Propriétés/EffetsCode ATC
L01XL05
Mécanisme d'action
CARVYKTI est une immunothérapie anti-BCMA utilisant des lymphocytes T autologues génétiquement modifiés. Elle consiste à reprogrammer les lymphocytes T d'un patient à l'aide d'un transgène codant un récepteur antigénique chimérique (CAR) afin de reconnaître et d'éliminer les cellules exprimant le BCMA. Le BCMA est principalement exprimé à la surface des cellules de la lignée B du myélome multiple ainsi que des lymphocytes B au stade tardif et des plasmocytes. La protéine CAR dans CARVYKTI se compose de deux anticorps à domaine unique dirigés contre le BCMA, présentant une avidité élevée pour le BCMA humain, d'un domaine costimulateur 4-1BB et d'un domaine de signalisation cytoplasmique CD3-zeta (CD3ζ). Lors de la liaison aux cellules exprimant le BCMA, la protéine CAR transmet un signal qui favorise l'activation et l'expansion des lymphocytes T et l'élimination des cellules cibles.
Des expériences in vitro sur des cocultures ont montré que la cytotoxicité et la libération de cytokines (interféron gamma, [IFN-γ], facteur de nécrose tumorale [TNF-α], interleukine [IL]-2) à médiation par le ciltacabtagène autoleucel sont dépendantes du BCMA.
Pharmacodynamique
Après une perfusion unique de CARVYKTI, l'expansion des lymphocytes T CAR-positifs s'accompagne de réductions du BCMA soluble dans le sérum, du gradient M et/ou des chaînes légères libres dans le sérum. Une augmentation des concentrations d'IL-6, d'IL-10, d'IFN-γ et du récepteur alpha de l'IL-2 a été observée chez tous les patients après la perfusion, avec un pic aux jours 7 à 14. En général, les concentrations sériques de toutes les cytokines sont revenues aux valeurs initiales dans les 2 à 3 mois suivant la perfusion.
Immunogénicité
L'immunogénicité de CARVYKTI a été évaluée avant la perfusion et à plusieurs reprises après la perfusion, à l'aide d'un test validé pour la détection des anticorps de liaison dirigés contre CARVYKTI. Dans l'étude MMY2001, 19 patients sur 97 (19,6%) ont été positifs aux anticorps anti-CAR. Dans l'étude MMY3002, 37 patients sur 176 (21%) ont été positifs aux anticorps anti-CAR.
Il n'a pas été clairement démontré que les anticorps anti-CAR affectent la cinétique d'expansion initiale et de persistance de CARVYKTI, son efficacité ou sa sécurité.
Efficacité clinique
Étude MMY2001
MMY2001 était une étude ouverte évaluant CARVYKTI pour le traitement de patients atteints de myélome multiple récidivant ou réfractaire ayant déjà reçu un inhibiteur du protéasome, un agent immunomodulateur et un anticorps anti-CD38 et dont la maladie avait progressé pendant ou après le dernier traitement. Au total, 113 patients ont subi une leucaphérèse. CARVYKTI a été préparé pour chaque patient. Seize patients n'ont pas été traités par CARVYKTI (n = 12 après leucaphérèse et n = 4 après la chimiothérapie lymphodéplétive), en raison d'un retrait du consentement (n = 5), d'une progression de la maladie (n = 2) ou en raison d'un décès (n = 9).
Sur les 97 patients traités, le délai médian entre le jour suivant la réception du matériel de leucaphérèse dans l'établissement de préparation et la libération du produit pour la perfusion a été de 29 jours (fourchette: 23 à 64 jours), et le délai médian entre la leucaphérèse initiale et la perfusion de CARVYKTI a été de 47 jours (fourchette: 41 à 167 jours).
Après la leucaphérèse et avant la perfusion de CARVYKTI, 73 des 97 patients traités (75%) ont reçu un traitement de transition. Les principes actifs les plus souvent utilisés pour le traitement de transition (≥20% des patients) ont été la dexaméthasone: 62 patients (64%), le bortézomib: 26 patients (27%), le cyclophosphamide: 22 patients (23%) et le pomalidomide: 21 patients (22%).
CARVYKTI a été administré en perfusion intraveineuse unique 5 à 7 jours après le début d'une chimiothérapie lymphodéplétive (cyclophosphamide 300 mg/m2 par voie intraveineuse une fois par jour et fludarabine 30 mg/m2 par voie intraveineuse une fois par jour pendant 3 jours). Nonante-sept patients ont reçu CARVYKTI à une dose médiane de 0,71 × 106 lymphocytes T CAR-positifs viables/kg (fourchette: 0,51 à 0,95 × 106 cellules/kg). Tous les patients ont été hospitalisés pour la perfusion de CARVYKTI et pour une période d'au moins 10 jours après celle-ci.
Sur les 113 patients ayant subi une leucaphérèse, 58% étaient de sexe masculin, 74% étaient caucasiens et 15% étaient afro-américains. L'âge médian des patients étaient de 62 ans (fourchette: 29 à 78 ans). Les patients avaient reçu 5 (intervalle: 3 à 18) lignes de traitements antérieures en médiane, et 88% des patients avaient reçu antérieurement une greffe de cellules souches autologues (GCSA). Nonante-neuf pour cent des patients étaient réfractaires à leur dernière ligne de traitement antérieure, et 89% étaient réfractaires à un inhibiteur du protéasome (IP), à un agent immunomodulateur et à un anticorps anti-CD38.
Sur les 97 patients traités, 59% étaient des hommes, 71% étaient caucasiens et 18% étaient noirs ou afro-américains. L'âge médian des patients était de 61 ans (fourchette: 43 à 78 ans). Les patients avaient reçu en médiane 6 (fourchette: 3 à 18) lignes de traitement antérieures, et 90% des patients avaient reçu antérieurement une greffe de cellules souches autologues (GCSA). Nonante-neuf pour cent des patients étaient réfractaires à la dernière ligne de traitement antérieure, et 88% étaient réfractaires à un inhibiteur du protéasome (IP), à un agent immunomodulateur et à un anticorps anti-CD38.
Les patients avec atteinte significative connue du système nerveux central (SNC), passée ou présente, y compris un myélome multiple avec manifestation touchant le SNC, une greffe de cellules souches allogéniques dans les 6 mois précédant l'aphérèse ou un traitement en cours par immunosuppresseurs, une clairance de la créatinine < 40 ml/min, une concentration absolue de lymphocytes < 300/µl, une augmentation des transaminases hépatiques à plus de 3 fois la limite supérieure de la normale, une fraction d'éjection < 45% ou une infection grave active ont été exclus de la participation à l'étude.
Les résultats d'efficacité ont été fondés sur le taux de réponse global, qui a été évalué par un comité d'évaluation indépendant selon les critères de l'IMWG (voir tableau 5).
Tableau 5: résultats d'efficacité pour l'étude MMY2001
|
|
Tous les patients traités (N = 97)
|
Tous les patients avec leucaphérèse (N = 113)
| |
Taux de réponse global (sCRa + VGPR + PR), n (%)
|
95 (97,9)
|
95 (84,1)
| |
IC à 95% (%)
|
(92,7; 99,7)
|
76,0; 90,3)
| |
Réponse complète stricte (sCRa) n (%)
|
80 (82,5)
|
80 (70,8)
| |
Très bonne réponse partielle (VGPR) n (%)
|
12 (12,4)
|
12 (10,6)
| |
Réponse partielle (PR) n (%)
|
3 (3,1)
|
3 (2,7)
| |
Durée de la réponse (DOR) b
| |
Nombre de répondants
DOR (mois): Médiane (IC à 95%):
|
95
NE (23,3; NE)
|
-
| |
Nombre de répondants avec sCRa
DOR lorsque la meilleure réponse est une sCRa (mois): médiane (IC à 95%)
|
80
NE (28,3; NE)
|
-
| |
Nombre de répondants avec VGPR ou mieux
DOR lorsque la meilleure réponse est une VGPR (mois): médiane (IC à 95%)
|
92
NE (24,4; NE)
|
-
| |
Délai de réponse (mois)
| |
Nombre de répondants
Médiane
Intervalle
|
95
0,95
(0,9; 10,7)
|
-
| |
Délai jusqu'à la sCRa (mois)
| |
Nombre de répondants avec sCRa
Médiane
Intervalle
|
80
2,89
(0,9; 17,8)
|
-
|
Remarque: basé sur une durée médiane de suivi de 27,7 mois
a Pour chaque rémission complète, il s'agissait d'une CR stricte
NE = non évaluable
b Le taux estimé de DOR était de 60,3% (IC à 95%: 49,6%; 69,5%) à 24 mois et de 51,2% (IC à 95%: 39,0%; 62,1%) à 30 mois.
Tableau 6: résumé de la fréquence de la négativité MRD
|
|
Tous les patients traités (N = 97)
|
Tous les patients avec leucaphérèse (N = 113)
| |
Proportion de patients avec négativité MRD, n (%)
|
56 (57,7)
|
56 (49,6)
| |
IC à 95% (%)
|
(47,3; 67,7)
|
(40,0; 59,1)
| |
Patients MRD-négatifs avec sCR n (%)a
|
42 (43,3)
|
42 (37,2)
| |
IC à 95% (%)
|
33,3; 53,7
|
(28,3; 46,8)
| |
|
Patients évaluables (N = 61)
| |
Proportion de patients avec négativité MRD, n (%)
|
56 (91,8)
|
-
| |
IC à 95% (%)
|
(81,9; 97,3)
|
-
|
MRD = maladie résiduelle minimale (minimal residual disease)
Remarque: basé sur une durée médiane de suivi de 27,7 mois.
a Seules les évaluations de la MRD (seuil de test de 10 -5) dans les 3 mois suivant l'obtention d'une CR/sCR jusqu'au décès/progression/traitement de suite (exclusivement) ont été prises en compte. Pour chaque rémission complète, il s'agissait d'une CR stricte.
Avec une durée médiane de suivi de 27,7 mois, la survie sans progression (Progression Free Survival, PFS) médiane n'a pas été atteinte (IC à 95%: 24,5, non évaluable). Le taux de PFS à 12 mois (IC à 95%) a été de 76,3% (66,5%, 83,6%). Le taux de PFS à 24 mois (IC à 95%) a été de 62,7% (52,2%, 71,5%).
Chez les patients présentant une sCR (pour chaque rémission complète, il s'agissait d'une CR stricte), la PFS médiane n'a pas été atteinte (IC à 95%: 30,1%, non évaluable), le taux de PFS estimé à 12 mois étant de 88,8% (IC à 95%: 79,5%, 94,0%). Le taux de PFS à 24 mois a été de 73,5% (IC à 95%: 62,3%, 81,9%).
La survie globale (Overall Survival, OS) médiane n'a pas été atteinte (IC à 95%: non évaluable, non évaluable). Le taux d'OS à 12 mois a été de 87,6% (IC à 95%: 79,2%, 92,8%). Le taux d'OS à 24 mois a été de 76,2% (IC à 95%: 66,5%, 83,5%).
La qualité de vie liée à la santé (Health-related quality of life, HRQoL) a été évaluée au début de l'étude (n = 63) et après la perfusion à l'aide du questionnaire EORTC QLQ-C30. La variation moyenne ajustée (IC à 95%) par rapport au résultat initial sur la sous-échelle de douleur du QLQ-C30 de l'EORTC a été de -1,9 (-8,5; -4,6) au jour 7, -9,9 (-16,5; -3,3) au jour 28, -6,3 (-12,9; -0,4) au jour 56, -9,4 (-16,3; -2,5) au jour 78 et -10,5 (-17,3; -3,8) au jour 100, ce qui, dans l'ensemble, indiquait une diminution de la douleur après la perfusion de CARVYKTI. Au jour 100, 72,2% des patients ont obtenu une amélioration cliniquement significative de leur score sur la sous-échelle de la douleur, 53,8% sur la sous-échelle de la fatigue, 57,7% sur la sous-échelle de la fonction physique et 53,7% sur la sous-échelle de l'état de santé général.
Étude MMY3002
MMY3002 est une étude de phase III randomisée, en ouvert, multicentrique visant à évaluer l'efficacité de CARVYKTI pour le traitement de patients présentant un myélome multiple récidivant et réfractaire au lénalidomide et ayant reçu auparavant au moins une ligne de traitement antérieure par un inhibiteur du protéasome et un agent immunomodulateur. Au total, 419 patients ont été randomisés et ont reçu soit une séquence de leucaphérère, traitement de transition, déplétion lymphocytaire et CARVYKTI (n = 208), soit le traitement standard qui comprenait, selon l'appréciation du médecin, soit du daratumumab, du pomalidomide et de la dexaméthasone, soit du bortézomib, du pomalidomide et de la dexaméthasone (n = 211).
Les patients présentant une atteinte significative connue ou identifiée à l'anamnèse du système nerveux central (SNC), les patients présentant des signes cliniques d'atteinte méningée du myélome multiple et les patients atteints de la maladie de Parkinson ou ayant des antécédents d'une autre maladie neurodégénérative ont été exclus de l'étude.
Sur les 419 patients ayant été randomisés (208 recevant CARVYKTI et 211 le traitement standard), 57% étaient de sexe masculin, 75% étaient caucasiens, 3% étaient noirs ou afro-américains et 7% étaient hispaniques ou latino-américains. L'âge médian des patients était de 61 ans (fourchette: 27 à 80 ans). Les patients avaient reçu en médiane 2 lignes de traitement antérieures (intervalle: 1 à 3), et 85% des patients avaient reçu antérieurement une greffe de cellules souches autologues (GCSA). Nonante-neuf pour cent des patients étaient réfractaires à leur dernière ligne de traitement antérieure. Quarante-huit pour cent étaient réfractaires à un inhibiteur du protéasome (IP) et 100% étaient réfractaires à un agent immunomodulateur. Chez les 208 patients qui ont été affectés au bras CARVYKTI, une leucaphérèse a été effectuée. Après la leucaphérèse et avant l'administration de CARVYKTI, les 208 patients randomisés ont reçu le traitement de transition prévu (traitement standard) conformément au protocole de l'étude. Sur ces 208 patients, 12 n'ont pas été traités par CARVYKTI en raison d'une progression de la maladie (n = 10) ou d'un décès (n = 2); chez 20 patients, une progression de la maladie est survenue avant la perfusion de CARVYKTI, mais ils ont pu recevoir CARVYKTI en tant que traitement complémentaire.
Chez les 176 patients ayant reçu CARVYKTI en tant que traitement d'étude, le délai médian entre le jour suivant la réception du matériel de leucaphérèse dans l'établissement de préparation et la libération du produit pour la perfusion a été de 44 jours (fourchette: 25 à 127 jours), et le délai médian entre la leucaphérèse initiale et la perfusion de CARVYKTI a été de 79 jours (fourchette: 45 à 246 jours).
CARVYKTI a été administré en perfusion intraveineuse unique 5 à 7 jours après le début d'une chimiothérapie lymphodéplétive (cyclophosphamide 300 mg/m2 par voie intraveineuse une fois par jour et fludarabine 30 mg/m2 par voie intraveineuse une fois par jour pendant 3 jours) à une dose médiane de 0,71 × 106 lymphocytes T CAR-positifs viables/kg (fourchette: 0,39 à 1,07 × 106 cellules/kg).
La valeur principale mesurée concernant l'efficacité était la survie sans progression (PFS), qui a été analysée à l'aide de l'ensemble d'analyse en intention de traiter (voir tableau 7 et figure 1). Après une période médiane de suivi de 15,9 mois, la PFS médiane a été de 11,8 mois (IC à 95%: 9,7; 13,8) dans le bras de comparaison et n'a pas été évaluable (IC à 95%: 22,8; non évaluable) dans le bras CARVYKTI (hazard ratio: 0,26 [IC à 95%: 0,18; 0,38]). Le taux estimé de PFS à 12 mois a été de 75,9% (IC à 95%: 69,4%; 81,1%) dans le bras CARVYKTI et de 48,6% (IC à 95%: 41,5%; 55,3%) dans le bras de comparaison. Dans le bras CARVYKTI, la durée médiane estimée de la réponse (DOR) n'a pas été atteinte. Dans le bras de comparaison, la durée médiane estimée de la réponse a été de 16,6 mois (IC à 95%: 12,9; non évaluable).
Tableau 7: résultats d'efficacité pour l'étude MMY3002 (ensemble d'analyse en intention de traiter)
|
|
CARVYKTI (n = 208)
|
Traitement de comparaison
(n = 211)
| |
Survie sans progressiona
|
|
| |
Nombre d'événements, n (%)
|
65 (31,3)
|
122 (57,8)
| |
Médiane, mois [IC à 95%]b
|
NA [22,8; NE]
|
11,8 [9,7; 13,8]
| |
Hazard ratio [IC à 95%]c
|
0,26 [0,18; 0,38]
| |
Valeur de pd
|
< 0,0001
| |
Taux d'une réponse complète ou mieuxa, % [IC à 95%]
|
73,1 [66,5; 79,0]
|
21,8 [16,4; 28,0]
| |
Valeur de pe
|
< 0,0001
| |
Taux de réponse global (ORR)a, % [IC à 95%]
|
84,6 [79,0; 89,2]
|
67,3 [60,5; 73,6]
| |
Valeur de pe
|
< 0,0001
| |
Taux global de MRD négative, % [IC à 95%]
|
60,6 [53,6; 67,3]
|
15,6 [11,0; 21,3]
| |
Valeur de pf
|
< 0,0001
| |
Survie globale (OS)
|
| |
Nombre d'événements, n (%)
|
39 (18,8)
|
47 (22,3)
| |
Médiane, mois [IC à 95%]b
|
NE [NE, NE]
|
26,7 [22,5, NE]
| |
Hazard ratio [IC à 95%]g
|
0,78 [0,50; 1,20]
| |
Vealeur de ph
|
0,2551
|
NE = non évaluable; IC = intervalle de confiance; MRD = maladie résiduelle minimale (minimal residual disease)
Remarque: basé sur une durée médiane de suivi de 15,9 mois
a Conformément au consensus du International Myeloma Working Group (IMWG), évalué à l'aide d'un algorithme assisté par ordinateur
b Estimation de Kaplan-Meier
c Basé sur un modèle stratifié à risques proportionnels de Cox prenant uniquement en compte les événements de PFS qui sont survenus plus de 8 semaines après la randomisation.Un hazard ratio < 1 indique un avantage pour le bras CARVYKTI. Pour toutes les analyses stratifiées, la stratification a été effectuée sur la base de la décision du médecin investigateur (PVd ou DPd), de la stadification ISS (I, II, III) et du nombre de lignes de traitement antérieures (1 vs 2 ou 3) qui ont été randomisées.
d Test du log-rank pondéré et stratifié (pondération de 0 dans la statistique du log-rank pour les 8 premières semaines après la randomisation et 1 après)
e Test stratifié du Chi carré de Cochran-Mantel-Haenszel
f Test exact de Fisher
g Basé sur un modèle stratifié à risques proportionnels de Cox. Un hazard ratio < 1 indique un avantage pour le bras CARVYKTI.
h Test du log-rank stratifié
Figure 1: courbe de Kaplan-Meier de la PFS dans l'étude MMY3002 (ensemble d'analyse en intention de traiter)
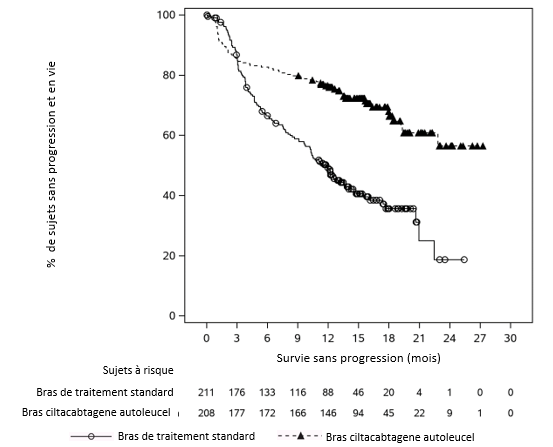
Remarque: l'ensemble d'analyse en intention de traiter est composé de patients qui ont été ransomisés dans l'étude.
Chez les 176 patients ayant reçu CARVYKTI en tant que traitement de l'étude, la PFS médiane n'a pas été évaluable (IC à 95%: non évaluable, non évaluable); le taux de PFS à 12 mois a été de 89,7%. L'ORR chez ces patients a été de 99,4% (IC à 95%: 96,9%; 100,0%). Les taux de CR/sCR ont été de 86,4% (IC à 95%: 80,4%; 91,1%).Chez les 20 patients qui ont présenté une progression précoce et rapide de la maladie et qui ont reçu CARVYKTI en tant que traitement complémentaire, la PFS médiane après la perfusion de CARVYKTI a été de 7,39 mois (IC à 95%: 1,61; non évaluable) avec un taux de PFS à 12 mois de 39,4% (IC à 95%: 18,6; 59,7), une ORR de 65% (IC à 95%: 40,8%; 84,6%) et une CR/sCR de 40% (IC à 95%: 19,1%; 63,9%).
Chez les 208 patients ayant été randomisés pour recevoir le traitement par CARVYKTI, le délai médian jusqu'à l'aggravation des symptômes du myélome multiple a été retardé (23,7 mois) comparé aux 211 patients ayant été randomisés pour recevoir le traitement standard (18,9 mois). Ceci a été mesuré à l'aide du questionnaire MySIm-Q (Multiple Myeloma Symptom and Impact Questionnaire).
|