CompositionPrincipes actifs
Tralokinumab
Le tralokinumab est produit dans des cellules de myélome de souris grâce à la technologie de l’ADN recombinant.
Excipients
Acétate de sodium trihydraté, acide acétique 99%, chlorure de sodium, polysorbate 80, eau pour préparations injectables.
1 ml de solution injectable contient 3 mg de sodium.
Indications/Possibilités d’emploiAdtralza® est indiqué dans le traitement de la dermatite atopique (DA) modérée à sévère de l’adulte, lorsque le traitement par des médicaments topiques délivrés sur ordonnance ne permet pas de contrôler adéquatement la maladie ou n'est pas recommandé.
Posologie/Mode d’emploiLe traitement doit être initié par un professionnel de la santé expérimenté dans le diagnostic et le traitement de la dermatite atopique.
Posologie usuelle
La dose recommandée d'Adtralza® pour les patients adultes est une dose initiale de 600 mg (quatre injections de 150 mg), suivie d’une dose de 300 mg (deux injections de 150 mg) administrés toutes les 2 semaines par injection sous-cutanée.
Chez les patients ayant obtenu une quérison complète ou presque complète de la peau, une posologie toutes les quatre semaines peut être envisagée, à la discrétion du médecin prescripteur.
Chez les patients ayant un poids corporel élevé (> 100 kg), une posologie toutes les deux semaines peut être plus appropriée (voir « Pharmacocinétique »).
Chez les patients qui n'ont pas montré de réponse après 16 semaines de traitement, l'arrêt du traitement doit être envisagé.
Adtralza® peut être utilisé avec ou sans corticostéroïdes topiques. L'utilisation d'inhibiteurs de la calcineurine topique avec Adtralza® est possible.
Afin d’assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est nécessaire chez les patients présentant un trouble de la fonction hépatique. Les données disponibles chez les patients présentant une insuffisance hépatique modérée ou sévère sont très limitées (voir « Pharmacocinétique »).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n'est nécessaire chez les patients présentant un trouble de la fonction rénale. Les données disponibles chez les patients présentant une insuffisance rénale sévère sont très limitées (voir « Pharmacocinétique »).
Patients âgés
Aucun ajustement posologique n’est recommandé chez les patients âgés (voir « Pharmacocinétique »).
Enfants et adolescents
La sécurité et l’efficacité d'Adtralza® chez les enfants âgés de moins de 18 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Mode d’administration
Voie sous-cutanée.
Adtralza® est administré par injection sous-cutanée dans la cuisse ou l’abdomen, à l’exception d’une zone de 5 centimètres autour du nombril. Si l'injection est réalisée par une autre personne, elle peut également être effectuée dans le haut du bras.
La dose initiale de 600 mg doit être administrée sous forme de quatre injections consécutives de 150 mg d'Adtralza® chacune, en choisissant des sites d’injection différents.
Il est recommandé de changer de site d’injection à chaque injection.
Adtralza® ne doit pas être administré aux endroits où la peau est sensible ou endommagée, ou présente des contusions ou de cicatrices.
Adtralza® peut être injecté par le patient lui-même ou par un soignant si le professionnel de santé juge cela approprié. Avant utilisation, le patient et/ou le personnel soignant doivent être formés à la préparation et à l'utilisation d'Adtralza® conformément aux instructions d'utilisation figurant dans la notice d'emballage.
Contre-indicationsHypersensibilité au principe actif ou à l’un des excipients selon la composition.
Mises en garde et précautionsRéactions d'hypersensibilité
En cas de survenue d’une réaction d’hypersensibilité systémique (immédiate ou retardée), l’administration d'Adtralza® doit être interrompue et un traitement approprié doit être instauré.
Dans un total de cinq études sur la dermatite atopique, aucun cas de réaction anaphylactique n'a été rapporté. Dans des études cliniques portant sur une autre indication, de très rares cas de réaction anaphylactique ont été rapportés après l'administration de tralokinumab.
Conjonctivite
Les patients traités par tralokinumab qui développent une conjonctivite non résolue avec un traitement standard doivent subir un examen ophtalmologique.
Infection par des helminthes
Les patients présentant des infections connues par des helminthes ont été exclus des études cliniques. L’influence du tralokinumab sur la réponse immunitaire contre les infections dues à des helminthes, par inhibition de la voie de signalisation de l’IL-13, n’est pas connue.
Les patients présentant des infections pré-existantes par des helminthes doivent être traités avant que l'administration d'Adtralza® ne soit initiée. Si des patients sont infectés au cours du traitement par tralokinumab et ne répondent pas au traitement anti-helminthique, le traitement par tralokinumab doit être interrompu jusqu’à la guérison de l’infection.
Vaccinations
Les vaccins vivants et vivants atténués ne doivent pas être administrés au cours du traitement par Adtralza®, car leur sécurité et leur efficacité cliniques n'ont pas été établies. L'intervalle de temps entre une vaccination par des vaccins vivants et le traitement par Adtralza® doit être conforme aux recommandations actuelles en matière de vaccination avant l’instauration du traitement par des agents immunomodulateurs. Des réponses immunitaires aux vaccins tétaniques et méningococciques non vivants ont été évaluées (voir « Interactions »).
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par 1 ml de solution injectable.
InteractionsEffets d'Adtralza® sur d'autres médicaments
La sécurité et l'efficacité de l'utilisation concomitante d'Adtralza® avec des vaccins vivants ou vivants atténués n’ont pas été étudiées.
Les réponses immunitaires aux vaccins non vivants ont été étudiées dans une étude dans laquelle des adultes souffrant de dermatite atopique reçu une dose initiale de 600 mg de tralokinumab (quatre injections de 150 mg), suivie de 300 mg de tralokinumab toutes les deux semaines en injection sous-cutanée. Après 12 semaines d'administration de tralokinumab, les patients ont reçu un vaccin combiné tétanos, diphtérie et coqueluche acellulaire, et un vaccin méningococcique, puis les réponses immunitaires ont été évaluées 4 semaines plus tard. Les réponses anticorps au vaccin tétanique et au vaccin méningococcique étaient similaires chez les patients traités par tralokinumab et les patients recevant le placebo.
Pour plus d'informations sur les vaccins vivants ou vivants atténués, voir « Mises en garde et précautions ».
Interactions pharmacocinétiques
Interactions avec les enzymes du cytochrome P450
On ne s’attend pas à ce que le tralokinumab soit métabolisé par les enzymes hépatiques ou éliminé par les reins. Aucune interaction n’est attendue entre le tralokinumab et les inhibiteurs, inducteurs ou substrats des enzymes métabolisant les médicaments. Aucun ajustement posologique n'est nécessaire (voir « Pharmacocinétique »).
Interactions avec d'autres médicaments
Les effets du tralokinumab sur la pharmacocinétique des substrats du CYP ont été étudiés chez des patients atteints de DA après une administration répétée. Les résultats n'ont pas montré d'effets cliniquement significatifs du tralokinumab sur la caféine (substrat du CYP1A2), la warfarine (substrat du CYP2C9), l'oméprazole (substrat du CYP2C19), le métoprolol (substrat du CYP2D6) et le midazolam (substrat du CYP3A). Le tralokinumab ne devrait donc pas influencer la pharmacocinétique des médicaments utilisés simultanément et métabolisés par les enzymes du cytochrome P450.
Grossesse, AllaitementGrossesse
Il n'existe pas de données suffisantes sur l'utilisation d'Adtralza® chez la femme enceinte.
Les études effectuées chez l’animal n'ont pas montré d'effets directs ou indirects sur la toxicité pour la reproduction (voir « Données précliniques »). Le tralokinumab ne doit pas être utilisé pendant la grossesse, sauf si les bénéfices potentiels dépassent les risques potentiels pour le fœtus.
Allaitement
On ne sait pas si le tralokinumab est excrété dans le lait maternel ou absorbé par voie systémique après ingestion. Une décision doit être prise quant à l'interruption de l'allaitement ou du traitement par Adtralza®. Il convient d'évaluer à la fois les bénéfices de l'allaitement pour l'enfant et les bénéfices du traitement pour la mère.
Fertilité
Les études animales n'ont montré aucun effets sur les organes reproducteurs mâles ou femelles ou sur la numération, la motilité et la morphologie des spermatozoïdes (voir « Données précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'influence d'Adtralza® sur l'aptitude à conduire ou à utiliser des machines n'a pas été spécifiquement étudiée.
Effets indésirablesRésumé du profil de sécurité
La sécurité d'Adtralza® a été évaluée dans cinq études randomisées, contrôlées versus placebo et en double aveugle réalisées chez des patients atteints de dermatite atopique modérée à sévère, à savoir trois études de phase III (ECZTRA 1, ECZTRA 2, ECZTRA 3), une étude de recherche de dose et une étude sur la réponse vaccinale.
Dans ces cinq études sur la dermatite atopique, 1 991 patients ont été traités par injections sous-cutanées de tralokinumab avec ou sans association de corticostéroïdes topiques. Au total, 807 patients ont été traités par tralokinumab pendant au moins un an.
Les patients présentant des idées/comportements suicidaires ont été exclus des études cliniques.
Les effets indésirables les plus fréquents ont été des infections des voies respiratoires supérieures (principalement des rhumes).
Dans les cinq études sur la dermatite atopique, la proportion de patients qui ont arrêté le traitement en raison d'événements indésirables pendant la phase initiale de traitement allant jusqu'à 16 semaines était de 2,3% dans le groupe tralokinumab et de 2,8% dans le groupe placebo.
Liste des effets indésirables
Les effets indésirables sont rangés par classe de système d’organes de la classification MedDRA et par fréquence selon la convention suivante :
« très fréquents » (≥1/10)
« fréquents » (≥1/100 à <1/10)
« occasionnels » (≥1/1000 à <1/100)
« rares » (≥1/10 000 à <1/1000)
« très rares » (<1/10 000)
Au sein de chaque groupe de fréquence, les effets indésirables sont classés par ordre de gravité décroissant. Les indications de fréquence sont basées sur la période initiale de traitement allant jusqu'à 16 semaines dans les cinq études.
|
Classe de système d’organes de MedDRA
|
Fréquence
|
Effet indésirable
| |
Infections et infestations
|
Très fréquent (23,4%)
|
Infections des voies respiratoires supérieures
| |
Fréquent
|
Conjonctivite
| |
Affections hématologiques et du système lymphatique
|
Fréquent
|
Éosinophilie
| |
Affections oculaires
|
Fréquent
|
Conjonctivite allergique
| |
Occasionnel
|
Kératite
| |
Troubles généraux et anomalies au site d’administration
|
Fréquent
|
Réactions au site d’injection
|
La sécurité à long terme d'Adtralza® a été évaluée dans les deux études en monothérapie jusqu’à 52 semaines et dans une étude en association aux corticostéroïdes topiques jusqu’à 32 semaines. Le profil de sécurité d'Adtralza® jusqu'à 52 semaines ou 32 semaines, était cohérent avec celui observé jusqu'à la semaine 16.
Description d’effets indésirables spécifiques et informations complémentaires
Conjonctivite et événements liés
La conjonctivite a été plus fréquente chez les patients atteints de dermatite atopique traités par tralokinumab que chez ceux ayant reçu le placebo (5,4% contre 1,9%) au cours de la période de traitement initiale de 16 semaines dans les cinq études. Chez la plupart des patients, elle était réversible ou en voie de disparition pendant la période de traitement.
Une kératite a été signalée chez 0,5% des patients traités par tralokinumab au cours de la période de traitement initiale et a été classée comme kératoconjonctivite chez la moitié d'entre eux. Tous les cas étaient non graves et de sévérité légère à modérée et aucune n'a entraîné l'arrêt du traitement.
Éosinophilie
Une éosinophilie a été signalé chez 1,3% des patients traités par tralokinumab et chez 0,3% des patients ayant reçu le placebo, au cours de la période de traitement initiale de 16 semaines dans l’ensemble des 5 études. Une augmentation initiale moyenne plus importante de leur taux d'éosinophiles par rapport à la valeur initiale a été observée chez les patients traités par tralokinumab que chez les patients ayant reçu le placebo. Toutefois, cette augmentation était transitoire chez les patients traités par tralokinumab et le taux moyen d'éosinophiles est revenu à sa valeur initiale pendant la suite du traitement. Le profil de sécurité chez les patients présentant une éosinophilie était comparable à celui observé chez tous les patients.
Infections
Dans les cinq études sur la dermatite atopique, des infections graves ont été rapportées chez 0,4% des patients traités par tralokinumab et chez 1,1% des patients ayant reçu le placebo pendant la période traitement initiale de 16 semaines.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe une possibilité d'immunogénicité avec le tralokinumab. L'incidence des anticorps anti-médicament (ADA) dépend en grande partie de la sensibilité et de la spécificité du test. En outre, l'incidence observée de la positivité des anticorps (y compris les anticorps neutralisants) dans un test peut être influencée par plusieurs facteurs, tels que la méthodologie du test, la manipulation des échantillons, le moment du prélèvement, les médicaments concomitants et la maladie sous-jacente. Pour ces raisons, toute comparaison entre l'apparition d'anticorps contre le tralokinumab et l'apparition d'anticorps contre d'autres médicaments biologiques doit être interprétée avec prudence.
Dans les études ECZTRA 1, ECZTRA 2, ECZTRA 3 et dans l'étude sur la réponse immunitaire après administration d'un vaccin, l'incidence des ADA jusqu'à 16 semaines était de 1,4% chez les patients traités par tralokinumab et de 1,3% chez les patients ayant reçu le placebo. Des anticorps neutralisants ont été détectés chez 0,1% des patients traités par tralokinumab et chez 0,2% des patients ayant reçu le placebo.
Sur l'ensemble des périodes de l'étude, l'incidence des ADA chez les patients traités par tralokinumab était de 4,6% ; 0,9% présentaient des ADA persistants et 1,0% des anticorps neutralisants.
Déclaration des effets indésirables suspectés
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'y a pas de traitement spécifique en cas de surdosage d'Adtralza®. Dans deux études cliniques sur le tralokinumab dans l'asthme, des doses intraveineuses uniques allant jusqu'à 30 mg/kg et des doses sous-cutanées multiples de 600 mg toutes les deux semaines pendant 12 semaines se sont révélées bien tolérées.
Propriétés/EffetsCode ATC
D11AH07
Mécanisme d’action
Le tralokinumab est un anticorps monoclonal IgG4 entièrement humain qui se lie spécifiquement à l'interleukine-13 (IL-13), une cytokine de type 2. L'activité biologique des IL-13 est neutralisée par le tralokinumab en bloquant leur interaction avec le complexe de récepteurs IL-13Rα1/IL-4Rα.
Chez les patients atteints de dermatite atopique, l'expression de la cytokine IL-13 dans le sérum ainsi que de la cytokine IL-13 et de ses récepteurs dans la peau est augmentée. La neutralisation de l'IL-13 s'effectue via le complexe de récepteurs IL-13-Rα et stimule les réactions inflammatoires. Cela contribue à l'apparition de démangeaisons et affecte l'expression des protéines nécessaires au maintien d'une barrière cutanée normale.
Pharmacodynamique
Dans les essais cliniques, le traitement par Adtralza® a été associé à une diminution des concentrations sanguines des biomarqueurs de l'immunité Th2 et Th22, par exemple TARC/CCL17 (Thymus and Activation-Regulated-Chemokin), périostine, IL-22, LDH et IgE sériques, par rapport aux valeurs initiales. Le traitement par Adtralza® a également été associé à une réduction de l'incidence de Staphylococcus aureus sur la peau lésionnelle.
Pendant le traitement, l'ajout de l'anticorps IgG4Tralokinumab entraîne une augmentation du taux d'IgG4 totales. Les effets à long terme de cette modification n'ont pas encore été suffisamment étudiés.
Efficacité clinique
L'efficacité et la sécurité d'Adtralza® en monothérapie et en association aux corticostéroïdes topiques (CST) ont été étudiées et évaluées dans trois études pivots randomisées, en double aveugle et contrôlées versus placebo (ECZTRA 1, ECZTRA 2, ECZTRA 3) menées sur 1 976 patients âgés de 18 ans ou plus souffrant de dermatite atopique (DA) modérée à sévère. L'investigateur a défini les paramètres suivants dans l'évaluation globale (Investigator's Global Assessment, IGA) : un score de 3 ou 4 (dermatite atopique modérée ou sévère), une valeur ≥16 dans le score d’indice de surface et de sévérité de l’eczéma (Eczema Area and Severity Index, EASI) au début de l'étude et une atteinte de la surface corporelle (BSA) d'au moins ≥10%.
Les patients éligibles inclus dans les trois études n'avaient auparavant pas suffisamment répondu aux médicaments topiques. La durée totale du traitement dans les études ECZTRA 1 et ECZTRA 2 était de 52 semaines et de 32 semaines dans l'étude ECZTRA 3.
ECZTRA 1, ECZTRA 2 et ECZTRA 3 - Traitement de la semaine 0 à la semaine 16
Dans les trois études, les patients ont reçu soit une dose initiale de 600 mg d'Adtralza® (quatre injections de 150 mg) au jour 1, suivie de 300 mg une fois toutes les deux semaines (Q2W) jusqu'à la semaine 16, soit un placebo équivalent. Le traitement a été administré en monothérapie dans les études ECZTRA 1 et ECZTRA 2 et en association aux corticostéroïdes topiques dans l'étude ECZTRA 3. Adtralza® a été administré par injection sous-cutanée (s.c.) dans toutes les études. L'étude ECZTRA 1 a porté sur 802 patients (199 dans le groupe placebo, 603 dans le groupe de traitement par 300 mg d'Adtralza® Q2W). L'étude ECZTRA 2 a porté sur 794 patients (201 dans le groupe placebo, 593 dans le groupe de traitement par 300 mg d'Adtralza® Q2W). L'étude ECZTRA 3 a inclus 380 patients (127 dans le groupe placebo + CST, 253 dans le groupe de traitement par 300 mg d'Adtralza® Q2W + CST).
ECZTRA 1 et ECZTRA 2 - Traitement semaines 16 à 52
Dans les études ECZTRA 1 et ECZTRA 2, les patients qui ont répondu au traitement initial de 16 semaines par Adtralza® (c'est-à-dire qui ont obtenu un IGA de 0 ou 1 ou un EASI de 75) ont été à nouveau randomisés dans le groupe de traitement par 300 mg d'Adtralza® Q2W, ou par 300 mg d'Adtralza® Q4W (alternance de 300 mg d'Adtralza® et de placebo Q2W) ou par placebo Q2W et traités jusqu'à la semaine 52. Les patients qui ont répondu au traitement initial de 16 semaines par placebo ont continué à recevoir le placebo.
Les patients qui n'avaient pas atteint l'IGA 0 ou 1 ni l'EASI 75 à la semaine 16 et les patients dont la réponse n'a pas été maintenue pendant la phase d'entretien ont été transférés dans le bras en ouvert pour recevoir 300 mg d'Adtralza® Q2W avec application optionnelle de corticostéroïdes topiques.
ECZTRA 3 - Traitement semaines 16 à 32
Dans l'étude ECZTRA 3, afin d'évaluer le maintien de la réponse au traitement, les patients ayant répondu au traitement initial de 16 semaines par Adtralza® + CST ont été à nouveau randomisés dans le groupe de traitement par 300 mg d'Adtralza® Q2W + CST ou 300 mg d'Adtralza® Q4W + CST (en alternance avec 300 mg de placebo Q2W + CST) et traités jusqu'à la semaine 32.
Les patients qui ont répondu au traitement initial de 16 semaines par placebo + CST ont continué à recevoir le placebo + CST. Les patients qui n'avaient pas atteint l'IGA 0 ou 1 ni l'EASI 75 à la semaine 16 ont poursuivi le traitement avec 300 mg d'Adtralza® Q2W + CST.
Critères d'évaluation
Dans les trois études pivots, les critères d'évaluation primaires étaient l'atteinte de l'IGA 0 ou 1 («guéri» ou «presque guéri») et une réduction d'au moins 75% du score EASI (EASI 75) entre l’inclusion et la semaine 16. Les critères d'évaluation secondaires comprenaient la réduction du prurit, définie par une amélioration d'au moins 4 points du score NRS (Numeric Rating Scale) du prurit maximal quotidien entre l’inclusion et la semaine 16, la réduction du score de gravité de la dermatite atopique SCORAD (SCORing Atopic Dermatitis) entre l’inclusion et la semaine 16, et la variation de la valeur de l'indice de qualité de vie en dermatologie (Dermatology Life Quality Index, DLQI) entre l’inclusion et la semaine 16. Les critères d'évaluation secondaires additionnels étaient une réduction d'au moins 50% et 90% de l'EASI (EASI 50 et EASI 90) et une réduction du score NRS pour le prurit maximal quotidien (moyenne hebdomadaire) entre l’inclusion à la semaine 16. Les autres critères d'évaluation étaient la variation de la valeur du score de mesure de l'eczéma par le patient (POEM) et du NRS des perturbations du sommeil liés à l'eczéma l’inclusion à la semaine 16. En outre, une évaluation générale de l'état de santé (SF 36) a été réalisée dans les études ECZTRA 1 et ECZTRA 2 de l’inclusion à la semaine 16.
Caractéristiques des patients à l’inclusion
Dans les études de monothérapie (ECZTRA 1 et ECZTRA 2), l'âge moyen dans tous les groupes de traitement était de 37,8 ans et le poids moyen de 76,0 kg, 40,7% étaient des femmes, 66,5% étaient caucasiens, 22,9% étaient asiatiques et 7,5% étaient noirs. Dans ces études, 49,9% des patients avaient un IGA de 3 (DA modérée) au début de l'étude, 49,7% des patients présentaient un score IGA de 4 (DA sévère) à l’inclusion, et 42,5% des patients avaient reçu auparavant des immunosuppresseurs systémiques (ciclosporine, méthotrexate, azathioprine et mycophénolate). Le score EASI était de 32,3, le score NRS de prurit maximal quotidien moyen était de 7,8, le score DLQI mpyen était de 17,3, le score SCORAD était de 70,4, le score POEM moyen était de 22,8 et les scores moyens des composantes physiques et psychologiques du SF 36 étaient respectivement de 43,4 et 44,3 (toujours par rapport au statut á l’inclusion).
Dans l'étude en association aux CST (ECZTRA 3), l'âge moyen dans les deux groupes de traitement était de 39,1 ans et le poids moyen de 79,4 kg, 45,0% étaient des femmes, 75,8% étaient caucasiens, 10,8% étaient asiatiques et 9,2% étaient noirs. Dans cette étude, 53,2% des patients avaient un score IGA de 3 à l’inclusion, 46,3% des patients avaient un score IGA de 4 à l’inclusion et 39,2% des patients avaient reçu un traitement antérieur par immunosuppresseur systémique. À l’inclusion, le score EASI était de 29,4, le score NRS de prurit maximal quotidien de 7,7, le score DLQI moyen de 17,5, le score SCORAD moyen de 67,6 et le score POEM moyen de 22,3.
Réponse clinique
Études en monothérapie (ECZTRA 1 et ECZTRA 2) - Période initiale de traitement, semaine 0 à 16
Dans ECZTRA 1 et ECZTRA 2, parmi les patients randomisés et traités par Adtralza®, une proportion significativement plus importante a obtenu un score IGA de 0 ou 1, un score EASI 75 et/ou une amélioration ≥ 4 points du score NRS de prurit maximal quotidien, entre l’inclusion et la semaine 16 par rapport au groupe placebo (voir tableau 1).
Dès la semaine 1, une amélioration significative du NRS pour le prurit quotidien le plus intense (pourcentage moyen de variation par rapport à la valeur au début de l'étude) a été observée chez les patients ayant reçu Adtralza® après randomisation, par rapport au placebo (voir figure 2). L'amélioration du NRS pour le prurit quotidien le plus intense a été accompagnée d'une amélioration du DLQI et des signes objectifs de la dermatite atopique, y compris le score SCORAD.
Tableau 1 : Résultats d'efficacité liés à la monothérapie par Adtralza® à la semaine 16 dans les études ECZTRA 1 et ECZTRA 2 (FAS)
|
Monothérapie
| |
|
ECZTRA 1
|
ECZTRA 2
| |
Semaine 16
|
Semaine 16
| |
Placebo
|
300 mg Adtralza® Q2W
|
Placebo
|
300 mg Adtralza® Q2W
| |
Nombre de patients randomisés et ayant reçu une dose (FAS)
|
197
|
601
|
201
|
591
| |
IGA 0 ou 1, % de répondeursa,b)
|
7,1
|
15,8#
|
10,9
|
22,2§
| |
EASI-50, % de répondeursa)
|
21,3
|
41,6§
|
20,4
|
49,9§
| |
EASI-75, % de répondeursa)
|
12,7
|
25,0§
|
11,4
|
33,2§
| |
EASI-90, % de répondeursa)
|
4,1
|
14,5§
|
5,5
|
18,3§
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)
|
-28,5
(± 3,66)
|
-51,3§
(± 1,92)
|
-22,2
(± 3,48)
|
-56,6§
(± 1,79)
| |
SCORAD 50, % de répondeursa)
|
11,7
|
26,0§
|
14,4
|
33,5§
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)c)
|
-20,3
(2,72)
|
-36,7§
(± 1,42)
|
-20,6
(2,62)
|
-40,6§
(1,34)
| |
SCORAD, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-14,7
(± 1,80)
|
-25,2§
(± 0,94)
|
-14,0
(± 1,79)
|
-28,1§
(± 0,92)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,d)
|
10,3
(20/194)
|
20,0#
(119/594)
|
9,5
(19/200)
|
25,0§
(144/575)
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-1,7
(± 0,21)
|
-2,6§
(± 0,11)
|
-1,6
(± 0,21)
|
-2,9§
(± 0,11)
| |
DLQI (amélioration > 4 points, % de répondeurs)a,d)
|
31,6
(60/190)
|
44,6#
(258/578)
|
27,3
(54/198)
|
56,3§
(325/577)
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-5,0
(± 0,59)
|
-7,1#
(± 0,31)
|
-4,9
(± 0,60)
|
-8,8§
(± 0,30)
|
MC=moindres carrés; ET=erreur-type, FAS=analyse de la population totale de l’étude (Full Analysis Set) – inclut tous les patients randomisés et ayant reçu une dose
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs.
b) Un répondeur était défini comme un patient présentant un score IGA de 0 ou 1 (« blanchi » ou « presque blanchi » sur l’échelle IGA de 0-4).
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement étaient considérées comme manquantes.
d) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
*p<0,05, #p<0,01, §p<0,001
Figure 1 : Pourcentage moyen de variation de l'EASI de la semaine 0 à 16 par rapport à la valeur à l’inclusion dans les études ECZTRA 1 et ECZTRA 2

Figure 2:Pourcentage moyen de variation de score NRS pour les prurits quotidiens les plus intenses par rapport à la valeur à l’inclusion dans les études ECZTRA 1 et ECZTRA 2

Les effets du traitement dans les sous-groupes (poids, âge, sexe, ethnie et traitement antérieur, y compris les immunosuppresseurs) dans les études ECZTRA 1 et ECZTRA 2 ont été cohérents avec les résultats obtenus dans le collectif global de l'étude.
Dans les deux études, parmi les patients ayant reçu 300 mg d'Adtralza® Q2W après la randomisation, ceux qui ont eu besoin d'un traitement de secours (corticostéroïdes topiques, corticostéroïdes systémiques, immunosuppresseurs non stéroïdiens) ont été moins nombreux que ceux qui ont été randomisés dans le groupe placebo (29,3% contre 45,3% dans les deux études combinées).
Études en monothérapie (ECZTRA 1 et ECZTRA 2) – période d’entretien (semaines 16 à 52)
Le tableau 2 présente les résultats d'efficacité de ce que l'on appelle les répondeurs.
Tableau 2: Résultats d’efficacité (IGA de 0 ou 1 ou EASI-75) à la semaine 52 chez les patients ayant répondu à 300 mg d’Adtralza® Q2W à la semaine 16
|
|
ECZTRA 1
|
ECZTRA 2
| |
|
Schéma de traitement de la semaine 16 à la semaine 52d)
|
Schéma de traitement de la semaine 16 à la semaine 52d)
| |
Évaluation à la semaine 52
|
Adtralza®
300 mg
Q2W
|
Adtralza®
300 mg
Q4W
|
Placebo
|
Adtralza®
300 mg
Q2W
|
Adtralza®
300 mg
Q4W
|
Placebo
| |
IGA 0/1a)
|
51,3%
(20/39)
|
38,9%
(14/36)
|
47,4%
(9/19)
|
59,3%c)
(32/54)
|
44,9%
(22/49)
|
25,0%
(7/28)
| |
EASI-75a)
|
59,6%b)
(28/47)
|
49,1%
(28/57)
|
33,3%
(10/30)
|
55,8%c)
(43/77)
|
51,4%
(38/74)
|
21,4%
(9/42)
|
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs. Le pourcentage est calculé par rapport au nombre de patients présentant une réponse à la semaine 16.
b) P = 0,004 par rapport au placebo
c) P < 0,001 par rapport au placebo
d) Tous les patients ont initialement été traités par 300 mg d’Adtralza® Q2W de la semaine 0 à la semaine 16.
Parmi les patients ayant reçu Adtralza® après la randomisation, mais qui n'avaient pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16 et qui sont passés au traitement dans le bras en ouvert pour recevoir 300 mg d'Adtralza® Q2W + CST optionnels, 20,8% dans ECZTRA 1 et 19,3% dans ECZTRA 2 avaient obtenu un IGA 0 ou 1 à la semaine 52, et 46,1% dans l'ECZTRA 1 et 39,3% dans l'ECZTRA 2 avaient obtenu un score EASI-75 à la semaine 52. La réponse clinique dépendait principalement de la poursuite du traitement par Adtralza® et non au traitement CST optionnel. Parmi les patients avec un score IGA de 2 ou EASI-50 à la semaine 16, une plus grande proportion avait obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 52 que les patients avec un score IGA de 3 ou 4 ou < EASI-50 à la semaine 16.
Étude de 32 semaines en association aux CST (ECZTRA 3) - période de traitement initiale de 0 à 16 semaines
Dans l'étude ECZTRA 3, parmi les patients ayant reçu 300 mg d'Adtralza® Q2W + CST après randomisation, un pourcentage significativement plus élevé a obtenu un score IGA de 0 ou 1, un score EASI-75 et/ou une amélioration du score NRS de prurit maximal quotidien ≥ 4 points entre l’inclusion et la semaine 16, par rapport au groupe placebo + CST (voir tableau 3).
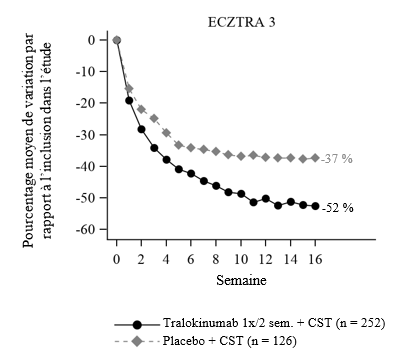
Dès la semaine 2, une amélioration significative du NRS de prurit maximal quotidien (pourcentage moyen de variation par rapport à la valeur à l’inclusion) a été observée chez les patients ayant reçu Adtralza® + CST après randomisation, par rapport au placebo + CST (voir figure 4). L'amélioration du score NRS de prurit maximal quotidien était associée à l'amélioration du DLQI et des signes objectifs de la dermatite atopique, y compris le score sur l'échelle SCORAD.
Tableau 3 : Résultats d’efficacité d’Adtralza® en association aux CST à la semaine 16 dans l’étude ECZTRA 3 (FAS)
|
ECZTRA 3 - Traitement en association
| |
|
Semaine 16
| |
Placebo + CST
|
300 mg Adtralza® Q2W + CST
| |
Nombre de patients randomisés et ayant reçu une dose (FAS)
|
126
|
252
| |
IGA 0 ou 1, % de répondeursa,b)
|
26,2
|
38,9*
| |
EASI-50, % de répondeursa
|
57,9
|
79,4§
| |
EASI-75, % de répondeursa)
|
35,7
|
56,0§
| |
EASI-90, % de répondeursa)
|
21,4
|
32,9*
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)
|
-55,3
(± 3,2)
|
-71,3§
(± 2,2)
| |
SCORAD 50, % de répondeursa)
|
38,1
|
61,1§
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)c)
|
-40,0
(± 2,6)
|
-55,9§
(± 1,8)
| |
SCORAD, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-26,8
(± 1,8)
|
-37,7§
(± 1,3)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,d)
|
34,1 (43/126)
|
45,4* (113/249)
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-2,9
(± 0,2)
|
-4,1§
(± 0,2)
| |
DLQI (amélioration > 4 points, % de répondeurs)a,d)
|
65,9 (81/123)
|
83,5§ (207/248)
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-8,8
(± 0,6)
|
-11,7§
(± 0,4)
|
MC=moindres carrés; ET=erreur-type, FAS=analyse de la population totale de l’étude (Full Analysis Set) – inclut tous les patients randomisés et ayant reçu une dose
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs.
b) Un répondeur était défini comme un patient présentant un score IGA de 0 ou 1 (« blanchi » ou « presque blanchi » sur l’échelle IGA de 0-4).
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement étaient considérées comme manquantes.
d) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
*p<0,05, #p<0,01, §p<0,001
Dans l'étude ECZTRA 3, les patients ayant reçu 300 mg d'Adtralza® Q2W de la semaine 0 à 16 ont utilisé 50% de CST en moins à la semaine 16 que les patients du groupe placebo.
Figure 3 : Pourcentage moyen de variation du score EASI dans l'étude ECZTRA 3 par rapport au début de l'étude
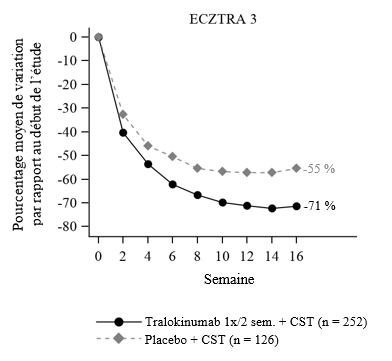
Figure 4 : Pourcentage moyen de variation du score NRS de prurit maximal quotidien (moyenne hebdomadaire) de la semaine 0 à 16 par rapport à l’inclusion dans l'étude ECZTRA 3
Étude de 32 semaines en association aux CST (ECZTRA 3) - période d’entretien de 16 à 32 semaines
Lors de l'utilisation de 300 mg d'Adtralza® Q2W + CST et de 300 mg d'Adtralza® Q4W + CST, un maintien élevé de l'efficacité clinique a été observé à la semaine 32 chez les patients ayant obtenu une réponse clinique à la semaine 16 (voir tableau 4).
Tableau 4 : Résultats d'efficacité à la semaine 32 chez les patients ayant présenté une réponse clinique à 300 mg d'Adtralza® + CST Q2W à la semaine 16
|
|
300 mg Adtralza® Q2W + CST
|
300 mg Adtralza® Q4W + CST
| |
IGA 0/1 à la semaine 32a)
% de répondeurs
|
89,6%
(43/48)
|
77,6%
(38/49)
| |
EASI-75 à la semaine 32a)
% de répondeurs
|
92,5%
(62/67)
|
90,8%
(59/65)
|
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs. Le pourcentage est calculé par rapport au nombre de patients présentant une réponse à la semaine 16.
Parmi tous les patients ayant obtenu soit un score IGA de 0 ou 1, soit un score EASI-75 à la semaine 16, le pourcentage moyen d'amélioration du score EASI par rapport à l’inclusion était de 93,5% à la semaine 32 pour les patients ayant continué à recevoir 300 mg d'Adtralza® Q2W + CST, et de 91,5% à la semaine 32 pour les patients ayant reçu 300 mg d'Adtralza® Q4W + CST.
Parmi les patients ayant reçu 300 mg d'Adtralza® Q2W + CST après la randomisation et n’ayant pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16, 30,5% ont obtenu un score IGA 0/1 à la semaine 32 et 55,8% un score EASI-75 lorsqu'ils ont été traités en continu avec 300 mg d'Adtralza® Q2W + CST pendant 16 semaines supplémentaires.
La poursuite de l’amélioration chez les patients n’ayant pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16 est survenue conjointement à l’amélioration du score NRS de prurit maximal quotidien et des signes cliniques objectifs de la dermatite atopique, incluant le score SCORAD.
Tableau 5: Résultats d’efficacité d‘Adtralza® en association aux CST aux semaines 16 et 32 dans l’étude ECZTRA 3 chez les patients initialement traités par Adtralza® Q2W + CST
|
|
Schéma de traitement des semaines 16 à 32d)
| |
Répondeurs à la semaine 16e)
|
Non-répondeurs à la semaine 16
| |
Patients randomisés
|
Adtralza® Q2W + CST
|
Adtralza® Q4W + CST
|
Adtralza® Q2W + CST
| |
N=69
|
N=69
|
N=95
| |
Semaine
|
W16
|
W32
|
W16
|
W32
|
W16
|
W32
| |
EASI-50, % de répondeursa)
|
100,0
|
98,6
|
97,1
|
91,3
|
63,2
|
76,8
| |
EASI-90, % de répondeursa)
|
58,0
|
72,5
|
60,9
|
63,8
|
1,1
|
34,7
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)b)
|
-90,5
(2,7)
|
-93,2
(2,3)
|
-89,3
(2,7)
|
-91,5
(2,3)
|
-46,9
(2,4)
|
-73,5
(2,0)
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)b)
|
-73,2
(2,1)
|
-79,2
(2,5)
|
-72,3
(2,1)
|
-73,3
(2,5)
|
-32,7
(1,8)
|
-54,5
(2,2)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,c)
|
63,2
|
70,6
|
64,2
|
61,2
|
27,4
|
38,9
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)b)
|
-5,0
(0,2)
|
-5,4
(0,2)
|
-4,6
(0,2)
|
-4,9
(0,2)
|
-3,0
(0,2)
|
-3,7
(0,2)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
b) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
c) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
d) Tous les patients ont initialement été traités par 300 mg Adtralza® Q2W + CST de la semaine 0 à la semaine 16. Ils ont ensuite été traités par 300 mg Adtralza® Q2W + CST ou 300 mg Adtralza® Q4W + CST.
e) Les répondeurs à la semaine 16 ont été identifiés comme les patients ayant obtenu un score IGA 0/1 et/ou EASI-75.
Qualité de vie/résultats rapportés par les patients (Patient-Reported Outcomes)
Dans les deux études en monothérapie (ECZTRA 1 et ECZTRA 2), l'utilisation de 300 mg d'Adtralza® Q2W a amélioré les symptômes rapportés par les patients, et l'impact de la DA sur le sommeil et la qualité de vie liée à la santé, mesuré par le score POEM,par le score NRS pour les troubles du sommeil liés à l'eczéma et par le SF 36, a été examiné à la semaine 16 par rapport au placebo. Parmi les patients traités par Adtralza®, une proportion plus élevée a présenté une réduction cliniquement significative du score POEM (définie comme une amélioration ≥ 4 points) entre l’inclusion et la semaine 16, par rapport au groupe placebo (voir tableau 6).
Tableau 6 : Autres résultats des critères d'évaluation liés à la monothérapie par Adtralza® à la semaine 16 dans les études ECZTRA 1 et ECZTRA 2
|
Monothérapie
| |
|
ECZTRA 1, Semaine 16
|
ECZTRA 2, Semaine 16
| |
|
Placebo
|
300 mg Adtralza® Q2W
|
Placebo
|
300 mg Adtralza® Q2W
| |
Patients randomisés
|
199
|
603
|
201
|
593
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-1,9
(0,2)
|
-2,6#
(0,1)
|
-1,5
(0,2)
|
-2,9§
(0,1)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-3,0
(0,7)
|
-7,6§
(0,4)
|
-3,7
(0,7)
|
-8,8§
(0,3)
| |
POEM (amélioration > 4 points, % de répondeurs)b)
|
18,0%
(35/194)
|
43,0 %§
(253/588)
|
22,1%
(44/199)
|
54,4 %§
(319/586)
| |
SF-36, composante physique, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
2,9
(0,6)
|
4,5*
(0,3)
|
3,2
(0,6)
|
5,8§
(0,3)
| |
SF-36, , composante physique, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
0,3
(0,8)
|
2,5*
(0,4)
|
0,5
(0,8)
|
3,5§
(0,4)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
b) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
Le pourcentage est calculé par rapport au nombre de patients avec un score POEM > 4 au début de l'étude.
*p < 0,05, #p < 0,01, §p < 0,001
Dans l'étude en association aux CST (ECZTRA 3), l'utilisation de 300 mg d'Adtralza® Q2W + CST a amélioré les symptômes rapportés par les patients et l'impact de la DA sur le sommeil et la qualité de vie liée à la santé, évalués par le score POEM et le score NRS pour les troubles du sommeil liés à l'eczéma, à la semaine 16 par rapport au placebo.
Parmi les patients traités par Adtralza®, une proportion plus élevée a obtenu une réduction cliniquement significative su score POEM (définie comme une amélioration ≥ 4 points) entre l’inclusion et la semaine 16, par rapport au groupe placebo (voir tableau 7).
Tableau 7 : Autres résultats des critères d'évaluation lors de l'utilisation d'Adtralza® avec CST à la semaine 16 dans l'étude ECZTRA 3
|
ECZTRA 3 - Traitement en association
| |
|
Woche 16
| |
Placebo + CST
|
300 mg Adtralza® Q2W + CST
| |
Patients randomisés
|
126
|
252
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-3,1
(0,2)
|
-4,3§
(0,2)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-7,8 (0,7)
|
-11,8§ (0,5)
| |
POEM (amélioration > 4 points, % de répondeurs)b)
|
59,3% (73/123)
|
78,4%§ (190/250)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
b) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
Le pourcentage est calculé par rapport au nombre de patients avec un score POEM > 4 au début de l'étude.
§p < 0,001
Chez les patients traités par 300 mg d'Adtralza® Q2W + CST avec une réponse à la semaine 16 et qui ont continué à recevoir 300 mg d'Adtralza® Q2W + CST ou Q4W + CST, des réductions cliniquement significatives du score POEM, du score DLQI et du score NRS pour les troubles du sommeil liés à l'eczéma ont été observées entre l’inclusion et la semaine 32.
Tableau 8: Autres critères d'évaluation lors de l'utilisation d'Adtralza® avec CST aux semaines 16 et 32 de l'étude ECZTRA 3 chez les patients ayant obtenu une réponse clinique à la semaine 16
|
|
Schéma de traitement des semaines 16 à 32a)
| |
|
Répondeurs à la semaine 16b)
| |
Adtralza® Q2W + CST
|
Adtralza® Q4W + CST
| |
Patients randomisés
|
N=69
|
N=69
| |
Semaine
|
W16
|
W32
|
W16
|
W32
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-14,0
(0,6)
|
-14,6
(0,6)
|
-13,9
(0,6)
|
-13,7
(0,6)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-15,2
(0,7)
|
-15,6
(0,7)
|
-14,1
(0,7)
|
-13,9
(0,8)
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-5,2
(0,2)
|
-5,5
(0,2)
|
-4,8
(0,2)
|
-5,2
(0,3)
| |
DLQI (amélioration > 4 points,
% de répondeursd)
|
98,5%
(65/66)
|
89,4%
(59/66)
|
100,0 %
(68/68)
|
83,8%
(57/68)
| |
POEM (amélioration > 4 points),
% de répondeursd
|
89,7%
(61/68)
|
88,2%
(60/68)
|
94,1 %
(64/68)
|
83,8%
(57/68)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Tous les patients ont initialement été traités par 300 mg Adtralza® Q2W + CST de la semaine 0 à la semaine 16. Ils ont ensuite été traités par 300 mg Adtralza® Q2W + CST ou 300 mg Adtralza® Q4W + CST.
b) Les répondeurs à la semaine 16 ont été identifiés comme les patients ayant obtenu un score IGA 0/1 et/ou EASI-75.
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
d) Nombre de répondeurs divisé par le nombre de patients ayant une valeur de référence > 4 pour chaque paramètre.
Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
PharmacocinétiqueAbsorption
Après injection sous-cutanée de tralokinumab, le durée médiane pour atteindre la concentration sérique maximale (tmax) était de 5 à 8 jours. La biodisponibilité absolue du tralokinumab après l'administration d'une dose sous-cutanée est de 76%.
Après une dose initiale de 600 mg, suivie d'une dose de 300 mg toutes les deux semaines, les concentrations à l'état d'équilibre ont été atteintes à la semaine 16. Dans les études cliniques (ECZTRA 1, 2 et 3), les concentrations à l'état d'équilibre étaient comprises entre 98,0 ± 41,1 µg/ml et 101,4 ± 42,7 µg/ml (moyennes ± écart-type) à la dose de 300 mg toutes les deux semaines.
Distribution
Sur la base de l'analyse de la cinétique de population, le volume de distribution a été estimé à environ 4,2 litres.
Métabolisme
Aucune étude de métabolisme spécifique n’a été menée car le tralokinumab est une protéine. Il est attendu que le tralokinumab se dégrade en petits peptides et acides aminés individuels.
Élimination
Le tralokinumab est éliminé par une voie protéolytique non saturable. La demi-vie est de 22 jours, ce qui correspond à l'estimation typique des anticorps monoclonaux humains IgG4 ciblant les cytokines solubles.
Linéarité/non-linéarité
L'exposition au tralokinumab augmente proportionnellement à la dose de tralokinumab sur une large gamme de doses étudiées (0,1 30 mg/kg).
Cinétique pour certains groupes de patients
Sexe
L'analyse de la pharmacocinétique de population n'a révélé aucune association entre le sexe et des effets cliniquement significatifs sur l'exposition systémique au tralokinumab.
Poids corporel
Les concentrations résiduelles de tralokinumab étaient plus faibles chez les sujets dont le poids corporel était plus élevé.
Troubles de la fonction hépatique
Le tralokinumab étant un anticorps monoclonal, il ne devrait pas être éliminé de manière significative par voie hépatique. Aucune étude clinique n'a été menée sur les effets d’un trouble de la fonction hépatique sur la pharmacocinétique du tralokinumab. L'analyse de la pharmacocinétique de population n’a pas mis en évidence d’impact d’une insuffisance hépatique légère sur la pharmacocinétique du tralokinumab. Aucun effet du tralokinumab n'a été observé sur la pharmacocinétique du tralokinumab en cas de trouble de la fonction hépatique modéré ou sévère. On ne dispose que de données très limitées concernant les patients présentant un trouble de la fonction hépatique modéré ou sévère.
Troubles de la fonction rénale
Le tralokinumab étant un anticorps monoclonal, il ne devrait pas être éliminé de manière significative par voie rénale. Aucune étude clinique n'a été menée pour évaluer sur les effets d’un trouble de la fonction rénale sur la pharmacocinétique du tralokinumab. L’analyse de pharmacocinétique de population n’a pas mis en évidence d’impact cliniquement significatif de troubles de la fonction rénale légère ou modérée sur l’exposition systémique au tralokinumab. On ne dispose que de données très limitées concernant les patients présentant un trouble de la fonction rénale modéré ou sévère.
Patients âgés
L'analyse de la pharmacocinétique de la population n'a révélé aucune association entre l'âge et des effets cliniquement significatifs sur l'exposition systémique au tralokinumab. L'étude a porté sur 109 patients âgés de plus de 65 ans.
Enfants et adolescents
La pharmacocinétique du tralokinumab chez les patients pédiatriques n’a pas encore été étudiée.
Polymorphismes génétiques
L'analyse de la pharmacocinétique de population n'a pas révélé de lien entre l'appartenance ethnique et des effets cliniquement significatifs sur l'exposition systémique d'Adtralza®.
Données précliniquesToxicité en cas d’administration répétée
Les données précliniques issues des études conventionnelles de toxicité par administration répétée (y compris les critères d'évaluation de la pharmacologie de sécurité) n'ont pas révélé de risque particulier pour l'homme.
Génotoxicité
Le potentiel mutagène du tralokinumab n'a pas été étudié. Il est toutefois peu probable que les anticorps monoclonaux modifient l'ADN ou les chromosomes.
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée avec le tralokinumab. L'évaluation des données disponibles concernant l'inhibition de l'IL 13 et les données toxicologiques animales avec le tralokinumab ne permettent pas de conclure à un potentiel de carcinogénicité accru du tralokinumab.
Toxicité sur la reproduction
Les données précliniques issues des études conventionnelles de toxicité sur la reproduction et le développement ne révèlent aucun risque particulier pour l'homme.
Des études pré- et postnatales élargies avec le tralokinumab chez le singe n'ont pas révélé d'effets indésirables sur la mère ou ses petits jusqu'à six mois après la naissance.
Chez les singes sexuellement matures traités chaque semaine à des doses allant jusqu'à 350 mg de tralokinumab par voie sous-cutanée (femelles) ou 600 mg (mâles), aucun effet n'a été observé sur les paramètres de fertilité, tels que les organes reproducteurs, le cycle menstruel et l'analyse des spermatozoïdes. L'exposition (ASC) des animaux dans ces études était environ 17 fois supérieure à l'exposition clinique.
Remarques particulièresIncompatibilités
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé à d’autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Stabilité après ouverture
Comme le médicament ne contient pas d'agent conservateur, les quantités résiduelles non utilisées du médicament dans la seringue préremplie doivent être jetées.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C).
Ne pas congeler.
Conserver les seringues préremplies dans leur carton afin de protéger le contenu de la chaleur et de la lumière directe du soleil.
Conserver hors de portée des enfants.
Si nécessaire, le médicament peut être conservé jusqu'à 14 jours dans son emballage d’origine jusqu’à une température de 30°C. Si le médicament doit être conservé plusieurs jours hors du réfrigérateur, la date de sortie du réfrigérateur peut être notée sur l’emballage.
Le médicament doit être utilisé ou jeté dans les 14 jours suivant sa sortie du réfrigérateur.
Protéger la seringue préremplie de la chaleur et de la lumière directe du soleil et ne pas l'agiter.
Remarques concernant la manipulation
Solution injectable limpide à opalescente, incolore à jaune pâle. Si la solution est trouble ou décolorée, si des particules en suspension sont visibles ou si la seringue préremplie est endommagée ou est tombée sur une surface dure, le médicament ne doit pas être utilisé.
Après avoir sorti la seringue préremplie du réfrigérateur, il convient d'attendre 30 minutes avant d'injecter Adtralza®, afin que la seringue préremplie puisse atteindre la température ambiante.
Les seringues préremplies sont à usage unique.
Numéro d’autorisation68229 (Swissmedic)
PrésentationAdtralza®, solution injectable en seringue préremplie de 1 ml de solution :
1 emballage multiple contenant au total 4 seringues préremplies (2 emballages individuels contenant chacun 2 seringues préremplies) [B]
Titulaire de l’autorisationLEO Pharmaceutical Products Sarath Ltd., Kloten
Domicile: Zurich
Mise à jour de l’informationAvril 2023
|