CompositionPrincipes actifs
Natalizumab, produit par génie génétique à partir de cellules de souris NS/0.
Excipients
Dihydrogénophosphate de sodium (monohydraté), hydrogénophosphate de disodium (heptahydraté), chlorure de sodium, polysorbate 80, eau pour préparation injectable.
Une seringue préremplie (150 mg/ml) contient 3,45 mg de sodium.
Indications/Possibilités d’emploiAu moment de la pose de l'indication ou avant le traitement, le risque de LEMP (LEMP= leucoencéphalopathie multifocale progressive) doit être pris en considération (voir «Mises en garde et précautions»).
Tysabri est indiqué en monothérapie comme traitement de fond des formes agressives de sclérose en plaques rémittente-récurrente (SEP-RR), pour les groupes de patients suivants:
·Patients présentant une forme agressive de la maladie malgré un traitement complet et bien conduit par au moins un traitement de fond (pour les exceptions et les informations sur les périodes de relais de traitement, voir les rubriques «Mises en garde et précautions» et «Pharmacodynamique»).
ou
·Patients présentant une sclérose en plaques rémittente-récurrente sévère d'évolution rapide, définie par 2 ou plusieurs poussées à caractère invalidant au cours d'une année, et présentant une ou plusieurs lésions rehaussées par le gadolinium à l'examen IRM cérébral ou une augmentation significative des lésions T2 par rapport à un examen IRM pratiqué récemment.
Tysabri est indiqué en monothérapie comme traitement de fond des formes actives de SEP rémittente-récurrente (SEP-RR) chez les patients ayant un statut des anticorps anti-JCV négatif.
Posologie/Mode d’emploiLe traitement doit être instauré et surveillé de manière continue par des médecins spécialistes, expérimentés dans la pose de diagnostics et dans le traitement des affections neurologiques, dans des centres disposant d'un accès rapide à l'IRM (imagerie par résonance magnétique).
Le traitement ne doit pas être réalisé à domicile.
L'administration doit être effectuée par un professionnel de la santé et les patients doivent faire l'objet d'une surveillance afin de détecter les signes et symptômes précoces d'une leucoencéphalopathie multifocale progressive (LEMP).
On devra disposer de possibilités de traitement des réactions d'hypersensibilité.
Les patients doivent être inclus dans un programme de monitoring prévu pour Tysabri.
Les patients traités par Tysabri doivent recevoir le passeport du patient ainsi que des explications claires au sujet des risques liés à Tysabri. Après deux ans de traitement, les patients devront une nouvelle fois être informés des risques liés à Tysabri, en particulier du risque accru de LEMP; ils recevront ainsi que leurs soignants des instructions concernant les signes et symptômes précoces de LEMP.
Certains patients peuvent avoir reçu des médicaments immunosuppresseurs (par exemple, mitoxantrone, cyclophosphamide, azathioprine) susceptibles de provoquer une immunosuppression prolongée, même après l'arrêt du traitement. Le médecin devra donc vérifier l'absence d'immunosuppression avant l'instauration du traitement par Tysabri (voir «Mises en garde et précautions»).
La poursuite du traitement devra être reconsidérée soigneusement chez les patients ne présentant aucun signe de bénéfice thérapeutique au-delà de 6 mois.
Les études contrôlées en double aveugle ont fourni des données sur la sécurité d'emploi et l'efficacité du natalizumab sur une période de 2 ans. La prolongation du traitement après ce délai ne devra être envisagée qu'après une réévaluation du rapport bénéfice-risque.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie usuelle
Adultes
La forme galénique sous-cutanée de Tysabri n'est pas destinée à l'administration intraveineuse. Tout changement de voie d'administration de Tysabri doit être effectué 4 semaines après la dose précédente de Tysabri.
La dose recommandée de Tysabri pour l'injection sous-cutanée est de 300 mg toutes les 4 semaines. Les deux injections (de 150 mg chacune) doivent être réalisées l'une après l'autre sans délai important entre les deux. La seconde injection doit être administrée au plus tard 30 minutes après la première.
Les patients doivent être surveillés pendant les injections sous-cutanées et pendant l'heure qui suit afin de détecter les signes et symptômes de réactions liées à l'injection, y compris de réactions d'hypersensibilité. Après les 6 premières doses, les patients doivent continuer d'être surveillés après les injections sous-cutanées selon l'appréciation clinique. Le temps d'observation d'une heure peut être réduit ou supprimé selon l'appréciation clinique, si les patients n'ont pas présenté de réactions liées à l'injection, y compris de réactions d'hypersensibilité.
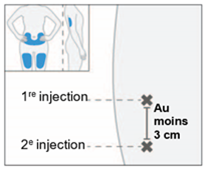
L'injection sous-cutanée doit être réalisée dans la cuisse, l'abdomen ou la face postérieure du bras. Elle ne doit pas être effectuée dans une zone du corps où la peau est irritée, rougie, meurtrie, infectée ou cicatricielle. Lors du retrait de la seringue du site d'injection, le piston doit être relâché tout en retirant l'aiguille tout droit. Le fait de relâcher le piston permet au protège-aiguille de recouvrir l'aiguille. La seconde injection doit être effectuée à au moins 3 cm du premier site d'injection.
Après deux ans de traitement, les patients doivent être nouvellement informés des facteurs de risque de LEMP, tels que la durée du traitement, l'utilisation d'immunosuppresseurs avant le traitement par Tysabri et la présence d'anticorps anti-JCV (voir «Mises en garde et précautions»).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Il n'a été mené aucune étude sur les effets d'une insuffisance hépatique.
Le mécanisme d'élimination et les résultats des études de pharmacocinétique de population suggèrent qu'il n'est pas nécessaire d'ajuster la posologie en cas de troubles de la fonction hépatique.
Patients présentant des troubles de la fonction rénale
Il n'a été mené aucune étude sur les effets d'une insuffisance rénale.
Le mécanisme d'élimination et les résultats des études de pharmacocinétique de population suggèrent qu'il n'est pas nécessaire d'ajuster la posologie en cas de troubles de la fonction rénale.
Patients âgés
Tysabri n'est pas indiqué chez les sujets âgés de plus de 65 ans en raison de l'absence de données dans cette population.
Enfants et adolescents
La sécurité et l'efficacité de Tysabri chez les enfants et adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune recommandation de posologie ne peut être prodiguée. Les données actuellement disponibles sont décrites dans les rubriques «Effets indésirables» et «Propriétés/Effets».
Mode d'administration
Pour les instructions concernant l'administration de Tysabri, solution injectable en seringue préremplie, voir «Remarques particulières».
Réadministration
Voir «Mises en garde et précautions».
Contre-indicationsHypersensibilité au natalizumab ou à l'un des excipients conformément à la composition.
Leucoencéphalopathie multifocale progressive (LEMP).
Patients présentant un risque accru d'infections opportunistes, y compris patients immunodéprimés (patients sous traitement immunosuppresseur ou patients immunodéprimés par des traitements antérieurs, par exemple mitoxantrone ou cyclophosphamide [voir aussi «Mises en garde et précautions» et «Effets indésirables»]).
En association avec d'autres traitements de fond.
Cancers diagnostiqués en évolution, à l'exception des carcinomes baso-cellulaires.
Mises en garde et précautionsLeucoencéphalopathie multifocale progressive (LEMP)
L'utilisation de Tysabri est associée à un risque accru de LEMP, une infection opportuniste provoquée par le virus John Cunningham (virus JC), qui peut avoir une issue fatale ou entraîner de graves handicaps. En raison du risque élevé de développement d'une LEMP, les risques et bénéfices d'un traitement par Tysabri devront être reconsidérés régulièrement par le ou la médecin spécialiste avec le patient dans le cadre des examens cliniques de contrôle. Les patients ainsi que leurs soignants doivent recevoir des instructions concernant les signes et symptômes précoces de LEMP et les patients doivent être surveillés régulièrement en conséquence.
Le virus JC (JCV) peut également provoquer une neuropathie des cellules granulaires (NCG) qui a été observée chez des patients traités par Tysabri. Les symptômes de la NCG due au virus JC ressemblent à ceux d'une LEMP (c.àd. syndrome cérébelleux) et le diagnostic ainsi que la prise en charge de la NCG due au virus JC doivent s'aligner sur les directives en vigueur relatives à la LEMP.
Les facteurs de risque suivants sont associés à un risque accru de développement d'une LEMP:
·Présence d'anticorps anti-JCV. Les tests de détection des anticorps anti-JCV ne doivent être réalisés qu'avec l'essai STRATIFY JCV.
·Durée du traitement, en particulier plus de deux ans.
·Traitement par un immunosuppresseur avant le traitement par Tysabri.
Après deux ans, tous les patients doivent être nouvellement informés du risque de LEMP associé à Tysabri.
Un algorithme d'évaluation du risque de LEMP résume le risque de LEMP selon les facteurs de risque susmentionnés et stratifie ce risque en fonction du taux d'anticorps anti-JCV mesuré (indice). Les médecins sont tenus de consulter les informations supplémentaires relatives à la prise en charge du risque de LEMP chez les patients traités par Tysabri dans le «Guide de prescription pour la prise en charge des patients».
Les patients dont le test d'anticorps anti-JCV est positif présentent un risque accru de développement d'une LEMP comparativement aux patients dont le test d'anticorps anti-JCV est négatif. Les patients qui présentent les trois facteurs de risque de LEMP (c'est-à-dire chez lesquels des anticorps anti-JCV ont été détectés et qui ont été traités par Tysabri pendant plus de deux ans et qui ont reçu un traitement immunosuppresseur au préalable) sont exposés à un risque significativement plus élevé de LEMP.
Chez les patients n'ayant pas reçu de traitement immunosuppresseur préalable et chez lesquels des anticorps anti-JCV ont été mis en évidence, le taux d'anticorps anti-JCV (indice) peut être utilisé pour stratifier plus précisément le risque de LEMP. Dans l'état actuel des connaissances, le risque de LEMP est plutôt faible si l'indice est inférieur ou égal à 0,9. Le risque de LEMP augmente chez les patients présentant un indice supérieur à 1,5 et ayant été traités par Tysabri pendant plus de 2 ans (voir «Guide de prescription pour la prise en charge des patients»).
Une administration à intervalle prolongé de Tysabri (environ toutes les 6 semaines [Q6W] en moyenne) chez les patients présentant des anticorps anti-JCV est associée à un risque de LEMP plus faible par rapport à une administration à l'intervalle autorisé (voir «Propriétés/Effets», Efficacité clinique, «Extension de l'intervalle posologique»). Comme l'efficacité d'une administration à intervalle prolongé par rapport au schéma posologique autorisé (toutes les 4 semaines, Q4W) n'a pas été formellement démontrée par une étude de non-infériorité, la prudence est de mise lorsque l'on a recours à une administration à intervalle prolongé. La diminution du risque de LEMP repose sur des données obtenues avec la voie d'administration intraveineuse. Aucune donnée clinique relative à la sécurité ou à l'efficacité de cette extension de l'intervalle de dose avec la voie d'administration sous-cutanée n'est disponible.
Chez les patients ayant un risque élevé (p.ex. présentant les trois facteurs de risque ou n'ayant pas reçu de traitement immunosuppresseur préalable, mais présentant un indice supérieur à 1,5 et ayant été traités par Tysabri pendant plus de 2 ans), le traitement par Tysabri ne doit être poursuivi que si le bénéfice est supérieur aux risques.
Les estimations du risque de LEMP déduites des données cliniques et des données issues des études d'observation ont correspondu aux données de post-commercialisation.
Patients atteints de SEP-RR active ayant un statut des anticorps anti-JCV négatif
Les patients ayant un statut des anticorps anti-JCV négatif ont un risque plus faible de développer une LEMP (voir également «Mises en garde et précautions», rubrique «Leucoencéphalopathie multifocale progressive (LEMP)» ci-dessus, et «Guide de prescription pour la prise en charge des patients»).
Les patients ne présentant pas d'anticorps anti-JCV peuvent néanmoins avoir un risque de LEMP, par exemple en raison d'une infection récente par le JCV, d'une fluctuation du statut des anticorps ou d'un résultat du test des anticorps anti-JCV faussement négatif. Dans une étude de phase IV ayant évalué le statut des anticorps à long terme pendant 18 mois, le statut des anticorps anti-JCV est passé de négatif à positif chez environ 11 % des patients par an. La recherche des anticorps doit être réalisée exclusivement avec le test STRATIFY JCV, un test de détection des anticorps anti-JCV en deux étapes (ELISA) avec un taux de faux négatifs de 3 %, qui a été validé pour l'utilisation chez les patients atteints de SEP.
Il est recommandé de répéter le test de détection des anticorps anti-JCV chez les patients ayant un statut des anticorps anti-JCV négatif, car ce dernier peut changer. Il est donc recommandé de refaire le test tous les 3 mois chez les patients atteints de formes actives de SEP-RR ayant un statut des anticorps anti-JCV négatif (voir «Test de détection des anticorps anti-JCV» ci-dessous).
La durée du traitement, notamment au-delà de 2 ans, est associée à un risque accru de LEMP.
Avant d'instaurer le traitement, le rapport bénéfice-risque d'un début précoce du traitement doit être discuté individuellement avec le patient, en tenant compte également d'autres effets indésirables graves tels que les infections opportunistes, les réactions d'hypersensibilité et les effets indésirables hépatiques (voir les paragraphes spécifiques sous «Mises en garde et précautions»).
Une éventuelle exacerbation de la maladie à l'arrêt du médicament doit être prise en considération.
Les patients doivent être informés des possibilités thérapeutiques, si leur statut des anticorps anti-JCV passe de négatif à positif au cours du traitement.
Test de détection des anticorps anti-JCV
Il est recommandé de réaliser un tel test avant le traitement par Tysabri ou chez les patients déjà sous Tysabri mais dont le statut des anticorps n'est pas connu. Les patients dont le test des anticorps anti-JCV est négatif peuvent tout de même présenter un risque de LEMP en raison d'une infection récente par le JCV, d'une fluctuation du statut des anticorps ou d'un résultat du test des anticorps anti-JCV faussement-négatif. Dans le cadre d'une étude de phase IV, au cours de laquelle le statut des anticorps anti-JCV a fait l'objet d'une analyse longitudinale pendant 18 mois, le sérostatut des anticorps anti-JCV est passé de négatif à positif chez environ 11 % des patients par an. Le test doit être réalisé exclusivement à l'aide d'un test de détection des anticorps anti-JCV qui a été validé pour l'analyse des patients atteints de SEP, tel que le STRATIFY JCV® ou le STRATIFY JCV® DxSelect™. Lors d'une forme agressive de SEP-RR, il est recommandé de répéter le test de détection des anticorps anti-JCV au moins tous les 6 mois chez 1) les patients dont le test d'anticorps anti-JCV est négatif et 2) les patients ayant un statut des anticorps anti-JCV positif, un indice plus faible et qui n'ont pas reçu de traitement immunosuppresseur préalable (dès que la durée du traitement de deux ans est atteinte), car le statut des anticorps ou l'indice peuvent changer. Chez les patients atteints de SEP-RR active et présentant un statut des anticorps anti-JCV négatif, il est recommandé de répéter le test tous les 3 mois (voir «Mises en garde et précautions» / Patients atteints de SEP-RR active ayant un statut des anticorps anti-JCV négatif).
Le test d'anticorps anti-JCV (ELISA) n'est cependant pas approprié pour poser le diagnostic d'une LEMP.
Pour de plus amples informations sur les tests de détection des anticorps anti-JCV, consulter le «Guide de prescription pour la prise en charge des patients».
Dépistage de la LEMP par IRM
On doit disposer d'un examen d'imagerie par résonance magnétique (IRM) récent (effectué généralement dans les 3 mois précédents) – il servira de document de référence – avant d'instaurer le traitement par Tysabri. Cet examen sera répété au moins une fois par an de manière systématique, afin de toujours disposer d'un document de référence actualisé. Les patients doivent être surveillés à intervalles réguliers.
Des contrôles IRM plus fréquents (p.ex. tous les 3 à 6 mois) doivent être envisagés chez les patients présentant un risque accru de LEMP. Il s'agit des groupes de patients suivants:
·patients présentant les trois facteurs de risque de LEMP (c.àd. chez lesquels des anticorps anti-JCV ont été détectés et ayant été traités par Tysabri pendant plus de deux ans et ayant reçu un traitement immunosuppresseur préalable),
ou
·patients qui présentant un indice d'anticorps anti-JCV supérieur à 1,5, n'ayant pas reçu de traitement immunosuppresseur préalable et ayant été traités par Tysabri pendant plus de 2 ans.
En cas de suspicion de LEMP ou de NCG due au JCV, l'administration de Tysabri doit être interrompue jusqu'à ce que le diagnostic de LEMP ou de NCG due au JCV soit exclu.
Le ou la médecin spécialiste devra examiner soigneusement le patient pour déterminer si les symptômes indiquent un dysfonctionnement neurologique et, si c'est le cas, il devra établir si ces symptômes sont caractéristiques d'une SEP ou évocateurs d'une LEMP ou d'une NCG due au JCV. En cas de doute, des examens complémentaires, y compris une IRM, de préférence avec produit de contraste (à comparer avec l'IRM réalisée avant le début du traitement), des tests pour mettre en évidence l'ADN du virus JC dans le LCR ainsi que des examens neurologiques répétés devront être envisagés (voir «Conduite éducative - Guide de prescription pour la prise en charge des patients»). Le traitement par Tysabri ne pourra être repris qu'après l'exclusion du diagnostic de LEMP et/ou de NCG due au JCV (le cas échéant après répétition des examens cliniques, de l'imagerie et/ou des examens de laboratoire au cas où les soupçons cliniques persistent).
Le médecin devra être particulièrement attentif à l'apparition de symptômes évocateurs d'une LEMP ou d'une NCG due au JCV que le patient pourrait ne pas remarquer (p.ex. symptômes cognitifs, psychiatriques ou signes d'un syndrome cérébelleux). Il faudra recommander aux patients d'informer leur conjoint ou le personnel soignant de leur traitement, ceux-ci pouvant remarquer des symptômes dont les patients ne sont pas conscients.
Des cas de LEMP asymptomatique ont également été rapportés. Une LEMP asymptomatique peut être dépistée par IRM et doit être confirmée par la présence d'ADN du JCV dans le liquide céphalo-rachidien ou par une biopsie cérébrale.
Par ailleurs, les membres du corps médical doivent être attentifs à tous les nouveaux signes (p. ex, à l'IRM) ou symptômes évocateurs d'une LEMP. Dans de tels cas, Tysabri doit être arrêté immédiatement. Si les premiers examens sont négatifs, mais que la suspicion clinique de LEMP subsiste, l'administration de Tysabri ne doit pas être poursuivie et les examens doivent être répétés.
Une LEMP est survenue aussi après l'arrêt de Tysabri chez des patients qui ne présentaient pas de signes d'une éventuelle LEMP au moment de l'arrêt du traitement. Après l'arrêt du traitement par Tysabri, les patients et les médecins doivent donc rester attentifs à tout nouveau signe ou symptôme d'une éventuelle LEMP pendant environ six mois et poursuivre les contrôles IRM pendant cette période.
En cas d'apparition d'une LEMP ou d'une NCG due au JCV, le traitement par Tysabri devra être interrompu définitivement.
Après reconstitution du système immunitaire chez les patients immunodéprimés ayant une LEMP, on a observé une amélioration de l'évolution.
LEMP et IRIS (Immune Reconstitution Inflammatory Syndrome)
Un IRIS apparaît chez presque tous les patients ayant développé une LEMP sous Tysabri, après l'arrêt ou l'élimination de Tysabri, par exemple par plasmaphérèse (PLEX) (voir «Pharmacocinétique»). Il est admis que l'IRIS est une conséquence de la reconstitution de la fonction immunitaire chez les patients atteints de LEMP. L'IRIS peut entraîner une aggravation rapide de l'état neurologique et de lourdes complications neurologiques et son évolution peut être fatale. Il faut alors aussi bien surveiller l'apparition d'un IRIS (entre quelques jours à quelques semaines après la plasmaphérèse chez les patients traités par Tysabri atteints de LEMP) qu'entreprendre un traitement adapté de l'inflammation qui y est liée pendant le rétablissement d'une LEMP.
Une analyse rétrospective du natalizumab, réalisée après son autorisation, n'a montré aucune différence en termes de taux de survie à 2 ans après le diagnostic de LEMP entre les patients ayant reçu un traitement par PLEX et ceux n'en ayant pas reçu. Le recours à une PLEX pour le traitement de la LEMP doit être évalué par le médecin. Le recours à une plasmaphérèse/échange plasmatique (PLEX) ou à des immunoglobulines intraveineuses (IgIV) peut compromettre la pertinence de l'interprétation des tests de détection des anticorps anti-JCV dans le sérum. La recherche d'anticorps anti-JCV ne doit pas être réalisée pendant un échange plasmatique et dans les 2 semaines suivant un tel échange en raison de l'élimination des anticorps du sérum ou dans les 6 mois suivant l'administration d'IgIV (c.àd. 6 mois = 5 demi-vies de l'immunoglobuline).
Infections y compris autres infections opportunistes
D'autres infections opportunistes ont été décrites sous Tysabri, notamment chez des patients atteints de la maladie de Crohn (indication non homologuée) qui étaient immunodéprimés ou lorsque des comorbidités significatives étaient présentes; toutefois, on ne peut pas exclure une augmentation du risque d'autres infections opportunistes sous Tysabri chez des patients ne présentant pas ces comorbidités. Des infections opportunistes ont également été décrites chez des patients souffrant de SEP et traités par Tysabri en monothérapie (voir «Effets indésirables»).
Tysabri augmente le risque d'encéphalite et de méningite à virus herpès simplex et à virus varicelle-zona. Chez des patients atteints de SEP et traités par Tysabri, de graves cas d'encéphalite et de méningite à virus herpès simplex et à virus varicelle-zona, menaçant le pronostic vital et en partie fatals, ont été rapportés lors de la surveillance post-commercialisation. La durée du traitement par Tysabri avant l'apparition de l'encéphalite ou de la méningite était comprise entre quelques mois et plusieurs années. En cas de survenue d'une encéphalite ou d'une méningite herpétique, Tysabri doit être arrêté et un traitement approprié de l'encéphalite ou de la méningite herpétique doit être administré (voir «Effets indésirables identifiés après la mise sur le marché/Infections»).
La nécrose rétinienne aiguë (ARN) est une infection virale fulminante de la rétine provoquée par des virus herpès (p.ex. virus varicelle-zona). L'ARN a été observée chez les patients traités par Tysabri et peut potentiellement provoquer une cécité. Les patients présentant des symptômes tels qu'une baisse de l'acuité visuelle, une rougeur ou une douleur oculaire doivent faire l'objet d'un examen de la rétine aux fins de dépistage de l'ARN. En cas de diagnostic clinique d'une ARN, l'arrêt du traitement par Tysabri doit être envisagé (voir «Effets indésirables identifiés après la mise sur le marché/Infections»).
Les prescripteurs doivent donc être avertis que d'autres infections opportunistes peuvent apparaître sous Tysabri et ils devront donc en tenir compte dans le diagnostic différentiel des éventuelles infections survenant sous Tysabri. En cas de suspicion d'infection opportuniste, le traitement par Tysabri devra être suspendu jusqu'à ce que des examens complémentaires permettent d'exclure la présence d'une telle infection.
Hormis le risque de réactivation du virus JC Polyoma, aucune donnée sur la réactivation d'autres infections virales latentes (hépatite B ou C) n'est disponible.
La survenue d'une infection opportuniste sous Tysabri doit conduire à l'arrêt définitif du traitement.
Conduite éducative
Tous les médecins prescrivant Tysabri doivent avoir pris connaissance du «Guide de prescription pour la prise en charge des patients».
Les médecins doivent discuter avec les patients des bénéfices et des risques du traitement par Tysabri et leur remettre les documents d'information destinés aux patients, y compris le passeport du patient. Les patients devront être informés qu'en cas d'apparition d'une quelconque infection, ils doivent prévenir leur médecin de leur traitement par Tysabri.
Les médecins doivent insister auprès de leurs patients sur l'importance de la prise ininterrompue de Tysabri, surtout durant les premiers mois du traitement (voir aussi «Hypersensibilité»).
Hypersensibilité
Des réactions d'hypersensibilité ont été associées à l'utilisation de Tysabri, y compris des réactions systémiques graves lors d'une perfusion intraveineuse (voir «Effets indésirables»).
Lors de l'administration sous-cutanée du natalizumab, aucune réaction d'hypersensibilité grave n'a été observée, mais les données sont limitées (voir «Propriétés/Effets» et «Pharmacocinétique»).
En cas d'administration intraveineuse, les réactions d'hypersensibilité sont généralement survenues pendant la perfusion ou dans l'heure suivant la fin de la perfusion. Ces réactions peuvent cependant également survenir plus de quatre heures après la perfusion. Le risque de réactions d'hypersensibilité a été le plus important lors des premières perfusions. Les patients recevant de nouveau une perfusion de Tysabri après une période d'exposition très brève (une ou deux perfusions) et un intervalle sans traitement relativement long (trois mois ou plus) ont également présenté un risque accru de réactions d'hypersensibilité. Il importe toutefois d'envisager ce risque lors de chaque administration (voir «Posologie/Mode d'emploi»).
Les patients doivent être surveillés pendant les injections sous-cutanées et pendant l'heure qui suit, afin de détecter les signes et symptômes de réactions liées à l'injection, y compris de réactions d'hypersensibilité (voir «Posologie/Mode d'emploi» et «Effets indésirables»). Les ressources nécessaires à la prise en charge d'éventuelles réactions d'hypersensibilité devront être disponibles.
Les patients présentant des symptômes de réactions d'hypersensibilité, telles que de l'urticaire, doivent en informer immédiatement leur médecin.
Avant chaque administration, il convient de vérifier si l'injection précédente a été suivie de symptômes indésirables révélant éventuellement une réaction allergique/d'hypersensibilité, tels que des douleurs ou des troubles thoraciques, une détresse respiratoire, des variations significatives de la tension artérielle, un angio-œdème, des réactions cutanées et/ou un prurit.
Le traitement par Tysabri devra être interrompu et un traitement approprié devra être instauré dès les premiers signes ou symptômes d'hypersensibilité.
Les patients ayant présenté une réaction d'hypersensibilité doivent arrêter définitivement le traitement par Tysabri.
Traitement associé par immunosuppresseurs
L'efficacité et la sécurité d'emploi de Tysabri en association avec d'autres traitements immunosuppresseurs ou anticancéreux n'ont pas été totalement établies. L'utilisation concomitante de ces médicaments avec Tysabri est susceptible de majorer le risque d'infections, y compris les infections opportunistes, et est par conséquent contre-indiquée (voir «Contre-indications»).
Au cours des études cliniques de phase III réalisées dans la SEP, le traitement concomitant des poussées par des corticoïdes sur une courte période n'a pas été associé à une augmentation du taux d'infections. Des cures courtes de corticoïdes peuvent être administrées en association avec Tysabri.
Traitement antérieur par immunosuppresseurs ou immunomodulateurs
Les patients ayant reçu un traitement antérieur par immunosuppresseurs présentent un risque accru de LEMP.
Il n'a pas été réalisé d'études évaluant l'efficacité et la tolérance de Tysabri administré en relais d'un traitement de fond ayant un effet immunosuppresseur. On ne sait pas si le risque de LEMP est plus élevé chez les patients passant d'un de ces traitements à Tysabri; par conséquent, ces patients doivent être surveillés plus fréquemment (c'est-à-dire de la même manière que les patients passant d'un médicament immunosuppresseur à Tysabri), voir «Dépistage à l'IRM de la LEMP».
Chez ces patients, il faudra veiller à laisser un délai suffisant pour permettre la reconstitution du système immunitaire. Avant de débuter le traitement par Tysabri, les médecins devront évaluer chaque cas de façon individuelle afin de mettre en évidence une éventuelle immunosuppression (voir «Contre-indications»).
En cas de relais par Tysabri d'un autre traitement de fond, la demi-vie et le mode d'action de l'autre traitement doivent être pris en compte afin d'éviter un effet additif sur le système immunitaire et de minimiser le risque de réactivation de la maladie. Il est recommandé de réaliser une numération-formule sanguine (NFS, incluant les lymphocytes) avant l'instauration du traitement par Tysabri pour s'assurer de la résolution des effets immunitaires du traitement antérieur (cytopénie).
Tysabri peut être initié immédiatement après l'arrêt de l'interféron bêta ou de l'acétate de glatiramère, à condition qu'il n'y ait pas d'anomalies significatives imputables au traitement, par exemple une neutropénie ou une lymphopénie.
En relais du diméthyl fumarate, la fenêtre thérapeutique doit être suffisante pour que le taux de lymphocytes retrouve sa valeur normale avant le début du traitement par Tysabri.
Après l'arrêt du fingolimod, le taux de lymphocytes revient progressivement dans les valeurs normales en un à deux mois après l'arrêt du traitement. La fenêtre thérapeutique doit être suffisante pour que le taux de lymphocytes retrouve sa valeur normale avant le début du traitement par Tysabri.
Le tériflunomide est éliminé lentement du plasma. Sans une procédure d'élimination accélérée, la clairance plasmatique du tériflunomide peut durer de quelques mois à deux ans. Il est recommandé soit de procéder à une élimination accélérée du tériflunomide, conformément à ce qui est précisé dans l'information professionnelle du produit, soit de respecter une fenêtre thérapeutique d'au moins 3,5 mois. Il convient d'être prudent lors du passage d'un traitement par le tériflunomide à Tysabri, compte tenu des effets cumulatifs potentiels sur le système immunitaire.
L'alemtuzumab possède des effets immunosuppresseurs importants et prolongés. Compte tenu du fait que la durée réelle de ces effets est inconnue, il n'est pas recommandé d'initier un traitement par Tysabri après administration d'alemtuzumab, sauf si les bénéfices escomptés sont nettement supérieurs aux risques encourus par le patient.
Vaccins et tests cutanés in vivo
Dans une étude d'immunisation ouverte randomisée chez des patients atteints de SEP récurrente, une comparaison entre les patients d'un groupe de contrôle non traité (n= 23) et des patients traités pendant 6 mois par Tysabri (n= 19) n'a montré aucune différence significative au niveau du doublement du taux d'anticorps contre un néoantigène (keyhole limpet hemocyanin, KLH) ou un antigène de rappel (toxoïde du tétanos). Les moyennes de chacun des taux d'anticorps étaient toutefois nettement plus basses sous Tysabri que dans le groupe de contrôle. Un patient sous Tysabri était non-répondeur au toxoïde du tétanos, deux patients sous Tysabri étaient non-répondeurs au KLH. Des vaccins vivants n'ont pas été testés.
On ne dispose d'aucune donnée sur la valeur significative des tests cutanés (p.ex. la tuberculine) pendant et après le traitement par Tysabri.
Immunogénicité
Une aggravation de la maladie ou la survenue d'évènements liés à l'injection peuvent faire suspecter le développement d'anticorps anti-natalizumab. Dans ces cas et chez les patients qui ont été exposés courtement au Tysabri puis n'ont plus reçu de traitement pendant un intervalle relativement long, on recherchera la présence d'anticorps. Les patients ayant reçu initialement peu de traitements, notamment 1 à 2 administrations de Tysabri, puis ayant eu une période prolongée sans traitement, présentent un risque accru de développer des anticorps et/ou des réactions d'hypersensibilité lors de la reprise du traitement. En cas de résultat positif des anticorps confirmé par un second test effectué au moins 6 semaines plus tard, le traitement devra être arrêté et ne sera pas repris, car la présence d'anticorps persistants est associée à une diminution de l'efficacité de Tysabri et à une incidence accrue de réactions d'hypersensibilité (voir «Effets indésirables»).
Effets indésirables hépatiques
De rares cas d'effets indésirables graves spontanés faisant état d'une lésion hépatique ont été signalés après l'introduction sur le marché.
Les lésions hépatiques peuvent survenir à n'importe quel moment durant le traitement, même juste après la première dose. Dans certains cas, la réaction est réapparue après la reprise du traitement par Tysabri. Chez certains patients ayant précédemment présenté des tests hépatiques perturbés, les valeurs pathologiques des enzymes hépatiques se sont péjorées sous Tysabri.
Chez les patients avec insuffisance hépatique, on surveillera régulièrement la fonction hépatique. Ils devront en outre être informés qu'ils doivent contacter leur médecin s'ils présentent des signes ou des symptômes indiquant une affection du foie, p.ex. ictère ou vomissements.
Tysabri sera interrompu en cas d'atteinte hépatique significative.
Arrêt du traitement par Tysabri
En cas de décision d'arrêt du traitement, le médecin doit être averti qu'en raison de sa demi-vie de 16 ±4 jours (voir «Pharmacocinétique»), Tysabri reste présent dans le sang et a des effets pharmacodynamiques (par exemple, augmentation des lymphocytes) pendant environ 12 semaines après la dernière administration. L'instauration d'autres traitements au cours de cette période sera donc nécessairement associée à une exposition concomitante au natalizumab. Au cours des essais cliniques, une exposition concomitante à l'interféron ou à l'acétate de glatiramère durant cette période n'a pas été associée à des problèmes de sécurité. Il n'existe actuellement aucune donnée sur l'exposition concomitante de Tysabri et d'immunosuppresseurs chez les patients souffrant de SEP. L'utilisation de ces médicaments peu après l'arrêt de Tysabri peut alors se traduire par un effet immunosuppresseur additif. Cet élément devra être pris soigneusement en considération, au cas par cas, et l'instauration d'une période de washout (sans traitement) pourrait être appropriée. Au cours des essais cliniques, le traitement des poussées par corticothérapie sur une courte période n'a pas été associé à une augmentation de la fréquence des infections.
Teneur en sodium
Une seringue préremplie (150 mg/ml) contient 3,45 mg de sodium. Une dose complète (300 mg/2 ml) contient 6,9 mg de sodium.
Enfants et adolescents
La sécurité et l'efficacité de Tysabri chez les enfants et les adolescents jusqu'à l'âge de 18 ans n'ont pas été établis. Le bénéfice du traitement chez des enfants et adolescents par Tysabri doit être évalué contre le risque de développer une LEMP ou d'autres infections opportunistes (voir «Effets indésirables» et «Propriétés/Effets»).
InteractionsVoir «Contre-indications».
Grossesse, allaitementFemmes en âge de procréer
L'arrêt de Tysabri devra être envisagé pour toute femme qui tomberait enceinte durant le traitement par le natalizumab.
Grossesse
Il n'existe pas de données pertinentes concernant l'administration de natalizumab chez la femme enceinte. Des études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir «Données précliniques»). Le risque potentiel pour l'être humain n'est pas connu.
Après la mise sur le marché, des cas de thrombocytopénie et d'anémie ont été rapportés chez des nourrissons dont les mères avaient été exposées au natalizumab pendant la grossesse (surtout après le premier trimestre). L'anémie des nouveau-nés était plus prononcée que la baisse physiologique attendue de l'hémoglobine après la naissance et a nécessité un traitement spécifique dans certains cas. Chez les nouveau-nés dont les mères ont été exposées au natalizumab pendant la grossesse, il est recommandé de surveiller les paramètres hématologiques, notamment la numération plaquettaire, l'hémoglobine et l'hématocrite après la naissance et éventuellement jusqu'à la normalisation des valeurs.
Le natalizumab ne doit pas être administré au cours de la grossesse, sauf si l'état clinique de la patiente nécessite un traitement par Tysabri.
Allaitement
Tysabri passe dans le lait maternel. L'effet du natalizumab sur les nouveau-nés/nourrissons n'est pas connu. C'est pourquoi les femmes traitées par Tysabri devraient sevrer leur enfant.
Fertilité
Données sur la fertilité voir «Données précliniques».
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été réalisée avec Tysabri sur les effets sur l'aptitude à conduire des véhicules et à utiliser des machines. Des vertiges ayant cependant été fréquemment rapportés, il devrait être conseillé aux patients présentant cet effet indésirable de ne pas conduire des véhicules ou d'utiliser des machines jusqu'à la disparition des vertiges.
Effets indésirablesLe profil de sécurité du natalizumab administré par voie sous-cutanée était largement conforme au profil de sécurité connu du natalizumab administré par voie intraveineuse, à l'exception de l'effet indésirable «douleur au site d'injection». La fréquence globale de la «douleur au site d'injection» était de 4 % (3/71) chez les patients de l'étude ayant reçu 300 mg de natalizumab toutes les 4 semaines par voie sous-cutanée (voir «Propriétés/Effets», Efficacité clinique et «Pharmacocinétique»).
Dans des études contrôlées contre placebo conduites chez 1617 patients souffrant de SEP et traités par le natalizumab (perfusion intraveineuse) pendant un maximum de 2 ans (placebo: 1135), des événements indésirables conduisant à l'arrêt du traitement se sont produits chez 5,8 % des patients sous natalizumab (placebo: 4,8 %). Pendant ces études d'une durée de deux ans, 43,5 % des patients traités par le natalizumab ont signalé des effets indésirables liés au médicament (placebo: 39,6 %).
Les effets indésirables observés avec la plus grande incidence dans les études cliniques contrôlées contre placebo, menées chez des patients atteints de SEP traités par l'administration intraveineuse de natalizumab à la posologie recommandée, ont été des vertiges, des nausées, une urticaire et une rigidité en rapport avec les perfusions.
Les effets indésirables associés au médicament, rapportés lors de l'administration intraveineuse de natalizumab, avec une incidence supérieure de 0,5 % par rapport au placebo, sont présentés ci-dessous.
Très fréquents (≥1/10), fréquents (≥1/100 à < 1/10), occasionnels (≥1/1000 à < 1/100), rares (≥1/10 000 à < 1/1000).
Dans chaque groupe de fréquence, les effets indésirables sont présentés selon un ordre de gravité décroissante.
Infections et infestations
Très fréquents: Infections urinaires, rhinopharyngite.
Occasionnels: Infections opportunistes, LEMP, herpès (encéphalite/méningite à virus herpès simplex ou à virus varicelle-zona).
Affections du système immunitaire
Fréquents: Urticaire.
Occasionnels: Réaction anaphylactique/anaphylactoïde pendant la perfusion ou dans l'heure suivant la fin de la perfusion.
Affections du système nerveux
Très fréquents: Céphalées, vertiges.
Affections gastro-intestinales
Très fréquents: Nausées.
Fréquents: Vomissements.
Affections hépatobiliaires
Cas isolés: Hyperbilirubinémie, augmentation des transaminases.
Affections musculosquelettiques et du tissu conjonctif
Très fréquents: Arthralgie.
Troubles généraux et anomalies au site d'administration
Très fréquents: Fatigue.
Fréquents: Rigidité, fièvre.
Réactions liées à la perfusion
Très fréquents: Réactions liées à la perfusion (23,1 %; placebo: 18,7 %) telles que vertiges, nausées, vomissements, urticaire, frissons, flush et rigidité.
Description d'effets indésirables spécifiques et informations complémentaires
Réactions d'hypersensibilité
Les réactions d'hypersensibilité sont survenues généralement dans l'heure suivant la fin des injections sous-cutanées.
Lors de l'administration sous-cutanée du natalizumab, aucune réaction d'hypersensibilité grave n'a été observée, mais les données sont limitées (voir «Propriétés/Effets», Efficacité clinique et «Pharmacocinétique»).
Immunogénicité
Fréquents: Anticorps anti-natalizumab chez 10 % des patients, dont anticorps anti-natalizumab persistants (un test positif et un second test positif au moins 6 semaines plus tard) chez environ 6 % des patients qui ont reçu du natalizumab per voie intraveineuse. La présence d'anticorps persistants est associée à une diminution importante de l'efficacité de Tysabri et à une augmentation de l'incidence des réactions d'hypersensibilité. Les autres réactions liées à la perfusion et associées à la présence d'anticorps persistants peuvent être une rigidité, des nausées, des vomissements et un flush.
Au cours de l'étude DELIVER de 32 semaines, menée chez des patients atteints de SEP n'ayant jamais été exposés au natalizumab, des anticorps anti-natalizumab persistants se sont développés chez 1 patient (4 %) parmi les 26 patients ayant reçu le natalizumab par voie sous-cutanée. Des anticorps n'ont été détectés qu'à une seule reprise chez 5 autres patients (19 %). Au cours de l'étude REFINE de 60 semaines, menée chez des patients atteints de SEP, aucun patient (136 patients) passé de l'administration intraveineuse de natalizumab à l'administration sous-cutanée n'a présenté d'anticorps neutralisants anti-médicament détectables pendant l'étude (voir «Propriétés/Effets»).
Si, après environ 6 mois de traitement, des anticorps persistants sont suspectés (p.ex. du fait d'une diminution de l'efficacité ou d'une hypersensibilité), ceux-ci peuvent être mis en évidence et confirmés par un nouveau test réalisé 6 semaines après le premier test positif. Le traitement doit être interrompu chez les patients développant des anticorps persistants.
Infections
Dans le cadre d'études cliniques, des cas de LEMP ont été décrits chez des patients atteints de SEP traités par Tysabri. La LEMP a été associée à un handicap ou au décès (voir «Mises en garde et précautions»).
Lors d'études cliniques réalisées chez des patients atteints de sclérose en plaques, le taux d'infections a été d'environ 1,5 par année-patient chez les patients traités par Tysabri ainsi que chez les patients des groupes sous placebo. Un cas de diarrhée à Cryptosporidium et des cas d'autres infections opportunistes, dont certains d'évolution fatale, ont également été rapportés. Le type d'infections a été similaire chez les patients traités par Tysabri et chez les patients ayant reçu le placebo. La plupart des patients n'ont pas interrompu leur traitement par Tysabri pendant les infections et un traitement approprié a permis de guérir les infections.
Effets sur les examens de laboratoire
Dans le cadre d'études cliniques contrôlées, menées pendant 2 ans chez des patients atteints de SEP, le traitement par le natalizumab a été associé à une augmentation des taux circulants de lymphocytes, de monocytes, d'éosinophiles, de basophiles et d'érythrocytes nucléés.
Aucune augmentation du nombre de neutrophiles n'a été observée. Une baisse légère et le plus souvent passagère des taux d'hémoglobine (diminution moyenne de 0,6 g/dl), d'hématocrite (diminution moyenne de 2 %) et des globules rouges (diminution moyenne du nombre de cellules de 0,1 × 106/l) a été observée pendant le traitement par le natalizumab. Tous ces paramètres hématologiques modifiés sont généralement revenus aux valeurs initiales observées avant le début du traitement, dans les 16 semaines suivant la dernière administration du natalizumab, et ces modifications n'ont pas été associées à des symptômes cliniques.
Effets indésirables identifiés après la mise sur le marché
Depuis la commercialisation, une éosinophilie (nombre d'éosinophiles > 1500/mm³) sans symptômes cliniques a été rapportée. Lorsque le traitement par Tysabri a été interrompu dans de tels cas, le nombre élevé d'éosinophiles s'est normalisé.
Depuis la commercialisation, des thrombocytopénies et des thrombopénies immunitaires (ITP) ont été rapportées. Elles ont été en partie réversibles lors de l'administration de stéroïdes.
Dans le cadre d'études observationnelles post-commercialisation, de rares cas d'anémie sévère et d'anémie hémolytique ont été rapportés chez des patients traités par Tysabri.
Infections
Des cas de LEMP, parmi lesquels des cas asymptomatiques au début du traitement, ont été rapportés après la commercialisation chez des patients ayant reçu Tysabri en monothérapie. Certains cas ont été observés jusqu'à 6 mois après l'arrêt de Tysabri en monothérapie (voir rubrique «Mises en garde et précautions»). Le test de détection des anticorps anti-JCV dans le sérum fournit des informations complémentaires permettant de stratifier le risque du traitement par Tysabri (voir rubrique «Mises en garde et précautions»). Les patients dont le test des anticorps anti-JCV est négatif peuvent tout de même présenter un risque de LEMP en raison, par exemple, d'une infection récente par le JCV, d'une fluctuation du statut des anticorps ou d'un test dont le résultat est un faux négatif. Dans le cadre d'une étude de phase IV ayant évalué le statut des anticorps à long terme pendant 18 mois, le statut des anticorps anti-JCV est passé d'un résultat négatif à un résultat positif chez environ 11 % des patients par an. Les tests doivent être réalisés uniquement avec l'essai STRATIFY JCV, un test de détection des anticorps anti-JCV à deux étapes (ELISA), dont la spécificité est de 97 % et qui a été validé pour l'utilisation chez les patients atteints de SEP. Des cas de NCG due au JCV ont également été rapportés après la commercialisation de Tysabri. Les symptômes d'une NCG due au JCV peuvent être similaires à ceux d'une LEMP (voir rubrique «Mises en garde et précautions»).
Chez les patients atteints de sclérose en plaques traités par le natalizumab, de graves cas d'encéphalite et de méningite consécutifs à une infection par le virus herpès-simplex ou par le virus varicelle-zona, menaçant le pronostic vital et en partie d'évolution fatale, ont été rapportés après la commercialisation (voir rubrique «Mises en garde et précautions»).
Depuis la commercialisation, des cas de nécrose rétinienne aiguë (ARN) sont survenus plus fréquemment chez les patients traités par le natalizumab. Dans certains cas, il s'agissait de patients présentant des infections herpétiques du système nerveux central (SNC) (p.ex. méningite et encéphalite herpétique). Des cas graves d'ARN, touchant l'un des yeux ou les deux, ont provoqué une cécité chez certains patients. Ces cas ont été traités par exemple par des antiviraux et parfois par une intervention chirurgicale (voir rubrique «Mises en garde et précautions»).
Population pédiatrique
Les évènements indésirables graves ont été évalués chez 621 enfants et adolescents atteints de SEP inclus dans une méta-analyse (voir rubrique «Efficacité clinique»). Dans les limites de ces données, aucun nouveau signal de pharmacovigilance n'a été identifié dans cette population de patients. Un cas de méningite herpétique a été rapporté dans la méta-analyse. Il n'a pas été identifié de cas de LEMP dans la méta-analyse; cependant, des cas de LEMP ont été rapportés dans la population d'enfants et adolescents traités par le natalizumab depuis sa commercialisation.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucun cas de surdosage n'a été rapporté.
Propriétés/EffetsCode ATC
L04AG03
Mécanisme d'action
Le natalizumab est un inhibiteur sélectif des molécules d'adhésion. Il se fixe sur la sous-unité α4 des intégrines humaines, fortement exprimée sur la surface de tous les leucocytes, à l'exception des neutrophiles. Le natalizumab se lie spécifiquement à l'intégrine α4β1 en bloquant l'interaction de cette dernière avec son récepteur, la molécule VCAM-1 (vascular cell adhesion molecule-1), avec le ligand ostéopontine, ou avec un variant d'épissage de la fibronectine, le CS-1 (connecting segment-1). Le natalizumab bloque l'interaction de l'intégrine α4β7 avec la molécule MadCAM-1 (mucosal addressin cell adhesion molecule-1). L'inhibition de ces interactions moléculaires empêche la migration des leucocytes mononucléaires à travers l'endothélium vers les tissus parenchymateux inflammatoires. Un autre mécanisme d'action du natalizumab pourrait consister à supprimer les réactions inflammatoires en cours dans les tissus pathologiques en inhibant les interactions des leucocytes exprimant la sous-unité intégrine α4 avec leurs ligands dans la matrice extracellulaire et sur les cellules parenchymateuses. Le natalizumab pourrait donc agir en supprimant l'activité inflammatoire dans la zone malade et en inhibant le recrutement ultérieur de cellules immunitaires dans les tissus inflammatoires.
Dans la SEP, les lésions semblent apparaître lorsque les lymphocytes T activés traversent la barrière hémato-encéphalique (BHE). Cette migration leucocytaire à travers la BHE implique l'interaction entre les molécules d'adhésion présentes sur les cellules inflammatoires et les récepteurs des cellules endothéliales de la paroi vasculaire. L'interaction entre l'intégrine α4β1 et ses cellules cibles est une composante importante de l'inflammation pathologique cérébrale, laquelle diminue avec l'inhibition de ces interactions. Dans les conditions normales, la VCAM-1 n'est pas exprimée dans le parenchyme cérébral mais, en présence de cytokines proinflammatoires, cette expression est stimulée sur les cellules endothéliales et probablement les cellules gliales proches des sites inflammatoires. Dans le contexte de l'inflammation du système nerveux central (SNC) associée à la SEP, c'est l'interaction de l'intégrine α4β1 avec les VCAM-1, CS-1 et ostéopontine qui sert de médiateur à l'adhésion ferme et à la migration transendothéliale des leucocytes dans le parenchyme cérébral, pouvant perpétuer la cascade inflammatoire dans le tissu du SNC. Le blocage des interactions moléculaires entre l'intégrine α4β1 et ses cellules cibles diminue l'activité inflammatoire cérébrale chez le sujet atteint de SEP et inhibe le recrutement ultérieur de cellules immunitaires dans les tissus inflammatoires, diminuant la formation ou l'extension des lésions de SEP.
La CE50 du natalizumab se liant à l'intégrine α4β1 est estimée à 2,04 mg/l selon un modèle pharmacocinétique/pharmacodynamique de population. Il n'y avait pas de différence en termes de liaison à l'intégrine α4β1 entre l'administration sous-cutanée et l'administration intraveineuse de 300 mg de natalizumab toutes les 4 semaines. La PD moyenne (saturation de l'alpha-4 sur les lymphocytes mononucléés) était similaire entre les schémas d'administration par voie intraveineuse toutes les 6 semaines (Q6W) et toutes les 4 semaines (Q4W), avec une différence au niveau du pourcentage moyen de saturation de l'alpha-4 allant de 9 % à 16 %.
Pharmacodynamique
Voir «Mécanisme d'action».
Efficacité clinique
Étude clinique AFFIRM
L'efficacité en monothérapie a été évaluée au cours d'une étude de 2 ans (étude AFFIRM) randomisée, en double insu, contrôlée versus placebo, conduite chez des patients atteints de SEP rémittente-récurrente ayant présenté au moins 1 poussée clinique durant l'année précédant l'inclusion dans l'étude, et dont le score EDSS (Kurtzke Expanded Disability Status Scale, échelle d'évaluation du handicap) était compris entre 0 et 5. L'âge médian était de 37 ans (de 18 à 50 ans) et la durée médiane de la maladie de 5 ans. Les patients ont été randomisés selon un rapport 2:1 et répartis dans des bras de traitement Tysabri 300 mg par voie iv (n= 627) ou placebo (n= 315) toutes les 4 semaines et ils recevaient jusqu'à 30 perfusions. Des examens neurologiques étaient effectués toutes les 12 semaines et au moment des suspicions de poussées. Les examens IRM (recherche de lésions rehaussées par gadolinium avec pondération en T1 et recherche de lésions hyperintenses en T2) étaient effectués tous les ans.
Les caractéristiques et les résultats de l'étude sont présentés dans le tableau ci-dessous.
|
Étude AFFIRM: Caractéristiques et principaux résultats
| |
Type de l'étude
|
Essai en monothérapie, randomisé, en double insu, contrôlé versus placebo, sur des groupes parallèles, d'une durée de 120 semaines.
| |
Patients
|
SEP RR (critères de McDonald)
| |
Traitement
|
Placebo / Natalizumab 300 mg i.v. toutes les 4 semaines
| |
Critère d'évaluation principal à un an
|
Fréquence des poussées
| |
Critère d'évaluation principal à deux ans
|
Progression sur le score EDSS
| |
Critères d'évaluation secondaires
|
Variables dérivées du taux de poussées / variables dérivées de l'IRM
| |
Patients
|
Placebo
|
Natalizumab
| |
Randomisés
|
315
|
627
| |
Ayant terminé la 1re année
|
296
|
609
| |
Ayant terminé les 2 années
|
285
|
589
| |
Âge, années, médiane (intervalle)
|
37 (19-50)
|
36 (18-50)
| |
Nombre d'années de SEP, médiane (intervalle)
|
6,0 (0-33)
|
5,0 (0-34)
| |
Nombre d'années depuis le diagnostic, médiane (intervalle)
|
2,0 (0-23)
|
2,0 (0-24)
| |
Nombre de poussées au cours des 12 derniers mois, médiane (intervalle)
|
1,0 (0-5)
|
1,0 (0-12)
| |
Score EDSS initial, médiane (intervalle)
|
2 (0-6,0)
|
2 (0-6,0)
| |
RÉSULTATS
| |
Fréquence annuelle des poussées
| |
À un an (critère principal d'évaluation)
|
0,805
|
0,261
| |
À deux ans
|
0,733
|
0,235
| |
Un an
|
Rapport de taux: 0,33 IC95 % 0,26; 0,41
| |
Deux ans
|
Rapport de taux: 0,32 IC95 % 0,26; 0,40
| |
Patients sans poussée
| |
À un an
|
53 %
|
76 %
| |
À deux ans
|
41 %
|
67 %
| |
Handicap
| |
Pourcentage de progression1 (confirmation à 12 semaines, critère d'évaluation principal)
|
29 %
|
17 %
| |
|
Rapport de risque: 0,58, IC95 % 0,43; 0,73, p< 0,001
| |
Pourcentage de progression
(confirmation à 24 semaines)
|
23 %
|
11 %
| |
|
Rapport de risque: 0,46, IC95 % 0,33; 0,64, p< 0,001
| |
IRM (0-2 ans)
| |
Variation médiane du volume des lésions hyperintenses en T2 (en %)
|
+8,8 %
|
-9,4 %
(p< 0,001)
| |
Nombre moyen de lésions hyperintenses en T2 nouvelles ou d'extension récente
|
11,0
|
1,9
(p< 0,001)
| |
Nombre moyen de lésions hypo-intenses en T1
|
4,6
|
1,1
(p< 0,001)
| |
Nombre moyen de lésions rehaussées par le Gd
|
1,2
|
0,1
(p< 0,001)
| |
1
L'évolution du handicap était définie par une augmentation, pendant au moins 12 ou 24 semaines, d'au moins 1,0 point de l'EDSS pour les patients ayant initialement un EDSS ≥1,0, ou une augmentation, pendant au moins 12 ou 24 semaines, d'au moins 1,5 points de l'EDSS pour les patients ayant un EDSS initial = 0.
|
Dans le sous-groupe de patients pour lesquels un traitement d'une SEP récurrente-rémittente d'évolution agressive a été indiqué (patients avec au moins 2 poussées et 1 ou plusieurs lésion(s) rehaussée(s) par le gadolinium), la fréquence annuelle des poussées a été de 0,282 dans le groupe traité par Tysabri (n= 148) et de 1,455 dans le groupe placebo (n= 61) (p< 0,001). Le rapport de risque de progression du handicap était de 0,36 (IC à 95 %: 0,17; 0,76) p= 0,008. Ces résultats ont été obtenus à partir d'une analyse post-hoc et doivent donc être interprétés avec précaution. Il n'existe pas de données disponibles sur la sévérité des poussées avant l'inclusion des patients dans l'étude.
Tysabri Observational Program (TOP)
Une analyse intermédiaire des résultats (à la date de mai 2015) de l'étude Tysabri Observational Program (TOP) en cours, une étude de phase IV multicentrique à un seul bras (n= 5770), a montré une diminution significative (p< 0,0001) maintenue du taux annualisé de poussées chez les patients étant passés de l'interféron bêta (n= 3255) ou de l'acétate de glatiramère (n= 1384) à Tysabri par voie intraveineuse. Les scores EDSS moyens sont restés stables pendant 5 ans. De façon concordante avec les résultats d'efficacité observés après le relais par Tysabri de l'interféron bêta ou de l'acétate de glatiramère, il a été observé chez les patients étant passés du fingolimod à Tysabri (n= 147) une diminution significative du taux annualisé de poussées, qui est resté stable pendant deux ans, et les scores EDSS moyens sont restés comparables aux scores initiaux jusqu'à l'année 2. La taille limitée de l'effectif et la durée plus courte de traitement par Tysabri dans ce sous-groupe de patients doivent être prises en compte pour interpréter ces données.
Extension de l'intervalle posologique
Une analyse rétrospective prédéfinie (programme de prescription TOUCH aux États-Unis, n= 15 120) a montré qu'une administration à intervalle prolongé (Extended Interval Dosing ou EID, environ toutes les 6 semaines) de Tysabri par voie intraveineuse chez les patients présentant des anticorps anti-JCV est associée à un risque plus faible de LEMP par rapport à l'administration à intervalle standard autorisée (rapport de risque= 0,06, IC à 95 %= 0,01-0,22). La plupart de ces patients avaient été traités pendant 1 an ou plus selon l'intervalle d'administration autorisé avant de passer à l'EID.
Des modèles et des simulations montrent que le risque d'activité de la SEP pourrait être plus élevé chez les patients qui sont passés à des intervalles posologiques prolongés après avoir reçu la posologie autorisée pendant 1 an ou plus, chez ceux dont les intervalles entre les doses sont de 7 semaines ou plus.
L'efficacité de Tysabri administré toutes les 6 semaines (Q6W) par rapport à l'administration Q4W autorisée n'a pas été formellement démontrée par une étude de non-infériorité.
Aucune donnée clinique relative à la sécurité ou à l'efficacité de cette extension de l'intervalle posologique n'est disponible pour l'administration sous-cutanée.
Étude clinique REFINE (voie d'administration sous-cutanée, population prétraitée par le natalizumab [en perfusion intraveineuse] pendant au minimum 12 mois)
L'efficacité et la sécurité de Tysabri administré par voie sous-cutanée ont été évaluées au cours d'une étude de phase II (REFINE, 101MS206) randomisée, en aveugle et en groupes parallèles, visant à évaluer la sécurité, la tolérance et l'efficacité de plusieurs schémas thérapeutiques du natalizumab (300 mg par voie intraveineuse toutes les 4 semaines, 300 mg par voie sous-cutanée toutes les 4 semaines, 300 mg par voie intraveineuse toutes les 12 semaines, 300 mg par voie sous-cutanée toutes les 12 semaines, 150 mg par voie intraveineuse toutes les 12 semaines et 150 mg par voie sous-cutanée toutes les 12 semaines) chez des sujets adultes (n= 290) atteints de sclérose en plaques rémittente-récurrente sur une période de 60 semaines.
Le critère d'évaluation principal de cette étude était le nombre cumulé de lésions IRM actives uniques combinées (Combined Unique Active, CUA) (somme des nouvelles lésions Gd+ à l'IRM cérébrale et des lésions hyperintenses en T2, nouvelles ou ayant augmenté de volume, et qui n'étaient pas associées au Gd+ à l'examen pondéré en T1). Le nombre cumulé moyen de lésions CUA dans le groupe traité par 300 mg par voie sous-cutanée toutes les 4 semaines était faible (0,02) et comparable à celui du groupe traité par 300 mg par voie intraveineuse toutes les 4 semaines (0,23). Le nombre moyen cumulé de lésions CUA dans les groupes traités toutes les 12 semaines était significativement plus élevé que celui dans les groupes traités toutes les 4 semaines, ce qui a entraîné l'arrêt prématuré de tous les groupes traités toutes les 12 semaines.
Étude clinique DELIVER (voie d'administration sous-cutanée, population naïve de natalizumab)
L'efficacité et la sécurité du natalizumab administré par voie sous-cutanée chez des patients atteints de SEP et naïfs de natalizumab ont été évaluées au cours d'une étude de détermination de dose de phase I (DELIVER), randomisée et en ouvert. Douze patients atteints de SEP-RR et 14 patients atteints de SEP secondaire progressive ont été inclus dans les groupes recevant le traitement par voie sous-cutanée.
Un critère d'évaluation exploratoire de cette étude comprenait le nombre de nouvelles lésions Gd+ à l'IRM cérébrale, entre l'inclusion et la semaine 32. Aucun des patients traités par le natalizumab n'a présenté de lésions Gd+ après l'inclusion, quel que soit le stade de la maladie (SEP-RR ou SEP secondaire progressive), la voie d'administration attribuée ou la présence de lésions Gd+ à l'inclusion.
Dans la population globale de patients atteints de SEP-RR et de SEP secondaire progressive, au total 2 patients du groupe traité par 300 mg de natalizumab par voie sous-cutanée ont présenté des poussées, comparativement à 3 patients du groupe ayant reçu 300 mg de natalizumab par voie intraveineuse. La petite taille de la population ainsi que la variabilité inter- et intra-patients a empêché de faire des comparaisons pertinentes des données d'efficacité entre les groupes.
Pédiatrie
Une méta-analyse post-commercialisation a inclus les données de 621 enfants et adolescents atteints de SEP traités par Tysabri (âge médian 17 ans, âge: 7 à 18 ans, 91 % des patients âgés de 14 ans et plus). Cette méta-analyse a montré chez un sous-groupe limité de patients pour lesquels des données étaient disponibles avant traitement (158 sur les 621 patients) une réduction du TAP de 1,466 avant traitement (IC à 95 %: 1,337; 1,604) à 0,110 (IC à 95 %: 0,094; 0,128).
PharmacocinétiqueAbsorption
La pharmacocinétique du natalizumab après administration sous-cutanée a été évaluée au cours de 2 études (DELIVER, REFINE). L'étude DELIVER (101MS102) était une étude de détermination de dose, randomisée et en ouvert, visant à évaluer la pharmacocinétique du natalizumab administré par voie sous-cutanée et intramusculaire chez des patients atteints de sclérose en plaques (n= 76) (voir «Efficacité clinique» pour la description de l'étude REFINE [101MS206]). Après administration sous-cutanée de 300 mg de natalizumab, la concentration plasmatique maximale (Cmax) de natalizumab a été atteinte avec un retard de 5,8 jours (intervalle de 2 à 7,9 jours); la cinétique du natalizumab a ensuite correspondu à celle observée lors de l'administration intraveineuse. La Cmax moyenne chez les sujets atteints de SEP-RR était de 35,44 µg/ml (intervalle de 22,0 à 47,8 µg/ml), soit 33 % de la concentration maximale atteinte après administration intraveineuse.
L'administration sous-cutanée de doses multiples de 300 mg toutes les 4 semaines a conduit à une Cmin comparable à celle atteinte après l'administration intraveineuse de 300 mg toutes les 4 semaines. Lors de l'administration aussi bien intraveineuse que sous-cutanée de natalizumab toutes les 4 semaines, les valeurs de Cmin ont conduit à des liaisons à l'intégrine α4β1 comparables.
La biodisponibilité du natalizumab après administration sous-cutanée était estimée à 84 %, comme l'a montré l'analyse pharmacocinétique de population actualisée. L'absorption dans la circulation systémique depuis le site d'injection est une absorption de premier ordre avec un retard estimé par le modèle de 3 heures. Aucune covariable n'a été identifiée pour l'absorption. Des paramètres PK d'élimination (Cl, Vss et t½) identiques et les mêmes groupes de covariables que ceux décrits dans l'analyse pharmacocinétique de population actualisée ont été retrouvés pour les voies d'administration intraveineuse et sous-cutanée.
Distribution
Le volume de distribution médian à l'état d'équilibre était de 5,96 l (5,59–6,38 l; intervalle de confiance à 95 %).
Métabolisme
Voir «Absorption».
Élimination
Une analyse (administration intraveineuse) de la pharmacocinétique de population comprend 12 études portant sur 1781 individus ayant reçu des doses comprises entre 1 et 6 mg/kg de natalizumab ou des doses fixes de 150/300 mg en monothérapie. La clairance linéaire médiane estimée était de 6,08 ml/h (5,75-6,33 ml/h, intervalle de confiance à 95 %) pour la population évaluée. La demi-vie médiane estimée était de 28,2 jours. L'analyse de population avec 1781 patients a examiné les effets de covariables sélectionnées, telles que le poids, l'âge, le sexe, la présence d'anticorps anti-natalizumab et la formulation, sur les propriétés pharmacocinétiques. Seuls le poids, la présence d'anticorps anti-natalizumab et la formulation utilisée dans les études de phase II ont modifié la cinétique du natalizumab. L'effet du poids n'a pas été totalement proportionnel, puisqu'une variation de ±43 % du poids a conduit à une variation d'uniquement -33 % à 30 % de la clairance. La présence d'anticorps anti-natalizumab persistants a augmenté la clairance du natalizumab d'environ 2,45 fois, ce qui correspond à la diminution des concentrations sériques de natalizumab observée chez les patients porteurs de ce type d'anticorps (voir «Effets indésirables»).
Les conséquences d'une plasmaphérèse (échange plasmatique) sur la clairance et la pharmacodynamique du natalizumab ont été examinées lors d'une étude portant sur 12 patients atteints de SEP. L'élimination estimée du principe actif après 3 séances de plasmaphérèse (sur une période de 5-8 jours) était d'environ 70-80 %. Cette valeur était par contre d'environ 40 % dans des études antérieures au cours desquelles les mesures ont été conduites sur une durée d'observation semblable après l'interruption du traitement par le principe actif, mais toutefois sans plasmaphérèse. L'importance d'une plasmaphérèse pour la restauration de la migration des lymphocytes et, en fin de compte, son utilité clinique ne sont pas connues. Une analyse rétrospective du natalizumab, réalisée après son autorisation, n'a montré aucune différence en termes de survie à 2 ans après le diagnostic de LEMP entre les patients ayant reçu un traitement par PLEX et ceux n'en ayant pas reçu (voir aussi «Mises en garde et précautions», LEMP et IRIS (Immune Reconstitution Inflammatory Syndrome)).
Cinétique pour certains groupes de patients
Les paramètres pharmacocinétiques du natalizumab administré par voie sous-cutanée n'ont pas été étudiés chez les patients pédiatriques atteints de SEP.
Les paramètres pharmacocinétiques du natalizumab n'ont pas été étudiés chez les patients atteints d'insuffisance rénale ou hépatique.
Données précliniquesLes données précliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicologie après administration répétée, de génotoxicité, de carcinogénicité et de toxicité sur la reproduction n'ont révélé aucun risque particulier pour l'homme.
En raison de l'activité pharmacologique du natalizumab, une modification de la circulation des lymphocytes sous forme d'une augmentation des globules blancs et d'une hypersplénie a été observée dans la plupart des études in vivo. Ces modifications étaient réversibles et n'ont pas semblé provoquer d'effets toxiques.
Carcinogénicité
Dans les études conduites chez la souris, l'administration de natalizumab n'a pas provoqué de croissance ni d'apparition de métastases de tumeurs de type mélanome ou leucémie lymphoblastique.
Le natalizumab n'a exercé aucun effet clastogène ou mutagène dans les tests d'Ames ou dans les tests d'aberrations chromosomiques de cellules humaines. Par ailleurs, le natalizumab n'a présenté aucun effet lors d'essais in vitro relatifs à la prolifération ou à la cytotoxicité de lignées tumorales intégrine α4-positives.
Toxicité sur la reproduction
Dans une étude utilisant des doses plus élevées que chez l'être humain, une réduction de la fertilité des cobayes femelles a été observée; le natalizumab n'a pas altéré la fertilité masculine. On considère comme peu probable que le natalizumab, administré à des doses inférieures à la dose maximale recommandée, altère la fertilité de l'être humain.
L'effet du natalizumab sur la reproduction a été évalué dans 5 études: 3 chez le cobaye et 2 chez le singe cynomolgus. Ces études n'ont montré aucun signe de tératogénicité ni aucun effet sur le développement des nouveau-nés. Une étude chez le cobaye a montré une faible diminution de la survie des nouveau-nés. Dans une étude chez le singe, le nombre d'avortements spontanés dans le lot traité par 30 mg/kg de natalizumab a été le double de celui observé dans le lot témoin apparié. Ceci a été expliqué par la fréquence élevée d'avortements spontanés observée dans les lots traités de la première cohorte d'animaux et qui n'a pas été observée dans la seconde cohorte. Aucun effet sur les taux d'avortements n'a été observé dans les autres études. Une étude chez la femelle cynomolgus gravide a mis en évidence des modifications fœtales attribuées au natalizumab, notamment une faible anémie, une diminution des plaquettes, une hypersplénie ainsi qu'une diminution du poids du foie et du thymus. Ces modifications ont été associées à une augmentation de l'hématopoïèse extra-médullaire splénique, ainsi qu'à une atrophie du thymus et à une diminution de l'hématopoïèse hépatique. Le taux des plaquettes était également diminué chez les nouveau-nés de mères traitées par le natalizumab jusqu'à la mise bas, cependant on n'a pas observé d'anémie chez ces nouveau-nés. Toutes ces altérations observées à des doses supérieures à celles utilisées chez l'homme ont disparu après la clairance du natalizumab.
Chez le singe cynomolgus traité par le natalizumab jusqu'à la parturition, de faibles taux de natalizumab ont été observés dans le lait maternel de certains animaux.
Remarques particulièresIncompatibilités
Tysabri pour injection sous-cutanée ne doit pas être dilué.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Les seringues préremplies peuvent être conservées à température ambiante dans leur emballage d'origine pendant au maximum 24 heures. Les seringues préremplies ne doivent pas être remises au réfrigérateur. Ne pas utiliser de sources de chaleur externes telles que l'eau chaude pour réchauffer les seringues préremplies.
Remarques particulières concernant le stockage
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
Conserver les seringues préremplies dans leur emballage d'origine afin de les protéger de la lumière.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
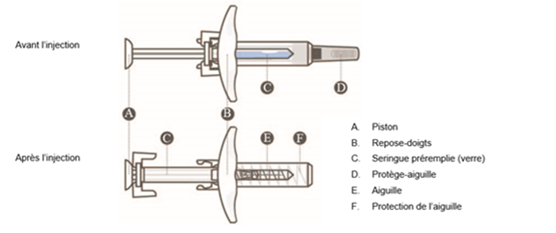
La seringue préremplie est dotée d'un système de protection de l'aiguille qui s'active automatiquement lorsque le piston est complètement enfoncé. Lorsque l'on relâche le piston, la protection de l'aiguille recouvre l'aiguille.
1. Sortir la boîte du réfrigérateur et la laisser revenir à température ambiante avant de réaliser les injections. Le temps de réchauffement recommandé est de 30 minutes.
·Ne pas utiliser de sources de chaleur externes telles que l'eau chaude pour réchauffer les seringues préremplies.
2. Retirer les DEUX seringues préremplies du plateau. Vérifier que le médicament contenu dans chaque seringue préremplie est une solution incolore à légèrement jaune, légèrement opalescente et essentiellement exempte de particules visibles. Il est possible d'observer des bulles d'air dans la fenêtre d'affichage. Ceci est normal et n'affecte pas la dose administrée.
·Ne pas utiliser les seringues préremplies si:
·leur date de péremption est dépassée.
·la couleur et la limpidité du liquide ne correspondent pas à ce qui est mentionné ci-dessus, ou si le liquide contient des particules en suspension.
·des signes de dommages (fissures, éclats, etc.) sont visibles.
·Si l'une des circonstances décrites ci-dessus s'applique, contacter immédiatement la pharmacie.
3. Une dose complète équivaut à deux seringues administrées l'une après l'autre en l'espace de 30 minutes.

4. Utiliser une technique d'asepsie (propre et exempte de germes) et une surface de travail plane.
5. Choisir comme premier site d'injection sous-cutanée la cuisse, l'abdomen ou la face postérieure de la partie supérieure du bras.
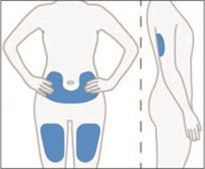
·Ne pas injecter dans une zone du corps où la peau est irritée, rougie, meurtrie, infectée ou cicatricielle.
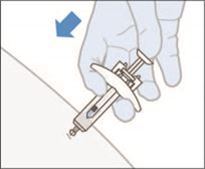
6. Effectuer la première injection en suivant le procédé standard recommandé pour les injections sous-cutanées.
7. Pousser lentement et régulièrement le piston jusqu'à ce que la seringue soit complètement vide. Ne pas retirer le piston.
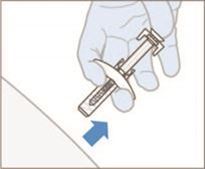
8. Lors du retrait de la seringue du site d'injection, relâcher le piston TOUT EN retirant l'aiguille tout droit. Lorsque l'on relâche le piston, la protection de l'aiguille recouvre l'aiguille.
·Ne pas toucher l'aiguille pour éviter toute blessure accidentelle par piqûre d'aiguille.
·Ne pas remettre le protège-aiguille sur l'aiguille pour éviter toute blessure accidentelle par piqûre d'aiguille.
9. Réaliser les injections l'une après l'autre sans délai important entre les deux. Dans le cas où la seconde injection ne pourrait pas être administrée immédiatement après la première, la seconde injection doit être administrée au plus tard 30 minutes après la première. La seconde injection doit être effectuée à une distance d'au moins 3 cm du premier site d'injection.
Les patients doivent rester en observation pendant les injections sous-cutanées et pendant l'heure suivant la dernière injection. Arrêter rapidement l'injection en cas d'apparition de signes ou symptômes évoquant une réaction d'hypersensibilité (voir «Mises en garde et précautions»).
10.Éliminer la seringue usagée conformément aux exigences locales.
Numéro d’autorisation68008 (Swissmedic).
PrésentationLa dose de 300 mg de Tysabri pour injection sous-cutanée se trouve dans deux seringues préremplies à usage unique contenant chacune 150 mg/1 ml. B
Titulaire de l’autorisationBiogen Switzerland SA, 6340 Baar.
Mise à jour de l’informationNovembre 2024
|