CompositionPrincipe actif
Edaravone.
Excipients
Solution pour perfusion
Hydrogénosulfite de sodium (E 222) 20 mg/100 ml, chlorhydrate de L-cystéine monohydraté, chlorure de sodium, hydroxyde de sodium, acide phosphorique et eau pour préparations injectables.
Teneur totale en sodium 340,8 mg/100 ml.
Suspension buvable
Hydrogénosulfite de sodium (E 222) 5 mg/5 ml, chlorhydrate de L-cystéine monohydraté, hydroxyde de sodium, acide phosphorique, poly(alcool vinylique), gomme xanthane, sorbitol (E 420) 400 mg/ml, émulsion de siméticone et eau purifiée.
Teneur totale en sodium par dose: 1,10 mg/5 ml.
Indications/Possibilités d’emploiRADICAVA est utilisé dans le traitement de la sclérose latérale amyotrophique (SLA).
Posologie/Mode d’emploiRemarques générales:
L'instauration et le suivi du traitement par RADICAVA doivent avoir lieu dans des centres spécialisés par des médecins expérimentés dans le traitement de patients souffrant de SLA.
Le traitement par RADICAVA ne doit être instauré qu'en présence d'une SLA dont le diagnostic est cliniquement assuré, cliniquement vraisemblable ou «vraisemblable et appuyé par les analyses de laboratoire/l'EMG».
L'efficacité de RADICAVA n'a jusqu'à présent été démontrée que lors d'une instauration du traitement dans une phase initiale de la maladie (les patients peuvent soit encore travailler, soit n'ont au moins pas besoin d'aide pour les activités de la vie quotidienne). S'il existe des signes d'efficacité de RADICAVA lors d'une instauration du traitement par RADICAVA chez des patients à un stade avancé de la maladie, les données à ce sujet sont cependant très limitées dans l'ensemble.
On ne dispose pas de suffisamment de données en faveur d'une poursuite d'un traitement par RADICAVA chez des patients présentant un trouble marqué de la fonction respiratoire (% de référence approximatif de CVF ≤50%) ou chez des patients présentant une altération fonctionnelle marquée. Par conséquent, l'arrêt de RADICAVA doit être envisagé en cas de dégradation marquée de l'état général et/ou des capacités fonctionnelles et/ou des symptômes pulmonaires.
Posologie/Mode d'emploi
Solution pour perfusion
RADICAVA, solution pour perfusion est destiné uniquement à une administration en perfusion intraveineuse.
La posologie recommandée de RADICAVA est une perfusion intraveineuse de 60 mg administrée sur une période de 60 minutes selon le schéma suivant:
·Un premier cycle de 14 jours de traitement avec administration quotidienne, suivi d'une période de 14 jours sans administration
·Ensuite, des cycles de traitement de 10 jours répartis sur une période de 14 jours, suivis d'une période de 14 jours sans administration
La dose de 60 mg de solution pour perfusion RADICAVA est administrée en utilisant consécutivement deux poches pour perfusion intraveineuse de 30 mg/100 ml sur une durée totale de 60 minutes (le débit de perfusion est d'environ 1 mg par minute [3,33 ml par minute]).
La perfusion doit être immédiatement interrompue au premier signe ou symptôme d'une réaction d'hypersensibilité (voir Mises en garde et précautions).
Suspension buvable
RADICAVA, suspension buvable est destiné uniquement à une administration par voie orale.
La posologie recommandée de RADICAVA, suspension buvable est de 5 mg (105 mg), administrés par voie orale ou par sonde d'alimentation [sonde nasogastrique ou sonde de gastrostomie percutanée endoscopique (PEG)] selon le schéma suivant:
·Un premier cycle de traitement de 14 jours à raison d'une dose par jour, suivi d'une période de 14 jours sans traitement. Utiliser pour cela la boîte prévue pour le début du traitement.
·Par la suite, des cycles de 10 jours de traitement s'étendant sur des périodes de 14 jours, suivies de périodes de 14 jours sans traitement.
RADICAVA, suspension buvable doit être pris le matin; avant la prise, le patient doit rester à jeun toute la nuit et ne rien manger ni boire (à part de l'eau) pendant au moins 1 heure après la prise.
Si le patient ne peut pas rester à jeun toute la nuit, d'autres options d'administration de RADICAVA, suspension buvable sont possibles, en fonction de la prise des repas (voir Pharmacocinétique). Voir Tableau 1 pour les conditions spécifiques de jeûne.
Tableau 1: Administration de RADICAVA, suspension buvable selon le type de nourriture consommée
|
Type de nourriture/complément alimentaire calorique consommé
|
Durée de jeûne avant et après la prise de RADICAVA, suspension buvable selon le type de repas
| |
Repas riche en matières grasses (800-1000 calories, 50% de matières grasses)
|
8 heures avant la prise et 1 heure après la prise
| |
Repas pauvre en matières grasses (400-500 calories, 25% de matières grasses)
|
4 heures avant la prise et 1 heure après la prise
| |
Complément alimentaire calorique (250 calories, par ex. boisson protéinée)
|
2 heures avant la prise et 1 heure après la prise
|
Avant chaque administration, secouer vigoureusement le flacon de RADICAVA, suspension buvable pendant au moins 30 secondes.
RADICAVA, suspension buvable est administré directement dans la bouche. Utiliser pour ce faire la seringue graduée de 5 ml fournie dans l'emballage.
Administration par sonde d'alimentation
Si le patient ne peut pas déglutir ou n'est pas en mesure, pour quelque raison que ce soit, de prendre la suspension par voie orale, RADICAVA, suspension buvable peut être administré via une sonde nasogastrique ou une sonde de gastrostomie percutanée endoscopique (PEG) (voir Remarques particulières).
Pour de plus amples informations sur l'administration, voir Remarques concernant la manipulation.
Passage de RADICAVA, solution pour perfusion à RADICAVA, suspension buvable
Les patients recevant la dose de 60 mg de RADICAVA, solution pour perfusion peuvent passer à 5 ml (105 mg) de RADICAVA, suspension buvable, avec la même fréquence d'administration. Après le passage à RADICAVA, suspension buvable, les patients doivent suivre les recommandations posologiques pour RADICAVA, suspension buvable concernant la prise alimentaire.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est requis chez les patients présentant une insuffisance hépatique légère, modérée ou sévère.
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est requis chez les patients présentant une insuffisance rénale légère ou modérée (débit de filtration glomérulaire estimé (DFGe) ≥30 ml/min/1,73 m²).
Les effets d'une insuffisance rénale sévère sur la pharmacocinétique de RADICAVA n'ont pas été étudiés; il est cependant peu probable que l'exposition à l'édaravone ait des effets notables chez les patients présentant un DFGe <30 ml/min/1,73 m² ne requérant pas de traitement par suppléance rénale. La pharmacocinétique de RADICAVA n'a pas été étudiée chez les patients sous traitement par suppléance rénale, et l'utilisation de RADICAVA au sein de cette population de patients n'est pas recommandée.
Patients âgés
Sur les 184 patients atteints de SLA qui ont reçu RADICAVA, solution pour perfusion dans le cadre de 3 études cliniques contrôlées contre placebo, 53 patients au total avaient plus de 65 ans, dont 2 qui avaient plus de 75 ans. Sur les 185 patients atteints de SLA qui ont reçu RADICAVA, suspension buvable dans le cadre d'une étude clinique en ouvert, 65 patients au total avaient plus de 65 ans, dont 6 qui avaient plus de 75 ans.
Aucune différence significative n'a été observée sur le plan de la sécurité ou de l'efficacité entre ces patients et les patients plus jeunes. Néanmoins, une plus grande sensibilité chez certains patients âgés ne peut être exclue.
Enfants et adolescents
La sécurité et l'efficacité de RADICAVA chez les enfants et les adolescents n'ont pas été établies.
Contre-indicationsRADICAVA est contre-indiqué chez les patients présentant dans leurs antécédents une hypersensibilité à l'édaravone ou à l'un de ses composants. Des réactions d'hypersensibilité et des réactions anaphylactiques ont été observées en lien avec RADICAVA, solution pour perfusion (voir Mises en garde et précautions).
Mises en garde et précautionsRéactions d'hypersensibilité
Des réactions d'hypersensibilité (rougeur, papules et érythème polymorphe) et des cas d'anaphylaxie (urticaire, diminution de la pression artérielle et dyspnée) ont été signalés dans des rapports de pharmacovigilance spontanés après la mise sur le marché de RADICAVA, solution pour perfusion.
Les patients doivent être observés avec attention afin d'identifier toute réaction d'hypersensibilité. En cas de réactions d'hypersensibilité, il convient d'arrêter immédiatement le traitement par RADICAVA, d'instaurer un traitement standard et de surveiller le patient jusqu'à disparition de la complication (voir Contre-indications).
Réactions allergiques aux sulfites
RADICAVA, solution pour perfusion et RADICAVA, suspension buvable contiennent de l'hydrogénosulfite de sodium (E 222), un sulfite pouvant causer des réactions allergiques, notamment des réactions d'hypersensibilité et des bronchospasmes sévères, des symptômes d'anaphylaxie et des épisodes asthmatiques menaçant le pronostic vital ou moins sévères. La prévalence globale de la sensibilité aux sulfites dans la population générale est inconnue. La sensibilité aux sulfites est plus fréquente chez les personnes asthmatiques que chez les personnes non asthmatiques.
Sodium
RADICAVA, solution pour perfusion contient 340,8 mg de sodium pour 100 ml, soit 17% de l'apport alimentaire quotidien maximal recommandé par l'OMS.
La dose quotidienne maximale de ce médicament correspond à 34% de l'apport alimentaire quotidien maximal recommandé par l'OMS.
RADICAVA, solution pour perfusion est considéré comme riche en sodium. Il convient d'en tenir compte, notamment chez les patients suivant un régime pauvre en sodium.
RADICAVA, suspension buvable contient 1,104 mg de sodium par dose de 5 ml, soit moins de 1 mmol de sodium (23 mg) par dose de 5 ml, c'est-à-dire qu'il est essentiellement «sans sodium».
Sorbitol
RADICAVA, suspension buvable contient 2 g de sorbitol par dose de 5 ml, soit 400 mg/ml.
L'effet additif des produits administrés concomitamment contenant du sorbitol (ou du fructose) et l'apport alimentaire de sorbitol (ou de fructose) doit être pris en compte.
La teneur en sorbitol dans les médicaments à usage oral peut affecter la biodisponibilité d'autres médicaments à usage oral administrés de façon concomitante.
Les patients présentant une intolérance héréditaire au fructose (IHF) ne doivent pas recevoir ce médicament, sauf en cas de nécessité uniquement.
InteractionsIl est peu probable que la pharmacocinétique de l'édaravone soit significativement affectée par les inhibiteurs des enzymes CYP, des UGT ou des principaux transporteurs.
Les données in vitro indiquant un éventuel potentiel d'induction du CYP3A4 et un potentiel d'inhibition des deux transporteurs BCRP et OAT3 après administration orale, des études sur les interactions médicamenteuses in vivo ont été menées pour examiner ces interactions potentielles avec l'édaravone par voie orale. L'administration concomitante de 120 mg d'édaravone (dose supérieure à la dose recommandée de 105 mg pour la suspension buvable) et de sildenafil (substrat du CYP3A4), de rosuvastatine (substrat de la BCRP) et de furosémide (substrat de l'OAT3) n'a entraîné aucune variation de la Cmax et de l'ASC de ces substances.
Des études in vitro ont démontré que, aux doses cliniques recommandées, que ce soit par voie intraveineuse ou par voie orale, l'édaravone et ses métabolites ne devraient pas inhiber significativement les enzymes du cytochrome P450 (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4), UGT1A1, UGT2B7, ou les autres transporteurs (P-gp, OATP1B1, OATP1B3, OAT1, OCT2, MATE1 et MATE2-K) ou entraîner une induction de CYP1A2 ou CYP2B6 chez l'être humain.
Les données in vitro indiquent que l'édaravone n'est pas un substrat de l'OATP1B1 ou de l'OATP1B3.
Grossesse, allaitementGrossesse
Seules des données insuffisantes sont disponibles sur le risque associé à l'utilisation de RADICAVA pour le développement chez les femmes enceintes.
Aucun effet tératogène n'a été observé dans les études chez l'animal. Toutefois, des effets néfastes sur le développement ont été observés (voir Données précliniques).
Sauf en cas de nécessité absolue, RADICAVA ne doit pas être utilisé pendant la grossesse, chez les mères qui allaitent ou chez les patientes en âge de procréer qui n'utilisent pas de méthode de contraception sûre.
Allaitement
Il n'existe aucune donnée clinique sur la concentration d'édaravone ou de ses métabolites dans le lait maternel. Des études expérimentales menées chez l'animal ont montré le passage de l'édaravone et de ses métabolites dans le lait (voir Données précliniques).
L'intérêt de l'allaitement pour le développement et la santé de l'enfant doivent être évalués au regard de la nécessité clinique pour la mère de recevoir RADICAVA. Les effets indésirables potentiels de RADICAVA sur l'enfant représentent un risque potentiel pour l'enfant.
Fertilité
Il n'existe pas de données cliniques sur un effet potentiel de RADICAVA sur la fertilité. Dans les études expérimentales menées chez l'animal, une diminution de la fertilité a été observée (voir Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'édaravone n'a aucune influence sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesLa fréquence des réactions indésirables au médicament déterminée lors d'études cliniques sur RADICAVA, solution pour perfusion contrôlées contre placebo chez des patients atteints de sclérose latérale amyotrophique est indiquée au moyen des catégories standard suivantes:
Très fréquents (≥1/10), fréquents (≥1/100, <1/10), occasionnels (≥1/1000, <1/100), rares (≥1/10 000, <1/1000), très rares (<1/10 000).
Les réactions indésirables les plus fréquentes survenues chez les patients traités par RADICAVA, solution pour perfusion étaient la contusion (15%), les troubles de la marche (13%) et les céphalées (10%).
Infections et infestations:
Fréquents: infection à Tinea.
Affections du système nerveux:
Fréquents: céphalée.
Affections respiratoires, thoraciques et médiastinales:
Fréquents: insuffisance respiratoire, affections des voies respiratoires, hypoxie.
Affections de la peau et du tissu sous-cutané:
Fréquents: eczéma, dermatite.
Affections du rein et des voies urinaires:
Fréquents: glycosurie.
Troubles généraux et anomalies au site d'administration:
Très fréquents: troubles de la marche (13%).
Lésions, intoxications et complications liées aux procédures:
Très fréquents: contusion (15%).
Les fréquences indiquées pour l'infection à Tinea, les céphalées, l'insuffisance respiratoire/les affections des voies respiratoires, l'hypoxie, l'eczéma, la dermatite sont basées respectivement sur un groupe de termes privilégiés similaires.
Effets indésirables supplémentaires en lien avec RADICAVA, suspension buvable
Dans une étude en ouvert menée chez des patients atteints de SLA (n = 185) qui ont été traités pendant 6 mois par RADICAVA, suspension buvable, 7,6% des patients ont rapporté un épuisement, 3,8% des sensations vertigineuses, 3,2% des nausées, 2,7% une diminution de l'appétit et 2,2% une sécheresse de la peau.
Effets indésirables après commercialisation
Des réactions d'hypersensibilité (rougeur, papules et érythème polymorphe) et des cas d'anaphylaxie (urticaire, diminution de la pression artérielle et dyspnée) ont été signalés dans des rapports de pharmacovigilance spontanés après la mise sur le marché de RADICAVA, solution pour perfusion
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucune information concernant le surdosage n'est disponible. En cas de surdosage, un traitement symptomatique et de soutien est recommandé.
Propriétés/EffetsCode ATC
N07XX14 édaravone
Mécanisme d'action
RADICAVA est un capteur de radicaux libres avec effet neuroprotecteur potentiel.
Pharmacodynamique
Le mécanisme par lequel RADICAVA exerce son effet thérapeutique chez les patients atteints de SLA est inconnu.
Efficacité clinique sur l'intervalle QT/QTc
L'effet de doses thérapeutiques et suprathérapeutiques d'édaravone administrées par voie intraveineuse sur l'intervalle QT/QTc a été examiné dans le cadre d'une étude croisée à trois bras, randomisée, en simple aveugle et contrôlée contre placebo.
27 sujets masculins sains à jeun ont reçu pour cela pendant 60 minutes une perfusion intraveineuse unique de 60 mg (dose thérapeutique) ou de 300 mg (dose suprathérapeutique) ou une solution saline à 0,9% (p/v) (placebo).
Le critère principal était le lien entre la variation de l'intervalle QT corrigé selon Fridericia (QTcF) par rapport à la situation initiale (ΔQTcF), corrigée de la valeur comparative relevée sous placebo (variation de QTcF ajustée par placebo par rapport à la valeur initiale [ΔΔQTcF]), et la concentration d'édaravone.
Cette étude a montré qu'il n'y avait pas de variation cliniquement significative de l'intervalle QTcF après l'administration de l'édaravone et en a conclu que l'édaravone n'avait pas d'effet cliniquement significatif sur l'allongement de l'intervalle QTcF à une exposition s'élevant à environ cinq fois la dose thérapeutique recommandée.
Efficacité clinique
L'efficacité de RADICAVA, suspension buvable repose sur une étude comparative de la biodisponibilité de RADICAVA, solution pour perfusion et de RADICAVA, suspension buvable chez des sujets en bonne santé (voir Pharmacocinétique).
L'efficacité de RADICAVA, solution pour perfusion dans le traitement de la SLA a été établie dans une étude de six mois randomisée, contrôlée contre placebo, en double aveugle, menée chez des patients japonais atteints de SLA qui vivaient de façon autonome et répondaient aux critères suivants au moment de la sélection:
1.Capacité fonctionnelle permettant d'être encore autonome dans la plupart des activités de la vie quotidienne (définie par un score d'au moins deux points pour chaque élément de l'échelle d'évaluation de la capacité fonctionnelle dans la SLA [ALSFRS-R])
2.Fonction respiratoire normale (définie en tant que pourcentage de capacité vitale forcée prédite (CVF) ≥80%)
3.SLA certaine ou probable selon les critères d'El Escorial révisés
4.Durée de la maladie de ≤2 ans
L'étude a inclus 69 patients dans le groupe traité par édaravone et 68 patients dans le groupe placebo. Les caractéristiques initiales étaient similaires dans ces deux groupes et plus de 90% des patients de chaque groupe étaient traités par riluzole.
L'édaravone a été administrée en perfusion intraveineuse de 60 mg sur une période de 60 minutes selon le schéma suivant:
·Un premier cycle de 14 jours de traitement avec administration quotidienne, suivi d'une période de 14 jours sans administration (cycle 1)
·Des cycles de traitement subséquents de 10 jours répartis sur une période de 14 jours, suivis d'une période de 14 jours sans administration (cycles 2-6)
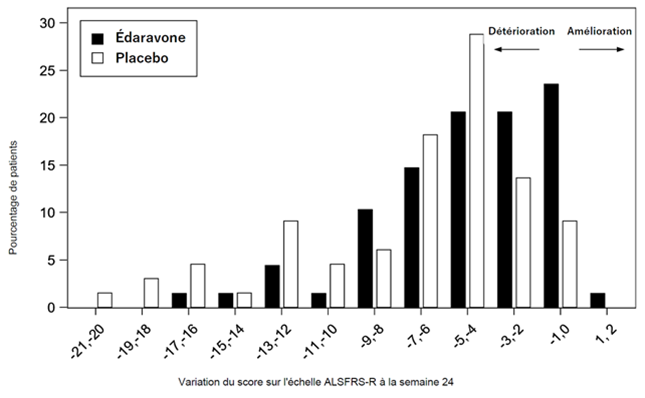
Le critère d'efficacité principal était la variation des scores totaux sur l'échelle d'évaluation ALSFRS-R entre le début de l'étude et la semaine 24 comparée entre les groupes de traitement. La diminution du score total sur l'échelle ALSFRS-R par rapport au début de l'étude a été significativement inférieure chez les patients traités par RADICAVA par rapport à ceux recevant le placebo (voir tableau 2). La distribution de la variation des scores sur l'échelle ALSFRS-R entre le début de l'étude et la semaine 24 est présentée en lien avec le pourcentage respectif de patients à la figure 1.
Tableau 2: Analyse de la variation des scores à l'échelle ALSFRS-R entre le début de l'étude et la semaine 24
|
Traitement
|
Évolution par rapport à la valeur initiale
Moyenne des moindres carrés ± ET (IC à 95%)
|
Différences entre les traitements (RADICAVA – placebo [IC à 95%])
|
Valeur p
| |
RADICAVA 60 mg
|
−5,01±0,64
|
2,49 (0,99, 3,98)
|
0,0013
| |
Placebo
|
-7,50±0,66
|
Figure 1: Distribution de la variation des scores sur l'échelle ALSFRS-R entre le début de l'étude et la semaine 24
Capacité vitale forcée (CVF)
Les variations du pourcentage de CVF entre la situation initiale et la semaine 24 s'élevaient à -15,6% sous RADICAVA et -20,4% sous placebo (des pourcentages en baisse indiquent une détérioration de la CVF).
Échelle d'évaluation de la qualité de vie (ALSAQ-40)
RADICAVA a entraîné une réduction significative de la dégradation de la qualité de vie mesurée à l'aide de l'échelle ALSAQ-40. Le nombre de points entre la situation initiale et la semaine 24 a évolué de 17,3 points sous RADICAVA et de 26,0 points sous placebo (un nombre de points en augmentation indique une dégradation de la qualité de vie).
Échelle modifiée de Norris (total)
RADICAVA a entraîné une réduction significative de la dégradation des fonctions mesurées au moyen de l'échelle modifiée de Norris entre la situation initiale et la semaine 24, avec une variation de -15,9 points sous RADICAVA et de 20,8 points sous placebo (un nombre de points en baisse indique une dégradation fonctionnelle).
Force de préhension et de pincement
Aucun effet n'a été observé sur la force de préhension et de pincement pour RADICAVA.
PharmacocinétiqueAbsorption
Solution pour perfusion
La concentration plasmatique maximale (Cmax) de l'édaravone était atteinte à la fin de la perfusion (60 mg d'édaravone sur 60 min.) Une tendance à une augmentation plus que proportionnelle à la dose de l'aire sous la courbe (ASC) et de la Cmax de l'édaravone a été observée. L'édaravone ne s'accumule pas dans le plasma lors d'une administration de doses multiples.
Suspension buvable
Il a été montré que RADICAVA, suspension buvable présente, lors d'une administration à jeun par voie orale à une posologie de 105 mg, une ASC identique à celle de RADICAVA, solution pour perfusion (60 mg, administré par voie intraveineuse sur 60 minutes) et que la Cmax n'est pas inférieure à celle de RADICAVA, solution pour perfusion.
L'édaravone est rapidement absorbée, la concentration maximale est atteinte en un délai médian d'environ 0,5 heure (intervalle: 0,25−0,75 heure) lors d'une administration orale à jeun. Le taux d'absorption totale de l'édaravone s'élève à au moins 77% et la biodisponibilité orale absolue à environ 57% (à la suite de l'effet de premier passage) lors d'une comparaison entre 105 mg d'édaravone en suspension buvable et 60 mg d'édaravone en formulation intraveineuse. Administré une fois par jour à des sujets en bonne santé, RADICAVA, suspension buvable n'a pas montré d'accumulation et la Cmax comme l'ASC de l'édaravone étaient plus que proportionnelles à la dose dans l'intervalle posologique de 30 à 300 mg.
Une dose unique de 105 mg d'édaravone, administrée par voie orale ou au moyen d'une sonde de PEG, a montré chez les patients atteints de SLA une pharmacocinétique comparable à celle observée chez les sujets en bonne santé, et aucune différence n'était identifiable entre l'administration orale et l'administration par sonde de PEG. Une étude pharmacocinétique menée chez des adultes en bonne santé recevant l'édaravone en suspension buvable à une posologie de 105 mg a montré une pharmacocinétique comparable après une administration intragastrique via une sonde d'alimentation et après une administration par voie orale.
Effet de la prise de nourriture:
Après administration orale d'édaravone chez des sujets en bonne santé, la Cmax et l'ASC ont diminué respectivement de 82% et 61%, lorsque la prise avait lieu avec un repas riche en matières grasses (800-1000 calories, 50% de matières grasses), par rapport à l'administration à jeun. Après administration orale d'édaravone 4 heures après un repas riche en matières grasses, la Cmax et l'ASC avaient diminué de respectivement 44% et 24%. Après administration orale d'édaravone 2 heures après un repas pauvre en matières grasses (400-500 calories, 25% de matières grasses), la Cmax et l'ASC avaient diminué de respectivement 45% et 21%.
Après administration orale d'édaravone chez des sujets en bonne santé 1 heure avant ou 8 heures après un repas riche en matières grasses, 4 heures après un repas pauvre en matières grasses ou 2 heures après un complément alimentaire calorique (250 calories, par ex. une boisson protéinée), la Cmax et l'ASC n'étaient pas significativement plus basses (respectivement moins de 20% et 10% de variation de la Cmax et de l'ASC).
Distribution
L'édaravone se lie aux protéines sériques humaines (92%), principalement à l'albumine, indépendamment de la concentration dans l'intervalle de 0,1 à 50 μmol/l.
Métabolisme
L'édaravone est métabolisée en un sulfoconjugué et en un glucuronoconjugué qui ne sont pas actifs sur le plan pharmacologique. Plusieurs isoformes d'uridine diphosphoglucuronosyltransférase (UGT) (UGT1A1, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B7 et UGT2B17) participent à la glucuronoconjugaison de l'édaravone. Dans le plasma humain, l'édaravone est principalement détectée sous la forme du sulfoconjugué, que l'on présume formé à partir de sulfotransférases.
En raison du métabolisme de premier passage, RADICAVA, suspension buvable entraîne des expositions 1,3 et 1,7 fois plus élevées pour respectivement les métabolites sulfate et glucuronide par rapport à RADICAVA, solution pour perfusion.
Élimination
La demi-vie moyenne d'élimination terminale de l'édaravone est d'environ 9 heures après une administration par voie intraveineuse ou orale. Les demi-vies des métabolites s'élèvent à 3-6 heures. La clairance totale de l'édaravone est estimée à 35,9 l/h après l'administration par voie intraveineuse. Dans des études menées chez des volontaires en bonne santé d'origine japonaise et caucasienne, l'édaravone a été principalement excrétée dans l'urine sous la forme de son glucuronoconjugué (60-80% de la dose en l'espace de 48 heures). Environ 6 à 8% de la dose a été retrouvée dans l'urine sous la forme du sulfoconjugué, et seulement < 1% de la dose a été retrouvée dans l'urine sous forme inchangée. Des études in vitro indiquent que le sulfoconjugué de l'édaravone est hydrolysé pour revenir à l'état d'édaravone, qui est ensuite convertie en glucuronoconjugué dans le rein humain puis excrétée par voie urinaire.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La pharmacocinétique de l'édaravone a été examinée dans le cadre d'une étude menée auprès de 14 sujets présentant des troubles légers à modérés de la fonction hépatique et dans le cadre d'une étude menée auprès de 6 sujets présentant des troubles sévères de la fonction hépatique.
Dans ces deux études, seul un effet léger à non cliniquement significatif a été observé sur l'exposition systémique à l'édaravone et ses sulfoconjugués présents sous forme de métabolites, après une perfusion intraveineuse unique de 30 mg administrée pendant 60 minutes, par rapport aux sujets ayant une fonction hépatique normale.
Les moyennes géométriques des moindres carrés (moyenne des LS) de la Cmax et de l'ASC du temps zéro à l'infini avec extrapolation de la phase terminale (ASC0-∞) après l'administration d'édaravone inchangée étaient supérieures de 1,20 et 1,07 fois chez les sujets avec trouble léger de la fonction hépatique, de 1,24 et 1,14 fois chez les sujets avec trouble modéré de la fonction hépatique et de 1,20 et 1,19 fois chez les sujets avec trouble sévère de la fonction hépatique à celles des sujets présentant une fonction hépatique normale.
Les moyennes géométriques des LS de la Cmax et de l'ASC0-∞ de sulfoconjugués inactifs étaient supérieures de 1,16 et 1,26 fois chez les sujets avec trouble léger de la fonction hépatique, de 0,96 et 1,31 fois chez les sujets avec trouble modéré de la fonction hépatique et de 0,99 et 1,58 fois chez les sujets avec trouble sévère de la fonction hépatique à celles des sujets présentant une fonction hépatique normale.
L'effet d'un trouble de la fonction hépatique sur la pharmacocinétique de l'édaravone est considéré si faible qu'aucun ajustement posologique n'est nécessaire chez ces patients (voir Posologie/Mode d'emploi − Patients présentant des troubles de la fonction hépatique).
Troubles de la fonction rénale
La pharmacocinétique de l'édaravone a été étudiée chez des sujets présentant un trouble léger à modéré de la fonction rénale.
Seul un effet léger à non cliniquement significatif a été observé sur l'exposition systémique à l'édaravone et ses sulfoconjugués présents sous forme de métabolites après une perfusion intraveineuse unique de 30 mg chez des sujets présentant un DFGe de 30-89 ml/min/1,73m², par rapport aux sujets ayant une fonction rénale normale. Lors d'un trouble plus sévère de la fonction rénale, la moyenne t½ de l'édaravone inchangée montrait une tendance à la prolongation.
Après une perfusion intraveineuse unique de 30 mg d'édaravone pendant 60 minutes chez des sujets présentant un trouble léger de la fonction rénale, un trouble modéré de la fonction rénale et une fonction rénale normale, la Cmax moyenne et l'ASC0-∞ de l'édaravone inchangée étaient respectivement supérieures du facteur 1,15 et 1,20 chez les sujets avec un trouble léger de la fonction rénale (DFGe 60-89 ml/min/1,73m²) par rapport aux sujets présentant une fonction rénale normale, et du facteur 1,25 et 1,29 chez les sujets avec un trouble modéré de la fonction rénale (DFGe 30-59 ml/min/1,73m²) par rapport aux sujets présentant une fonction rénale normale.
La Cmax moyenne et l'ASC0-∞ de l'édaravone du sulfoconjugué étaient respectivement supérieures du facteur 1,41 et 1,50 chez les sujets avec un trouble léger de la fonction rénale par rapport aux sujets présentant une fonction rénale normale, et du facteur 1,41 et 1,97 chez les sujets avec un trouble modéré de la fonction rénale par rapport aux sujets présentant une fonction rénale normale. L'augmentation de l'exposition au sulfoconjugué a été considérée comme faible et non cliniquement significative.
Il a été admis qu'un trouble léger à moyen de la fonction rénale n'a pas d'effet cliniquement significatif sur la pharmacocinétique de l'édaravone (voir Posologie/Utilisation chez certains groupes de patients – Patients présentant un trouble de la fonction rénale).
Patients âgés
Aucun effet lié à l'âge sur la pharmacocinétique de l'édaravone n'a été démontré (voir Posologie/Utilisation chez certains groupes de patients).
Patients de sexe masculin et féminin
Aucun effet du sexe sur la pharmacocinétique de l'édaravone n'a été démontré.
Appartenance ethnique
Aucune différence significative associée à l'appartenance ethnique n'a été démontrée pour la Cmax et l'ASC de l'édaravone entre les sujets d'origine japonaise et les sujets d'origine caucasienne.
Données précliniquesLes données précliniques issues des études conventionnelles sur la pharmacologie de sécurité, la génotoxicité et la cancérogénicité n'ont pas révélé de risque particulier pour l'homme.
Toxicité en cas d'administration répétée
Voie intraveineuse (solution pour perfusion)
Des études sur la toxicité en cas d'administration répétée en bolus intraveineux chez le rat et le chien sur une durée de 26 semaines maximum ont mis en évidence le SNC (sédation, hypoactivités) et le système hématopoïétique (anémie régénérative) comme organes cibles. Les résultats étaient transitoires ou réversibles après une période de régénération. L'exposition à l'édaravone chez les animaux des études de toxicité était de manière générale plus élevée par rapport à celle observée chez les patients qui ont reçu RADICAVA à la posologie/utilisation recommandée.
Les marges de sécurité étaient multipliées par environ 2,8 pour l'ASC24h et par 32 pour la C0 chez le rat ainsi que par 66 pour l'ASC24h et par 277 pour la C0 chez le chien.
Une neurotoxicité a été observée après une perfusion i.v. continue sur 24 heures chez le chien et le singe, mais pas chez le rat. Aucun résultat neurotoxicologique n'a été observé chez le chien, qui présente une plus haute sensibilité à l'édavarone que le singe, lors d'une administration intermittente d'édaravone (injection en bolus et perfusion i.v. en continu sur 2 heures) comparable au mode d'administration chez l'homme (60 mg/60 min par voie i.v.). Dans l'étude de 28 jours menée sur des chiens à qui l'édaravone a été administrée par perfusion i.v. continue sur 24 heures − et donc un mode d'administration qui se distingue de l'utilisation clinique prévue pour la SLA − la marge de sécurité était multipliée par 12 pour l'ASC24h et par 0,6 pour la Css.
Voie orale (suspension buvable)
Des études sur la toxicité en cas d'administration orale répétée d'une durée allant jusqu'à 26 semaines chez le rat et jusqu'à 39 semaines chez le chien ont été menées. Les résultats des études sur la toxicité en cas d'administration orale répétée étaient comparables à ceux des études sur la toxicité de l'administration en bolus intraveineux chez le rat et le chien, ainsi que de l'administration par perfusion intraveineuse chez le chien. Les marges de sécurité étaient multipliées par environ 14 pour l'ASC24h et par 21 pour la Cmax chez le rat ainsi que par 13 pour l'ASC24h et par 10 pour la Cmax chez le chien. La neurotoxicité n'a été observée que dans l'étude sur la toxicité en cas d'administration répétée sur 39 semaines menée chez le chien. Les marges de sécurité pour la neurotoxicité étaient multipliées par environ 13 pour l'ASC24h et par 10 pour la Cmax.
Toxicité sur la reproduction
Fertilité
L'administration intraveineuse d'édaravone (0, 3, 20 ou 200 mg/kg) avant et pendant l'accouplement à des rats et des rates jusqu'au 7e jour de gestation n'a pas eu d'effet sur la fertilité. Une interruption du cycle œstral et du comportement d'accouplement a cependant été observée aux plus hautes doses testées. Aux doses plus faibles (≤20 mg/kg/jour 120 mg/m2), équivalant à jusqu'à 2 à 3 fois la dose de 105 mg ou 60 mg recommandée chez l'homme sur la base de la surface corporelle, aucun effet sur la fonction reproductive n'a été observé.
Toxicité sur le développement
Dans les études chez l'animal, l'administration de l'édaravone à des rattes et lapines gravides à des doses cliniquement pertinentes a entraîné une toxicité pour la reproduction. La plupart de ces effets sont apparus à des doses qui étaient également associées à une toxicité maternelle.
Aucune analyse toxicocinétique n'a été réalisée dans les études sur la reproduction, la marge de sécurité a donc été déterminée sur la base de la surface corporelle.
Chez la ratte, l'administration intraveineuse d'édaravone (0, 3, 30 ou 300 mg/kg/jour) pendant la période d'organogenèse a entraîné une diminution du poids des fœtus à toutes les doses. Le poids de la progéniture, née de manière naturelle, était réduit à la plus haute dose testée administrée à la mère. Une toxicité maternelle a aussi été observée à la dose moyenne et à la plus haute dose testée. Aucun effet indésirable sur la fonction reproductrice de la progéniture n'a été démontré. La dose sans effet toxique pour le développement embryofœtal n'a pas été identifiée. La dose faible (3 mg/kg/jour≈18 mg/m2) est inférieure à la dose recommandée chez l'homme sur la base de la surface corporelle.
Chez la lapine, l'administration intraveineuse d'édaravone (0, 3, 20 ou 100 mg/kg/jour) pendant la période de l'organogenèse a entraîné une mortalité embryofœtale à la plus haute dose testée, qui était associée à une toxicité maternelle. La dose la plus élevée sans effet toxique sur le développement embryofœtal est d'environ quatre à six fois la dose de 105 mg ou 60 mg recommandée chez l'homme sur la base de la surface corporelle (mg/m2).
Développement pré- et postnatal
L'effet de l'édaravone sur la progéniture (0, 3, 20, ou 200 mg/kg/jour) administrée à des rates par injection intraveineuse du 17e jour de gestation à la lactation a été étudié dans deux études. Dans la première étude, une mortalité de la progéniture a été observée à la dose la plus élevée et une activité accrue a été observée à la dose intermédiaire et à la dose la plus élevée. Dans la deuxième étude, il y a eu une augmentation de la mortinatalité, de la mortalité de la progéniture et un retard du développement physique (ouverture vaginale) à la plus haute dose testée.
La fonction reproductrice de la progéniture n'a été affectée dans aucune des deux études. Dans les deux études, une toxicité maternelle a été observée à toutes les doses testées, sauf à la plus faible. La dose sans effet toxique sur le développement (3 mg/kg/jour≈18 mg/m2) est inférieure à la dose recommandée chez l'homme sur la base de la surface corporelle.
L'édaravone et ses métabolites sont excrétés dans le lait de rates allaitantes.
Remarques particulièresIncompatibilités
Solution pour perfusion
Aucun autre médicament ne doit être injecté dans la poche pour perfusion ou mélangé à RADICAVA, solution pour perfusion.
La solution pour perfusion RADICAVA ne doit pas être administrée par l'intermédiaire de la même ligne intraveineuse que les préparations de nutrition parentérale et/ou avec des perfusions d'acides aminés.
Suspension buvable
Chez les patients chez qui l'administration par voie orale n'est pas possible ou non souhaitée, RADICAVA, suspension buvable peut être administré via une sonde d'alimentation. Les sondes en polychlorure de vinyle, en polyuréthane ou en silicone conviennent à cet effet. Aucune donnée n'est disponible concernant les sondes en latex; celles-ci ne doivent pas être utilisées.
La sonde doit être rincée avec environ 30 ml d'eau avant et après chaque administration au moyen d'une seringue pour cathéter.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Solution pour perfusion
Ne pas conserver au-dessus de 25 °C. Ne pas congeler.
Conserver hors de portée des enfants.
Conserver le produit dans son suremballage pour le protéger de toute dégradation oxydative jusqu'à son utilisation. Chaque poche est emballée dans un emballage secondaire. L'emballage secondaire contient un sachet absorbeur d'oxygène et un sachet indicateur d'oxygène. Lorsque la concentration en oxygène est correcte, la couleur de l'indicateur d'oxygène est rose. L'indicateur d'oxygène passe au bleu ou au violet lorsque l'oxygène a dépassé les valeurs autorisées.
Suspension buvable
Pharmacie: conserver au réfrigérateur (2-8 °C). Ne pas congeler.
Une fois la boîte sortie du réfrigérateur, noter la date de délivrance par la pharmacie sur le carton. Conserver à température ambiante (15-25 °C). Ne pas remettre au frais une fois sorti du réfrigérateur. Ne pas congeler. Éliminer les flacons non entamés au bout de 30 jours.
Patients: conserver à température ambiante (15-25 °C). Ne pas conserver au réfrigérateur ni congeler. Jeter tout flacon non entamé dans les 30 jours suivant la date de délivrance inscrite sur le carton.
Stable 15 jours à compter de l'ouverture. La date de la première ouverture doit être notée sur l'étiquette du flacon. Ceci est un produit multidose.
Tout contenu non utilisé 15 jours après l'ouverture d'un flacon doit être éliminé conformément à la réglementation en vigueur. Conserver le flacon bien fermé.
Conserver le flacon dans son carton pour le protéger de la lumière.
Conserver en position verticale.
Conserver hors de portée des enfants.
Remarques concernant la manipulation
Solution pour perfusion
Vérifier que la solution est claire et le récipient intact avant de procéder à l'administration.
Suspension buvable
Agiter vigoureusement le flacon pendant au moins 30 secondes avant l'utilisation.
Une croûte blanche peut se former sur le goulot ou les parois des flacons déjà entamés de RADICAVA, suspension buvable. Cela est normal; le médicament peut continuer à être utilisé selon la prescription.
L'emballage contient une notice avec des instructions détaillées sur l'utilisation et la manipulation.
Numéro d’autorisationSolution pour perfusion - 66492 (Swissmedic)
Suspension buvable - 68016 (Swissmedic)
PrésentationRADICAVA, solution pour perfusion
Poche pour perfusion 30 mg/100 ml (0,3 mg/ml); 2 poches par carton [B]
RADICAVA, suspension buvable
Boîte de 1 flacon multidose de 50 ml (10 doses de 5 ml). Contient également 1 adaptateur et une seringue graduée (5 ml) pour administration orale. [B]
Boîte de début de traitement avec 2 flacons multidoses de 35 ml (2x7 doses de 5 ml). Contient également 2 adaptateurs et 2 seringues graduées (5 ml) pour administration orale. [B]
Titulaire de l’autorisationMitsubishi Tanabe Pharma GmbH, Düsseldorf, succursale de Zurich, 8001 Zurich
Mise à jour de l’informationDécembre 2022
|