaines) ont examiné l'influence de différents facteurs sur le risque d'échec virologique (EVC). L'analyse des facteurs à la référence (Baseline Factors Analysis, BFA) a examiné les caractéristiques initiales (à l'inclusion) du virus et des participants à l'étude et le schéma d'administration; cela comprenait les facteurs au début de l'étude et les concentrations plasmatiques pronostiquées du principe actif après le début de l'étude chez des patients présentant un échec virologique confirmé (EVC) à l'aide d'un modèle de régression avec une procédure de sélection des covariables. Après un total de 4291 personnes-années, le taux d'incidence non ajusté de l'EVC était de 0,54 pour 100 personnes-années; 23 cas d'EVC ont été rapportés (1,4% des 1651 personnes incluses dans ces études).
La BFA a montré que les mutations de résistance à la rilpivirine à la référence (rapport de taux d'incidence IRR = 21,65; p < 0,0001), le sous-type A6/A1 du VIH-1 (IRR = 12,87; p < 0,0001) et l'indice de masse corporelle (IRR = 1,09 par unité d'augmentation; p = 0,04; IRR = 3,97 de ≥30 kg/m²; p = 0,01) étaient associés à l'EVC. D'autres variables, notamment l'administration Q4W ou Q8W, le sexe féminin ou les mutations de résistance CAB/INSTI n'étaient pas significativement associées à un EVC. Une combinaison d'au moins 2 des facteurs clés suivants, présents à l'inclusion, a été associée à un risque accru d'EVC: mutations associées à une résistance à la rilpivirine, sous-type A6/A1 du VIH-1 ou IMC ≥30 kg/m2 (tableau 16).
|
Tableau 16: Résultats virologiques selon la présence des facteurs initiaux principaux: mutations associées à une résistance à la rilpivirine, sous-type A6/A11 du VIH-1 et IMC ≥30 kg/m2
| |
Facteur initial (nombre)
|
Succès virologiques2
|
Échec virologique confirmé (%)3
| |
0
|
844/970 (87,0)
|
4/970 (0,4)
| |
1
|
343/404 (84,9)
|
8/404 (2,0)4
| |
≥2
|
44/57 (77,2)
|
11/57 (19,3)5
| |
Total
(intervalle de confiance à 95%)
|
1231/1431 (86,0)
(84,1%, 87,8%)
|
23/1431 (1,6)6
(1,0%, 2,4%)
18/1224 (1,47)7
| |
1
Classification des sous-types A1 ou A6 du VIH-1 reposant sur la base de données de séquences du VIH de Los Alamos (juin 2020).
2 Sur la base de l'algorithme snapshot de la FDA: ARN < 50 copies/ml à la semaine 48 ATLAS, à la semaine 124 FLAIR, à la semaine 152 ATLAS-2M.
3 Défini par deux mesures consécutives de l'ARN du VIH > 200 copies/ml.
4 Valeur prédictive positive (VPP) < 1%; valeur prédictive négative (VPN) de 98,5%; sensibilité de 34,8%; spécificité de 71,9%.
5 VPP de 19,3%; VPN de 99,1%; sensibilité de 47,8%; spécificité de 96,7%.
6 Ensemble de données d'analyse contenant toutes les covariables non manquantes pour les facteurs d'entrée (sur un total de 1651 personnes).
7 Ensemble de données d'analyse contenant toutes les covariables non manquantes pour la modélisation multivariable, y compris les concentrations de médicaments.
|
La proportion de patients présentant un EVC était plus élevée chez les patients présentant au moins deux de ces facteurs de risque que chez les patients ne présentant aucun ou un seul facteur de risque; un EVC est survenu chez 6 patients sur 24 [25,0%, IC à 95% (9,8%; 46,7%)] avec injection bimensuelle et chez 5 patients sur 33 avec injection mensuelle [15,2%, IC à 95% (5,1%; 31,9%)].
Traitement de transition par voie orale avec d'autres TAR
Une analyse rétrospective des données groupées issues de 3 études cliniques (FLAIR, ATLAS-2M et LATTE-2/étude 200056) a inclus 29 patients qui ont reçu un traitement oral de transition pendant une durée moyenne de 59 jours (25e et 75e percentiles 53-135) par un TAR autre que cabotégravir plus rilpivirine (autre traitement oral de transition) pendant le traitement par injections intramusculaires (i.m.) de cabotégravir plus rilpivirine à longue durée d'action (LA). L'âge médian des patients était de 32 ans, 14% d'entre eux étaient des femmes, 31% étaient non-caucasiens, 97% ont reçu un schéma thérapeutique à base d'inhibiteur d'intégrase (INI) comme alternative de traitement oral de transition, 41% ont reçu un INNTI dans le cadre de leur traitement oral de transition alternatif (dont la rilpivirine dans 11 cas sur 12) et 62% ont reçu un INNTI. Trois patients ont interrompu le traitement pendant ou peu après la phase de transition orale pour des raisons non liées à la sécurité d'emploi. Chez la majorité (≥96%) des patients, la suppression virale (ARN du VIH-1 plasmatique < 50 copies/ml) a pu être maintenue. Aucun cas d'EVC (ARN du VIH-1 plasmatique ≥200 copies/ml) n'a été observé pendant la période de transition avec un traitement de transition oral alternatif et pendant la phase suivant le traitement de transition oral alternatif (jusqu'à 2 injections de cabotégravir plus rilpivirine après le traitement de transition oral).
Adolescents
Étude MOCHA
La sécurité, l'acceptabilité, la tolérance et la pharmacocinétique de la rilpivirine plus cabotégravir ont été examinées dans l'étude de phase I/II multicentrique, en ouvert, non comparative, en cours, MOCHA (IMPAACT 2017, étude 208580).
MOCHA S16, cohorte 1
55 adolescents infectés par le VIH-1 et virologiquement contrôlés, âgés de 12 à < 18 ans, ayant un poids corporel d'au moins 35 kg, ont été inclus dans l'un de 4 sous-groupes: 1R (rilpivirine) avec administration mensuelle, 1R avec administration tous les 2 mois, 1C (cabotégravir) avec administration mensuelle ou 1C avec administration tous les 2 mois.
Dans la cohorte 1R, en poursuivant le traitement en cours par cART, les participants (n = 25) ont reçu pendant au moins 4 semaines un comprimé de 25 mg de rilpivirine tous les jours, puis des injections mensuelles de rilpivirine pendant 3 mois (mois 1: injection de 900 mg, mois 2 et 3: injection de 600 mg) ou des injections de rilpivirine tous les 2 mois pendant 2 mois (mois 1 et 2: injection de 900 mg). Dans la cohorte 1C, en poursuivant le traitement en cours par cART, les participants (n = 30) ont reçu pendant au moins 4 semaines un comprimé de 30 mg de cabotégravir tous les jours, puis des injections mensuelles de cabotégravir pendant 3 mois (mois 1: injection de 600 mg, mois 2 et 3: injection de 400 mg) ou des injections de cabotégravir tous les 2 mois pendant 2 mois (mois 1 et 2: injection de 600 mg).
Au début de l'étude, l'âge médian des participants de la cohorte 1 (tant 1R que 1C) était de 15,0 ans, le poids corporel médian de 50,0 kg (plage de 37,4 à 98,5), 47,3% étaient de sexe féminin, 92,7% n'étaient pas blancs, et aucun participant n'avait un nombre de cellules CD4+ inférieur à 350 cellules par mm³.
Les objectifs principaux de l'étude à la semaine 16, qui consistaient en la validation de l'utilisation de la dose pour l'adulte par évaluation de la sécurité et de la pharmacocinétique chez des adolescents infectés par le VIH et virologiquement contrôlés, ont été atteints, de sorte que les participants ont pu passer dans la cohorte 2 (voir «Effets indésirables» et «Pharmacocinétique», «Cinétique pour certains groupes de patients»).
MOCHA S24, cohorte 2
La cohorte 2 incluait des participants appropriés ayant terminé la cohorte 1 ainsi que des participants appropriés qui n'avaient pas été inclus dans l'étude auparavant. Les participants de la cohorte 2 (n = 144) ont arrêté leur traitement antérieur par cART et ont reçu pendant au moins 4 semaines, tous les jours, un comprimé de 25 mg de rilpivirine et un comprimé de 30 mg de cabotégravir, puis des injections de rilpivirine tous les 2 mois (mois 1 et 2: injection de 900 mg, puis injection de 900 mg tous les 2 mois), et des injections de cabotégravir (mois 1 et 2: injection de 600 mg, puis injection de 600 mg tous les 2 mois).
Au début de l'étude, l'âge médian des participants de la cohorte 2 était de 15,0 ans, le poids corporel médian de 48,5 kg (plage de 35,2 à 100,9), 51,4% étaient de sexe féminin, 98,6% n'étaient pas blancs, et 4 participants avaient un nombre de cellules CD4+ inférieur à 350 cellules par mm³.
L'objectif principal de l'étude à la semaine 24, la confirmation de la sécurité de la rilpivirine injectable plus carbotégravir injectable chez des adolescents infectés par le VIH et virologiquement contrôlés, a été atteint (voir «Effets indésirables»). L'effet antiviral a été évalué en tant qu'objectif secondaire, 139 participants sur 141 pour lesquels les données étaient disponibles étant restés contrôlés virologiquement à la semaine 24 (taux d'ARN du VIH-1 dans le plasma < 50 copies/ml).
PharmacocinétiqueLes propriétés pharmacocinétiques de REKAMBYS ont été évaluées chez des adultes sains et chez des adultes infectés par le VIH-1.
|
Tableau 17: Paramètres pharmacocinétiques de population suite à l'administration de rilpivirine par voie orale une fois par jour et suite aux injections intramusculaires de REKAMBYS d'initiation, mensuelles ou tous les 2 mois chez les adultes
| |
Phase d'administration
|
Schéma posologique
|
Paramètres PK de la RPV plasmatique
Moyenne géométrique (5e; 95e percentile)
| |
ASC(0-tau)b
(ng•h/ml)
|
Cmax
(ng/ml)
|
Ctaub
(ng/ml)
| |
Instauration oralec
|
25 mg PO
une fois par jour
|
2083
(1125; 3748)
|
116
(49; 244)
|
79
(32; 177)
| |
Injection
d'initiationa,d
|
Dose initiale
de 900 mg en IM
|
44 842
(21 712; 87 575)
|
144
(94; 221)
|
42
(22; 79)
| |
Injection mensuellea,e
|
600 mg en IM
une fois par mois
|
68 324
(39 042; 118 111)
|
121
(68; 210)
|
86
(50; 147)
| |
Injection tous les 2 moisa,e
|
900 mg en IM
tous les 2 mois
|
132 450
(76 638; 221 783)
|
138
(81; 228)
|
69
(38; 119)
| |
a
Basé sur des estimations individuelles en post-hoc à partir du modèle pharmacocinétique de population pour la rilpivirine en IM (données compilées de FLAIR, ATLAS et ATLAS-2M).
b tau est l'intervalle entre les doses: 24 heures pour l'administration orale; 1 ou 2 mois pour les injections IM mensuelles ou les injections IM tous les 2 mois.
c Pour la rilpivirine orale, Ctau représente les données compilées observées des études FLAIR, ATLAS et ATLAS-2M, ASC(0-tau) et Cmax représentent les données pharmacocinétiques provenant des études de phase III de la rilpivirine orale.
d En cas d'administration après une instauration orale, la Cmax de l'injection d'initiation reflète principalement l'administration orale car la première injection a été administrée le même jour que la dernière dose orale. En cas d'administration sans instauration orale (directement par injection, n = 110), la moyenne géométrique (5e, 95e percentile) de la Cmax observée pour la rilpivirine (1 semaine après l'injection d'initiation) était de 68 ng/ml (28; 220) et la Ctau était de 49 ng/ml (18; 138).
e Données de la semaine 48.
|
Absorption
REKAMBYS présente une cinétique (flip-flop) limitée par l'absorption qui résulte d'une lente absorption dans la circulation systémique depuis le muscle fessier et qui entraîne des concentrations plasmatiques constantes de rilpivirine.
Après une dose intramusculaire unique, les concentrations plasmatiques de rilpivirine sont détectables le premier jour et atteignent un pic après 3 à 4 jours (médiane). Après l'administration d'une dose unique de REKAMBYS, la rilpivirine a été détectée dans le plasma pendant une période allant jusqu'à 52 semaines ou davantage. L'exposition à la rilpivirine à l'état d'équilibre est atteinte à environ 80% après l'administration d'injections tous les mois ou tous les 2 mois pendant 1 an.
Après des injections IM uniques et répétées de doses comprises entre 300 et 1200 mg, l'exposition plasmatique à la rilpivirine augmente de manière proportionnelle ou légèrement moins que proportionnelle à la dose.
Distribution
La liaison aux protéines plasmatiques de la rilpivirine, essentiellement à l'albumine, est d'environ 99,7% in vitro. D'après une analyse pharmacocinétique de population, le volume apparent type du compartiment central (Vc/F) pour la rilpivirine après une administration IM a été estimé à 132 l.
La rilpivirine est présente dans le liquide cérébrospinal (LCS). Chez des patients infectés par le VIH-1 traités par la rilpivirine IM plus le cabotégravir IM, le rapport médian des concentrations de rilpivirine dans le LCS et le plasma (n = 16) était compris entre 1,07 et 1,32% (intervalle: de non quantifiable à 1,69%).
Métabolisme
Biotransformation
Des essais in vitro indiquent que la rilpivirine subit principalement un métabolisme oxydatif médié par le système du cytochrome P450 3A (CYP3A).
Élimination
La demi-vie apparente moyenne de la rilpivirine après l'administration de REKAMBYS est limitée par la vitesse d'absorption et a été estimée entre 13 et 28 semaines.
La clairance plasmatique apparente (CL/F) de la rilpivirine après l'administration de REKAMBYS est estimée à 5,08 l/h.
Après administration d'une dose unique de 14C-rilpivirine orale, en moyenne 85% de la radioactivité ont été retrouvés dans les fèces et 6,1% dans l'urine. Dans les fèces, la proportion de rilpivirine inchangée représentait en moyenne 25% de la dose administrée. Seules des traces de rilpivirine inchangée (< 1% de la dose) ont été détectées dans l'urine.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La rilpivirine est principalement métabolisée et éliminée par le foie. Dans le cadre d'une étude ayant comparé 8 patients présentant des troubles légers de la fonction hépatique (score de Child-Pugh A) à 8 témoins appariés, et 8 patients présentant des troubles modérés de la fonction hépatique (score de Child-Pugh B) à 8 témoins appariés, la disponibilité de la rilpivirine orale lors d'une administration orale multiple était 47% plus élevée chez les patients présentant des troubles légers de la fonction hépatique et 5% plus élevée chez les patients présentant des troubles modérés de la fonction hépatique. REKAMBYS n'a pas été évalué chez les patients présentant des troubles sévères de la fonction hépatique (score de Child-Pugh C).
Troubles de la fonction rénale
La pharmacocinétique de la rilpivirine n'a pas été évaluée chez les patients présentant des troubles de la fonction rénale. L'élimination rénale de la rilpivirine est négligeable. Les effets de troubles de la fonction rénale sur l'élimination de la rilpivirine devraient donc être faibles. La rilpivirine étant fortement liée aux protéines plasmatiques, il est peu probable qu'elle soit éliminée de manière significative par hémodialyse ou par dialyse péritonéale.
Patients âgés
Aucun effet cliniquement pertinent de l'âge sur la disponibilité de la rilpivirine n'a été observé après administration intramusculaire. Les données pharmacocinétiques de la rilpivirine chez les patients de plus de 65 ans sont limitées.
Enfants et adolescents
Les analyses pharmacocinétiques de population n'ont mis en évidence aucune différence cliniquement significative au niveau de l'exposition entre les participants adolescents (âgés d'au moins 12 ans et pesant au moins 35 kg) et les participants adultes infectés par le VIH-1 et non infectés. Par conséquent, aucun ajustement posologique n'est nécessaire chez les adolescents pesant ≥35 kg.
|
Tableau 18: Paramètres pharmacocinétiques de population suite à l'administration de rilpivirine par voie orale une fois par jour et suite aux injections intramusculaires de REKAMBYS d'initiation, mensuelles ou tous les 2 mois chez les adolescents (âgés de 12 à < 18 ans et pesant ≥35 kg)
| |
Phase d'administration
|
Schéma posologique
|
Paramètres PK de la RPV plasmatique
Moyenne géométrique (5e; 95e percentile)
| |
ASC(0-tau)b
(ng•h/ml)
|
Cmax
(ng/ml)
|
Ctaub
(ng/ml)
| |
Instauration oralec
|
25 mg PO
une fois par jour
|
2389
(1259; 4414)
|
144
(81; 234)
|
76
(28; 184)
| |
Injection d'initiationa,d
|
Dose initiale de 900 mg en IM
|
35 259
(20 301; 63 047)
|
135
(86; 211)
|
37
(22; 59)
| |
Injection mensuellea,e
|
600 mg en IM
une fois par mois
|
84 280
(49 444; 156 987)
|
146
(85; 269)
|
109
(65; 202)
| |
Injection tous les 2 moisa,f
|
900 mg en IM
tous les 2 mois
|
110 686
(78 480; 151 744)
|
108
(68; 164)
|
62
(45; 88)
| |
a
Basé sur des estimations individuelles en post-hoc à partir du modèle pharmacocinétique de population pour la rilpivirine en IM (MOCHA, IMPAACT 2017).
b tau est l'intervalle entre les doses: 24 heures pour l'administration orale; 1 ou 2 mois pour les injections IM mensuelles ou les injections IM tous les 2 mois.
c Les valeurs des paramètres PK de la phase d'instauration orale représentent l'état d'équilibre.
d En cas d'administration après une instauration orale, la Cmax de l'injection d'initiation reflète principalement l'administration orale, car la première injection a été administrée le même jour que la dernière dose orale; cependant, l'ASCtau et la valeur de la Ctau à la semaine 4 reflètent la première injection.
e Chaque injection mensuelle: 11e injection IM de rilpivirine à action prolongée (40–44 semaines après l'injection d'initiation).
f Injection tous les 2 mois: 6e injection IM de rilpivirine à action prolongée (36–44 semaines après l'injection d'initiation).
|
Sexe
Aucune différence cliniquement pertinente en termes de disponibilité de la rilpivirine n'a été observée entre les hommes et les femmes après une administration intramusculaire.
Groupe ethnique
Aucun effet cliniquement pertinent de l'origine ethnique sur la disponibilité de la rilpivirine n'a été observé après une administration intramusculaire.
IMC
Aucun effet cliniquement pertinent de l'IMC sur la disponibilité de la rilpivirine n'a été observé après une administration intramusculaire.
Patients co-infectés par le VHB/VHC
Une analyse pharmacocinétique de population n'a révélé aucun élément indiquant un possible effet cliniquement pertinent d'une co-infection par le virus de l'hépatite B et/ou de l'hépatite C sur la disponibilité de la rilpivirine après la prise de rilpivirine orale.
Données précliniquesToxicité en cas d'administration répétée
Des études de toxicité animale ont été réalisées avec la rilpivirine administrée par voie orale chez la souris, le rat, le lapin, le chien et le singe cynomolgus. Les organes et systèmes cibles de la toxicité étaient la corticosurrénale et la biosynthèse des stéroïdes y étant reliée (souris, rat, chien, singe cynomolgus), les organes reproducteurs (femelles chez la souris, mâles et femelles chez le chien), le foie (souris, rat, chien), la thyroïde et l'hypophyse (rat), le rein (souris, chien), le système hématopoïétique (souris, rat, chien) et le système de la coagulation (rat).
Carcinogénicité et génotoxicité
Le potentiel carcinogène de la rilpivirine a été évalué chez la souris et le rat après administration orale par sonde sur une période allant jusqu'à 104 semaines. Des doses quotidiennes de 20, 60 et 160 mg/kg ont été administrées aux souris et des doses quotidiennes de 40, 200, 500 et 1500 mg/kg ont été administrées aux rats. Une augmentation de l'incidence des adénomes et des carcinomes hépatocellulaires a été observée chez la souris et le rat. Une augmentation de l'incidence des adénomes et/ou des carcinomes folliculaires de la thyroïde a été notée chez le rat. L'administration de la rilpivirine n'a pas entraîné d'augmentation statistiquement significative de l'incidence d'autres néoplasies bénignes ou malignes chez la souris ou le rat. Les anomalies hépatocellulaires observées chez la souris et le rat sont considérées comme spécifiques aux rongeurs et reliées à l'induction d'enzymes hépatiques. Il n'existe pas de mécanisme comparable chez l'être humain. Ces tumeurs ne sont donc pas pertinentes pour l'être humain. Les anomalies folliculaires sont considérées comme spécifiques aux rats et reliées à une augmentation de la clairance de la thyroxine, elles n'ont donc aucune pertinence pour l'être humain. Aux doses les plus faibles testées dans les études de carcinogénicité, la disponibilité systémique de la rilpivirine (sur la base de l'AUC) à la dose maximale recommandée chez l'être humain (DMRH) de 25 mg de rilpivirine par voie orale une fois par jour chez les patients infectés par le VIH-1 ou pour les injections IM de rilpivirine à la dose de 600 mg par mois ou de 900 mg tous les 2 mois correspondait respectivement à ≥17 fois (souris) et 2 fois (rat) la disponibilité chez l'être humain.
La rilpivirine a obtenu des résultats négatifs au test d'Ames de mutation réverse sur bactéries in vitro et au test de clastogénicité sur lymphome de souris in vitro en l'absence et en présence d'un système d'activation métabolique. La rilpivirine n'a pas induit d'aberrations chromosomiques lors du test du micronoyau in vivo après une administration orale chez la souris.
Toxicité sur la reproduction
Dans une étude menée chez le rat avec la rilpivirine à une dose orale allant jusqu'à 400 mg/kg/jour (dose toxique pour les mères), aucun effet sur le comportement d'accouplement ou la fertilité n'a été observé. Cette dose est associée à une exposition environ ≥28 fois supérieure à l'exposition chez l'être humain à la DMRH de 25 mg une fois par jour, ou à la dose intramusculaire de 600 mg ou 900 mg de rilpivirine sous forme de suspension injectable à libération prolongée. Les expérimentations animales n'ont révélé aucun signe de toxicité embryonnaire ou fœtale pertinente, ni aucun effet sur la fonction de reproduction. Aucune tératogénicité n'a été observée avec la rilpivirine chez le rat et le lapin. Les quantités disponibles aux valeurs de NOAEL pour l'embryon et le fœtus chez le rat et le lapin étaient respectivement ≥12 fois et ≥57 fois supérieures à la quantité disponible chez l'être humain à la DMRH de 25 mg une fois par jour, ou à la dose intramusculaire de 600 mg ou 900 mg de rilpivirine sous forme de suspension injectable à libération prolongée. Lors de l'évaluation du développement prénatal et postnatal chez le rat, la rilpivirine n'a eu aucune influence sur le développement de la progéniture pendant la lactation ou après son arrêt, lorsque les mères ont reçu une dose allant jusqu'à 400 mg/kg/jour.
Toxicité locale
Après une injection IM de rilpivirine chez le chien et le cochon nain pendant une période prolongée, un léger érythème de courte durée a été observé et des dépôts blancs au niveau des sites d'injection ont été notés lors de l'autopsie, accompagnés d'un gonflement et d'un changement de couleur des ganglions lymphatiques drainants. L'examen microscopique a montré une infiltration des macrophages et des dépôts éosinophiles au niveau des sites d'injection. Une réaction avec infiltration des macrophages a également été observée dans les ganglions lymphatiques drainants/régionaux. Ces observations ont été considérées comme une réaction aux dépôts plutôt que comme la manifestation d'une irritation locale.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments ou à un solvant.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Conserver REKAMBYS dans l'emballage d'origine jusqu'à son utilisation. Ne pas congeler.
Avant l'emploi, laisser le flacon atteindre la température ambiante (25 °C au max.). Le flacon peut rester dans son carton d'emballage à température ambiante pendant 6 heures au maximum; ne pas remettre au réfrigérateur. S'il n'est pas utilisé dans les 6 heures, il doit être jeté.
Dès que la suspension a été prélevée dans la seringue, l'injection doit être administrée dès que possible, mais elle peut être conservée dans la seringue pendant 2 heures au maximum. Au-delà de 2 heures, le médicament, la seringue et l'aiguille doivent être éliminés.
D'un point de vue microbiologique, la suspension doit être utilisée immédiatement. Si la suspension n'est pas utilisée immédiatement, le délai d'utilisation et les conditions de stockage relèvent de la responsabilité de l'utilisateur.
Conserver hors de portée des enfants.
Numéro d’autorisation67742 (Swissmedic)
PrésentationLa suspension injectable à libération prolongée de rilpivirine est disponible dans un flacon en verre.
Emballage contenant un flacon de 2 ml (600 mg)
Chaque emballage contient 1 flacon (600 mg), 1 seringue, 1 adaptateur pour flacon et 1 aiguille pour injection. [A].
Emballage contenant un flacon de 3 ml (900 mg)
Chaque emballage contient 1 flacon (900 mg), 1 seringue, 1 adaptateur pour flacon et 1 aiguille pour injection. [A].
Remarques concernant la manipulation
Tenir compte des remarques concernant la manipulation figurant à la fin de la présente information professionnelle destinée aux professionnels de la santé.
Titulaire de l’autorisationJanssen-Cilag AG, Zoug
Mise à jour de l’informationJuillet 2025
Les informations suivantes sont destinées exclusivement aux médecins ou aux professionnels de la santé et ne doivent être lues que par le médecin ou le professionnel de santé conjointement avec l'information professionnelle complète
Les informations suivantes sont destinées exclusivement aux médecins ou aux professionnels de la santé et ne doivent être lues que par le médecin ou le professionnel de santé conjointement avec l'information professionnelle complète
Injection de REKAMBYS 2 mL – Instructions d'utilisation:
|
Aperçu
Une dose complète nécessite deux injections:
2 ml de cabotégravir et 2 ml de rilpivirine.
Le cabotégravir et la rilpivirine sont des suspensions ne nécessitant pas de dilution ou de reconstitution supplémentaires. Les étapes de préparation sont les mêmes pour les deux médicaments. Suivre attentivement ces instructions lors de la préparation de la suspension injectable afin d'éviter les fuites.
Le cabotégravir et la rilpivirine doivent être administrés exclusivement par voie intramusculaire. Les deux injections doivent être administrées sur des sites d'injection distincts dans le muscle fessier.
|

Remarque: L'injection dans la face ventro-glutéale doit être privilégiée. L'ordre d'administration est sans importance.
| |
|

Remarques concernant le stockage
| |
• Conserver au réfrigérateur (2–8 °C).
|

| |
L'emballage contient
| |
·1 flacon de rilpivirine
·1 adaptateur pour flacon
·1 seringue
·1 aiguille pour injection (23 gauges, 1½ pouce)
Pour sélectionner la longueur appropriée de l'aiguille pour injection, il convient de tenir compte de la corpulence du patient et de l'appréciation clinique du médecin.
| |
|

| |
Matériel supplémentaire nécessaire
| |
·Gants non stériles
·2 tampons imbibés d'alcool
·2 compresses de gaze
·Récipient approprié résistant aux perforations
|

Il convient de s'assurer d'avoir l'emballage de cabotégravir à disposition avant de commencer.
| |
Préparation
| |
1. Inspecter le flacon
| |
|

|
·Vérifier que la date de péremption n'est pas dépassée.
·Inspecter immédiatement les flacons. Le produit ne doit pas être utilisé si des particules étrangères sont visibles.
|

| |
2. Attendre 15 minutes
| |
|

|
·Attendre au moins 15 minutes avant de réaliser l'injection, pour que le médicament puisse atteindre la température ambiante.
| |
3. Agiter vigoureusement
| |
|

|
·Tenir fermement le flacon et l'agiter vigoureusement pendant 10 secondes (voir illustration).
| |
4. Inspectez la suspension
| |
|

|
·Retourner le flacon et examiner la solution remise en suspension. Elle doit avoir un aspect homogène. Si la suspension n'est pas homogène, agiter de nouveau le flacon.
·Il est également normal de constater la présence de petites bulles d'air.
|

| |
5. Retirer le capuchon du flacon
| |
|

|
·Retirer le capuchon du flacon.
·Essuyer le bouchon en caoutchouc avec un tampon imbibé d'alcool.
|

| |
6. Ouvrir l'emballage contenant l'adaptateur pour flacon
| |
|

|
·Retirer la pellicule en papier de l'emballage contenant l'adaptateur pour flacon.
|

| |
7. Fixer l'adaptateur pour flacon
| |
|

|
·Placez le flacon sur une surface plane.
·Appuyer l'adaptateur pour flacon verticalement vers le bas sur le flacon (voir illustration).
·L'adaptateur doit être solidement emboîté avec un click.
| |
8. Retirez l'emballage
| |
|

|
·Puis soulever l'emballage de l'adaptateur pour flacon (voir illustration).
| |
9. Préparer la seringue
| |
|

|
·Sortir la seringue de son emballage.
·Aspirer 1 mL d'air dans la seringue. Cela facilitera ultérieurement le prélèvement du liquide.
| |
10. Fixer la seringue
| |
|
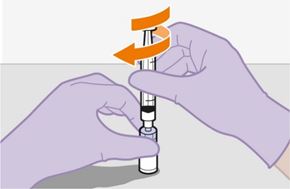
|
·Tenir fermement l'adaptateur pour flacon et le flacon (voir illustration).
·Appuyer fermement la seringue sur l'adaptateur pour flacon.
| |
11. Appuyer sur le piston
| |
|
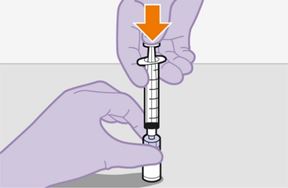
|
·Appuyer à fond sur le piston pour expulser l'air dans le flacon.
| |
12. Prélever lentement la dose
| |
|

|
·Retourner la seringue et le flacon afin de prélever lentement autant de liquide que possible dans la seringue. Il est possible qu'il y ait plus de liquide que nécessaire pour la dose.
|

| |
13. Retirer la seringue
| |
|
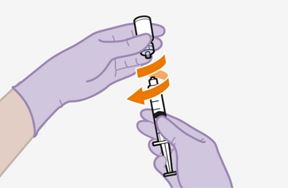
|
·Maintenez le piston de la seringue fermement en place comme illustré, afin d'éviter les fuites. Il est normal de sentir une certaine contrepression.
·Tout en tenant fermement l'adaptateur pour flacon, retirer la seringue de l'adaptateur pour flacon en la faisant tourner (voir illustration).
|

| |
14. Fixer l'aiguille
| |
|

|
·Ouvrir l'emballage contenant l'aiguille de manière à exposer la base de l'aiguille.
·Tenir la seringue verticalement vers le haut et la visser fermement sur l'aiguille.
·Retirer l'emballage de l'aiguille.
| |
Injection
| |
15. Préparer le site d'injection
| |
|

|
Les injections doivent être administrées dans le muscle fessier. Administrer les injections controlatéralement ou à 2 cm l'une de l'autre. Garder une distance d'au moins 2 cm par rapport aux sites d'injection précédents. Pour les injections, choisir l'un des sites suivants:
·Ventro-glutéal (recommandé)
·Dorso-glutéal (quadrant supéro-externe)
|

| |
16. Retirer le capuchon de protection
| |
|

|
·Rabattre le protège-aiguille vers le bas pour l'éloigner de l'aiguille.
·Retirer le capuchon de l'aiguille pour injection.
| |
17. Retirer l'excédent de liquide
| |
|

|
·Tenir la seringue avec l'aiguille pointée vers le haut. Appuyer sur le piston jusqu'à atteindre la graduation de 2 ml pour retirer l'excédent de liquide et les éventuelles bulles d'air.
|

| |
18. Tendre la peau
| |
|

|
Utiliser la technique d'injection «en Z» pour réduire au maximum la fuite du médicament liquide par le site d'injection.
·Tendre la peau au niveau du site d'injection en la déplaçant de 2,5 cm environ.
·Maintenir la peau dans cette position pour réaliser l'injection.
| |
19. Enfoncer l'aiguille
| |
|

|
·Enfoncer l'aiguille sur toute sa longueur ou jusqu'à atteindre le muscle.
| |
20. Injecter la dose
| |
|

|
·Toujours en maintenant la peau tendue, pousser lentement le piston vers le bas jusqu'à la butée.
·S'assurer que la seringue est vide.
·Retirer l'aiguille et relâcher immédiatement la peau tendue.
| |
21. Vérifier le site d'injection
| |
|

|
·Appuyer une compresse de gaze sur le site d'injection.
·Un petit pansement peut être appliqué en cas de saignement.
|

| |
22. Sécuriser l'aiguille
| |
|

|
·Rabattre le protège-aiguille sur l'aiguille.
·Exercer prudemment une pression sur une surface dure pour verrouiller le protège-aiguille.
·Un déclic est audible lorsque le protège-aiguille se verrouille.
| |
Après l'injection
| |
23. Éliminer en toute sécurité
| |
|

|
·Éliminer les aiguilles, seringues, flacons et adaptateurs pour flacon usagés selon la législation en vigueur relative à la sécurité et la protection de la santé.
| |
Répéter les étapes décrites pour le 2e médicament
| |
|

|
Si le cabotégravir n'a pas encore été injecté, suivez les instructions spécifiques pour la préparation et l'injection de ce médicament.
| |
Questions et réponses
| |
1. Combien de temps le médicament peut-il être conservé hors du réfrigérateur?
Il est préférable d'injecter le médicament dès qu'il a atteint la température ambiante. Le flacon peut cependant rester dans son carton d'emballage à température ambiante (25 °C au max.) pendant 6 heures au maximum; ne pas remettre au réfrigérateur. Si le flacon n'est pas utilisé dans les 6 heures, il doit être jeté.
2. Combien de temps le médicament peut-il rester dans la seringue?
Il est préférable d'injecter le médicament (à température ambiante) le plus rapidement possible après l'avoir prélevé dans la seringue. Le médicament peut cependant être conservé dans la seringue pendant 2 heures au maximum avant l'injection. Au-delà de 2 heures, le médicament, la seringue et l'aiguille doivent être éliminés.
3. Pourquoi faut-il injecter de l'air dans le flacon?
L'injection de 1 mL d'air dans le flacon facilite le prélèvement de la dose dans la seringue. En l'absence d'air, une petite quantité de liquide risquerait de refluer accidentellement dans le flacon et la quantité présente dans la seringue serait alors inférieure à celle prévue.
4. L'ordre dans lequel les médicaments sont injectés est-il important?
Non, l'ordre n'a aucune importance.
5. Est-il sûr de réchauffer plus rapidement le flacon à température ambiante?
Il est préférable de laisser le flacon atteindre naturellement la température ambiante. Cependant, il est possible d'accélérer quelque peu la durée du réchauffement avec la chaleur des mains. Il convient toutefois de veiller à ne pas réchauffer le flacon à une température supérieure à 25 °C. N'utiliser aucune autre méthode de réchauffement.
6. Pourquoi l'administration ventro-glutéale est-elle recommandée?
L'injection ventro-glutéale dans le muscle moyen fessier est recommandée, car celui-ci n'est pas situé à proximité des principaux nerfs et des gros vaisseaux sanguins. L'injection dorso-glutéale dans le muscle grand fessier est acceptable, si elle est privilégiée par le professionnel de santé. L'injection ne doit être administrée dans aucun autre site.
|
Les informations suivantes sont destinées exclusivement aux médecins ou aux professionnels de la santé et ne doivent être lues que par le médecin ou le professionnel de santé conjointement avec l'information professionnelle complète.
Injection de REKAMBYS 3 mL – Instructions d'utilisation:
|
Aperçu
Une dose complète nécessite deux injections:
3 ml de cabotégravir et 3 ml de rilpivirine.
Le cabotégravir et la rilpivirine sont des suspensions ne nécessitant pas de dilution ou de reconstitution supplémentaires. Les étapes de préparation sont les mêmes pour les deux médicaments. Suivre attentivement ces instructions lors de la préparation de la suspension injectable afin d'éviter les fuites.
Le cabotégravir et la rilpivirine doivent être administrés exclusivement par voie intramusculaire. Les deux injections doivent être administrées sur des sites d'injection distincts dans le muscle fessier.
|

Remarque: L'injection dans la face ventro-glutéale doit être privilégiée. L'ordre d'administration est sans importance.
| |
|

Remarques concernant le stockage
| |
• Conserver au réfrigérateur (2–8 °C).
|

| |
L'emballage contient
| |
·1 flacon de rilpivirine
·1 adaptateur pour flacon
·1 seringue
·1 aiguille pour injection (23 gauges, 1½ pouce)
Pour sélectionner la longueur appropriée de l'aiguille pour injection, il convient de tenir compte de la corpulence du patient et de l'appréciation clinique du médecin.
| |
|

| |
Matériel supplémentaire nécessaire
| |
·Gants non stériles
·2 tampons imbibés d'alcool
·2 compresses de gaze
·Récipient approprié résistant aux perforations
|

Il convient de s'assurer d'avoir l'emballage de cabotégravir à disposition avant de commencer.
| |
Préparation
| |
1. Inspecter le flacon
| |
|

|
·Vérifier que la date de péremption n'est pas dépassée.
·Inspecter immédiatement les flacons. Le produit ne doit pas être utilisé si des particules étrangères sont visibles.
|

| |
2. Attendre 15 minutes
| |
|

|
·Attendre au moins 15 minutes avant de réaliser l'injection, pour que le médicament puisse atteindre la température ambiante.
| |
3. Agiter vigoureusement
| |
|

|
·Tenir fermement le flacon et l'agiter vigoureusement pendant 10 secondes (voir illustration).
| |
4. Inspectez la suspension
| |
|

|
·Retourner le flacon et examiner la solution remise en suspension. Elle doit avoir un aspect homogène. Si la suspension n'est pas homogène, agiter de nouveau le flacon.
·Il est également normal de constater la présence de petites bulles d'air.
|

| |
5. Retirer le capuchon du flacon
| |
|

|
·Retirer le capuchon du flacon.
·Essuyer le bouchon en caoutchouc avec un tampon imbibé d'alcool.
|

| |
6. Ouvrir l'emballage contenant l'adaptateur pour flacon
| |
|

|
·Retirer la pellicule en papier de l'emballage contenant l'adaptateur pour flacon.
|

| |
7. Fixer l'adaptateur pour flacon
| |
|

|
·Placez le flacon sur une surface plane.
·Appuyer l'adaptateur pour flacon verticalement vers le bas sur le flacon (voir illustration).
·L'adaptateur doit être solidement emboîté avec un click.
| |
8. Retirez l'emballage
| |
|

|
·Puis soulever l'emballage de l'adaptateur pour flacon (voir illustration).
| |
9. Préparer la seringue
| |
|

|
·Retirer la seringue de son emballage.
·Aspirer 1 mL d'air dans la seringue. Cela facilitera ultérieurement le prélèvement du liquide.
| |
10. Fixer la seringue
| |
|
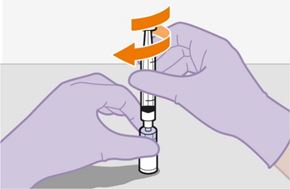
|
·Tenir fermement l'adaptateur pour flacon et le flacon (voir illustration).
·Appuyer fermement la seringue sur l'adaptateur pour flacon.
| |
11. Appuyer sur le piston
| |
|
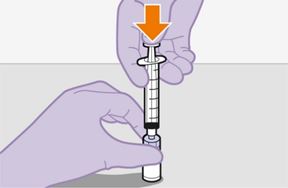
|
·Appuyer à fond sur le piston pour expulser l'air dans le flacon.
| |
12. Prélever lentement la dose
| |
|

|
·Retourner la seringue et le flacon et prélever lentement autant de liquide que possible dans la seringue. Il est possible qu'il y ait plus de liquide que nécessaire pour la dose.
|

| |
13. Retirer la seringue
| |
|
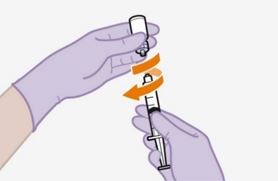
|
·Maintenez le piston de la seringue fermement en place comme illustré, afin d'éviter les fuites. Il est normal de sentir une certaine contrepression.
·Tout en tenant fermement l'adaptateur pour flacon, retirer la seringue de l'adaptateur pour flacon en la faisant tourner (voir illustration).
|

| |
14. Fixer l'aiguille
| |
|

|
·Ouvrir l'emballage contenant l'aiguille de manière à exposer la base de l'aiguille.
·Tenir la seringue verticalement vers le haut et la visser fermement sur l'aiguille.
·Retirer l'emballage de l'aiguille.
| |
Injection
| |
15. Préparer le site d'injection
| |
|

|
Les injections doivent être administrées dans le muscle fessier. Administrer les injections controlatéralement ou à 2 cm l'une de l'autre. Garder une distance d'au moins 2 cm par rapport aux sites d'injection précédents. Pour les injections, choisir l'un des sites suivants:
·Ventro-glutéal (recommandé)
·Dorso-glutéal (quadrant supéro-externe)
|

| |
16. Retirer le capuchon de protection
| |
|

|
·Rabattre le protège-aiguille vers le bas pour l'éloigner de l'aiguille.
·Retirer le capuchon de l'aiguille pour injection.
| |
17. Retirer l'excédent de liquide
| |
|

|
·Tenir la seringue avec l'aiguille pointée vers le haut. Appuyer sur le piston jusqu'à atteindre la graduation de 3 ml pour retirer l'excédent de liquide et les éventuelles bulles d'air.
|

| |
18. Tendre la peau
| |
|

|
Utiliser la technique d'injection «en Z» pour réduire au maximum la fuite de médicament liquide par le site d'injection.
·Tendre la peau au niveau du site d'injection en la déplaçant de 2,5 cm environ.
·Maintenir la peau dans cette position pour réaliser l'injection.
| |
19. Enfoncer l'aiguille
| |
|

|
·Enfoncer l'aiguille sur toute sa longueur ou jusqu'à atteindre le muscle.
| |
20. Injecter la dose
| |
|

|
·Toujours en maintenant la peau tendue, pousser lentement le piston vers le bas jusqu'à la butée.
·S'assurer que la seringue est vide.
·Retirer l'aiguille et relâcher immédiatement la peau tendue.
| |
21. Vérifier le site d'injection
| |
|

|
·Appuyer une compresse de gaze sur le site d'injection.
·Un petit pansement peut être appliqué en cas de saignement.
|

| |
22. Sécuriser l'aiguille
| |
|

|
·Rabattre le protège-aiguille sur l'aiguille.
·Exercer prudemment une pression sur une surface dure pour verrouiller le protège-aiguille.
·Un déclic est audible lorsque le protège-aiguille se verrouille.
| |
Après l'injection
| |
23. Éliminer en toute sécurité
| |
|

|
·Éliminer les aiguilles, seringues, flacons et adaptateurs pour flacon usagés selon la législation en vigueur relative à la sécurité et la protection de la santé.
| |
Répéter les étapes décrites pour le 2e médicament
| |
|

|
Si le cabotégravir n'a pas encore été injecté, suivez les instructions spécifiques pour la préparation et l'injection de ce médicament.
| |
Questions et réponses
| |
1. Combien de temps le médicament peut-il être conservé hors du réfrigérateur?
Il est préférable d'injecter le médicament dès qu'il a atteint la température ambiante. Le flacon peut cependant rester dans son carton d'emballage à température ambiante (25 °C au max.) pendant 6 heures au maximum; ne pas remettre au réfrigérateur. Si le flacon n'est pas utilisé dans les 6 heures, il doit être jeté.
2. Combien de temps le médicament peut-il rester dans la seringue?
Il est préférable d'injecter le médicament (à température ambiante) le plus rapidement possible après l'avoir prélevé dans la seringue. Le médicament peut cependant être conservé dans la seringue pendant 2 heures au maximum avant l'injection. Au-delà de 2 heures, le médicament, la seringue et l'aiguille doivent être éliminés.
3. Pourquoi faut-il injecter de l'air dans le flacon?
L'injection de 1 mL d'air dans le flacon facilite le prélèvement de la dose dans la seringue. En l'absence d'air, une petite quantité de liquide risquerait de refluer accidentellement dans le flacon et la quantité présente dans la seringue serait alors inférieure à celle prévue.
4. L'ordre dans lequel les médicaments sont injectés est-il important?
Non, l'ordre n'a aucune importance.
5. Est-il sûr de réchauffer plus rapidement le flacon à température ambiante?
Il est préférable de laisser le flacon atteindre naturellement la température ambiante. Cependant, il est possible d'accélérer quelque peu la durée du réchauffement avec la chaleur des mains. Il convient toutefois de veiller à ne pas réchauffer le flacon à une température supérieure à 25 °C. N'utiliser aucune autre méthode de réchauffement.
6. Pourquoi l'administration ventro-glutéale est-elle recommandée?
L'injection ventro-glutéale dans le muscle moyen fessier est recommandée, car celui-ci n'est pas situé à proximité des principaux nerfs et des gros vaisseaux sanguins. L'injection dorso-glutéale dans le muscle grand fessier est acceptable, si elle est privilégiée par le professionnel de santé. L'injection ne doit être administrée dans aucun autre site.
|
|