CompositionPrincipes actifs
Immunoglobuline humaine normale pour perfusion sous-cutanée (IgSC).
Teneur minimale en immunoglobuline G (IgG) ≥95 %.
Distribution des sous-classes d'IgG (valeurs approx.):
IgG1 .......... 70 %
IgG2 .......... 25 %
IgG3 ............ 3 %
IgG4 ............ 2 %
La teneur maximale en IgA est de 300 µg/ml.
Excipients
Maltose
Polysorbate 80
Eau pour préparations injectables
Indications/Possibilités d’emploiTraitement de substitution chez les adultes, enfants et adolescents (0-18 ans) atteints de:
·Déficits immunitaires primaires (DIP) avec altération de la production d'anticorps.
·Déficits immunitaires secondaires (DIS) chez les patients souffrant d'infections sévères ou récurrentes, dont le traitement antimicrobien est inefficace et qui présentent une insuffisance prouvée en anticorps spécifiques (PSAF, proven specific antibody failure)* ou un taux d'IgG sérique <4 g/l.
*PSAF = incapacité à atteindre une augmentation d'au moins deux fois le titre d'anticorps IgG avec les vaccins à polysaccharides pneumococciques et antigènes polypeptidiques.
Posologie/Mode d’emploiTraitement de substitution
Le traitement de substitution doit être instauré et surveillé par un médecin spécialiste expérimenté dans le traitement de l'immunodéficience.
La dose et le schéma posologique dépendent de l'indication.
Le médicament doit être administré par voie sous-cutanée.
Pour une thérapie de substitution, la dose doit être éventuellement individualisée pour chaque patient en fonction de la réponse pharmacocinétique et clinique. Cutaquig peut être administré à intervalles réguliers, allant d'une administration quotidienne jusqu'à une administration toutes les deux semaines. Les schémas posologiques suivants sont fournis à titre indicatif:
Traitement de substitution des déficits immunitaires primaires (DIP)
Le schéma posologique devrait permettre d'atteindre un taux minimal d'IgG (mesuré avant l'injection suivante) de 5 à 6 g/l et doit avoir pour objectif de se situer dans l'intervalle de référence de l'IgG sérique pour le groupe d'âge. Une dose initiale (de charge) d'au minimum 0,2 à 0,5 g/kg (1,2 à 3,0 ml/kg) de poids corporel (PC) peut être nécessaire. Celle-ci peut être répartie sur plusieurs jours avec une dose journalière maximale de 0,1 à 0,15 g/kg PC.
Lorsque des taux d'IgG stables ont été atteints, des doses d'entretien seront administrées à intervalles répétés pour parvenir à une dose mensuelle cumulée de l'ordre de 0,4-0,8 g/kg PC (2,4 à 4,8 ml/kg PC). Il peut être nécessaire d'administrer chaque dose unique à des sites d'injection différents.
Les taux résiduels devraient être mesurés et évalués conjointement à l'incidence d'infection. Pour réduire le taux d'infection, il peut être nécessaire d'augmenter la dose et de viser un taux minimal plus élevé.
Traitement de substitution des déficits immunitaires secondaires
La dose unique recommandée sera administrée à intervalles répétés (environ une fois par semaine) pour parvenir à une dose mensuelle cumulative de l'ordre de 0,2-0,4 g/kg (1,2 à 2,4 ml/kg). Il peut être nécessaire d'administrer chaque dose unique à des sites d'injection différents.
Les taux résiduels d'IgG devraient être mesurés et évalués conjointement à l'incidence des infections. La dose devrait être ajustée si nécessaire pour obtenir une protection optimale contre les infections. Il peut être nécessaire d'augmenter la dose chez les patients présentant une infection persistante. Une diminution de la dose peut être envisagée lorsque le patient reste indemne d'infection.
Enfants et adolescents
La posologie chez les enfants et adolescents (0 à 18 ans) n'est pas différente de celle des adultes puisque la posologie pour chaque indication est calculée en fonction du poids corporel et ajustée au résultat clinique des indications pour le traitement de substitution.
Patients âgés
Étant donné que la dose est calculée en fonction du poids corporel et ajustée pour le résultat clinique dans les affections mentionnées ci-dessus, la dose chez les patients âgés n'est pas considérée comme étant différente de celle administrée aux patients âgés de 18 à 65 ans. Dans les études cliniques, Cutaquig a été évalué chez 17 patients âgés de plus de 65 ans. L'étude clinique n'a pas inclus un nombre suffisant de patients âgés de plus de 65 ans pour déterminer si les patients âgés répondent différemment des patients plus jeunes. Dans l'ensemble, aucune différence de sécurité ou d'efficacité n'est à attendre entre les patients âgés et les patients plus jeunes.
Mode d'administration
Administration sous-cutanée uniquement.
La perfusion par voie sous-cutanée pour le traitement à domicile doit être instaurée et supervisée par un personnel médical spécialisé, qui est expérimenté dans l'instruction des patients pour le traitement à domicile. Le patient et/ou sa personne accompagnante doivent être formés aux techniques de perfusion, à l'utilisation d'un dispositif de perfusion si nécessaire, à la manipulation aseptique, à la tenue d'un journal de traitement ainsi qu'à l'identification des effets indésirables graves et aux mesures correspondantes à prendre en cas de survenue de tels effets.
Cutaquig peut être injecté dans des sites tels que l'abdomen, la cuisse, la partie supérieure du bras et la face latérale de la hanche.
Vitesse de perfusion
La vitesse de perfusion et le volume de perfusion par site d'injection doivent être adaptés en fonction de la tolérance du patient.
Chez les patients n'ayant jamais été traités par IgSC, il est recommandé d'utiliser une vitesse initiale de perfusion de 15 ml/heure/site de perfusion. Chez les patients ayant déjà reçu un traitement par IgSC et qui changent pour Cutaquig, il est recommandé d'utiliser la vitesse de perfusion précédente lors des perfusions initiales. La vitesse de perfusion peut, si la tolérance est bonne, être augmentée progressivement pour les perfusions suivantes d'environ 10 ml/heure/site d'injection toutes les 2 à 4 semaines chez les adultes (≥40 kg) et d'un maximum de 10 ml/heure/site d'injection toutes les 4 semaines chez les patients pédiatriques (< 40 kg).
Par la suite, si le patient tolère bien les perfusions initiales à la dose complète par site de perfusion et à la vitesse de perfusion maximale, une augmentation de la vitesse de perfusion peut être envisagée, jusqu'à atteindre une vitesse de perfusion maximale de 67,5 ml/h/site de perfusion chez les adultes et de 25 ml/h/site de perfusion chez les enfants.
Plusieurs dispositifs de perfusion peuvent être utilisés simultanément.
Volume de perfusion par site
La quantité de produit perfusée à un site précis varie. Chez les enfants et adolescents, le site de perfusion peut être changé tous les 5 à 15 ml. Chez les adultes, des doses de plus de 30 ml peuvent être fractionnées selon les préférences du patient. Il n'y a pas de limite au nombre de sites de perfusion. La distance entre les sites de perfusion devrait être d'au moins 5 cm.
Contre-indicationsHypersensibilité à la substance active ou à l'un des autres composants mentionnés (voir rubrique «Composition»).
Cutaquig ne doit pas être injecté par voie intravasculaire.
De même, en cas de thrombocytopénie sévère et autres troubles de l'hémostase, il ne doit pas être administré par voie intramusculaire.
Chez les patients déficients en IgA, présentant des anticorps anti-IgA et une hypersensibilité connue à un traitement par globuline humaine.
Mises en garde et précautionsIl est vivement recommandé à chaque administration de Cutaquig de consigner le nom et le numéro du lot du produit afin de pouvoir faire le lien entre le patient et le lot du produit.
Ce médicament contient au maximum 90 mg de maltose par ml en tant qu'autre composant. L'interférence du maltose dans les dosages de glucose sanguin peut entraîner des résultats faussement élevés de glucose et, par conséquent, une administration inappropriée d'insuline, qui peut entraîner une hypoglycémie grave ou le décès. De même, des cas de véritable hypoglycémie peuvent rester non traités, si l'état hypoglycémique est masqué par des taux de glucose faussement élevés (voir rubrique «Interactions»). Concernant l'insuffisance rénale aiguë, voir ci-dessous.
Cutaquig est destiné à être administré par voie sous-cutanée uniquement. En cas d'administration accidentelle de Cutaquig dans un vaisseau sanguin, les patients peuvent développer un choc.
La vitesse de perfusion recommandée dans la rubrique «Mode d'administration/Vitesse de perfusion» doit être respectée scrupuleusement. Pendant toute la durée de la perfusion, les patients doivent être surveillés étroitement afin d'observer minutieusement l'apparition de symptôme.
Certains effets indésirables peuvent survenir plus fréquemment chez les patients qui reçoivent de l'immunoglobuline humaine normale pour la première fois ou, dans de rares cas, lorsque le produit d'immunoglobuline humaine normale est changé ou lorsque la perfusion précédente a été administrée il y a longtemps.
Les complications potentielles peuvent souvent être évitées en:
·commençant par injecter lentement le produit.
·s'assurant que les patients soient surveillés attentivement pendant toute la durée de la perfusion pour constater l'apparition éventuelle de symptôme. En particulier, les patients n'ayant jamais reçu d'immunoglobuline humaine normale, ainsi que les patients qui ont changé d'immunoglobuline ou dont la dernière injection remonte à une longue période, devraient être surveillés pendant la première perfusion et pendant la première heure suivant la première perfusion, afin de détecter d'éventuels effets indésirables.
Tous les autres patients doivent rester sous observation pendant au moins 20 minutes après l'administration.
En cas d'effet indésirable, soit la vitesse d'administration doit être réduite, soit l'injection doit être arrêtée. Si une réaction allergique ou anaphylactique est suspectée, l'injection doit être stoppée immédiatement. Le traitement nécessaire dépend de la nature et de la sévérité de la réaction indésirable.
En cas de choc, le traitement médical standard de déchoquage adapté doit être appliqué.
Hypersensibilité
Les vraies réactions allergiques sont rares. Elles peuvent apparaître notamment chez les patients qui présentent des anticorps anti-IgA. Ces patients doivent être traités avec une précaution particulière. Les patients ayant des anticorps anti-IgA, pour lesquels le traitement avec des IgG par voie sous-cutanée reste la seule option, ne doivent être traités par Cutaquig que sous supervision médicale étroite.
Dans de rares cas, les immunoglobulines humaines normales peuvent entraîner une chute de la pression artérielle associée à une réaction anaphylactique, même chez des patients ayant bien toléré un traitement antérieur par une immunoglobuline humaine normale.
Thromboembolie
Les patients devraient être suffisamment hydratés avant de recevoir des immunoglobulines. Des événements thromboemboliques artériels et veineux, y compris infarctus du myocarde, accident vasculaire cérébral, thrombose veineuse profonde et embolie pulmonaire ont été associés à l'utilisation d'immunoglobulines. La prudence est de mise chez les patients présentant des facteurs de risque préexistants pour des événements thrombotiques (tels qu'un âge avancé, une hypertension, un diabète sucré et des antécédents de maladies vasculaires ou d'épisodes thrombotiques dans l'anamnèse, des troubles thrombophiliques acquis ou héréditaires, des immobilisations prolongées, hypovolémie sévère, maladies qui augmentent la viscosité sanguine).
Les patients devraient être informés des premiers signes d'événements thromboemboliques, tels qu'un essoufflement, une douleur et un gonflement d'une extrémité, des déficits neurologiques focaux ainsi que des douleurs dans la poitrine et, dès l'apparition de ce type de symptômes, d'aller consulter immédiatement leur médecin.
Syndrome de méningite aseptique (SMA)
Des cas de syndrome de méningite aseptique sont survenus en association avec un traitement par immunoglobulines sous cutanées; les symptômes débutent en général plusieurs heures jusqu'à 2 jours après le traitement. L'arrêt du traitement par les immunoglobulines peut conduire à la résolution du SMA en quelques jours sans séquelles.
Les patients doivent être informés des premiers symptômes qui incluent des maux de tête sévères, une raideur de la nuque, une somnolence, de la fièvre, une photophobie, des nausées et des vomissements.
Altération du fonctionnement des reins/Insuffisance rénale
Des cas d'effets indésirables rénaux sévères ont été rapportés chez des patients recevant des immunoglobulines et en particulier les produits contenant du saccharose (Cutaquig ne contient pas de saccharose). Ces effets regroupent une insuffisance rénale aiguë, une nécrose tubulaire aiguë, une néphropathie tubulaire proximale et une néphrose osmotique. Les facteurs qui augmentent le risque de complications rénales incluent notamment une insuffisance rénale préexistante, un diabète sucré, une hypovolémie, la prise concomitante de médicaments néphrotoxiques, un âge supérieur à 65 ans, une septicémie, une hyperviscosité et une paraprotéinémie.
Hémolyse
Les IgG peuvent contenir des anticorps de groupes sanguins pouvant agir comme des hémolysines et capable d'induire le recouvrement in vivo des globules rouges par des immunoglobulines, provoquant ainsi une réaction antiglobuline directe positive (test de Coombs) et, dans de rares cas, une hémolyse. Les patients traités par produits à base d'immunoglobuline doivent être surveillés afin de déceler tous signes cliniques et symptômes d'hémolyse.
Sécurité virale
Les mesures habituelles de prévention des infections dues à l'utilisation de médicaments préparés à partir de sang ou de plasma humain comprennent la sélection des donneurs, la recherche des marqueurs spécifiques d'infection sur chaque don et sur les mélanges (pool) de plasma et l'inclusion dans le procédé de fabrication d'étapes efficaces pour l'inactivation/élimination des virus. Malgré ces mesures, le risque de transmission d'agents infectieux ne peut pas être totalement exclu lorsque des médicaments préparés à partir de sang ou de plasma humain sont administrés. Ceci s'applique également à tous les virus inconnus ou émergents ou aux autres types d'agents pathogènes.
Les mesures prises sont considérées comme efficaces contre les virus enveloppés tels que le virus de l'immunodéficience humaine (VIH), le virus de l'hépatite B (VHB) et le virus de l'hépatite C (VHC).
Les mesures prises peuvent être d'une efficacité limitée contre les virus non enveloppés tels que le virus de l'hépatite A (VHA) et le parvovirus B19.
Des données cliniques rassurantes révèlent l'absence de transmission du virus de l'hépatite A ou du parvovirus B19 par les immunoglobulines. Il est également admis que la concentration en anticorps contribue de façon importante à la sécurité virale.
Ce médicament contient 33,1 mg de sodium par flacon de 48 ml, ce qui équivaut à 1,7 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium par adulte.
InteractionsVaccins vivants atténués
L'administration d'immunoglobulines peut altérer pendant une période d'au moins 6 semaines à 3 mois l'efficacité des vaccins contenant des virus vivants atténués, comme les vaccins contre la rougeole, la rubéole, les oreillons et la varicelle. Après administration de ce médicament, un intervalle de 3 mois doit s'écouler avant une vaccination avec des vaccins contenant des virus vivants atténués. Dans le cas de la rougeole, cette diminution d'efficacité peut persister jusqu'à 1 an. Pour cette raison, les patients, qui se sont fait vacciner contre la rougeole, devraient se faire contrôler leur taux d'anticorps.
Mesure de la glycémie
Cutaquig contient du maltose, qui peut être faussement interprété comme du glucose par certains dispositifs de mesure de la glycémie. Pour éviter ces valeurs faussement élevées de glucose, seuls des systèmes de tests spécifiques au glucose doivent être utilisés chez les patients diabétiques
Interférence avec les tests sérologiques
Après administration d'immunoglobuline, l'augmentation transitoire de différents anticorps transférés passivement dans le sang des patients peut entraîner des résultats faussement positifs lors des tests sérologiques.
La transmission passive d'anticorps anti-érythrocytaires, par exemple A, B, D, peut fausser certains tests sérologiques (par exemple, numération des réticulocytes, haptoglobine et test de Coombs).
Enfants et adolescents
Les interactions mentionnées s'appliquent aux adultes ainsi qu'aux enfants et aux adolescents.
Grossesse, allaitementGrossesse
La sécurité de ce médicament au cours de la grossesse n'ayant pas été étudiée par des études cliniques contrôlées, il ne devrait dès lors être administré qu'avec précaution chez la femme enceinte et la femme qui allaite. Il a été démontré que les produits à base d'immunoglobulines traversent la barrière placentaire, surtout pendant le troisième trimestre. L'expérience clinique avec les immunoglobulines indique qu'aucun effet nocif n'est à attendre sur le déroulement de la grossesse, le fœtus et le nouveau-né.
Allaitement
Les immunoglobulines passent dans le lait maternel et peuvent contribuer à protéger le nouveau-né contre les agents pathogènes qui pénètrent à travers les muqueuses.
Fertilité
L'expérience clinique avec les immunoglobulines indique qu'aucun effet nocif n'est à attendre sur la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'aptitude à conduire et à utiliser des machines peut être altérée par certains effets indésirables associés à Cutaquig. Les patients qui présentent des effets indésirables pendant le traitement devraient attendre que ces effets se soient dissipés avant de conduire ou d'utiliser des machines.
Effets indésirablesRésumé du profil de sécurité
Des effets indésirables tels que frissons, maux de tête, étourdissements, fièvre, vomissements, réactions allergiques, nausées, arthralgies, pression artérielle basse et douleur lombaire modérée peuvent survenir occasionnellement.
Rarement, les immunoglobulines humaines normales peuvent provoquer une chute brutale de la pression artérielle et, dans des cas isolés, un choc anaphylactique, même si le patient n'a présenté aucun signe d'hypersensibilité lors d'une administration précédente.
Des réactions locales aux sites de perfusion: gonflement, douleur, rougeur, induration, sensation de chaleur locale, démangeaisons, ecchymose et éruption cutanée peuvent survenir fréquemment. La fréquence de ces réactions diminue normalement au fur et à mesure que le traitement se poursuit.
Pour des informations de sécurité concernant les agents pathogènes transmissibles, voir rubrique «Mises en garde et précautions».
Liste des effets indésirables
Les données de sécurité clinique sur Cutaquig chez les patients atteints de DIP sont basées sur l'étude pivot de Phase III en ouvert, à bras unique, prospective, multicentrique (n = 75; 4462 perfusions), sur ceux de l'étude d'extension de Phase III prospective, en ouvert, à bras unique, multicentrique (n = 27; 2777 perfusions) ainsi que ceux de l'étude de Phase III ouverte, à trois bras, multicentrique (n = 64; 1338 perfusions).
Le tableau présenté ci-dessous correspond à la classification par systèmes d'organes MedDRA (CSO et «terme préféré»).
Les fréquences par patient ont été évaluées selon les catégories suivantes: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100); rares (≥1/10000 à < 1/1000); très rares (< 1/10000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Fréquence des effets indésirables (FEI) par patient et par perfusion dans l'étude clinique avec Cutaquig:
|
Classe de systèmes d'organes MedDRA (CSO)
|
Effet indésirable
|
Fréquence/perfusion
|
Fréquence/patient
| |
Affections du système nerveux
|
Maux de tête
|
Occasionnels
|
Fréquents
| |
|
Étourdissement (vertige)
|
Rares
|
Occasionnels
| |
Affections gastro-intestinales
|
Nausée
|
Occasionnels
|
Fréquents
| |
|
Distension abdominale
|
Rares
|
Fréquents
| |
|
Douleurs abdominales
|
Rares
|
Fréquents
| |
|
Vomissement
|
Rares
|
Fréquents
| |
|
Haut-le-cœur
|
Rares
|
Occasionnels
| |
Affections hépatobiliaires
|
Hypertransaminasémie
|
Rares
|
Occasionnels
| |
Affections de la peau et du tissu sous-cutané
|
Éruption cutanée
|
Rares
|
Occasionnels
| |
|
Réaction cutanée
|
Rares
|
Occasionnels
| |
Affections musculo-squelettiques et systémiques
|
Myalgie
|
Rare
|
Fréquents
| |
|
Arthralgie
|
Rares
|
Occasionnels
| |
Troubles généraux et anomalies au site d'administration
|
Réaction au site d'injection
|
Très fréquents (23.3 %)
|
Très fréquents (75 %)
| |
|
Pyrexie
|
Rares
|
Fréquents
| |
|
Frissons
|
Rares
|
Fréquents
| |
|
Fatigue
|
Occasionnels
|
Fréquents
| |
|
Douleurs thoraciques
|
Rares
|
Occasionnels
| |
|
Syndrome pseudo-grippal
|
Rares
|
Occasionnels
| |
|
Malaise
|
Rares
|
Occasionnels
| |
|
Douleur
|
Rares
|
Occasionnels
| |
Investigations
|
Présence d'hémoglobine libre
|
Rares
|
Fréquents
| |
|
Test de Coombs positif
|
Rares
|
Occasionnels
| |
|
Présence d'hémoglobine libre
|
Rare
|
Fréquents
| |
|
Diminution de l'haptoglobine
|
Rares
|
Occasionnels
| |
|
Augmentation de l'hémoglobine
|
Rares
|
Occasionnels
| |
|
Augmentation de la créatine sanguine
|
Rares
|
Occasionnels
|
*MedDRA [Dictionnaire médical pour les activités dans le cadre de l'autorisation de mise sur le marché des médicaments].
Les effets indésirables suivants ont été observés durant l'utilisation de Cutaquig après sa mise sur le marché. Ces effets indésirables rapportés de façon spontanée étant issus d'une population de taille indéterminée, il n'est pas toujours possible d'estimer avec précision leur fréquence ou d'établir un lien de causalité avec l'exposition au médicament.
Les réactions déjà rapportées au cours des études cliniques avec Cutaquig ne sont pas incluses dans cette liste:
|
Classe de systèmes d'organes MedDRA (CSO) par ordre:
|
Effet indésirable (EI)
| |
Affections du système immunitaire
|
Hypersensibilité (p.ex. erythème, urticaire)
| |
Affections vasculaires
|
Thromboembolie, thrombose (p.ex. thrombose veineuse profonde, accident vasculaire cérébral), hypertension
| |
Affections de la peau et du tissu sous-cutané
|
Prurit
| |
Affections musculosquelettiques et du tissu conjonctif
|
Douleurs dorsales
|
D'autres effets indésirables ont été signalés après le début de la commercialisation de produits à base d'immunoglobulines: œdème du visage, tremblements, pâleur, bronchospasme, dyspnée, toux, diarrhée, rougeur du visage, sensation de chaleur, sensation de froid, asthénie, douleur au site de perfusion, sensation de gorge serrée, méningite aseptique.
Informations sur la sécurité virale: voir rubrique «Mises en garde et précautions».
Enfants et adolescents
On s'attend à ce que la fréquence, le type et la gravité des effets indésirables chez les enfants et adolescents soient les mêmes que chez les adultes.
Déclaration des effets indésirables suspectés
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes conséquences d'un surdosage ne sont pas connues.
Propriétés/EffetsCode ATC
J06BA01
Classe pharmacothérapeutique: Antisérums et immunoglobulines: Immunoglobuline humaine normale, pour administration extravasculaire
Mécanisme d'action
L'immunoglobuline humaine normale contient principalement des immunoglobulines G (IgG) présentant un large spectre d'anticorps contre les agents infectieux.
L'immunoglobuline humaine normale contient des anticorps IgG présents dans la population normale. La production a lieu à partir de plasma mélangé provenant d'au moins 1000 dons. Les sous-classes d'IgG sont présentes dans une répartition à peu près identique à celle du plasma humain natif. Des doses appropriées de Cutaquig peuvent ramener à une valeur normale des taux pathologiquement bas d'immunoglobuline G.
Pharmacodynamique
Des doses adéquates de ce médicament peuvent entraîner une augmentation des taux anormalement bas d'immunoglobuline G vers une valeur normale en cas de déficits immunitaires primaires et secondaires et aider ainsi à lutter contre les infections.
Efficacité clinique
Lors de la première étude clinique, un total de 75 patients (37 adultes, 12 jeunes enfants [≥2 et < 6 ans], 14 enfants plus âgés [≥6 et < 12 ans] et 12 adolescents [≥12 et < 17 ans]) atteints d'un syndrome de déficit immunitaire primaire a été traité par Cutaquig pendant une durée allant jusqu'à 64 semaines. La dose moyenne administrée chaque semaine à chaque patient était de 0,187 g/kg chez les patients adultes, de 0,150 g/kg chez les jeunes enfants, de 0,164 g/kg chez les enfants plus âgés et de 0,170 g/kg chez les adolescents. Au total, 4462 injections hebdomadaires de Cutaquig ont été administrées.
Aucune infection bactérienne grave n'a été rapportée ni pendant la période de wash-in/wash-out, ni pendant la période d'efficacité chez les patients recevant Cutaquig dans le cadre de l'étude clinique.
Cutaquig a été évalué chez 38 patients pédiatriques (26 enfants [entre 2 et < 12 ans] et 12 adolescents [entre 12 et < 16 ans]) présentant un déficit immunitaire primaire. Aucune exigence relative à la dose pédiatrique n'a été nécessaire pour atteindre les taux sériques d'IgG souhaités.
L'étude d'extension était une étude de suivi de la sécurité de Phase III, prospective, ouverte, à 1 bras, multicentrique, qui a été menée chez 27 patients (17 adultes, 2 jeunes enfants [≥2 et < 6 ans], 4 enfants plus âgés [≥6 et < 12 ans], 4 adolescents [≥12 et < 17 ans]) présentant un déficit immunitaire primaire. Outre les 21 patients de l'étude pivot, 6 patients ont été nouvellement recrutés. Les patients ayant participé à l'étude pivot auparavant ont été suivis sur une durée allant jusqu'à 4,5 ans et les patients nouvellement recrutés pendant 12 mois. Les patients ont reçu Cutaquig une fois par semaine (25 patients) ou toutes les deux semaines (2 patients). La dose moyenne de Cutaquig effectivement administrée par patient a été de 0,127 g/kg chez les jeunes enfants, 0,210 g/kg chez les enfants plus âgés, 0,160 g/kg chez les adolescents et 0,166 g/kg chez les adultes. Les patients ont reçu un total de 2777 perfusions (2740 une fois par semaine et 37 toutes les deux semaines). Une infection bactérienne sévère (IBS) sous la forme d'une bactériémie/septicémie a été rapportée.
Afin de surveiller la sécurité, la tolérance et l'efficacité de Cutaquig, une étude de Phase III prospective, ouverte, à trois bras, multicentrique a été menée chez 64 patients atteints de, âgés de 5 à 74 ans (59 adultes, 1 jeune enfant [≥2 et < 6 ans], 2 enfants plus âgés [≥6 et < 12 ans] et 2 adolescents [≥12 et < 17 ans]).
À l'issue de la période de stabilisation de 4 semaines, les patients ont débuté une période de traitement avec un suivi de 24 semaines et ont été répartis dans les 3 cohortes suivantes:
·Cohorte 1: évaluation de l'augmentation du volume par site jusqu'à un maximum de 100 ml/site de perfusion.
·Cohorte 2: évaluation de l'augmentation de la vitesse de perfusion jusqu'à un maximum de 100 ml/heure et par site de perfusion repectivement jusqu'à la vitesse de perfusion maximale permise par la pompe à perfusion utilisée.
·Cohorte 3: évaluation de l'administration de Cutaquig toutes les deux semaines, à une dose équivalant à deux fois la dose hebdomadaire définie selon le poids.
Un critère d'évaluation principal supplémentaire était composé de la comparaison des taux résiduels d'IgG entre les perfusions hebdomadaires et les perfusions toutes les deux semaines, ainsi que de l'évaluation de la sécurité et de la tolérance des perfusions lors de l'augmentation des volumes et des vitesses de perfusion sur chaque site de perfusion.
Globalement, les patients ont reçu un total de 1338 perfusions (386 dans la Cohorte 1, 396 dans la Cohorte 2, 556 dans la Cohorte 3). Dans la Cohorte 1 (n = 15 adultes), le volume maximal moyen atteint par site a été de 69,4 ml/site de perfusion, avec un volume maximal de 108 ml/site de perfusion. Un tiers des patients (5/15; 33,3 %) a atteint au moins 90 % du volume maximal autorisé de 100 ml/site de perfusion, un tiers a atteint entre 50 % et < 90 % du volume maximal autorisé, et un tiers a atteint moins de 50 % du volume maximal autorisé. La vitesse maximale médiane atteinte par patient a été de 56,9 ml/heure (plage: entre 34,0 ml/heure et 94,7 ml/heure.
Dans la Cohorte 2 (n = 15; 13 adultes, 1 enfant plus âgé [≥6 et < 12 ans], 1 adolescent [≥12 et < 17 ans]), la vitesse maximale moyenne atteinte par site a été de 42,1 ml/heure/site, avec une vitesse maximale de 67,5 ml/heure/site de perfusion. Chez 73,3 % des patients, une vitesse maximale par site < 50 % du volume maximal autorisé de 100 ml/h/site de perfusion a été atteinte. Les 26,7 % des patients restants ont atteint entre 50 % et 75 % du volume maximal autorisé. La vitesse maximale médiane atteinte par patient a été de 135,0 ml/h (plage: entre 51,4 ml/h et 192,0 ml/h).
Dans la Cohorte 3 (n = 34; 31 adultes, 1 jeune enfant [≥2 et < 6 ans], 1 enfant plus âgé [≥6 et < 12 ans], 1 adolescent [≥12 et < 17 ans]), une diminution des taux résiduels moyens (ET) d'IgG a été observée avec le schéma d'administration toutes les deux semaines (9,927 [2,0146] g/l) par comparaison avec l'administration hebdomadaire (10,364 [1,9632] g/l) (p = 0,0017; limite inférieure de l'IC unilatéral à 97,5 % = -0,799). La vitesse maximale médiane atteinte par patient a été de 93,5 ml/h (plage: entre 24,3 ml/h et 145,9 ml/h).
La dose moyenne de Cutaquig effectivement administrée en fonction du poids a été de 0,143 g/kg dans la Cohorte 1, 0,157 g/kg dans la Cohorte 2 et 0,256 g/kg dans la Cohorte 3, respectivement.
Aucune infection bactérienne sévère n'a été rapportée au cours de l'étude et le taux global d'infections bactériennes sévères a été de 0,00 personne-année (limite supérieure de l'IC à 98 % [méthode alternative] = 0,135 [0,614 dans la Cohorte 1, 0,602 dans la Cohorte 2 et 0,244 dans la Cohorte 3]).
Enfants et adolescents
Aucune différence n'a été observée dans les propriétés pharmacodynamiques entre les adultes ainsi que les enfants et les adolescents.
PharmacocinétiqueLors d'une étude clinique de phase III, une sous-étude pharmacocinétique (PK) a été menée chez 37 patients atteints de déficits immunitaires primaires (DIP). Des échantillons de sang pour l'étude PK ont été prélevés avant le passage à Cutaquig (profil IgIV: PKIV), après la 11e injection de Cutaquig (premier profil SC: PKSC1) et après la 28e injection de Cutaquig (deuxième profil SC: PKSC2). L'objectif de la sous-étude PK était de comparer les ASC (Aire sous la courbe) après administration i.v. et s.c., en utilisant un facteur de correction de dose (FCD) de 1,5. À partir d'un modèle PK de population, les paramètres PK ont été estimés et des simulations ont été effectuées.
Absorption
Après l'administration par voie sous-cutanée de Cutaquig, le taux sérique maximal est atteint après environ 2 jours.
Distribution
En cas d'administration sous-cutanée, l'immunoglobuline normale se répartit d'abord dans le tissu sous-cutané local, puis diffuse lentement dans la circulation sanguine du receveur et dans l'espace extravasculaire.
Métabolisme
Du fait de l'absorption progressive, l'administration de IgSC conduit à des profils plus plats et à des fluctuations plus faibles à l'état d'équilibre (Steady State) par rapport au traitement par IgIV: la Cmax moyenne était plus faible après IgSC (13,2±3,4 g/l et 13,5±3,7 g/l pour PKSC1 et PKSC2, respectivement) par rapport à la concentration à la fin de la perfusion par IgIV (18,0±4,5 g/l). En conséquence, les taux sériques moyens d'IgG et les taux résiduels de sous-classe d'IgG étaient plus élevés après le traitement SC (11,5 et 11,7 g/l pour PKSC1 et PKSC2, respectivement, la marge globale étant de 6,5 à 18,9 g/l), par rapport à ceux à la fin du traitement par IgIV (10,1 g/l; amplitude de variation: 6,5 g/l à 14,3 g/l).
La biodisponibilité SC a été calculée à 75 %, ce qui correspond à un facteur de correction de dose de 1,3 pour atteindre une ASC d'exposition identique après traitement par IgSC corrélé au poids par rapport au traitement par IgIV.
Une modélisation basée sur la PK et une simulation à partir des données recueillies dans l'étude clinique sur l'administration hebdomadaire de Cutaquig ont montré qu'une dose ajustée en fonction du poids, sans facteur de correction de dose pour la biodisponibilité SC plus basse, serait suffisante pour maintenir une exposition systémique aux IgG dans l'intervalle thérapeutique, pour un intervalle entre les injections allant jusqu'à 1 semaine, y compris pour une administration plus fréquente (par ex. des doses quotidiennes). Des intervalles de dose plus longs (en particulier pour les niveaux de base d'IgG faibles) augmentent le risque de tomber à un taux résiduel d'IgG sous 5 g/l.
Exemple: En considérant un taux basal d'IgG de 4,0 g/l et un facteur de conversion de dose de 1,0 pour passer du traitement par IgIV au traitement par IgSC, il a été estimé que le pourcentage de patients tombant sous le taux résiduel d'IgG de 5 g/l atteindrait 4 % avec une dose toutes les 2 semaines contre 1,4 % avec des intervalles de dose inférieurs ou égaux à une semaine.
Élimination
Les IgG et les complexes IgG sont dégradés dans les cellules du système réticulo-endothélial. La demi-vie médiane des IgG après administration de Cutaquig chez les patients atteints de DIP a été estimée à environ 16 jours [9,2-36,3], le calcul a été effectué dans le modèle PK de population en supposant qu'il n'y a pas de production endogène d'IgG.
Enfants et adolescents
Aucune différence cliniquement pertinente n'a été observée concernant les paramètres pharmacocinétiques entre les patients adultes et pédiatriques de l'étude DIP.
La modélisation basée sur la pharmacocinétique et la simulation réalisée sur les données recueillies dans l'étude clinique sur l'administration hebdomadaire de Cutaquig indiquent qu'une dose ajustée en fonction du poids serait suffisante pour maintenir des taux systémiques d'IgG dans l'intervalle thérapeutique quel que soit l'âge.
Données précliniquesLes immunoglobulines sont des constituants normaux du plasma humain. Les données précliniques, qui reposent sur les études conventionnelles de pharmacologie sur la sécurité et la tolérance locale, laissent conclure qu'il n'y a pas de danger particulier pour l'homme. Étant donné que l'expérience clinique n'apporte pas de preuves d'un potentiel carcinogène ou mutagène des immunoglobulines, aucune étude expérimentale n'a été réalisée chez les espèces hétérologues.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, le médicament ne doit pas être mélangé à d'autres médicaments.
Influence sur les méthodes de diagnostic
Voir rubrique «Interactions».
Stabilité
Cutaquig est stable entre 2°C et 8°C jusqu'à la date de péremption imprimée sur l'emballage et l'étiquette sous la mention «EXP».
Stabilité après ouverture
Cutaquig doit être utilisé immédiatement après l'ouverture du flacon.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Conserver dans l'emballage d'origine.
Conserver le récipient dans son carton pour le protéger de la lumière.
Tenir hors de la portée des enfants.
Pendant sa durée de conservation, le produit emballé peut être conservé à température ambiante (jusqu'à 25°C maximum) pendant une durée maximale de 9 mois. Dans ce cas, la durée de conservation du produit expire après 9 mois. Au début du stockage à température ambiante, la nouvelle date de péremption doit être notée sur l'emballage extérieur.
Remarques concernant la manipulation
Avant administration, le produit doit être amené à température ambiante ou à température corporelle.
Avant l'administration, les médicaments à usage parentéral doivent être inspectés visuellement pour s'assurer de l'absence de particules et de coloration anormale.
Ne pas utiliser de solutions troubles ou qui présentent des dépôts.
Guide d'utilisation
1. Préparez le nombre de flacons de Cutaquig nécessaire
·Si les flacons sont conservés au réfrigérateur, laissez-les atteindre la température ambiante avant la perfusion ce qui peut durer au moins 90 minutes.
·Ne réchauffez pas les flacons avec une source extérieure de chaleur et ne les placez pas au micro-ondes.
·N'agitez pas les flacons, pour éviter la formation de mousse.
2. Préparation de l'injection
·Préparez une zone de travail propre en utilisant des lingettes antiseptiques ou une solution désinfectante (Figure 1).
·Rassemblez le matériel de perfusion:
·Pompe à perfusion (facultatif) et seringue(s) compatible(s)
·Aiguille ou système de transfert ventilé (pour prélever le produit du flacon)
·Aiguilles pour perfusion sous-cutanée
·Alcool et lingettes alcoolisées/antiseptiques
·Compresse ou pansement et adhésif transparent
·Conteneur pour objets tranchants
·Journal de traitement et stylo
·Lavez-vous soigneusement les mains et attendez jusqu'à ce qu'elles soient bien sèches (Figure 2). Utilisez un gel désinfectant comme on vous l'a montré pendant la formation.
·Si nécessaire, programmez la pompe conformément aux instructions du manuel de l'utilisateur et comme vous l'a montré votre professionnel de la santé.
3. Vérification et ouverture des flacons
·Suivez les étapes suivantes pour chaque flacon:
·Vérifiez que la dose indiquée sur l'étiquette est la bonne et correspond à celle indiquée sur l'ordonnance,
·Vérifiez l'aspect de la solution (elle doit être transparente et incolore à jaune pâle ou brun clair),
·Assurez vous que le capuchon protecteur est bien présent et qu'il n'est pas cassé,
·Vérifiez la date de péremption et le numéro de lot.
·N'utilisez pas la solution si elle est trouble ou contient des particules.
·Retirez le capuchon protecteur.
·Désinfectez le bouchon en caoutchouc à l'aide d'une lingette antiseptique et laissez-le sécher (Figure 3).
4. Préparation et remplissage de la seringue
·Ouvrez l'emballage de la seringue et de l'aiguille stériles ou le système d'injection ventilé.
·Si vous utilisez une aiguille, fixez-la à la seringue en la vissant.Si vous utilisez un système de perfusion ventilé, fixez-le au flacon.
·Si vous utilisez une aiguille, reculez le piston de la seringue pour la remplir d'une quantité d'air approximativement égal à la quantité de solution à prélever du flacon.Cette étape n'est pas nécessaire si vous utilisez un système de perfusion ventilé.
·Si vous utilisez une aiguille, insérez-la dans le flacon et retournez-le. Injectez l'air dans le flacon - assurez-vous que la pointe de l'aiguille n'est pas immergée dans la solution pour éviter la formation de mousse.Si vous utilisez un système de perfusion ventilé, connectez la seringue au système d'injection.
·Ensuite, assurez-vous ensuite que l'aiguille reste toujours dans la solution et aspirez lentement Cutaquig (Figure 4).
·Retirez l'aiguille du flacon ou détachez la seringue du système de perfusion.
·Il peut être nécessaire de répéter cette procédure si vous devez utiliser plus d'un flacon pour obtenir la dose calculée.
·Lorsque vous avez terminé, retirez l'aiguille et placez-la dans le conteneur pour objets tranchants.
·Passez immédiatement à l'étape suivante, car la solution d'immunoglobulines doit être utilisée rapidement.
5. Préparation de la pompe à perfusion et de la tubulure (facultatif)
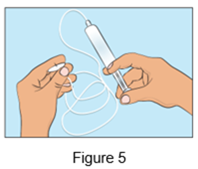
·Fixez la seringue remplie à la/ ou aux aiguilles pour perfusion sous-cutanée et faites avancer lentement le piston de la seringue pour remplir la tubulure de Cutaquig et évacuer l'air (Figure 5); veuillez ne pas purger complètement l'/les aiguille(s) pour perfusion sous-cutanée.
·Suivez les instructions du fabricant pour la préparation de la pompe à perfusion.
6. Choix des sites de perfusion et insertion des aiguilles pour perfusion sous-cutanée
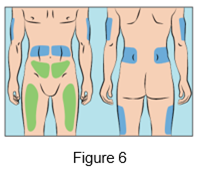
·Cutaquig peut être perfusé dans les zones suivantes: abdomen, cuisse, partie haute du bras et/ou de la cuisse/hanche (Figure 6).
·Les sites de perfusion doivent être espacés d'au moins 5 cm.
·Utilisez des sites d'injection différents de ceux que vous avez utilisés lors de l'administration précédente.
·Évitez d'insérer l'aiguille dans des cicatrices, des tatouages, des vergetures ou des zones de peau blessée/inflammée/rouge.
·Nettoyez la peau du (des) site(s) de perfusion choisi(s) avec une lingette antiseptique et laissez la peau sécher.
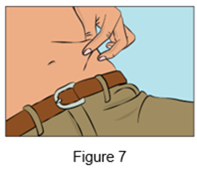
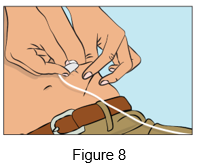
·Pincez la peau entre le pouce et l'index autour du site d'injection (Figure 7) et retirez délicatement la coiffe de l'aiguille et insérez l'aiguille dans la peau (Figure 8). L'angle de l'aiguille dépendra du type d'aiguille pour perfusion sous-cutanée utilisé.
7. Vérification de la perfusion
·La solution ne doit pas être injectée dans un vaisseau sanguin.
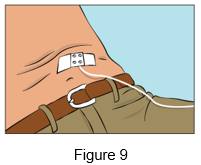
·Fixez l'aiguille en place en la recouvrant d'une compresse stérile et d'un adhésif ou d'un pansement transparent (Figure 9).
8. Démarrage de la perfusion
·Commencez la perfusion. Si vous utilisez une pompe à perfusion, suivez les instructions du fabricant.
9. Consignation de la perfusion
·Sur chaque flacon de Cutaquig se trouve une étiquette détachable qui indique des informations sur le numéro de lot. Collez cette étiquette dans votre journal de traitement du patient ou dans votre journal de perfusions. Inscrivez les informations concernant la dose, la date, l'heure, le site de perfusion ainsi que toute infection, tout effet indésirable ou autre commentaire en lien avec cette perfusion.
10. Lorsque la perfusion est terminée
·Retirez l'aiguille et placez-la immédiatement dans le conteneur pour objets tranchants.
·Si nécessaire, appuyez une petite compresse à l'endroit où l'aiguille a été insérée et placez un pansement.
·Jetez immédiatement tout le matériel à usage unique et tout reste de produit non utilisé. Videz le(s) flacon(s) comme recommandé par votre professionnel de la santé et conformément aux exigences locales.
Rangez et conservez dans un lieu sûr tout le matériel réutilisable (p.ex. la pompe) jusqu'à la prochaine perfusion.
Les restes de médicaments non utilisés ou les déchets doivent être éliminés conformément à la réglementation locale.
Numéro d’autorisation68222 (Swissmedic)
Présentation6 ml, 12 ml, 24 ml ou 48 ml de solution dans un flacon (verre de type I) muni d'un bouchon en caoutchouc bromobutyle – conditionnements de 1, 10 ou 20 flacons. (B)
Toutes les présentations peuvent ne pas être commercialisées.
Titulaire de l’autorisationOctapharma AG,
Seidenstrasse 2
8853 Lachen
Mise à jour de l’informationDécembre 2023
|