Propriétés/EffetsCode ATC
D11AH07
Mécanisme d’action
Le tralokinumab est un anticorps monoclonal IgG4 entièrement humain qui se lie spécifiquement à l'interleukine-13 (IL-13), une cytokine de type 2. L'activité biologique des IL-13 est neutralisée par le tralokinumab en bloquant leur interaction avec le complexe de récepteurs IL-13Rα1/IL-4Rα.
Chez les patients atteints de dermatite atopique, l'expression de la cytokine IL-13 dans le sérum ainsi que de la cytokine IL-13 et de ses récepteurs dans la peau est augmentée. La neutralisation de l'IL-13 s'effectue via le complexe de récepteurs IL-13-Rα et stimule les réactions inflammatoires. Cela contribue à l'apparition de démangeaisons et affecte l'expression des protéines nécessaires au maintien d'une barrière cutanée normale.
Pharmacodynamique
Dans les essais cliniques, le traitement par Adtralza® a été associé à une diminution des concentrations sanguines des biomarqueurs de l'immunité Th2 et Th22, par exemple TARC/CCL17 (Thymus and Activation-Regulated-Chemokin), périostine, IL-22, LDH et IgE sériques, par rapport aux valeurs initiales. Le traitement par Adtralza® a également été associé à une réduction de l'incidence de Staphylococcus aureus sur la peau lésionnelle.
Pendant le traitement, l'ajout de l'anticorps IgG4Tralokinumab entraîne une augmentation du taux d'IgG4 totales. Les effets à long terme de cette modification n'ont pas encore été suffisamment étudiés.
Efficacité clinique
L'efficacité et la sécurité d'Adtralza® en monothérapie et en association aux corticostéroïdes topiques (CST) ont été étudiées et évaluées dans trois études pivots randomisées, en double aveugle et contrôlées versus placebo (ECZTRA 1, ECZTRA 2, ECZTRA 3) menées sur 1 976 patients âgés de 18 ans ou plus souffrant de dermatite atopique (DA) modérée à sévère. L'investigateur a défini les paramètres suivants dans l'évaluation globale (Investigator's Global Assessment, IGA) : un score de 3 ou 4 (dermatite atopique modérée ou sévère), une valeur ≥16 dans le score d’indice de surface et de sévérité de l’eczéma (Eczema Area and Severity Index, EASI) au début de l'étude et une atteinte de la surface corporelle (BSA) d'au moins ≥10%.
Les patients éligibles inclus dans les trois études n'avaient auparavant pas suffisamment répondu aux médicaments topiques. La durée totale du traitement dans les études ECZTRA 1 et ECZTRA 2 était de 52 semaines et de 32 semaines dans l'étude ECZTRA 3.
ECZTRA 1, ECZTRA 2 et ECZTRA 3 - Traitement de la semaine 0 à la semaine 16
Dans les trois études, les patients ont reçu soit une dose initiale de 600 mg d'Adtralza® (quatre injections de 150 mg) au jour 1, suivie de 300 mg une fois toutes les deux semaines (Q2W) jusqu'à la semaine 16, soit un placebo équivalent. Le traitement a été administré en monothérapie dans les études ECZTRA 1 et ECZTRA 2 et en association aux corticostéroïdes topiques dans l'étude ECZTRA 3. Adtralza® a été administré par injection sous-cutanée (s.c.) dans toutes les études. L'étude ECZTRA 1 a porté sur 802 patients (199 dans le groupe placebo, 603 dans le groupe de traitement par 300 mg d'Adtralza® Q2W). L'étude ECZTRA 2 a porté sur 794 patients (201 dans le groupe placebo, 593 dans le groupe de traitement par 300 mg d'Adtralza® Q2W). L'étude ECZTRA 3 a inclus 380 patients (127 dans le groupe placebo + CST, 253 dans le groupe de traitement par 300 mg d'Adtralza® Q2W + CST).
ECZTRA 1 et ECZTRA 2 - Traitement semaines 16 à 52
Dans les études ECZTRA 1 et ECZTRA 2, les patients qui ont répondu au traitement initial de 16 semaines par Adtralza® (c'est-à-dire qui ont obtenu un IGA de 0 ou 1 ou un EASI de 75) ont été à nouveau randomisés dans le groupe de traitement par 300 mg d'Adtralza® Q2W, ou par 300 mg d'Adtralza® Q4W (alternance de 300 mg d'Adtralza® et de placebo Q2W) ou par placebo Q2W et traités jusqu'à la semaine 52. Les patients qui ont répondu au traitement initial de 16 semaines par placebo ont continué à recevoir le placebo.
Les patients qui n'avaient pas atteint l'IGA 0 ou 1 ni l'EASI 75 à la semaine 16 et les patients dont la réponse n'a pas été maintenue pendant la phase d'entretien ont été transférés dans le bras en ouvert pour recevoir 300 mg d'Adtralza® Q2W avec application optionnelle de corticostéroïdes topiques.
ECZTRA 3 - Traitement semaines 16 à 32
Dans l'étude ECZTRA 3, afin d'évaluer le maintien de la réponse au traitement, les patients ayant répondu au traitement initial de 16 semaines par Adtralza® + CST ont été à nouveau randomisés dans le groupe de traitement par 300 mg d'Adtralza® Q2W + CST ou 300 mg d'Adtralza® Q4W + CST (en alternance avec 300 mg de placebo Q2W + CST) et traités jusqu'à la semaine 32.
Les patients qui ont répondu au traitement initial de 16 semaines par placebo + CST ont continué à recevoir le placebo + CST. Les patients qui n'avaient pas atteint l'IGA 0 ou 1 ni l'EASI 75 à la semaine 16 ont poursuivi le traitement avec 300 mg d'Adtralza® Q2W + CST.
Critères d'évaluation
Dans les trois études pivots, les critères d'évaluation primaires étaient l'atteinte de l'IGA 0 ou 1 («guéri» ou «presque guéri») et une réduction d'au moins 75% du score EASI (EASI 75) entre l’inclusion et la semaine 16. Les critères d'évaluation secondaires comprenaient la réduction du prurit, définie par une amélioration d'au moins 4 points du score NRS (Numeric Rating Scale) du prurit maximal quotidien entre l’inclusion et la semaine 16, la réduction du score de gravité de la dermatite atopique SCORAD (SCORing Atopic Dermatitis) entre l’inclusion et la semaine 16, et la variation de la valeur de l'indice de qualité de vie en dermatologie (Dermatology Life Quality Index, DLQI) entre l’inclusion et la semaine 16. Les critères d'évaluation secondaires additionnels étaient une réduction d'au moins 50% et 90% de l'EASI (EASI 50 et EASI 90) et une réduction du score NRS pour le prurit maximal quotidien (moyenne hebdomadaire) entre l’inclusion à la semaine 16. Les autres critères d'évaluation étaient la variation de la valeur du score de mesure de l'eczéma par le patient (POEM) et du NRS des perturbations du sommeil liés à l'eczéma l’inclusion à la semaine 16. En outre, une évaluation générale de l'état de santé (SF 36) a été réalisée dans les études ECZTRA 1 et ECZTRA 2 de l’inclusion à la semaine 16.
Caractéristiques des patients à l’inclusion
Dans les études de monothérapie (ECZTRA 1 et ECZTRA 2), l'âge moyen dans tous les groupes de traitement était de 37,8 ans et le poids moyen de 76,0 kg, 40,7% étaient des femmes, 66,5% étaient caucasiens, 22,9% étaient asiatiques et 7,5% étaient noirs. Dans ces études, 49,9% des patients avaient un IGA de 3 (DA modérée) au début de l'étude, 49,7% des patients présentaient un score IGA de 4 (DA sévère) à l’inclusion, et 42,5% des patients avaient reçu auparavant des immunosuppresseurs systémiques (ciclosporine, méthotrexate, azathioprine et mycophénolate). Le score EASI était de 32,3, le score NRS de prurit maximal quotidien moyen était de 7,8, le score DLQI mpyen était de 17,3, le score SCORAD était de 70,4, le score POEM moyen était de 22,8 et les scores moyens des composantes physiques et psychologiques du SF 36 étaient respectivement de 43,4 et 44,3 (toujours par rapport au statut á l’inclusion).
Dans l'étude en association aux CST (ECZTRA 3), l'âge moyen dans les deux groupes de traitement était de 39,1 ans et le poids moyen de 79,4 kg, 45,0% étaient des femmes, 75,8% étaient caucasiens, 10,8% étaient asiatiques et 9,2% étaient noirs. Dans cette étude, 53,2% des patients avaient un score IGA de 3 à l’inclusion, 46,3% des patients avaient un score IGA de 4 à l’inclusion et 39,2% des patients avaient reçu un traitement antérieur par immunosuppresseur systémique. À l’inclusion, le score EASI était de 29,4, le score NRS de prurit maximal quotidien de 7,7, le score DLQI moyen de 17,5, le score SCORAD moyen de 67,6 et le score POEM moyen de 22,3.
Réponse clinique
Études en monothérapie (ECZTRA 1 et ECZTRA 2) - Période initiale de traitement, semaine 0 à 16
Dans ECZTRA 1 et ECZTRA 2, parmi les patients randomisés et traités par Adtralza®, une proportion significativement plus importante a obtenu un score IGA de 0 ou 1, un score EASI 75 et/ou une amélioration ≥ 4 points du score NRS de prurit maximal quotidien, entre l’inclusion et la semaine 16 par rapport au groupe placebo (voir tableau 1).
Dès la semaine 1, une amélioration significative du NRS pour le prurit quotidien le plus intense (pourcentage moyen de variation par rapport à la valeur au début de l'étude) a été observée chez les patients ayant reçu Adtralza® après randomisation, par rapport au placebo (voir figure 2). L'amélioration du NRS pour le prurit quotidien le plus intense a été accompagnée d'une amélioration du DLQI et des signes objectifs de la dermatite atopique, y compris le score SCORAD.
Tableau 1 : Résultats d'efficacité liés à la monothérapie par Adtralza® à la semaine 16 dans les études ECZTRA 1 et ECZTRA 2 (FAS)
|
Monothérapie
| |
|
ECZTRA 1
|
ECZTRA 2
| |
Semaine 16
|
Semaine 16
| |
Placebo
|
300 mg Adtralza® Q2W
|
Placebo
|
300 mg Adtralza® Q2W
| |
Nombre de patients randomisés et ayant reçu une dose (FAS)
|
197
|
601
|
201
|
591
| |
IGA 0 ou 1, % de répondeursa,b)
|
7,1
|
15,8#
|
10,9
|
22,2§
| |
EASI-50, % de répondeursa)
|
21,3
|
41,6§
|
20,4
|
49,9§
| |
EASI-75, % de répondeursa)
|
12,7
|
25,0§
|
11,4
|
33,2§
| |
EASI-90, % de répondeursa)
|
4,1
|
14,5§
|
5,5
|
18,3§
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)
|
-28,5
(± 3,66)
|
-51,3§
(± 1,92)
|
-22,2
(± 3,48)
|
-56,6§
(± 1,79)
| |
SCORAD 50, % de répondeursa)
|
11,7
|
26,0§
|
14,4
|
33,5§
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)c)
|
-20,3
(2,72)
|
-36,7§
(± 1,42)
|
-20,6
(2,62)
|
-40,6§
(1,34)
| |
SCORAD, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-14,7
(± 1,80)
|
-25,2§
(± 0,94)
|
-14,0
(± 1,79)
|
-28,1§
(± 0,92)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,d)
|
10,3
(20/194)
|
20,0#
(119/594)
|
9,5
(19/200)
|
25,0§
(144/575)
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-1,7
(± 0,21)
|
-2,6§
(± 0,11)
|
-1,6
(± 0,21)
|
-2,9§
(± 0,11)
| |
DLQI (amélioration > 4 points, % de répondeurs)a,d)
|
31,6
(60/190)
|
44,6#
(258/578)
|
27,3
(54/198)
|
56,3§
(325/577)
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-5,0
(± 0,59)
|
-7,1#
(± 0,31)
|
-4,9
(± 0,60)
|
-8,8§
(± 0,30)
|
MC=moindres carrés; ET=erreur-type, FAS=analyse de la population totale de l’étude (Full Analysis Set) – inclut tous les patients randomisés et ayant reçu une dose
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs.
b) Un répondeur était défini comme un patient présentant un score IGA de 0 ou 1 (« blanchi » ou « presque blanchi » sur l’échelle IGA de 0-4).
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement étaient considérées comme manquantes.
d) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
*p<0,05, #p<0,01, §p<0,001
Figure 1 : Pourcentage moyen de variation de l'EASI de la semaine 0 à 16 par rapport à la valeur à l’inclusion dans les études ECZTRA 1 et ECZTRA 2

Figure 2:Pourcentage moyen de variation de score NRS pour les prurits quotidiens les plus intenses par rapport à la valeur à l’inclusion dans les études ECZTRA 1 et ECZTRA 2

Les effets du traitement dans les sous-groupes (poids, âge, sexe, ethnie et traitement antérieur, y compris les immunosuppresseurs) dans les études ECZTRA 1 et ECZTRA 2 ont été cohérents avec les résultats obtenus dans le collectif global de l'étude.
Dans les deux études, parmi les patients ayant reçu 300 mg d'Adtralza® Q2W après la randomisation, ceux qui ont eu besoin d'un traitement de secours (corticostéroïdes topiques, corticostéroïdes systémiques, immunosuppresseurs non stéroïdiens) ont été moins nombreux que ceux qui ont été randomisés dans le groupe placebo (29,3% contre 45,3% dans les deux études combinées).
Études en monothérapie (ECZTRA 1 et ECZTRA 2) – période d’entretien (semaines 16 à 52)
Le tableau 2 présente les résultats d'efficacité de ce que l'on appelle les répondeurs.
Tableau 2: Résultats d’efficacité (IGA de 0 ou 1 ou EASI-75) à la semaine 52 chez les patients ayant répondu à 300 mg d’Adtralza® Q2W à la semaine 16
|
|
ECZTRA 1
|
ECZTRA 2
| |
|
Schéma de traitement de la semaine 16 à la semaine 52d)
|
Schéma de traitement de la semaine 16 à la semaine 52d)
| |
Évaluation à la semaine 52
|
Adtralza®
300 mg
Q2W
|
Adtralza®
300 mg
Q4W
|
Placebo
|
Adtralza®
300 mg
Q2W
|
Adtralza®
300 mg
Q4W
|
Placebo
| |
IGA 0/1a)
|
51,3%
(20/39)
|
38,9%
(14/36)
|
47,4%
(9/19)
|
59,3%c)
(32/54)
|
44,9%
(22/49)
|
25,0%
(7/28)
| |
EASI-75a)
|
59,6%b)
(28/47)
|
49,1%
(28/57)
|
33,3%
(10/30)
|
55,8%c)
(43/77)
|
51,4%
(38/74)
|
21,4%
(9/42)
|
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs. Le pourcentage est calculé par rapport au nombre de patients présentant une réponse à la semaine 16.
b) P = 0,004 par rapport au placebo
c) P < 0,001 par rapport au placebo
d) Tous les patients ont initialement été traités par 300 mg d’Adtralza® Q2W de la semaine 0 à la semaine 16.
Parmi les patients ayant reçu Adtralza® après la randomisation, mais qui n'avaient pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16 et qui sont passés au traitement dans le bras en ouvert pour recevoir 300 mg d'Adtralza® Q2W + CST optionnels, 20,8% dans ECZTRA 1 et 19,3% dans ECZTRA 2 avaient obtenu un IGA 0 ou 1 à la semaine 52, et 46,1% dans l'ECZTRA 1 et 39,3% dans l'ECZTRA 2 avaient obtenu un score EASI-75 à la semaine 52. La réponse clinique dépendait principalement de la poursuite du traitement par Adtralza® et non au traitement CST optionnel. Parmi les patients avec un score IGA de 2 ou EASI-50 à la semaine 16, une plus grande proportion avait obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 52 que les patients avec un score IGA de 3 ou 4 ou < EASI-50 à la semaine 16.
Étude de 32 semaines en association aux CST (ECZTRA 3) - période de traitement initiale de 0 à 16 semaines
Dans l'étude ECZTRA 3, parmi les patients ayant reçu 300 mg d'Adtralza® Q2W + CST après randomisation, un pourcentage significativement plus élevé a obtenu un score IGA de 0 ou 1, un score EASI-75 et/ou une amélioration du score NRS de prurit maximal quotidien ≥ 4 points entre l’inclusion et la semaine 16, par rapport au groupe placebo + CST (voir tableau 3).
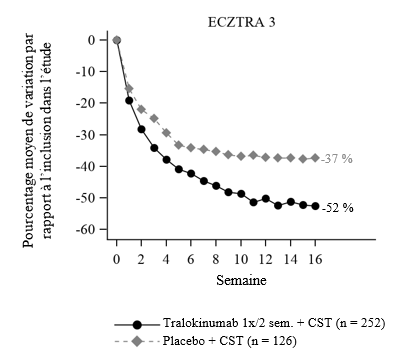
Dès la semaine 2, une amélioration significative du NRS de prurit maximal quotidien (pourcentage moyen de variation par rapport à la valeur à l’inclusion) a été observée chez les patients ayant reçu Adtralza® + CST après randomisation, par rapport au placebo + CST (voir figure 4). L'amélioration du score NRS de prurit maximal quotidien était associée à l'amélioration du DLQI et des signes objectifs de la dermatite atopique, y compris le score sur l'échelle SCORAD.
Tableau 3 : Résultats d’efficacité d’Adtralza® en association aux CST à la semaine 16 dans l’étude ECZTRA 3 (FAS)
|
ECZTRA 3 - Traitement en association
| |
|
Semaine 16
| |
Placebo + CST
|
300 mg Adtralza® Q2W + CST
| |
Nombre de patients randomisés et ayant reçu une dose (FAS)
|
126
|
252
| |
IGA 0 ou 1, % de répondeursa,b)
|
26,2
|
38,9*
| |
EASI-50, % de répondeursa
|
57,9
|
79,4§
| |
EASI-75, % de répondeursa)
|
35,7
|
56,0§
| |
EASI-90, % de répondeursa)
|
21,4
|
32,9*
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)
|
-55,3
(± 3,2)
|
-71,3§
(± 2,2)
| |
SCORAD 50, % de répondeursa)
|
38,1
|
61,1§
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)c)
|
-40,0
(± 2,6)
|
-55,9§
(± 1,8)
| |
SCORAD, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-26,8
(± 1,8)
|
-37,7§
(± 1,3)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,d)
|
34,1 (43/126)
|
45,4* (113/249)
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-2,9
(± 0,2)
|
-4,1§
(± 0,2)
| |
DLQI (amélioration > 4 points, % de répondeurs)a,d)
|
65,9 (81/123)
|
83,5§ (207/248)
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-8,8
(± 0,6)
|
-11,7§
(± 0,4)
|
MC=moindres carrés; ET=erreur-type, FAS=analyse de la population totale de l’étude (Full Analysis Set) – inclut tous les patients randomisés et ayant reçu une dose
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs.
b) Un répondeur était défini comme un patient présentant un score IGA de 0 ou 1 (« blanchi » ou « presque blanchi » sur l’échelle IGA de 0-4).
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement étaient considérées comme manquantes.
d) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
*p<0,05, #p<0,01, §p<0,001
Dans l'étude ECZTRA 3, les patients ayant reçu 300 mg d'Adtralza® Q2W de la semaine 0 à 16 ont utilisé 50% de CST en moins à la semaine 16 que les patients du groupe placebo.
Figure 3 : Pourcentage moyen de variation du score EASI dans l'étude ECZTRA 3 par rapport au début de l'étude
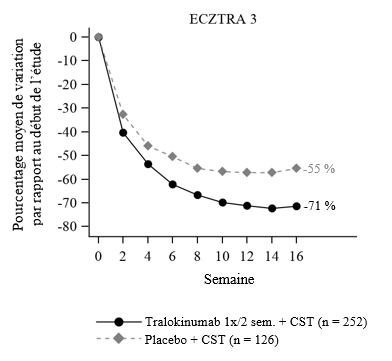
Figure 4 : Pourcentage moyen de variation du score NRS de prurit maximal quotidien (moyenne hebdomadaire) de la semaine 0 à 16 par rapport à l’inclusion dans l'étude ECZTRA 3
Étude de 32 semaines en association aux CST (ECZTRA 3) - période d’entretien de 16 à 32 semaines
Lors de l'utilisation de 300 mg d'Adtralza® Q2W + CST et de 300 mg d'Adtralza® Q4W + CST, un maintien élevé de l'efficacité clinique a été observé à la semaine 32 chez les patients ayant obtenu une réponse clinique à la semaine 16 (voir tableau 4).
Tableau 4 : Résultats d'efficacité à la semaine 32 chez les patients ayant présenté une réponse clinique à 300 mg d'Adtralza® + CST Q2W à la semaine 16
|
|
300 mg Adtralza® Q2W + CST
|
300 mg Adtralza® Q4W + CST
| |
IGA 0/1 à la semaine 32a)
% de répondeurs
|
89,6%
(43/48)
|
77,6%
(38/49)
| |
EASI-75 à la semaine 32a)
% de répondeurs
|
92,5%
(62/67)
|
90,8%
(59/65)
|
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs. Le pourcentage est calculé par rapport au nombre de patients présentant une réponse à la semaine 16.
Parmi tous les patients ayant obtenu soit un score IGA de 0 ou 1, soit un score EASI-75 à la semaine 16, le pourcentage moyen d'amélioration du score EASI par rapport à l’inclusion était de 93,5% à la semaine 32 pour les patients ayant continué à recevoir 300 mg d'Adtralza® Q2W + CST, et de 91,5% à la semaine 32 pour les patients ayant reçu 300 mg d'Adtralza® Q4W + CST.
Parmi les patients ayant reçu 300 mg d'Adtralza® Q2W + CST après la randomisation et n’ayant pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16, 30,5% ont obtenu un score IGA 0/1 à la semaine 32 et 55,8% un score EASI-75 lorsqu'ils ont été traités en continu avec 300 mg d'Adtralza® Q2W + CST pendant 16 semaines supplémentaires.
La poursuite de l’amélioration chez les patients n’ayant pas obtenu un score IGA de 0 ou 1 ou EASI-75 à la semaine 16 est survenue conjointement à l’amélioration du score NRS de prurit maximal quotidien et des signes cliniques objectifs de la dermatite atopique, incluant le score SCORAD.
Tableau 5: Résultats d’efficacité d‘Adtralza® en association aux CST aux semaines 16 et 32 dans l’étude ECZTRA 3 chez les patients initialement traités par Adtralza® Q2W + CST
|
|
Schéma de traitement des semaines 16 à 32d)
| |
Répondeurs à la semaine 16e)
|
Non-répondeurs à la semaine 16
| |
Patients randomisés
|
Adtralza® Q2W + CST
|
Adtralza® Q4W + CST
|
Adtralza® Q2W + CST
| |
N=69
|
N=69
|
N=95
| |
Semaine
|
W16
|
W32
|
W16
|
W32
|
W16
|
W32
| |
EASI-50, % de répondeursa)
|
100,0
|
98,6
|
97,1
|
91,3
|
63,2
|
76,8
| |
EASI-90, % de répondeursa)
|
58,0
|
72,5
|
60,9
|
63,8
|
1,1
|
34,7
| |
EASI, variation moyenne des MC en % par rapport à l’inclusion (ET)b)
|
-90,5
(2,7)
|
-93,2
(2,3)
|
-89,3
(2,7)
|
-91,5
(2,3)
|
-46,9
(2,4)
|
-73,5
(2,0)
| |
SCORAD, variation moyenne des MC en % par rapport à l’inclusion (ET)b)
|
-73,2
(2,1)
|
-79,2
(2,5)
|
-72,3
(2,1)
|
-73,3
(2,5)
|
-32,7
(1,8)
|
-54,5
(2,2)
| |
Score NRS de prurit (amélioration > 4 points, % de répondeurs)a,c)
|
63,2
|
70,6
|
64,2
|
61,2
|
27,4
|
38,9
| |
Score-NRS de prurit, variation moyenne des MC par rapport à l’inclusion (± ET)b)
|
-5,0
(0,2)
|
-5,4
(0,2)
|
-4,6
(0,2)
|
-4,9
(0,2)
|
-3,0
(0,2)
|
-3,7
(0,2)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
b) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
c) Le pourcentage est calculé par rapport au nombre de patients dont la valeur à l’inclusion était > 4.
d) Tous les patients ont initialement été traités par 300 mg Adtralza® Q2W + CST de la semaine 0 à la semaine 16. Ils ont ensuite été traités par 300 mg Adtralza® Q2W + CST ou 300 mg Adtralza® Q4W + CST.
e) Les répondeurs à la semaine 16 ont été identifiés comme les patients ayant obtenu un score IGA 0/1 et/ou EASI-75.
Qualité de vie/résultats rapportés par les patients (Patient-Reported Outcomes)
Dans les deux études en monothérapie (ECZTRA 1 et ECZTRA 2), l'utilisation de 300 mg d'Adtralza® Q2W a amélioré les symptômes rapportés par les patients, et l'impact de la DA sur le sommeil et la qualité de vie liée à la santé, mesuré par le score POEM,par le score NRS pour les troubles du sommeil liés à l'eczéma et par le SF 36, a été examiné à la semaine 16 par rapport au placebo. Parmi les patients traités par Adtralza®, une proportion plus élevée a présenté une réduction cliniquement significative du score POEM (définie comme une amélioration ≥ 4 points) entre l’inclusion et la semaine 16, par rapport au groupe placebo (voir tableau 6).
Tableau 6 : Autres résultats des critères d'évaluation liés à la monothérapie par Adtralza® à la semaine 16 dans les études ECZTRA 1 et ECZTRA 2
|
Monothérapie
| |
|
ECZTRA 1, Semaine 16
|
ECZTRA 2, Semaine 16
| |
|
Placebo
|
300 mg Adtralza® Q2W
|
Placebo
|
300 mg Adtralza® Q2W
| |
Patients randomisés
|
199
|
603
|
201
|
593
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-1,9
(0,2)
|
-2,6#
(0,1)
|
-1,5
(0,2)
|
-2,9§
(0,1)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-3,0
(0,7)
|
-7,6§
(0,4)
|
-3,7
(0,7)
|
-8,8§
(0,3)
| |
POEM (amélioration > 4 points, % de répondeurs)b)
|
18,0%
(35/194)
|
43,0 %§
(253/588)
|
22,1%
(44/199)
|
54,4 %§
(319/586)
| |
SF-36, composante physique, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
2,9
(0,6)
|
4,5*
(0,3)
|
3,2
(0,6)
|
5,8§
(0,3)
| |
SF-36, , composante physique, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
0,3
(0,8)
|
2,5*
(0,4)
|
0,5
(0,8)
|
3,5§
(0,4)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
b) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
Le pourcentage est calculé par rapport au nombre de patients avec un score POEM > 4 au début de l'étude.
*p < 0,05, #p < 0,01, §p < 0,001
Dans l'étude en association aux CST (ECZTRA 3), l'utilisation de 300 mg d'Adtralza® Q2W + CST a amélioré les symptômes rapportés par les patients et l'impact de la DA sur le sommeil et la qualité de vie liée à la santé, évalués par le score POEM et le score NRS pour les troubles du sommeil liés à l'eczéma, à la semaine 16 par rapport au placebo.
Parmi les patients traités par Adtralza®, une proportion plus élevée a obtenu une réduction cliniquement significative su score POEM (définie comme une amélioration ≥ 4 points) entre l’inclusion et la semaine 16, par rapport au groupe placebo (voir tableau 7).
Tableau 7 : Autres résultats des critères d'évaluation lors de l'utilisation d'Adtralza® avec CST à la semaine 16 dans l'étude ECZTRA 3
|
ECZTRA 3 - Traitement en association
| |
|
Woche 16
| |
Placebo + CST
|
300 mg Adtralza® Q2W + CST
| |
Patients randomisés
|
126
|
252
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-3,1
(0,2)
|
-4,3§
(0,2)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)a)
|
-7,8 (0,7)
|
-11,8§ (0,5)
| |
POEM (amélioration > 4 points, % de répondeurs)b)
|
59,3% (73/123)
|
78,4%§ (190/250)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
b) Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
Le pourcentage est calculé par rapport au nombre de patients avec un score POEM > 4 au début de l'étude.
§p < 0,001
Chez les patients traités par 300 mg d'Adtralza® Q2W + CST avec une réponse à la semaine 16 et qui ont continué à recevoir 300 mg d'Adtralza® Q2W + CST ou Q4W + CST, des réductions cliniquement significatives du score POEM, du score DLQI et du score NRS pour les troubles du sommeil liés à l'eczéma ont été observées entre l’inclusion et la semaine 32.
Tableau 8: Autres critères d'évaluation lors de l'utilisation d'Adtralza® avec CST aux semaines 16 et 32 de l'étude ECZTRA 3 chez les patients ayant obtenu une réponse clinique à la semaine 16
|
|
Schéma de traitement des semaines 16 à 32a)
| |
|
Répondeurs à la semaine 16b)
| |
Adtralza® Q2W + CST
|
Adtralza® Q4W + CST
| |
Patients randomisés
|
N=69
|
N=69
| |
Semaine
|
W16
|
W32
|
W16
|
W32
| |
DLQI, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-14,0
(0,6)
|
-14,6
(0,6)
|
-13,9
(0,6)
|
-13,7
(0,6)
| |
POEM, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-15,2
(0,7)
|
-15,6
(0,7)
|
-14,1
(0,7)
|
-13,9
(0,8)
| |
NRS pour les troubles du sommeil liés à l'eczéma, variation moyenne des MC par rapport à l’inclusion (± ET)c)
|
-5,2
(0,2)
|
-5,5
(0,2)
|
-4,8
(0,2)
|
-5,2
(0,3)
| |
DLQI (amélioration > 4 points,
% de répondeursd)
|
98,5%
(65/66)
|
89,4%
(59/66)
|
100,0 %
(68/68)
|
83,8%
(57/68)
| |
POEM (amélioration > 4 points),
% de répondeursd
|
89,7%
(61/68)
|
88,2%
(60/68)
|
94,1 %
(64/68)
|
83,8%
(57/68)
|
MC=moindres carrés; ET=erreur-type
Si nécessaire pour contrôler des symptômes intolérables de la dermatite atopique, les patients étaient autorisés à recevoir un traitement de secours à la discrétion de l’investigateur.
a) Tous les patients ont initialement été traités par 300 mg Adtralza® Q2W + CST de la semaine 0 à la semaine 16. Ils ont ensuite été traités par 300 mg Adtralza® Q2W + CST ou 300 mg Adtralza® Q4W + CST.
b) Les répondeurs à la semaine 16 ont été identifiés comme les patients ayant obtenu un score IGA 0/1 et/ou EASI-75.
c) Les données recueillies après l’instauration d’un traitement de secours ou l’arrêt définitif du traitement ont été exclues des analyses.
d) Nombre de répondeurs divisé par le nombre de patients ayant une valeur de référence > 4 pour chaque paramètre.
Les patients ayant reçu un traitement de secours ou dont les données étaient manquantes étaient considérés comme non-répondeurs dans les analyses.
|