Propriétés/EffetsCode ATC
L04AE01
Groupe pharmacothérapeutique: modulateurs des récepteurs de la spingosine-1-phosphate (S1P)
Mécanisme d'action
Le fingolimod est un modulateur du récepteur de la sphingosine-1-phosphate. Le principe actif est métabolisé par la sphingosine kinase en phosphate de fingolimod, son métabolite actif. Le phosphate de fingolimod se lie, dans des domaines de concentrations nanomolaires inférieurs, aux récepteurs 1, 3 et 4 de la sphingosine-1-phosphate (S1P) sur les lymphocytes et après un passage aisé de la barrière hémato-encéphalique, aux récepteurs S1P 1, 3 et 5 des cellules nerveuses dans le système nerveux central (SNC). En agissant comme antagoniste fonctionnel du S1PR sur les lymphocytes, le phosphate de fingolimod bloque la capacité des lymphocytes à quitter les ganglions lymphatiques de sorte qu'une redistribution et non pas une déplétion de lymphocytes ait lieu. Cette redistribution réduit l'infiltration par les lymphocytes pathogènes, y compris par les cellules proinflammatoires Th17, du SNC, dans lequel ils participent à l'inflammation des nerfs et à l'atteinte des tissus nerveux. Des expériences menées sur les animaux et des expérimentations in vitro ont démontré que les effets avantageux du fingolimod dans la sclérose en plaques pourraient aussi être attribués à une interaction avec les récepteurs S1P sur les cellules nerveuses. Chez les humains et les animaux, le fingolimod pénètre le SNC et l'on a démontré une réduction de l'astrogliose, de la démyélinisation et de la dégénérescence neuronale. En outre, le fingolimod élève les niveaux de BDNF (facteur neurotrophique issu du cerveau) dans le cortex, l'hippocampe et le corps strié du cerveau: il participe ainsi à la survie neuronale et améliore les fonctions motrices.
Pharmacodynamique
Système immunitaire
Effets sur le nombre de cellules immunitaires dans le sang. Dans les 4–6 heures suivant la première prise de 0.5 mg de fingolimod, le nombre de lymphocytes s'abaisse à un niveau équivalent à 75% de la valeur initiale. Si le traitement quotidien est poursuivi, le taux de lymphocytes s'abaisse encore pendant une période de deux semaines et atteint finalement sa valeur minimale d'environ 500 cellules/µl ou environ 30% de la valeur initiale. Dix-huit pourcent des patients ont au moins une fois atteint une valeur inférieure à 200 cellules/µl. Lors d'une administration quotidienne continue, les valeurs de lymphocytes restent basses. La plupart des lymphocytes T et B passent régulièrement par les organes lymphoïdes, c'est donc sur ces cellules que le fingolimod exerce ses effets les plus importants. Environ 15–20% des lymphocytes T ont un phénotype de cellules à mémoire-effectrices et jouent ainsi un rôle important dans la surveillance immunitaire périphérique. Ce sous-groupe de lymphocytes ne passant pas par les organes lymphoïdes, il n'est pas influencé par le fingolimod. Au cours des quelques jours qui suivent l'arrêt du fingolimod, on assiste à une augmentation du nombre de lymphocytes périphériques, qui généralement se normalise en un ou deux mois. Une administration continue de fingolimod conduit à une légère diminution du nombre de neutrophiles à environ 80% de la valeur initiale. Le fingolimod n'exerce aucune influence sur les monocytes.
Fréquence cardiaque et rythme cardiaque
Au début du traitement, le fingolimod provoque un ralentissement passager de la fréquence cardiaque et de la conduction auriculoventriculaire (voir «Effets indésirables»). Le ralentissement de la fréquence cardiaque est le plus manifeste 4–5 heures après la prise, le médicament déployant 70% de son effet chronotrope négatif le premier jour. La fréquence cardiaque rejoint le plus souvent les valeurs normales en l'espace d'un mois de traitement continu.
Le traitement par le fingolimod n'exerce aucune influence sur les réactions autonomes du muscle cardiaque, ni sur les variations diurnes de la fréquence cardiaque ou les réactions aux activités physiques.
Au début du traitement par le fingolimod, on assiste à une augmentation des extrasystoles d'origine atriale, mais non à l'augmentation du taux de fibrillation atriale/flutter atrial ou d'arythmies ventriculaires ou de battements ventriculaires ectopiques. Le traitement par le fingolimod n'entraîne aucune diminution du débit cardiaque.
Le ralentissement de la fréquence cardiaque provoqué par le fingolimod peut être combattu avec de l'atropine, de l'isoprénaline ou du salmétérol.
Potentiel d'allongement de l'intervalle QT
Dans une étude approfondie sur l'intervalle QT avec des doses de 1.25 ou 2.5 mg de fingolimod à l'état d'équilibre, alors que l'effet chronotrope négatif du fingolimod était encore présent, le traitement par le fingolimod a provoqué un allongement du QTcI avec une limite supérieure de l'IC à 90% de ≤13.0 ms. Il n'y avait pas de relation dose-effet ou exposition-effet entre le fingolimod et un allongement du QTcI. De même il n'y avait aucun signal cohérent en faveur d'une incidence plus élevée d'échappement du QTcI, ni absolue ni sous forme de modification par rapport à la valeur initiale, en relation avec un traitement par le fingolimod. Dans l'étude 1, des allongements du QTcF compris entre 30 et 60 ms sont cependant survenus chez 6.6% (placebo: 2.4%) des patients lors de la première administration de 0.5 mg de fingolimod et chez 13.9% (placebo: 6.7%) des patients lors d'administrations ultérieures. La pertinence clinique est inconnue.
Système respiratoire
Le traitement par une dose unique ou par des doses répétées de 0.5 mg ou 1.25 mg de fingolimod pendant 2 semaines n'est pas associé à une augmentation détectable de la résistance respiratoire mesurée au moyen du VEMS ou du DEF25–75. Des doses uniques de ≥5 mg (10 fois la dose recommandée) ont par contre provoqué une élévation dose-dépendante de la résistance respiratoire. Le traitement par des doses répétées de 0.5, 1.25 ou 5 mg de fingolimod n'a provoqué aucune modification de l'apport en oxygène, de la saturation en oxygène à l'effort et aucune élévation de la sensibilité des voies aériennes à la méthacholine. Les patients traités par le fingolimod ont développé une sensibilité normale aux bêta-agonistes inhalés.
Efficacité clinique
L'efficacité de fingolimod a été mise en évidence dans deux études utilisant une dose unique quotidienne de 0.5 mg et 1.25 mg de fingolimod chez les patients adultes atteints d'une forme récurrente-rémittente de la sclérose en plaques. Les participants aux deux études devaient avoir eu au moins 2 poussées cliniques dans les 2 ans précédant la randomisation, ou au moins 1 poussée clinique dans l'année précédant la randomisation et un score EDSS (Expanded Disability Status Scale) situé entre 0 et 5.5. Une troisième étude avec le même type de patients a été menée après l'inscription de fingolimod.
L'efficacité et la sécurité de la dose unique quotidienne de 0.25 mg et 0.5 mg de fingolimod (choix de la dose en fonction du poids corporel et des mesures de l'exposition) ont été évaluées chez les enfants et les adolescents âgés de 10 à 18 ans atteints d'une forme récurrente-rémittente de la sclérose en plaques.
Étude D2301 (FREEDOMS)
L'étude D2301 (FREEDOMS) était une étude de phase III de 2 ans, randomisée, en double aveugle et contrôlée par placebo chez des patients atteints de sclérose en plaques évoluant par poussées, qui n'avaient pas reçu d'interféron-bêta ou d'acétate de glatiramère pendant au moins 3 mois avant le début de l'étude, et n'avaient pas reçu de natalizumab pendant au moins 6 mois avant le début de l'étude.
L'âge médian était de 37 ans, la durée médiane de la maladie de 6.7 ans et le score EDSS initial médian était de 2.0. Les patients ont reçu après randomisation sur une période allant jusqu'à 24 mois, 0.5 mg de fingolimod (n = 425) ou 1.25 mg de fingolimod (n = 429) ou un placebo (n = 418). La durée moyenne de traitement avec la dose de 0.5 mg a été de 717 jours, de 715 jours avec la dose de 1.25 mg et la durée moyenne de prise de placebo de 718.5 jours.
Le critère d'évaluation principal était le taux annuel de poussées.
Le taux annualisé de poussées (ARR = annualized relapse rate) était significativement plus bas du point de vue statistique chez les patients traités par fingolimod que dans le groupe placebo. Le critère d'évaluation secondaire le plus important était le temps écoulé jusqu'à une progression du handicap persistante pendant 3 mois. La progression était mesurée à l'aide d'une élévation du score EDSS par rapport à la valeur de départ d'au moins 1 point (chez les patients dont la valeur EDSS de départ était de 5.5, au moyen d'une élévation d'au moins 0.5 point), qui persistait pendant au moins 3 mois. L'intervalle jusqu'à l'apparition d'une progression du handicap persistante sur 3 mois était significativement allongé du point de vue statistique lors du traitement par fingolimod en comparaison au traitement par placebo. À aucun moment une différence statistiquement significative entre la dose de 0.5 mg et la dose de 1.25 mg n'a été notée.
Les résultats de cette étude figurent dans le tableau 2 et la figure 1.
Tableau 2 Résultats cliniques et résultats de l'IRM dans l'étude FREEDOMS
|
|
Fingolimod 0.5 mg
|
Placebo
| |
Critères d'évaluation cliniques
|
N=425
|
N=418
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.18
(p<0.001*)
|
0.40
| |
Réduction relative (%)
|
54
|
| |
Proportion de patients toujours sans poussées au 24e mois (%)
|
70.4
(p<0.001*)
|
45.6
| |
Risque de progression du handicap
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.70 (0.52, 0.96)
(p=0.024*)
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 6 mois)
|
0.63 (0.44, 0.90)
(p=0.012*)
|
| |
Critères d'évaluation IRM
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=370
|
n=339
| |
Médiane (moyenne) du nombre pendant 24 mois
|
0.0 (2.5)
(p<0.001*)
|
5.0 (9.8)
| |
Nombre de lésions rehaussées au Gd
|
n=369 (mois 24)
|
n=332 (mois 24)
| |
Médiane (moyenne) du nombre au
| |
6e mois
|
0.0 (0.2)
|
0.0 (1.3)
| |
12e mois
|
0.0 (0.2)
|
0.0 (1.1)
| |
24e mois
|
0.0 (0.2)
(p<0.001* à
toutes les dates)
|
0.0 (1.1)
| |
Pourcentage de modification du volume total des lésions en T2
|
n=368
|
n=339
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-1.7 (10.6)
(p<0.001*)
|
8.6 (33.8)
| |
Modification du volume des lésions hypointenses en T1
|
n=346
|
n=305
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
0.0 (8.8)
(p=0.012*)
|
1.6 (50.7)
| |
Pourcentage de modification du volume cérébral
|
n=357
|
n=331
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-0.7 (-0.8)
(p<0.001*)
|
-1.0 (-1.3)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Montre une signification statistique par rapport au placebo avec un seuil de signification bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS
Figure 1 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée jusqu'au 24e mois - étude FREEDOMS (population ITT)
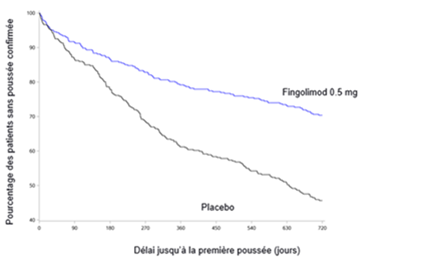
Les patients qui ont participé à l'étude FREEDOMS (D2301) se sont vus offrir la possibilité de participer à une étude de prolongation en aveugle D2301E1. Pour cette étude, 920 patients de l'étude principale traités par le fingolimod ont été sélectionnés (n = 331 ont poursuivi le traitement à la dose de 0.5 mg, 289 ont poursuivi le traitement à la dose de 1.25 mg, 155 sont passés du placebo à la dose de 0.5 mg et 145 sont passés du placebo à la dose de 1.25 mg). Au bout de 12 mois (mois 36), 856 patients (93%) étaient encore présents. On disposait de données de suivi pour un total de 811 patients (88.2%), sur au moins 18 mois pour la phase de prolongation.
Au mois 24 de l'étude de prolongation, le taux annualisé de poussées (Annualized Relapse Rate, ARR) pour les patients qui faisaient partie du groupe sous placébo dans l'étude principale et qui sont passés à la dose de 0.5 mg de fingolimod avait diminué de 55% (ARR 0.45, IC à 95% 0.32 à 0.62, p < 0.001). Chez les patients qui étaient déjà traités avec 0.5 mg de fingolimod dans l'étude principale, l'ARR est resté bas pendant toute la phase de prolongation (ARR = 0.10).
Entre les mois 24 et 36, le taux annualisé de poussées (ARR) a été de 0.17 pour les patients sous 0.5 mg de fingolimod dans l'étude principale et qui continuaient de recevoir 0.5 mg (0.21 dans l'étude principale). Chez les patients qui sont passés du placebo à 0.5 mg de fingolimod, l'ARR s'élevait à 0.22 (0.42 dans l'étude principale).
Étude D2309 (FREEDOMS II)
Dans l'étude de phase III de 2 ans répliquée, randomisée, en double aveugle et contrôlée contre placebo incluant 1083 patients présentant une sclérose en plaques récurrente-rémittente, des résultats comparables à ceux de l'étude D2301 ont été obtenus. Cette étude a été réalisée après l'autorisation de mise sur le marché du fingolimod.
L'âge médian était de 40.5 ans, la durée médiane de la maladie de 8.9 ans et le score EDSS initial médian de 2.5. Les résultats de cette étude sont présentés dans le tableau 3 et la figure 2.
Tableau 3 Résultats cliniques et résultats de l'IRM dans l'étude FREEDOMS II
|
|
0.5 mg Fingolimod
|
Placebo
| |
Critères d'évaluation cliniques
|
N=358
|
N=355
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.21
(p<0.001*)
|
0.40
| |
Réduction relative (en %)
|
48
|
| |
Proportion de patients toujours sans poussées au 24e mois (%)
|
71.5
(p<0.001*)
|
52.7
| |
Risque de progression du handicap†
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.83 (0.61, 1.12)
(p=0.227)
|
| |
Hazard Ratio (IC à 95%) (confirmé à 6 mois)
|
0.72 (0.48, 1.07)
(p=0.113)
|
| |
Critères d'évaluation IRM
| |
Pourcentage de modification du volume cérébral (en %)
|
n=266
|
n=249
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-0.7 (-0.9)
(p<0.001*)
|
-1.0 (-1.3)
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=264
|
n=251
| |
Médiane (moyenne) du nombre pendant 24 mois
|
0.0 (2.3)
(p<0.001*)
|
4.0 (8.9)
| |
Nombre de lésions rehaussées au Gd
|
n=269 (mois 24)
|
n=256 (mois 24)
| |
Médiane (moyenne) du nombre au
| |
6e mois
|
0.0 (0.2)
|
0.0 (1.1)
| |
12e mois
|
0.0 (0.2)
|
0.0 (1.3)
| |
24e mois
|
0.0 (0.4)
(p<0.001* à
toutes les dates)
|
0.0 (1.2)
| |
Pourcentage de modification du volume total des lésions en T2
|
n=262
|
n=247
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-7.1 (13.7)
(p<0.001*)
|
0.8 (25.1)
| |
Modification du volume des lésions hypodenses en T1
|
n=225
|
n=209
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-9.9 (12.6)
(p=0.372)
|
-8.5 (26.4)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Statistiquement significatif vs. placebo avec un niveau bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS.
† D'autres analyses ont montré que les résultats pour la population totale n'étaient pas statistiquement significatifs, du fait de progressions faussement positives dans le sous-groupe des patients avec un score EDSS initial = 0 (n = 62, 8.7% de la population de l'étude). Chez les patients affichant un score EDSS > 0 (n = 651; 91.3% de la population de l'étude), on a observé, pour une dose de 0.5 mg de fingolimod, une diminution cliniquement notable et statistiquement significative par rapport au placebo (HR = 0.70; IC (0.50, 0.98); p = 0.040), ce qui concorde avec l'étude FREEDOMS.
Figure 2 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée, jusqu'au 24e mois – étude FREEDOMS II (population ITT)
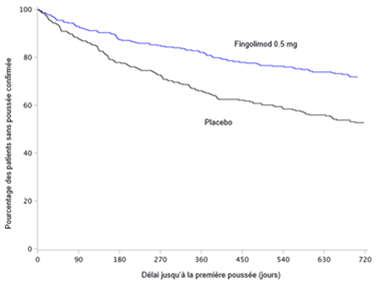
Aucune différence statistiquement significative n'a été constatée entre les doses de 0.5 mg et de 1.25 mg pour aucun des critères d'évaluation.
Étude D2302 (TRANSFORMS)
L'étude D2302 (TRANSFORMS) est une étude de phase III, randomisée, en double aveugle, double-dummy (ou double placebo), contrôlée par traitement actif (interféron bêta-1a, 30 microgrammes en intramusculaire, une fois par semaine) et menée sur un an chez les patients présentant une SEP évoluant par poussées et rémissions, qui n'avaient pas reçu de natalizumab pendant les 6 mois précédant le début de l'étude. Un traitement antérieur par interféron bêta ou par acétate de glatiramère était autorisé jusqu'au moment de la randomisation.
L'âge médian était de 36 ans, la durée médiane de la maladie était de 5.9 ans et le score EDSS initial médian était de 2.0. Après randomisation, les patients ont reçu, pendant une période allant jusqu'à 12 mois, 0.5 mg de fingolimod (n = 431) ou 1.25 mg de fingolimod (n = 426) ou 30 microgrammes d'interféron bêta-1a par voie intramusculaire une fois par semaine (n = 435). La valeur médiane pour la durée du traitement avec le médicament d'étude était de 365 jours (0.5 mg de fingolimod), 354 jours (1.25 mg de fingolimod) et 361 jours (interféron bêta-1a).
Les résultats de cette étude figurent dans le tableau 4 et la figure 3.
Tableau 4 Résultats cliniques et résultats IRM de l'étude TRANSFORMS
|
|
Fingolimod 0.5 mg
|
Interféron bêta-1a, 30 μg
| |
Critères d'évaluation cliniques
|
N=429
|
N=431
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.16
(p<0.001*)
|
0.33
| |
Réduction relative (%)
|
52
|
| |
Proportion de patients toujours sans poussées jusqu'au 12e mois (%)
|
82.5
(p<0.001*)
|
70.1
| |
Risque de progression du handicap
|
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.71 (0.42, 1.21)
(p=0.209)
|
| |
Critères d'évaluation IRM
|
|
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=380
|
n=365
| |
Médiane (moyenne) du nombre pendant 12 mois
|
0.0 (1.7)
(p=0.004*)
|
1.0 (2.6)
| |
Nombre de lésions rehaussées au Gd
|
n=374
|
n=354
| |
Médiane (moyenne) du nombre après 12 mois
|
0.0 (0.2)
(p<0.001*)
|
0.0 (0.5)
| |
Pourcentage de modification du volume cérébral
|
n=368
|
n=359
| |
Médiane (moyenne) de la modification pendant 12 mois en %
|
-0.2 (-0.3)
(p<0.001*)
|
-0.4 (-0.5)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Montre une signification statistique par rapport à l'interféron bêta-1a avec un seuil de signification bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS.
Figure 3 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée jusqu'au 12e mois - étude TRANSFORMS (population ITT)
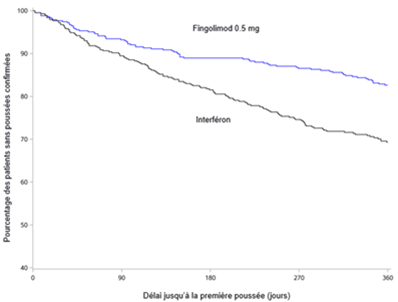
Pour aucun des critères d'évaluation des différences statistiquement significatives n'ont été constatées entre les doses à 0.5 mg et à 1.25 mg.
Les patients qui ont participé à l'étude TRANSFORMS (D2302) se sont vus offrir la possibilité de participer à une étude de prolongation en aveugle (D2302E1). Au total 1030 patients de l'étude principale traités par le fingolimod ont été sélectionnés (n = 357 ont poursuivi le traitement à la dose de 0.5 mg, 330 ont poursuivi le traitement à la dose de 1.25 mg, 167 sont passés de l'interféron bêta-1a à la dose de 0.5 mg et 176 sont passés de l'interféron bêta-1a à la dose de 1.25 mg). On disposait de données de suivi pour un total de 882 de ces patients (85.9%) sur au moins 12 mois pour la phase de prolongation.
Au mois 12 de l'étude de prolongation, chez les patients qui faisaient partie du groupe sous interféron bêta-1a i.m. dans l'étude principale et qui sont passés à la dose de 0.5 mg de fingolimod, l'ARR avait relativement diminué de 30% (ARR 0.70, p = 0.06). Chez les patients qui étaient déjà traités par 0.5 mg de fingolimod dans l'étude principale, l'ARR est resté bas pendant toute l'étude principale et la phase de prolongation (ARR = 0.18 jusqu'au mois 24).
Entre les mois 12 et 24, l'ARR a été de 0.20 (0.19 dans l'étude principale) pour les patients sous 0.5 mg de fingolimod dans l'étude principale et qui continuaient de recevoir 0.5 mg. Chez les patients qui sont passés de l'interféron bêta-1a à 0.5 mg de fingolimod, l'ARR s'élevait à 0.33 (0.48 dans l'étude principale).
Dans l'ensemble, les résultats des études D2301 (FREEDOMS) et D2302 (TRANSFORMS) ont montré une réduction homogène du taux annualisé de poussées par rapport à la préparation utilisée pour la comparaison dans les sous-groupes définis en fonction du sexe, de l'âge, du traitement antérieur de la SEP, de l'activité de la maladie ou du degré de handicap initial.
Étude D2311 (PARADIGMS) chez les enfants et les adolescents à partir de 10 ans
L'étude D2311 (PARADIGMS) est une étude à groupes parallèles en double aveugle, randomisée, contrôlée par substance active et multicentrique, d'une durée flexible allant jusqu'à 24 mois pour l'évaluation de l'efficacité et de la sécurité du fingolimod (n = 107) en comparaison avec l'interféron bêta-1a (n = 107) chez les enfants et les adolescents atteints de SEP récurrente-rémittente âgés de 10 à moins de 18 ans. Un traitement préalable avec de l'interféron bêta, du fumarate de diméthyle ou de l'acétate de glatiramère était autorisé jusqu'au moment de la randomisation. Les patients présentant au moins une poussée clinique l'année précédente ou au moins 2 poussées cliniques dans les 2 ans précédant la randomisation ou la détection de ≥1 lésion rehaussée au Gd en IRM dans les 6 mois précédant la randomisation et avec un score EDSS situé entre 0 et 5.5 ont été inclus. Des examens neurologiques ont été réalisés lors de la sélection ainsi que par la suite tous les 3 mois et lors d'une poussée présumée. Les évaluations des résultats de l'IRM ont eu lieu lors de la sélection et par la suite tous les 6 mois pendant toute la durée de l'étude. Le critère d'évaluation principal était le taux annuel de poussées.
L'âge médian était de 16 ans, la durée médiane de la maladie depuis l'apparition du premier symptôme de 1.5 ans et le score EDSS initial médian était de 1.5. Les patients ont été randomisés et ont reçu du fingolimod ou de l'interféron bêta-1a, qui a été administré par voie intramusculaire une fois par semaine pour une durée allant jusqu'à 24 mois. Dans la classe d'âge ≥10 et ≤12 ans (n = 13 sous fingolimod), ainsi que dans la catégorie de poids ≤40 kg (n = 9 sous fingolimod), peu de patients ont été inclus, de sorte que dans ces groupes de patients seules des données limitées sont disponibles pour évaluer l'efficacité et la sécurité. La durée médiane du traitement par le médicament à l'étude était de 634 jours sous fingolimod et de 547 jours sous interféron bêta-1a.
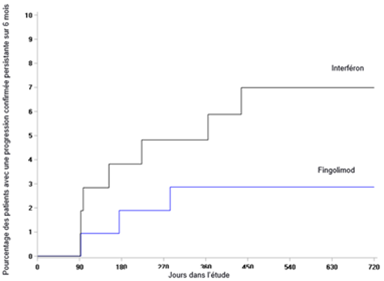
Le critère d'évaluation principal, le taux annuel de poussées (Annualized Relapse Rate, ARR), était significativement plus faible d'un point de vue statistique chez les patients traités par le fingolimod en comparaison avec ceux qui ont reçu de l'interféron bêta-1a (réduction relative de l'ARR de 81.9%). Le critère d'évaluation secondaire le plus important, le nombre annualisé de nouvelles lésions en T2 ou de lésions en T2 agrandies jusqu'au mois 24 était en outre significativement plus faible d'un point de vue statistique chez les patients traités par le fingolimod en comparaison avec ceux qui ont reçu de l'interféron bêta-1a, de même que le nombre de lésions en T1 rehaussées au Gd par examen jusqu'au mois 24. En outre, le taux annualisé d'atrophie cérébrale entre l'inclusion et le mois 24 était réduit de façon statistiquement significative par le fingolimod. Une analyse post-hoc supplémentaire a montré que l'intervalle jusqu'à l'apparition d'une progression du handicap persistante sur 3 mois était allongé de façon statistiquement significative par le fingolimod en comparaison avec l'interféron bêta-1a. Les résultats de cette étude sont présentés dans le tableau 5 et les figures 4 et 5.
Tableau 5 Résultats cliniques et résultats de l'IRM dans l'étude PARADIGMS
|
|
Fingolimod
0.25 mg ou 0.5 mg
|
Interféron bêta-1a
30 µg
| |
Critères d'évaluation cliniques
|
N=107
|
N=107#
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.122
(p<0.001*)
|
0.675
| |
Réduction relative (%)
|
81.9
|
| |
Proportion de patients toujours sans poussées
jusqu'au 24e mois (%)
|
85.7
(p<0.001*)
|
38.8
| |
Risque de progression du handicap
|
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.23 (0.08, 0.66)
(p=0.007*)
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 6 mois)
|
0.20 (0.04; 0.93)
(p=0.040**)
|
| |
Critères d'évaluation IRM
|
|
| |
Nombre annualisé de lésions en T2, nouvelles ou récemment agrandies
|
n=106
|
n=102
| |
Moyenne ajustée
|
4.393
(p<0.001*)
|
9.269
| |
Réduction relative (%)
|
52.6
|
| |
Nombre de lésions en T1 rehaussées au Gd par examen jusqu'au mois 24
|
n=106
|
n=101
| |
Moyenne ajustée
|
0.436
(p<0.001*)
|
1.282
| |
Réduction relative (%)
|
66.0
|
| |
Taux annualisé d'atrophie cérébrale entre l'inclusion et le mois 24
|
n=96
|
n=89
| |
Moyenne des moindres carrés
|
-0.48
(p=0.014*)
|
-0.80
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées sur la population totale d'analyse. Les données évaluables ont été utilisées pour les analyses de l'IRM.
# Un patient randomisé ayant reçu 30 µg d'interféron bêta-1a en injection intramusculaire une fois par semaine était incapable d'avaler la dose supplémentaire nécessaire de placebo dans le cadre de la procédure double-dummy et a été retiré de l'étude. Le patient a été exclu de la population totale d'analyse et de la population de sécurité.
* Montre une signification statistique par rapport à l'interféron bêta-1a IM, avec un seuil de signification bilatéral de 0.05.
** Analyse post-hoc, modèle à risques proportionnels de Cox. p = 0.180 dans le test du log-rank.
Détermination des valeurs de p: ARR agrégé: par régression binomiale négative, ajustée au traitement, au pays, au stade pubertaire (le facteur de stratification dans le système de dialogue vocal interactif, SVI) et au nombre de poussées durant les 2 années précédentes (offset: durée dans l'étude); Pourcentage des patients sans poussée: sur la base d'une estimation de Kaplan-Meier; Risque de progression du handicap: sur la base du modèle à risques proportionnels de Cox, ajusté au traitement, au pays, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de poussées dans les 2 années précédentes; Nombre annualisé de lésions en T2, nouvelles ou récemment agrandies: par régression binomiale négative, ajustée au traitement, à la région, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de lésions en T2 initial (offset: durée dans l'étude); Nombre de lésions rehaussées au Gd par examen: par régression binomiale négative avec le nombre cumulé de lésions en T1 rehaussées au Gd pour tous les examens IRM programmés réalisés durant l'étude depuis l'initiation comme variable réponse, ajustée au traitement, au pays, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de lésions en T1 rehaussées au Gd initial (offset: nombre d'examens IRM); Taux annualisé d'atrophie cérébrale: au moyen d'une analyse ANCOVA, ajusté au traitement, à la région, au stade pubertaire (le facteur de stratification dans le SVI) et au volume cérébral total initial.
Figure 4 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée, jusqu'au 24e mois – étude PARADIGMS- (population totale d'analyse)
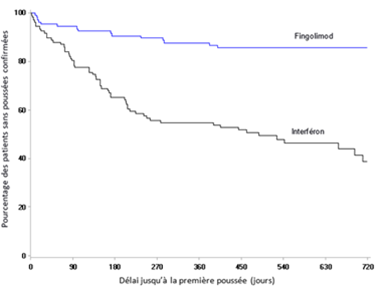
Figure 5 Graphique de Kaplan-Meier du délai jusqu'à la constitution d'une progression confirmée du handicap persistante sur 6 mois - étude PARADIGMS- (population totale d'analyse)
|