CompositionPrincipes actifs
Risankizumab, produit à partir de cellules CHO (cellules ovariennes de hamster chinois) génétiquement modifiées.
Excipients
Skyrizi 150 mg, solution injectable en stylo prérempli:
Acétate de sodium trihydraté, acide acétique 99 %, tréhalose dihydraté, polysorbate 20 et eau pour préparations injectables q.s. ad solutionem pro 1 ml corresp. 0,21 mg de sodium.
Skyrizi 150 mg, solution injectable en seringue préremplie:
Acétate de sodium trihydraté, acide acétique 99 %, tréhalose dihydraté, polysorbate 20 et eau pour préparations injectables q.s. ad solutionem pro 1 ml corresp. 0,21 mg de sodium.
Skyrizi 75 mg, solution injectable en seringue préremplie:
Succinate disodique hexahydraté, acide succinique, 34,0 mg de sorbitol (E420), polysorbate 20 et eau pour préparations injectables q.s.ad solutionem pro 0,83 ml corresp. 0,15 mg de sodium.
Indications/Possibilités d’emploiSKYRIZI est indiqué dans le traitement du psoriasis en plaques modéré à sévère chez les patients adultes ayant présenté une réponse insuffisante à d'autres traitements systémiques tels que la ciclosporine, le méthotrexate (MTX) ou la PUVA (psoralène et UV-A) ou présentant une contre-indication ou une intolérance à de tels traitements.
Posologie/Mode d’emploiSKYRIZI est destiné à être utilisé sous la surveillance d'un médecin expérimenté dans le diagnostic et le traitement du psoriasis en plaques. Avant le début du traitement, le médecin doit vérifier que les patients ont compris que SKYRIZI est un traitement innovant dont l'expérience est limitée au-delà d'un an et les risques à long terme inconnus.
Après une formation adaptée à la technique de l'injection sous-cutanée et à l'élimination des déchets, les patients peuvent s'injecter eux-mêmes SKYRIZI si le médecin le juge approprié. Le médecin doit toutefois veiller à ce que le patient bénéficie d'un suivi adapté. Les patients et leurs soignants doivent être informés que les instructions d'utilisation figurent dans la notice d'emballage correspondante.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Posologie recommandée
La dose recommandée est de 150 mg en injection sous-cutanée les semaines 0 et 4, puis toutes les 12 semaines par la suite.
Il convient d'envisager l'arrêt du traitement chez les patients qui n'ont pas obtenu de réponse après 16 semaines de traitement. Certains patients obtenant initialement une réponse partielle peuvent présenter une nouvelle amélioration avec la poursuite du traitement au-delà de 16 semaines.
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale ou hépatique
Aucune étude spécifique n'a été menée pour évaluer l'effet d'une insuffisance hépatique ou rénale sur la pharmacocinétique de SKYRIZI. En règle générale, on ne s'attend pas à ce que ces maladies aient une influence significative sur la pharmacocinétique des anticorps monoclonaux. C'est pourquoi un ajustement posologique ne devrait pas être nécessaire (voir «Pharmacocinétique»).
Patients âgés
Aucun ajustement posologique est nécessaire (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de SKYRIZI chez les enfants et les adolescents de moins de 18 ans n'ont pas encore été établies.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Infection active cliniquement significative (par ex. tuberculose active).
Mises en garde et précautionsInfections
SKYRIZI peut accroître le risque d'infections. Lors des études cliniques, des infections ont été observées pendant le traitement de 16 semaines chez 22,1% des patients du groupe SKYRIZI et chez 14,7% des patients du groupe placebo. Les taux d'infections graves ont été de 0,4% (5/1'306) dans le groupe SKYRIZI et de 0,3% (1/300) dans le groupe placebo. Chez les patients présentant une infection active cliniquement significative, le traitement par SKYRIZI ne doit pas être instauré tant que l'infection n'a pas régressé ou qu'elle n'est pas traitée de façon adéquate. Ont été exclus des études cliniques les patients qui étaient séropositifs pour le VHC ou le VIH, qui avaient obtenu un résultat positif au test de dépistage de l'hépatite B et qui avaient des antécédents d'infections chroniques ou récidivantes.
Chez les patients ayant des infections chroniques ou des antécédents d'infections récidivantes connus, l'infection doit avoir régressé ou être traitée de façon adéquate avant l'instauration de SKYRIZI.
Les patients doivent être informés de la nécessité de consulter un médecin s'ils développent des signes ou des symptômes d'infection cliniquement significative. Si un patient développe une infection de ce type ou s'il ne répond pas à un traitement standard, il doit être placé sous surveillance étroite et le traitement par SKYRIZI doit être interrompu tant que l'infection n'a pas régressé.
Tuberculose
Dans les études cliniques de phase III sur le psoriasis, aucun des 72 patients atteints de tuberculose (TB) latente ayant reçu pendant l'étude SKYRIZI et une prophylaxie antituberculeuse adéquate n'a développé de tuberculose active au cours de la période de surveillance moyenne de 61 semaines. Il convient donc d'effectuer un dépistage de la tuberculose chez les patients avant l'instauration du traitement. Chez les patients atteints de TB latente, un traitement antituberculeux doit être instauré avant l'administration de SKYRIZI. SKYRIZI ne doit pas être utilisé chez les patients atteints de TB active.
Les patients traités par SKYRIZI doivent faire l'objet d'une surveillance pendant et après le traitement afin de déceler d'éventuels signes et symptômes de TB active.
Vaccinations
Il faut envisager d'administrer tous les vaccins adéquats avant le début du traitement par SKYRIZI, conformément aux directives actuelles en matière de vaccination. Les vaccins vivants ne doivent pas être administrés en concomitance avec SKYRIZI. Aucune donnée n'est disponible sur la réponse aux vaccins vivants ou inactivés. Conformément aux directives de vaccination actuelles concernant les médicaments immunosuppresseurs, il convient de respecter un délai suffisant entre la vaccination par des vaccins vivants et le début du traitement. Les informations professionnelles de médicaments immunosuppresseurs fournissent des renseignements sur leur utilisation en concomitance avec certains vaccins.
Maladies malignes
Les études cliniques avec une durée de suivi de plus d'un an n'ont montré aucune augmentation du risque de maladies malignes.
Les patients atteints de psoriasis précédemment traités par UV doivent être examinés pour dépister d'éventuelles tumeurs cutanées avant et pendant le traitement par SKYRIZI.
Traitement concomitant par d'autres immunosuppresseurs systémiques ou par photothérapie
La sécurité et l'efficacité de SKYRIZI en association avec des immunosuppresseurs, y compris des médicaments biologiques, ou avec une photothérapie n'ont pas été évaluées.
Hypersensibilité
En cas de réaction d'hypersensibilité grave, l'administration de SKYRIZI doit être immédiatement interrompue et des mesures thérapeutiques appropriées doivent être prises.
Autres composants dont l'effet est connu
SKYRIZI, solution injectable en stylo préremplie ou en seringue préremplie à 150 mg et SKYRIZI, solution injectable en seringue préremplie à 75 mg contiennent moins de 1 mmol de sodium (23 mg) par dose de 150 mg, c.-à-d. qu'ils sont essentiellement «sans sodium».
SKYRIZI, solution injectable en seringue préremplie à 75 mg contient 68,0 mg de sorbitol par dose de 150 mg.
InteractionsOn ne s'attend pas à ce que SKYRIZI soit métabolisé par des enzymes hépatiques ou excrété par voie rénale. Il ne devrait donc pas y avoir d'interactions médicamenteuses entre SKYRIZI et les inhibiteurs ou inducteurs des enzymes métabolisant des principes actifs.
D'après les résultats d'une étude d'interactions médicamenteuses menée chez des patients atteints de psoriasis en plaques et les résultats des analyses pharmacocinétiques de population, le risankizumab ne devrait pas provoquer d'interactions médicamenteuses et ne devrait pas être influencé par d'autres médicaments (voir «Pharmacocinétique»).
Aucun ajustement posologique n'est nécessaire lors de l'administration concomitante de risankizumab et de substrats du cytochrome P450.
Grossesse, allaitementFemmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode de contraception fiable pendant le traitement et durant 20 semaines au moins après le traitement.
Grossesse
On ne possède pas suffisamment de données sur l'utilisation de SKYRIZI chez les femmes enceintes pour pouvoir décrire les risques associés au médicament. Des études menées sur les animaux ne démontrent aucun effet nocif direct ou indirect en lien avec une toxicité pour la reproduction (voir «Données précliniques»). Par mesure de précaution, l'utilisation de SKYRIZI pendant la grossesse doit être évitée.
Allaitement
On ne possède aucune donnée concernant le passage du risankizumab dans le lait maternel chez l'être humain, les effets sur l'enfant allaité ou les effets sur la production de lait. La décision d'interrompre l'allaitement ou de renoncer à la prise de SKYRIZI doit donc être prise en prenant en compte les avantages de l'allaitement pour l'enfant et ceux du traitement pour la mère.
Fertilité
Aucune donnée clinique concernant l'effet du risankizumab sur la fertilité n'est disponible.
Les expérimentations animales n'ont indiqué aucun effet direct ou indirect sur la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée.
Effets indésirablesÉtudes cliniques
Dans le cadre du développement clinique, 2'234 patients au total ont été traités par SKYRIZI au cours d'études sur le psoriasis en plaques, ce qui correspond à une exposition de 2'167 patients-années. Parmi ces patients, 1'208 patients atteints de psoriasis ont reçu SKYRIZI pendant au moins un an. Au total, 1'590 patients ont reçu 150 mg de SKYRIZI après la randomisation ou après 16 semaines de traitement par placebo, ce qui correspond à une exposition de 1'688 patients-années. Parmi ces patients, 1'091 ont reçu SKYRIZI pendant au moins un an.
Les données issues des études contrôlées par placebo et par comparateur actif ont été combinées afin d'évaluer la sécurité de SKYRIZI sur une période allant jusqu'à 16 semaines. Dans le groupe SKYRIZI à 150 mg, les données de 1'306 patients ont été évaluées. Dans le groupe SKYRIZI, 2,4% des patients ont développé des événements indésirables graves (9,9 événements pour 100 patients-années), comparativement à 4,0% dans le groupe placebo (17,4 événements pour 100 patients-années), 5,0% dans le groupe ustékinumab (18,4 événements pour 100 patients-années) et 3,0% dans le groupe adalimumab (14,7 événements pour 100 patients-années).
Les effets indésirables de SKYRIZI rapportés lors des études cliniques (tableau 1) sont indiqués ci-dessous par classes de systèmes d'organes MedDRA, selon la convention suivante: très fréquents (≥1/10); fréquents (<1/10, ≥1/100); occasionnels (<1/100, ≥1/1'000); rares (<1/1'000, ≥1/10'000); très rares (<1/10'000).
Tableau 1: Effets indésirables observés au cours des études cliniques
|
Classe de systèmes d'organes
|
Fréquence
|
Effets indésirables
| |
Infections et infestations
|
Très fréquents
|
Infection des voies aériennes supérieuresa (13%)
| |
Fréquents
|
Teigneb
| |
Occasionnels
|
Folliculite
| |
Affections du système nerveux
|
Fréquents
|
Céphaléesc
| |
Troubles généraux et anomalies au site d'administration
|
Fréquents
|
Fatigued
Réactions au site d'injectione
| |
a
Y compris infection des voies respiratoires (virales, bactériennes ou non spécifiées), sinusite (aiguë également), rhinite, rhinopharyngite, pharyngite (virale également), amygdalite
b Y compris Tinea pedis, Tinea cruris, Tinea corporis, Tinea versicolor, Tinea manuum, onychomycose
c Y compris céphalées, céphalées de tension, céphalées sinusales
d Y compris fatigue, asthénie
e Y compris ecchymose, érythème, hématome, saignement, irritation, douleur, démangeaison, réaction et gonflement au site d'injection
|
Description de certains effets indésirables
Infections
Au cours des 16 premières semaines, 22,1% des patients du groupe SKYRIZI ont développé des infections (90,8 événements pour 100 patients-années), comparativement à 14,7% dans le groupe placebo (56,5 événements pour 100 patients-années), 20,9% dans le groupe ustékinumab (87,0 événements pour 100 patients-années) et 24,3% dans le groupe adalimumab (104,2 événements pour 100 patientsannées). Dans la majorité des cas, ces infections n'ont pas été graves, ont été légèrement à modérément sévères et n'ont pas nécessité l'arrêt de SKYRIZI.
Dans l'ensemble du programme de recherche sur le psoriasis, y compris l'exposition de longue durée à SKYRIZI, le taux d'infections (75,5 événements pour 100 patients-années) a été comparable à celui observé au cours des 16 premières semaines de traitement.
Sécurité à long terme
La fréquence des effets indésirables jusqu'à la semaine 52 incluse a coïncidé avec le profil de sécurité observé lors des 16 premières semaines de traitement. Le taux d'événements indésirables graves pour 100 patients-années, ajusté selon l'exposition, a atteint jusqu'à la semaine 52 incluse 9,4% chez les patients sous SKYRIZI et 10,9% chez les patients sous ustékinumab. Chez les patients exposés à SKYRIZI pendant un maximum de 77 semaines, aucun nouvel effet indésirable n'a été rapporté par rapport aux 16 premières semaines de traitement.
Immunogénicité
Comme toutes les protéines thérapeutiques, SKYRIZI possède également une immunogénicité potentielle. La détection d'anticorps dépend fortement de la sensibilité et de la spécificité du test. De plus, l'incidence de positivité pour les anticorps (y compris pour les anticorps neutralisants) d'un test peut être influencée par plusieurs facteurs, notamment la méthode de test, la manipulation des échantillons, le moment du prélèvement des échantillons, les médicaments concomitants et la maladie sous-jacente. C'est pourquoi une comparaison de l'incidence des anticorps dirigés contre le risankizumab avec l'incidence des anticorps dirigés contre d'autres agents biologiques doit être interprétée avec prudence.
Parmi les patients qui ont reçu SKYRIZI pendant 52 semaines au plus à la dose clinique recommandée dans le cadre des études cliniques sur le psoriasis, 24% (263/1'079) des patients évalués pendant le traitement ont développé des anticorps contre le principe actif et 14% (150/1'079) ont développé des anticorps neutralisants.
Chez la plupart des patients, les anticorps dirigés contre le risankizumab, y compris les anticorps neutralisants, n'étaient pas associés à des modifications de la réponse clinique ou de la sécurité. Chez environ 1% des patients traités par SKYRIZI, des titres d'anticorps élevés étaient associés à des concentrations plus faibles de risankizumab et une réponse clinique réduite.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageEn cas de surdosage, il est recommandé de surveiller tout signe ou symptôme d'effet indésirable chez le patient et de commencer immédiatement un traitement symptomatique approprié.
Propriétés/EffetsCode ATC
L04AC
Le risankizumab, inhibiteur de l'interleukine 23, est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1). Le risankizumab est produit à partir d'une lignée cellulaire de mammifère à l'aide de la technologie de l'ADN recombinant.
Mécanisme d'action
Le risankizumab est un anticorps monoclonal humanisé de type immunoglobuline G1 (IgG1) qui se lie sélectivement avec une grande affinité à la sous-unité p19 de la cytokine humaine interleukine 23 (IL-23) et en inhibe l'interaction avec le complexe récepteur de l'IL-23. L'IL-23 est une cytokine présente naturellement qui participe aux réactions inflammatoires et immunitaires. L'IL-23 favorise la prolifération, le maintien et l'activation des cellules Th17, qui produisent l'IL-17A, l'IL-17F et l'IL-22 de même que d'autres cytokines pro-inflammatoires et jouent un rôle central dans la pathogenèse des maladies auto-immunes inflammatoires telles que le psoriasis. Chez les patients atteints de psoriasis en plaques, on observe une régulation positive de l'expression de l'IL-23 dans les lésions cutanées par rapport aux zones non touchées. Le risankizumab bloquant la liaison de l'IL-23 à son récepteur, il inhibe ainsi la transduction du signal cellulaire IL-23 dépendante et la libération des cytokines pro-inflammatoires.
Le risankizumab ne se lie pas à l'IL-12 humain, qui possède comme l'IL-23 une sous-unité p40.TE
Pharmacodynamique
Dans une étude menée chez des patients atteints de psoriasis, une diminution de l'expression des gènes associés à l'axe IL-23/IL-17 a été observée dans la peau après une dose unique de risankizumab. En outre, des réductions de l'épaisseur de l'épiderme, de l'infiltration de cellules inflammatoires et de l'expression des marqueurs du psoriasis dans les lésions psoriasiques ont été observées.
Efficacité clinique
L'efficacité et la sécurité de SKYRIZI ont été évaluées chez 2'109 patients atteints de psoriasis en plaques modéré à sévère dans le cadre de quatre études multicentriques randomisées à double insu, contrôlées par placebo et/ou par comparateur actif (ULTIMMA-1, ULTIMMA-2, IMMHANCE et IMMVENT). Dans les études ULTIMMA-1 et ULTIMMA-2, l'ustékinumab a été utilisé comme comparateur actif. L'étude IMMHANCE a évalué l'arrêt, puis la reprise du traitement par le risankizumab chez les patients ayant obtenu une réponse. Dans l'étude IMMVENT, l'adalimumab a été utilisé comme comparateur actif. À l'issue des études, les patients pouvaient être inclus dans la phase de suivi en ouvert LIMITLESS. Les patients inclus étaient âgés d'au moins 18 ans et étaient atteints de psoriasis en plaques avec une surface cutanée atteinte (body surface area, BSA) ≥10%, un score d'évaluation globale statique par un médecin (static Physician Global Assessment, sPGA) ≥3, un psoriasis de grade de 0 à 4 et un score PASI (Psoriasis Area and Severity Index) ≥12. Les patients atteints de psoriasis érythrodermique, de psoriasis en gouttes ou de psoriasis pustuleux étaient exclus.
Au total, les patients avaient un score PASI initial médian de 17,8 et une surface cutanée atteinte moyenne de 20,0%. Chez 19,3% des patients, le score sPGA initial correspondait à une maladie sévère. Chez 9,8% des patients des études au total, l'anamnèse a révélé une arthrite psoriasique diagnostiquée.
Sur l'ensemble des études, 30,9% des patients n'avaient reçu auparavant aucun traitement systémique biologique ou non biologique, 38,1% avaient reçu une photothérapie, 48,3% un traitement systémique non biologique et 42,1% un traitement biologique (23,7% de tous les patients dans ces études ont reçu au moins un inhibiteur du TNF-alpha) pour leur psoriasis. Afin d'éviter tout biais dans l'évaluation de l'efficacité de SKYRIZI dans le traitement du psoriasis, l'utilisation concomitante d'un médicament antipsoriasique systémique ou topique (à l'exception des corticostéroïdes topiques sur le visage, les aisselles et/ou les organes génitaux) ou d'une photothérapie était proscrite pendant les études.
ULTIMMA-1 et ULTIMMA-2
Les études ULTIMMA-1 et ULTIMMA-2 ont inclus 997 patients (598 patients dans le groupe SKYRIZI 150 mg, 199 patients dans les groupes ustékinumab 45 mg ou 90 mg [selon le poids corporel initial] et 200 patients dans le groupe placebo). Le traitement a été administré les semaines 0 et 4, puis toutes les 12 semaines par la suite. Les résultats sont présentés dans le tableau 2 et la figure 1.
Tableau 2: Résultats obtenus chez les adultes atteints de psoriasis en plaques dans les études d'efficacité ULTIMMA 1 et ULTIMMA 2
|
|
ULTIMMA-1
|
ULTIMMA-2
| |
|
SKYRIZI
(N=304)
n (%)
|
Ustékinumab
(N=100)
n (%)
|
Placebo
(N=102)
n (%)
|
SKYRIZI
(N=294)
n (%)
|
Ustékinumab
(N=99)
n (%)
|
Placebo
(N=98)
n (%)
| |
Score sPGA «sans» ou «minime» (0 ou 1)
| |
Semaine 16
|
267 (87,8)a
|
63 (63,0)
|
8 (7,8)
|
246 (83,7)a
|
61 (61,6)
|
5 (5,1)
| |
Semaine 52
|
262 (86,2)
|
54 (54,0)
|
--
|
245 (83,3)
|
54 (54,5)
|
--
| |
Score sPGA «sans» (0)
| |
Semaine 16
|
112 (36,8)
|
14 (14,0)
|
2 (2,0)
|
150 (51,0)
|
25 (25,3)
|
3 (3,1)
| |
Semaine 52
|
175 (57,6)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
PASI 75
| |
Semaine 12
|
264 (86,8)
|
70 (70,0)
|
10 (9,8)
|
261 (88,8)
|
69 (69,7)
|
8 (8,2)
| |
Semaine 52
|
279 (91,8)
|
70 (70,0)
|
--
|
269 (91.5)
|
76 (76,8)
|
--
| |
PASI 90
| |
Semaine 16
|
229 (75,3)a
|
42 (42,0)
|
5 (4,9)
|
220 (74,8)a
|
47 (47,5)
|
2 (2,0)
| |
Semaine 52
|
249 (81,9)
|
44 (44,0)
|
--
|
237 (80,6)
|
50 (50,5)
|
--
| |
PASI 100
| |
Semaine 16
|
109 (35,9)
|
12 (12,0)
|
0 (0,0)
|
149 (50,7)
|
24 (24,2)
|
2 (2,0)
| |
Semaine 52
|
171 (56,3)
|
21 (21,0)
|
--
|
175 (59,5)
|
30 (30,3)
|
--
| |
Dans toutes les comparaisons de SKYRIZI à l'ustékinumab et au placebo, une valeur p < 0,001 a été atteinte, sauf pour le score PASI 75 la semaine 52 dans l'étude ULTIMMA-2 (p = 0,001).
a Co-critères d'évaluation principaux par rapport au placebo
|
Figure 1: Évolution du pourcentage de variation moyenne par rapport à l'inclusion du score PASI dans les études ULTIMMA-1 et ULTIMMA-2
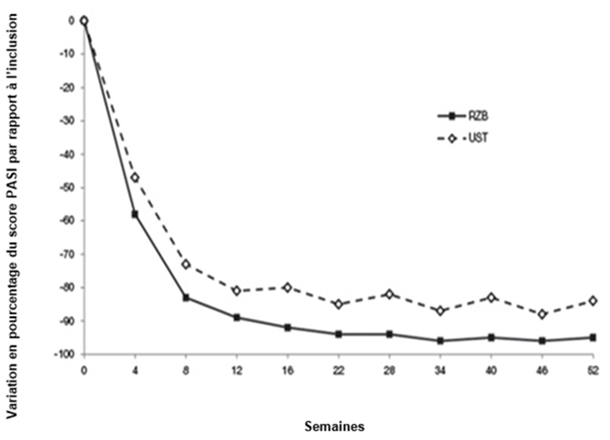
RZB = risankizumab
UST = ustékinumab
p < 0,001 à chaque point dans le temps
Une analyse en fonction de l'âge, du sexe, de l'origine ethnique, du poids corporel, du score PASI initial, de la présence concomitante d'arthrite psoriasique, des antécédents de traitement systémique non biologique, des antécédents de traitement biologique et de l'absence de réponse antérieure aux médicaments biologiques n'a montré aucune différence entre ces sous-groupes pour ce qui est de la réponse à SKYRIZI.
Chez les patients traités par SKYRIZI, une amélioration du psoriasis au niveau du cuir chevelu, des ongles, de la paume des mains et de la plante des pieds a été observée les semaines 16 et 52.
IMMHANCE
L'étude IMMHANCE a inclus 507 patients (407 patients dans le groupe SKYRIZI 150 mg et 100 patients dans le groupe placebo). Le traitement a été administré les semaines 0 et 4, puis toutes les 12 semaines par la suite.
La semaine 16, SKYRIZI s'est avéré supérieur au placebo sur les co-critères d'évaluation principaux, à savoir score sPGA «sans» ou «minime» (83,5% SKYRIZI vs. 7,0% placebo) et PASI 90 (73,2% SKYRIZI vs. 2,0% placebo). La semaine 16, un plus grand nombre de patients sous SKYRIZI n'avaient plus de lésions cutanées (score sPGA 0 [46,4% SKYRIZI vs. 1,0% placebo] ou score PASI 100 [47,2% SKYRIZI vs. 1,0% placebo]). En outre, les patients sous SKYRIZI avaient une plus grande probabilité d'obtenir un score PASI 75 que ceux sous placebo (88,7% SKYRIZI vs. 8,0% placebo).
Aucun des 31 patients de l'étude IMMHANCE atteints d'une tuberculose (TB) latente et qui n'avaient reçu aucune prophylaxie pendant l'étude n'a présenté une TB active pendant la période de suivi moyenne sous SKYRIZI de 55 semaines. Toutefois, les patients présentant une TB latente doivent recevoir un traitement antituberculeux avant l'instauration du traitement par SKYRIZI (voir «Mises en garde et précautions»).
IMMVENT
L'étude IMMVENT a inclus 605 patients (301 patients dans le groupe SKYRIZI et 304 patients dans le groupe adalimumab). Les patients randomisés dans le groupe SKYRIZI ont reçu une dose de 150 mg les semaines 0 et 4, puis toutes les 12 semaines par la suite. Les patients randomisés dans le groupe adalimumab ont reçu 80 mg la semaine 0, 40 mg la semaine 1, puis 40 mg toutes les deux semaines jusqu'à la semaine 15. A partir de la semaine 16, en fonction de leur réponse, les patients traités par adalimumab ont poursuivi le traitement ou ont changé de traitement:
·< PASI 50: changement par SKYRIZI,
·PASI 50 à < PASI 90: nouvelle randomisation pour continuer le traitement avec 40 mg d'adalimumab toutes les 2 semaines ou changement par SKYRIZI,
·PASI 90: poursuite du traitement avec 40 mg d'adalimumab toutes les 2 semaines.
Dans l'étude IMMVENT, les patients sous SKYRIZI avaient obtenu à la semaine 16 des résultats semblables à ceux rapportés dans les autres études cliniques (tableau 3 et figure 2).
Tableau 3: Résultats obtenus à la semaine 16 chez les adultes atteints de psoriasis en plaques dans l'étude d'efficacité IMMVENT
|
|
SKYRIZI
(N=301)
n (%)
|
Adalimumab
(N=304)
n (%)
| |
Score sPGA «sans» ou «minime»a
|
252 (83,7)
|
183 (60,2)
| |
PASI 75
|
273 (90,7)
|
218 (71,7)
| |
PASI 90a
|
218 (72,4)
|
144 (47,4)
| |
PASI 100
|
120 (39,9)
|
70 (23,0)
| |
Dans toutes les comparaisons, une valeur p < 0,001 a été atteinte.
a Co-critères d'évaluation principaux
|
Chez les patients sous adalimumab ayant obtenu à la semaine 16 un score PASI 50 à < PASI 90 et de nouveau randomisés, une différence entre ceux qui sont passés à SKYRIZI et ceux qui ont poursuivi le traitement par adalimumab a été observée en termes de taux de réponse PASI 90 (49,1% vs. 26,8%) dès la quatrième semaine suivant la nouvelle randomisation. Au total, 66,0% (35/53) des patients ont obtenu un score PASI 90 après 28 semaines de traitement par SKYRIZI, comparativement à 21,4% (12/56) chez ceux qui ont continué de recevoir l'adalimumab. Des autres paramètres de réponse ont été également supérieurs après le relais par SKYRIZI: 39,6% ont obtenu un score PASI 100, 39,6% un score sPGA «sans» et 73,6% un score sPGA «sans» ou «minime» après le changement par SKYRIZI, comparativement à 7,1% ayant obtenu un score PASI 100, 7,1% un score sPGA «sans» et 33,9% un score sPGA «sans» ou «minime» avec la poursuite du traitement par adalimumab.
Figure 2: Évolution du score PASI 90 après la nouvelle randomisation dans l'étude IMMVENT
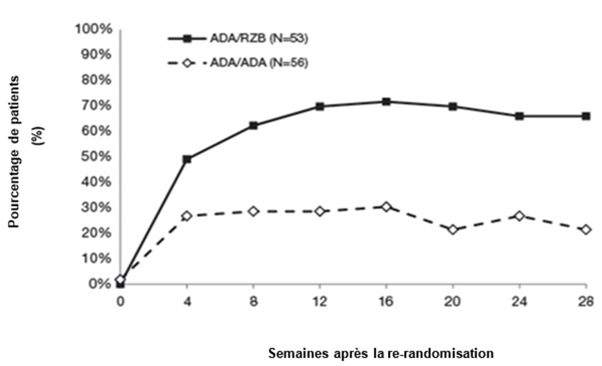
ADA/ADA: patients affectés initialement au groupe adalimumab et qui ont continué de recevoir l'adalimumab
ADA/RZB: patients affectés initialement au groupe adalimumab et qui sont passés à SKYRIZI
p < 0,05 la semaine 4 et p < 0,001 à chaque point dans le temps à partir de la semaine 8
Obtention et maintien de la réponse
Dans une analyse combinée des patients ayant reçu SKYRIZI dans le cadre des études ULTIMMA-1 et ULTIMMA-2 et qui présentaient un score PASI 100 à la semaine 16, le taux de réponse est resté à 79,8% (206/258) chez les patients qui ont poursuivi le traitement par SKYRIZI jusqu'à la semaine 52. Parmi les patients ayant obtenu un score PASI 90 à la semaine 16, 88,4% (398/450) ont maintenu leur réponse jusqu'à la semaine 52.
Après une nouvelle randomisation, les patients ayant reçu initialement SKYRIZI dans le cadre de l'étude IMMHANCE et dont le score sPGA à la semaine 28 était «sans» ou «minime» ont continué d'être traités par SKYRIZI toutes les 12 semaines jusqu'à la semaine 88 incluse (N=111) ou ont arrêté le traitement (N=225).
À la semaine 104 (soit 16 semaines après la dernière administration de SKYRIZI), 81,1% (90/111) des patients ayant poursuivi le traitement par SKYRIZI ont obtenu un score sPGA «sans» ou «minime», comparativement à 7,1% (16/225) des patients ayant arrêté le traitement par SKYRIZI. Chez les patients ayant arrêté le traitement par SKYRIZI, une perte du score sPGA «sans» ou «minime» a été observée dès 12 semaines après une dose manquée.
À la semaine 104, 63,1% (70/111) des patients ayant poursuivi le traitement par SKYRIZI ont obtenu un score sPGA «sans», comparativement à 2,2% (5/225) des patients ayant arrêté le traitement par SKYRIZI.
Une augmentation des scores sPGA «sans» et PASI 100 a été constatée entre la semaine 28 et la semaine 88 chez les patients ayant poursuivi le traitement par SKYRIZI.
Parmi les patients ayant obtenu un score sPGA «sans» ou «minime» à la semaine 28 et ayant présenté une rechute (sPGA ≥3) après l'arrêt du traitement par SKYRIZI, 83,7% (128/153) ont de nouveau obtenu un score sPGA «sans» ou «minime» 16 semaines après la reprise du traitement.
Qualité de vie / Événements rapportés par les patients
Un nombre significativement supérieur de patients traités par SKYRIZI ont obtenu un score DLQI (Dermatology Life Quality Index) de 0 ou 1 [aucun effet sur la qualité de vie liée à la santé] à la semaine 16 comparativement aux patients sous placebo, adalimumab ou ustékinumab. L'amélioration s'est maintenue jusqu'à la semaine 52 dans les études ULTIMMA-1 et ULTIMMA-2.
Dans les études ULTIMMA-1 et ULTIMMA-2, une amélioration significativement supérieure des symptômes du psoriasis (démangeaisons, douleurs, rougeur et sensation de brûlure, mesurés au moyen du score PSS [Psoriasis Symptom Score]) a été observée comparativement au placebo à la semaine 16. Comparativement à l'ustékinumab et au placebo, une proportion significativement supérieure de patients sous SKYRIZI a obtenu un score PSS de 0 (absence de symptômes) à la semaine 16. Jusqu'à la semaine 52, 55,7% (333/598) des patients sous SKYRIZI n'ont rapporté aucune démangeaison, douleur, rougeur ou sensation de brûlure.
PharmacocinétiqueAbsorption
Dans une plage posologique de 18 à 300 mg et de 0,25 à 1 mg/kg par voie sous-cutanée et de 200 à 1'200 mg et de 0,01 à 5 mg/kg par voie intraveineuse, le risankizumab a montré une pharmacocinétique linéaire avec une augmentation de l'exposition proportionnelle à la dose.
Après une administration sous-cutanée de risankizumab, la concentration plasmatique maximale a été obtenue 3 à 14 jours après l'administration, avec une biodisponibilité absolue estimée à 89%. Avec le schéma posologique pour les patients atteints de psoriasis (150 mg les semaines 0 et 4, puis toutes les 12 semaines par la suite), les concentrations maximales à l'état d'équilibre et les concentrations minimales dans le plasma sont estimées à 12 et 2 µg/ml.
Avec la seringue préremplie, la bioéquivalence entre une dose unique à 150 mg de risankizumab et deux injections à 75 mg a été démontrée. La bioéquivalence entre la seringue préremplie de risankizumab 150 mg et le stylo prérempli a également été démontrée.
Distribution
Chez un patient atteint de psoriasis avec un poids corporel de 90 kg, le volume de distribution à l'état d'équilibre (Vss) était de 11,2 l, ce qui évoque une distribution du risankizumab limitée primaire à l'espace vasculaire et interstitiel.
Métabolisme
Comme l'IgG endogène, les anticorps IgG monoclonaux thérapeutiques sont généralement décomposés en peptides et acides aminés de plus petite taille par métabolisme catabolique. On ne s'attend pas à ce que le risankizumab soit métabolisé via les enzymes du cytochrome P450.
Élimination
Chez un patient atteint de psoriasis avec un poids corporel de 90 kg, la clairance systémique (CL) du risankizumab atteignait 0,31 l/jour et la demi-vie d'élimination terminale 28 jours.
Le risankizumab étant un anticorps monoclonal IgG1, il ne devrait pas subir de filtration glomérulaire rénale ni être excrété sous forme de molécule intacte dans les urines.
Interactions médicamenteuses
Une étude d'interactions médicamenteuses a été réalisée chez des patients atteints de psoriasis en plaques afin d'évaluer l'effet d'une administration répétée de risankizumab sur la pharmacocinétique de substrats tests sensibles au cytochrome P450 (CYP). L'exposition à la caféine (substrat du CYP1A2), à la warfarine (substrat du CYP2C9), à l'oméprazole (substrat du CYP2C19), au métoprolol (substrat du CYP2D6) et au midazolam (substrat du CYP3A4) a été semblable avant et après le traitement par risankizumab, ce qui suggère l'absence d'interactions médicamenteuses cliniquement significatives via cette enzyme.
Les analyses pharmacocinétiques de population ont montré que l'exposition au risankizumab n'était pas influencée par les médicaments concomitants (metformine, atorvastatine, lisinopril, amlodipine, ibuprofène, acide acétylsalicylique et lévothyroxine) pris par certains patients atteints de psoriasis en plaques participant aux études cliniques (voir «Interactions»).
Cinétique pour certains groupes de patients
Insuffisance rénale ou hépatique
Aucune étude spécifique n'a été menée pour évaluer l'effet d'une insuffisance rénale ou hépatique sur la pharmacocinétique du risankizumab. Les analyses pharmacocinétiques de population n'ont montré aucune influence significative du taux de créatinine sérique, de la clairance de la créatine ou des paramètres de la fonction hépatique (ALAT/ASAT/bilirubine) sur la clairance du risankizumab chez les patients atteints de psoriasis.
Le risankizumab étant un anticorps monoclonal IgG1, il est principalement éliminé par catabolisme intracellulaire et, comme on pouvait s'y attendre, n'est pas métabolisé par les enzymes hépatiques du cytochrome P450 et n'est pas éliminé par voie rénale (voir «Posologie/Mode d'emploi»).
Patients âgés
Sur les 2'234 patients atteints de psoriasis en plaques traités par SKYRIZI, 243 avaient au moins 65 ans et 24 avaient au moins 75 ans. Parmi les patients traités par SKYRIZI, il n'y a généralement eu aucune différence entre les patients plus jeunes et plus âgés en termes d'exposition, de sécurité et d'efficacité du risankizumab (voir «Posologie/Mode d'emploi»).
Enfants et adolescents
La pharmacocinétique du risankizumab chez les enfants et les adolescents n'a pas été étudiée.
Poids corporel
La clairance et le volume de distribution du risankizumab augmentent avec le poids corporel. Aucune corrélation n'a été observée entre le poids corporel et des modifications cliniquement significatives de l'efficacité et de la sécurité du risankizumab; aucun ajustement posologique en fonction du poids n'est donc nécessaire.
Sexe ou origine ethnique
Chez les patients adultes atteints de psoriasis en plaques, le sexe ou l'origine ethnique n'ont eu aucun effet significatif sur la clairance du risankizumab. Dans une étude clinique sur la pharmacocinétique, aucune différence significative relativement à l'exposition au rizankizumab entre les sujets chinois ou japonais et les sujets d'origine caucasienne n'a été observée.
Données précliniquesDans les études sur la toxicité d'une administration répétée dans lesquelles des critères d'évaluation de la pharmacologie de sécurité ont aussi été évalués, ainsi que dans une étude sur la toxicité pour la reproduction et le développement menée chez le singe cynomolgus à des doses allant jusqu'à 50 mg/kg/semaine (ce qui correspond à une exposition environ 70 fois supérieure à l'exposition clinique à la dose maximale recommandée chez l'être humain [MRHD, maximum recommended human dose]), les données précliniques n'ont mis en évidence aucun risque particulier pour l'être humain.
Mutagénicité
Aucune étude n'a été effectuée pour évaluer la mutagénicité de SKYRIZI.
Carcinogénicité
Aucune étude n'a été effectuée pour évaluer la cancérogénicité de SKYRIZI. Dans une étude de 26 semaines sur la toxicité chronique chez le singe cynomolgus à des doses allant jusqu'à 50 mg/kg/semaine (environ 70 fois l'exposition clinique à la MRHD), aucune lésion prénéoplasique ou néoplasique n'a été observée.
Influence sur la fertilité
Les études de SKYRIZI chez le singe cynomolgus à des doses allant jusqu'à 50 mg/kg/semaine (environ 70 fois l'exposition clinique à la MRHD) n'ont montré aucun signe d'effets nocifs directs ou indirects sur la fertilité masculine ou féminine. Dans l'étude de 26 semaines sur la toxicité d'une administration répétée, l'examen histopathologique n'a montré aucun résultat significatif sur les organes reproducteurs des singes cynomolgus mâles ou femelles. Dans une étude de 26 semaines sur l'administration répétée à des singes cynomolgus mâles, aucun effet sur les paramètres de la fertilité masculine n'a été observé.
Pharmacologie et/ou toxicologie chez les animaux
Dans une étude de toxicologie de 26 semaines à des doses sous-cutanées hebdomadaires allant jusqu'à 50 mg/kg chez des singes cynomolgus mâles et femelles, aucun effet indésirable n'a été observé à des expositions correspondant à environ 70 fois l'exposition clinique à la MRHD.
Une autre étude a été menée sur la toxicité pour le développement pré- et postnatal chez le singe cynomolgus. Des femelles cynomolgus gestantes ont reçu à partir du 20e jour de gestation des doses sous-cutanées hebdomadaires de risankizumab de 5 ou de 50 mg/kg. Les singes cynomolgus (mères et jeunes animaux) ont été observés pendant 6 mois (180 jours) après la naissance. Ces doses ont entraîné des expositions correspondant à environ 70 fois l'exposition clinique à la dose maximale recommandée chez l'être humain (MRHD). Aucun décès et/ou malformation congénitale liés au médicament n'ont été observés chez les fœtus ou les jeunes animaux. Lors de l'évaluation des paramètres externes, viscéraux, squelettiques et neuro-comportementaux et des critères d'évaluation d'immunotoxicologie développementale, aucun effet sur la croissance et le développement n'a été rapporté. Les concentrations sériques moyennes chez les jeunes animaux ont augmenté de façon dose-dépendante, représentant environ 20 à 90% des concentrations maternelles correspondantes. La plupart des femelles cynomolgus adultes et tous les jeunes animaux du groupe traités par risankizumab jusqu'à 91 jours après la naissance avaient des concentrations sériques mesurables postpartum. Les concentrations sériques étaient inférieures à la limite de détection 180 jours après la naissance.
Remarques particulièresIncompatibilités
Aucune étude de tolérance n'ayant été effectuée, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C). Ne pas congeler. Conserver dans l'emballage d'origine pour protéger le contenu de la lumière. Tenir le médicament hors de portée des enfants.
Remarques concernant la manipulation
La solution contenue dans le stylo prérempli à 150 mg et la seringue préremplie à 150 mg est incolore à jaune et transparente à légèrement opalescente. La solution contenue dans la seringue préremplie à 75 mg est incolore à jaunâtre et transparente à légèrement opalescente.
La solution contenue dans le stylo prérempli à 150 mg et la seringue préremplie à 150 mg ainsi que dans la seringue préremplie à 75 mg peut comporter quelques particules de produit translucides à blanches. SKYRIZI ne doit pas être utilisé si la solution est trouble ou décolorée ou si elle contient de grosses particules.
Les patients peuvent s'injecter SKYRIZI eux-mêmes après avoir été formés à la technique d'injection sous-cutanée. Les patients doivent lire le mode d'emploi avant toute utilisation.
En cas d'utilisation de la seringue préremplie SKYRIZI 75 mg, il convient d'informer les patients qu'ils doivent prévoir 2 injections (2 seringues préremplies) pour s'administrer la dose complète de 150 mg. Chaque stylo prérempli et chaque seringue préremplie sont destinés à une utilisation unique.
Les patients ne doivent pas réaliser l'injection dans les zones où la peau est sensible, présente une ecchymose, est rouge, indurée ou dans une lésion psoriasique. SKYRIZI ne peut être injecté sur la face externe du bras que par un professionnel de santé ou un soignant.
Lors de l'utilisation du stylo prérempli, il convient de sortir la boîte du réfrigérateur avant l'injection et de la laisser reposer à température ambiante (30 à 90 minutes, à l'abri de la lumière directe du soleil), sans sortir le stylo prérempli de son emballage.
Pour une injection plus agréable, les patients utilisant la seringue préremplie à 75 mg ou 150 mg peuvent sortir la boîte du réfrigérateur avant l'injection et la laisser reposer à température ambiante (15 à 30 minutes, à l'abri de la lumière directe du soleil), sans sortir la seringue préremplie de son emballage.
Le médicament non utilisé ou les déchets doivent être éliminés conformément aux directives locales.
Numéro d’autorisation66944, 68118 (Swissmedic)
PrésentationSKYRIZI 150 mg, solution injectable en stylo prérempli
Chaque boîte contient 1 stylo prérempli. (B)
SKYRIZI 150 mg, solution injectable en seringue préremplie
Chaque boîte contient 1 seringue préremplie avec un dispositif de protection d'aiguille. (B)
SKYRIZI 75 mg, solution injectable en seringue préremplie
Chaque boîte contient 2 seringues préremplies avec un dispositif de protection d'aiguille et 2 compresses d'alcool. (B)
Titulaire de l’autorisationAbbVie AG, 6330 Cham, Suisse.
Mise à jour de l’informationMai 2021
|