CompositionPrincipes actifs
Sémaglutide.
Analogue du glucagon-like peptide-1 (GLP-1) humain, produit par génie génétique par la technologie de l'ADN recombinant dans des cellules de Saccharomyces cerevisiae.
Excipients
Wegovy FixDose
Dinatrii phosphas dihydricus, Natrii chloridum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile.
Le médicament contient 0.078557 mmol de sodium par dosage (0.25 mg, 0.5 mg, 1.0 mg), resp. 0.11785 mmol de sodium par dose (1.7 mg, 2.4 mg).
Wegovy Multi FixDose
Dinatrii phosphas dihydricus, Propylenglycolum, Phenolum, Acidum hydrochloridum/Natrii hydroxidum (q.s. ad pH), Aqua ad iniectabile q.s. ad solutionem pour 0.37 ml resp. 0.75 ml.
Le médicament contient 0.00591 mmol (0.136 mg) de sodium par dosage (0.25 mg, 0.5 mg) resp. 0.01197 mmol (0.275 mg) de sodium par dosage (1 mg, 1.7 mg, 2.4 mg).
Indications/Possibilités d’emploiRégulation du poids
Wegovy est utilisé en complément d'un régime hypocalorique et d'une activité physique accrue pour réguler le poids chez:
·les patients adultes dont l'indice de masse corporelle (IMC) initial est de
·≥30 kg/m2 (obésité) ou
·≥27 kg/m2 à < 30 kg/m2 (surpoids) en présence d'au moins une comorbidité due au poids.
·les patients adolescents, à partir de 12 ans, atteints d'obésité selon les valeurs limites acceptées au niveau international* et ayant un poids corporel supérieur à 60 kg
*Obésité (percentile IMC ≥95) selon les tableaux de croissance de l'IMC par rapport au sexe et à l'âge (CDC.gov) (cf. Tableau 1).
Maladie cardiovasculaire établie
Wegovy est indiqué pour réduire le risque d'événements cardiovasculaires graves chez les adultes atteints de maladie cardiovasculaire établie et présentant un IMC ≥27 kg/m2. Le traitement doit être un complément au traitement standard chez les patients atteints de maladie cardiovasculaire établie (cf. rubrique «Efficacité clinique»).
Tableau 1: Seuils d'IMC pour l'obésité (percentile IMC ≥95) par sexe et par âge chez les patients pédiatriques âgés de 12 ans et plus (critères CDC)
|
Âge (années)
|
Percentile IMC 95 % (kg/m2)
| |
Homme
|
Femme
| |
12
|
24.2
|
25.2
| |
12.5
|
24.7
|
25.7
| |
13
|
25.1
|
26.3
| |
13.5
|
25.6
|
26.8
| |
14
|
26.0
|
27.2
| |
14.5
|
26.4
|
27.7
| |
15
|
26.8
|
28.1
| |
15.5
|
27.2
|
28.5
| |
16
|
27.5
|
28.9
| |
16.5
|
27.9
|
29.3
| |
17
|
28.2
|
29.6
| |
17.5
|
28.6
|
30.0
|
Posologie/Mode d’emploiPosologie usuelle
Adultes
La dose d'entretien de 2.4 mg une fois par semaine est atteinte avec une dose initiale de 0.25 mg. La dose doit, pour réduire la probabilité de symptômes gastro-intestinaux, être augmentée sur une période de 16 semaines jusqu'à la dose d'entretien de 2.4 mg une fois par semaine (cf. Tableau 2). Il faut, en cas d'apparition de symptômes gastro-intestinaux importants, envisager de suspendre l'augmentation de la dose jusqu'à l'amélioration des symptômes.
Des doses hebdomadaires supérieures à 2.4 mg ne sont pas recommandées.
Tableau 2: Calendrier d'augmentation des doses
|
Augmentation de la dose
|
Dose hebdomadaire
| |
Semaine 1 à 4
|
0.25 mg
| |
Semaine 5 à 8
|
0.5 mg
| |
Semaine 9 à 12
|
1 mg
| |
Semaine 13 à 16
|
1.7 mg
| |
Dose d'entretien
|
2.4 mg
|
Adultes dans l'indication «Régulation du poids»
Si, après 28 semaines de traitement, les patients n'ont pas perdu au moins 5 % de leur poids corporel initial, il convient de décider si le traitement doit être poursuivi en tenant compte du profil de bénéfice et de risque de chaque patient.
Adolescents dans l'indication «Régulation du poids»
Pour les adolescents de 12 ans ou plus, le même schéma de dosage que pour les adultes doit être appliqué (cf. Tableau 2). La dose doit être augmentée jusqu'à 2.4 mg (dose d'entretien) ou jusqu'à ce que la dose maximale tolérée soit atteinte.
Il convient de décider si le traitement doit être poursuivi en tenant compte du profil bénéfice-risque de chaque patient si l'IMC des patients ne s'est pas amélioré d'au moins 5 % après 28 semaines de traitement.
Des doses hebdomadaires supérieures à 2.4 mg ne sont pas recommandées.
Il est recommandé, pour assurer la traçabilité des médicaments préparés par biotechnologie, de documenter le nom et le numéro de lot à chaque traitement.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance hépatique. L'expérience de l'utilisation du sémaglutide chez les patients présentant une insuffisance hépatique grave est limitée. La prudence est de mise lors du traitement de ces patients par le sémaglutide (cf. «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance rénale légère, modérée ou sévère. L'expérience de l'utilisation du sémaglutide chez les patients présentant une insuffisance rénale sévère est limitée. L'utilisation du sémaglutide n'est pas recommandée chez les patients atteints d'insuffisance rénale terminale (cf. «Pharmacocinétique»).
Patients âgés (≥65 ans)
Un ajustement de la dose n'est pas nécessaire chez les personnes plus âgées.
Enfants et adolescents
Le sémaglutide n'est pas autorisé chez les enfants de moins de 12 ans. Le sémaglutide est autorisé pour le traitement des adolescents à partir de 12 ans dans l'indication «Régulation du poids».
Patients souffrant d'autres maladies sous-jacentes
Patients atteints de diabète de type 2
Wegovy ne doit pas être utilisé en combinaison avec d'autres agonistes des récepteurs du GLP-1.
Une réduction de la dose d'insuline ou de sécrétagogue d'insuline (comme les sulfonylurées) utilisés simultanément afin de réduire le risque d'hypoglycémie doit être considérée lorsque le traitement par le sémaglutide est initié.
Prise retardée
Si une dose est omise, elle doit être administrée dès que possible et dans les 5 jours suivant la date de la dose prévue initialement. Si plus de 5 jours se sont écoulés, la dose omise doit être ignorée et la dose suivante doit être administrée le jour prévu. Dans tous les cas, les patients pourront reprendre leur programme de dosage régulier une fois par semaine. Si plusieurs doses ont été omises, il faut envisager de reprendre le traitement avec une dose plus faible.
Mode d'administration
Wegovy est administré une fois par semaine, à n'importe quel moment de la journée, avec ou sans repas.
Wegovy doit être injecté par voie sous-cutanée dans l'abdomen, la cuisse ou le bras. Le site d'injection peut être modifié sans ajustement de la dose. Wegovy ne doit pas être administré par voie intraveineuse ou intramusculaire. Le jour de l'administration hebdomadaire peut être changé si nécessaire, tant que l'intervalle entre deux doses est d'au moins 3 jours (> 72 heures). La dose hebdomadaire doit être poursuivie une fois le nouveau jour d'administration choisi.
Lors de l'administration de Wegovy, le stylo doit être pressé fermement contre la peau jusqu'à ce que la barre jaune ne bouge plus. L'injection dure environ de 5 à 10 secondes.
Les patients doivent lire attentivement les instructions de la notice d'emballage avant d'utiliser Wegovy.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsRisque de tumeurs thyroïdiennes à cellules C
Des études précliniques sur les agonistes des récepteurs du GLP-1 chez les rongeurs suggèrent que les agonistes des récepteurs du GLP-1 pourraient être associés à un risque accru d'hyperplasie focale des cellules C thyroïdiennes et de tumeurs des cellules C (cf. «Données précliniques»).
Un lien entre les agonistes des récepteurs du GLP-1 et les tumeurs thyroïdiennes à cellules C, y compris le carcinome médullaire de la thyroïde (CMT) chez les humains n'est pas connu. Les patients atteints de CMT et les patients ayant des antécédents de syndrome de néoplasie endocrinienne multiple de type 2 (MEN 2) n'ont pas été traités par le sémaglutide dans les études cliniques. Il est par conséquent nécessaire d'évaluer soigneusement les bénéfices et les risques avant d'entreprendre un traitement avec Wegovy dans ce collectif spécifique. La valeur clinique d'une surveillance de routine du taux de calcitonine sérique n'a pas été démontrée.
Effets sur le tractus gastro-intestinal
L'utilisation d'agonistes des récepteurs du GLP-1 peut être associée à des effets secondaires gastro-intestinaux pouvant provoquer une déshydratation, ce qui peut pour sa part entraîner une détérioration de la fonction rénale. Les patients doivent être informés du risque potentiel de déshydratation lié aux effets indésirables gastro-intestinaux et prendre des précautions contre les pertes de liquide.
Pancréatite aiguë
Une pancréatite aiguë a été observée lors de l'utilisation d'agonistes des récepteurs du GLP-1. Les patients devraient être informés des symptômes caractéristiques d'une pancréatite aiguë. Wegovy doit être arrêté en cas de potentielle pancréatite. Le traitement par Wegovy ne devra pas être repris si cette dernière devait être confirmée. Les patients ayant des antécédents de pancréatite n'ont pas été examinés dans les études cliniques sur le sémaglutide. La prudence est de mise chez les patients ayant déjà souffert de pancréatite.
Une élévation des enzymes pancréatiques ne constitue pas à elle seule un facteur prédictif de pancréatite aiguë en l'absence d'autres signes et symptômes de pancréatite aiguë.
Patients atteints de diabète de type 2
Wegovy ne doit pas être utilisé comme substitut de l'insuline chez les patients diabétiques.
Hypoglycémie chez les patients souffrant d'obésité ou de surpoids et de diabète de type 2
L'insuline et les sulfonylurées peuvent provoquer une hypoglycémie. Les patients traités par Wegovy en association avec une sulfonylurée ou de l'insuline peuvent présenter un risque accru d'hypoglycémie. Le risque d'hypoglycémie peut être réduit en diminuant la dose de sulfonylurée ou d'insuline au début du traitement par un agoniste des récepteurs du GLP-1. L'administration supplémentaire de Wegovy chez des patients déjà traités à l'insuline n'a pas été étudiée.
Rétinopathie diabétique chez les patients souffrant d'obésité ou de surpoids et de diabète de type 2
Un risque accru de survenue de complications de rétinopathie diabétique a été observé chez les patients atteints de rétinopathie diabétique sous insuline et sémaglutide. Une amélioration rapide du contrôle glycémique a été associée à une aggravation temporaire de la rétinopathie diabétique. Les patients atteints de rétinopathie diabétique utilisant le sémaglutide doivent être surveillés de près et traités conformément aux directives cliniques. Il n'y a pas d'expérience avec le sémaglutide 2.4 mg chez les patients atteints de diabète de type 2 souffrant d'une rétinopathie diabétique non contrôlée ou potentiellement instable.
Insuffisance rénale aiguë
Après la commercialisation, chez des patients traités par des agonistes des récepteurs du GLP-1, une insuffisance rénale aiguë et une aggravation d'une insuffisance rénale chronique pouvant parfois nécessiter une hémodialyse, ont été rapportées. Certains de ces événements ont été signalés chez des patients ne présentant aucune affection rénale sous-jacente. La plupart des événements signalés sont survenus chez des patients qui présentaient déjà des nausées, des vomissements, des diarrhées et une déshydratation. La fonction rénale doit être surveillée lors de l'instauration ou de l'ajustement d'un traitement par Wegovy chez des patients rapportant de sévères réactions indésirables gastro-intestinales.
Populations non examinées
Il n'y a pas d'expérience chez les patients souffrant d'insuffisance cardiaque de stade IV de la NYHA. L'expérience est limitée chez les patients âgés de 85 ans et plus.
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsDes études in vitro ont montré que le sémaglutide a un très faible potentiel d'inhibition ou d'induction des enzymes CYP et d'inhibition des transporteurs de substances actives.
Comme d'autres agonistes des récepteurs du GLP-1, le sémaglutide peut retarder la vidange gastrique et éventuellement influencer l'absorption de médicaments administrés simultanément par voie orale. Aucun effet cliniquement significatif sur la vitesse de vidange gastrique n'a été observé avec le sémaglutide 2.4 mg. Dans les études cliniques visant à examiner l'effet du sémaglutide 1.0 mg sur l'absorption de médicaments administrés simultanément par voie orale à l'état d'équilibre, aucune interaction médicamenteuse cliniquement pertinente n'a été constatée avec le sémaglutide pour les médicaments étudiés. Aucun ajustement de la dose n'est par conséquent nécessaire en cas d'administration simultanée avec le sémaglutide.
Effet de sémaglutide sur d'autres médicaments
Contraceptifs oraux
Une diminution de l'efficacité des contraceptifs oraux par le sémaglutide n'est pas attendue car le sémaglutide n'a pas modifié l'exposition totale à l'éthinylestradiol et au lévonorgestrel de manière cliniquement significative lorsqu'un contraceptif oral combiné (0.03 mg d'éthinylestradiol/0.15 mg de lévonorgestrel) a été utilisé conjointement avec le sémaglutide. L'exposition à l'éthinylestradiol n'a pas été affectée; une augmentation de 20 % de l'exposition au lévonorgestrel à l'état d'équilibre a été observée. La Cmax n'a été influencée pour aucun des principes actifs.
Atorvastatine
Après l'administration d'une dose unique d'atorvastatine (40 mg), le sémaglutide n'a pas modifié l'exposition totale à l'atorvastatine. La Cmax de l'atorvastatine a été diminuée de 38 %. Cela a été considéré comme cliniquement non significatif.
Digoxine
Après l'administration d'une dose unique de digoxine (0.5 mg), le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la digoxine.
Metformine
Après administration de metformine 500 mg deux fois par jour pendant 3.5 jours, la sémaglutide n'a pas modifié l'exposition globale ou la Cmax de la metformine.
Warfarine et autres dérivés de la coumarine
Le sémaglutide n'a pas modifié l'exposition totale ou la Cmax de la R- et S-warfarine après une dose unique de warfarine (25 mg). Aussi l'effet pharmacodynamique de la warfarine tel que mesuré par le rapport normalisé international (International Normalised Ratio, INR) n'a pas été affecté de manière cliniquement significative. Toutefois, des cas de diminution de l'INR ont été rapportés lors de l'utilisation concomitante d'acénocoumarol et de sémaglutide. Lors de l'instauration du traitement par sémaglutide chez des patients sous warfarine ou autres dérivés de la coumarine, il est donc recommandé de surveiller fréquemment l'INR.
Grossesse, allaitementGrossesse
Une toxicité sur la reproduction a été montrée dans les études animales (cf. «Données précliniques»). Les données sur l'utilisation du sémaglutide chez les femmes enceintes sont limitées. Le sémaglutide ne doit par conséquent pas être utilisé pendant la grossesse. Il est recommandé aux femmes en âge de procréer d'utiliser une méthode de contraception fiable pendant le traitement par sémaglutide. Si une patiente souhaite tomber enceinte ou si une grossesse survient, la prise de sémaglutide doit être arrêtée. Le sémaglutide doit être arrêté au moins 2 mois avant une grossesse prévue du fait de sa longue demi-vie (cf. «Pharmacocinétique»).
Allaitement
Le sémaglutide a été excrété dans le lait maternel des rates allaitantes. Un risque pour l'enfant allaité ne peut pas être exclu. Le sémaglutide ne doit pas être utilisé pendant l'allaitement.
Fertilité
On ne sait pas si le sémaglutide a un effet sur la fertilité humaine. Le sémaglutide n'a pas affecté la fertilité des rats mâles. Un allongement de l'œstrus et une faible diminution du nombre d'ovulations ont été observés chez les rates à des doses entraînant une perte de poids maternelle.
Effet sur l’aptitude à la conduite et l’utilisation de machinesLe sémaglutide n'a aucun effet, ou un effet négligeable, sur l'aptitude à conduire ou à utiliser des machines. Une sensation de vertige peut toutefois survenir, en particulier pendant la période d'augmentation de la dose. La prudence est de mise dans la circulation routière et lors de l'utilisation de machines en cas de vertiges.
Patients atteints de diabète sucré de type 2
Lorsque le sémaglutide est utilisé en association avec une sulfonylurée ou de l'insuline, les patients devraient être incités à prendre des mesures pour éviter l'hypoglycémie lors de la conduite d'un véhicule ou de l'utilisation de machines (cf. «Mises en garde et précautions»).
Effets indésirablesRésumé du profil de sécurité
2 650 patients adultes ont été traités avec Wegovy dans 4 études de phase 3a. La durée des études a été de 68 semaines. Comme avec d'autres agonistes des récepteurs du GLP-1, les effets indésirables les plus fréquemment rapportés étaient des troubles gastro-intestinaux, notamment des nausées, des diarrhées, une constipation et des vomissements.
Liste tabellaire des effets secondaires
Le Tableau 3 présente les effets indésirables chez les adultes rapportés dans les études de phase 3a. Les fréquences sont basées sur les données groupées des études de phase 3a et les rapports de post-marketing chez les patients atteints de diabète sucré de type 2.
Liste des effets indésirables
Les effets indésirables sont rangés par classe de système d'organes de la classification MedDRA et par fréquence selon la convention suivante: «très fréquents» (≥1/10), «fréquents» (≥1/100 à < 1/10), «occasionnels» (≥1/1000 à < 1/100), «rares» (≥1/10'000 à < 1/1000), «très rares» (< 1/10'000), «fréquence inconnue» (ne peut être estimée sur la base des données disponibles).
Tableau 3: Effets indésirables chez les adultes tirés d'études de phase 3 contrôlées et de rapports post-marketing
|
Classe d'organe systémique selon MedDRA
|
Très fréquents
|
Fréquents
|
Occasionnels
|
Rares
|
Fréquence inconnue
| |
Affections du système immunitaire
|
|
|
|
Réaction anaphylactique
|
| |
Troubles du métabolisme et de la nutrition
|
|
Hypoglycémie chez des patients atteints de diabète de type 2a
|
|
|
| |
Affections du système nerveux
|
Céphaléesb
|
Sensation vertigineuseb
Dysgueusieb,c
Dysesthésiea,c
|
|
|
| |
Affections oculaires
|
|
Rétinopathie diabétique chez les patients atteints de diabète de type 2a
|
|
|
| |
Affections cardiaques
|
|
|
Fréquence cardiaque accruea,c
|
|
| |
Affections gastro-intestinales
|
Vomissementsa,b
Diarrhéesa,b
Constipationa,b
Nauséesa,b
Douleurs abdominalesb,c
|
Gastriteb,c
Reflux gastro-œsophagienb
Dyspepsieb
Éructationb
Flatulenceb
Ventre gonfléb
|
Pancréatite aiguëa
Vidange gastrique retardée
|
|
Obstruction intestinalec,d,e
| |
Affections hépatobiliaires
|
|
Cholélithiasea
|
Amylase accruec
Lipase accruec
Cholécystite
|
|
| |
Affections de la peau et du tissu sous-cutané
|
|
Chute des cheveuxa
|
|
|
| |
Affections du rein et des voies urinaires
|
|
|
|
|
Insuffisance rénale aiguë
| |
Troubles généraux et anomalies au site d'administration
|
Fatigueb,c
|
Réactions au site d'injectionc
|
|
|
| |
a)
Voir la description suivante des effets indésirables sélectionnés
b) Principalement pendant l'escalade posologique
c) Groupement des termes privilégiés
d) D'après les rapports post-marketing
e) Terme groupé couvrant les événements indésirables obstruction intestinale, iléus et obstruction de l'intestine grêle
|
Description d'effets indésirables spécifiques et informations complémentaires
Affections gastro-intestinales
Très fréquents – nausées (43.9 %), diarrhées (29.7 %), vomissements (24.5 %), constipation (24.2 %)
Les événements les plus fréquemment rapportés ont eu lieu pendant l'escalade posologique. Des nausées sont survenues chez 43.9 % des patients sous traitement par Wegovy (16.1 % sous placebo), des diarrhées chez 29.7 % (15.9 % sous placebo) et des vomissements chez 24.5 % (6.3 % sous placebo) sur une période de 68 semaines. La plupart des événements étaient de gravité légère à moyenne et de courte durée. Une constipation légère à modérée et de longue durée est apparue chez 24.2 % des patients sous traitement par Wegovy (11.1 % sous placebo).
Les événements gastro-intestinaux ont entraîné l'arrêt définitif du traitement par Wegovy (0.7 % sous placebo) chez 4.3 % des patients.
Occasionnels – pancréatite aiguë
La fréquence des cas confirmés de pancréatite aiguë rapportés dans les essais cliniques de phase 3a était de 0.2 % pour Wegovy et < 0.1 % pour le placebo.
Dans l'étude SELECT, c'est-à-dire l'étude des résultats cardiovasculaires, la fréquence des cas confirmés de pancréatite aiguë était de 0.2 % pour Wegovy et de 0.3 % pour le placebo.
Fréquents – lithiase biliaire aiguë/cholécystite
Une cholélithiase a été rapportée chez 1.6 % des patients sous Wegovy et a entraîné une cholécystite chez 0.6 % d'entre eux. Une cholélithiase et une cholécystite ont été rapportées respectivement chez 1.1 % et 0.3 % des patients sous placebo.
Affections de la peau et du tissu sous-cutané
Fréquents – chute de cheveux
Une chute de cheveux a été rapportée chez 2.5 % des patients sous Wegovy et chez 1.0 % des patients sous placebo. Les événements étaient principalement de nature légère et ont diminué chez la plupart des patients lorsque de la poursuite du traitement. La chute de cheveux a été rapportée plus fréquemment chez les patients ayant une perte de poids plus importante (≥20 %).
Affections cardiaques
Occasionnels – augmentation de la fréquence cardiaque
Dans les études de phase 3a, une augmentation moyenne de la fréquence cardiaque de 3 battements par minute (bpm) a été observée sous Wegovy par rapport à une valeur initiale moyenne de 72 bpm. La proportion de sujets présentant une augmentation maximale du pouls au repos de ≥20 battements par minute par rapport à la valeur initiale à n'importe quel moment pendant la période de traitement était de 26.0 % dans le groupe Wegovy contre 15.6 % dans le groupe placebo.
Affections du système nerveux
Fréquents – dysesthésie
Une analyse des études de phase 3a contrôlées par placebo (STEP 1-4) a montré une augmentation de l'incidence des dysesthésies. Des événements liés au tableau clinique d'une altération des sensations cutanées, tels que dysesthésie, paresthésie, hyperesthésie, brûlures, allodynie et sensibilité cutanée ont été rapportés chez 2.1 % des patients traités par Wegovy et chez 1.2 % des patients traités par placebo. Les événements étaient d'intensité légère à modérée et la plupart des patients se sont rétablis en poursuivant le traitement.
Immunogénicité
En fonction des propriétés potentiellement immunogènes des médicaments contenant des protéines ou des peptides, les patients peuvent former des anticorps anti-sémaglutide suite à un traitement par Wegovy. La détection de la formation d'anticorps dépend fortement de la sensibilité et de la spécificité du test. L'incidence observée de la positivité des anticorps (y compris les anticorps neutralisants) dans un test peut par ailleurs être influencée par plusieurs facteurs, y compris la méthodologie du test, la manipulation des échantillons, le moment du prélèvement, la médication concomitante et la maladie sous-jacente. L'incidence des anticorps anti-sémaglutide dans les études décrites ci-dessous ne peut par conséquent être directement comparée à l'incidence des anticorps dans d'autres études ou avec d'autres médicaments.
Le pourcentage de patients dont le test de détection des anticorps dirigés contre le sémaglutide s'est révélé positif à n'importe quel moment après le début du traitement était faible (2.9 %). Sur les 50 patients traités par le sémaglutide ayant développé des anticorps au sémaglutide, 28 patients (1.6 % de la population totale de l'étude traitée par Wegovy) ont produit des anticorps ayant eu une réaction croisée avec le GLP-1 natif. Aucun patient n'avait d'anticorps neutralisants contre la sémaglutide à la fin de l'étude, ni d'anticorps contre le sémaglutide ayant un effet neutralisant endogène sur le GLP-1.
Troubles du métabolisme et de la nutrition
Fréquents – hypoglycémie chez les patients atteints de diabète de type 2
Chez les patients atteints de diabète de type 2 (étude STEP 2), une hypoglycémie cliniquement significative (définie par un glucose plasmatique < 3,0 mmol/l (54 mg/dl)) a été observée chez 6.2 % (0.1 événement/année-patient) des personnes traitées par Wegovy, contre 2.5 % (0.03 événement/année-patient) des personnes traitées par placebo. Un épisode (0.2 % des participants, 0.002 événement/année-patient) a été considéré comme grave. Le risque d'hypoglycémie a été plus élevé lorsque Wegovy a été utilisé en association avec une sulfonylurée.
Les hypoglycémies n'ont pas été systématiquement enregistrées ou rapportées dans les études cliniques menées avec Wegovy chez des patients non diabétiques de type 2.
Fréquents – rétinopathie diabétique chez des patients atteints de diabète de type 2
Peu d'épisodes de rétinopathie diabétique ont été observés (4.0 % des personnes traitées par Wegovy et 2.7 % des personnes traitées par placebo) chez les patients atteints de diabète de type 2 dans l'étude STEP 2.
Enfants et adolescents
Dans une étude clinique menée chez des adolescents âgés de 12 ans à 18 ans révolus présentant une obésité ou une surcharge pondérale avec au moins une maladie concomitante due au poids, 133 patients ont reçu Wegovy. La durée de l'étude a été de 68 semaines.
Dans l'ensemble, la fréquence, la nature et la gravité des effets indésirables chez les adolescents étaient comparables à celles observées chez les adultes. Avec 3.8 % des adolescents traités par Wegovy, la cholélithiase a été plus fréquente que chez les adultes, par rapport à 0 % des adolescents traités par placebo.
L'étude n'a révélé aucun effet sur la croissance ou le développement de la puberté.
Adultes ayant une maladie cardiovasculaire établie
Dans l'étude SELECT, le profil des effets indésirables chez les adultes atteints de maladie cardiovasculaire établie était similaire à celui observé dans les études de phase 3a sur la perte de poids.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageUn surdosage de sémaglutide peut s'accompagner de troubles gastro-intestinaux entraînant une déshydratation. Le patient doit être surveillé en cas de surdosage afin de déceler tout signe clinique et un traitement de soutien approprié doit être mis en place.
Propriétés/EffetsCode ATC
A10BJ06
Mécanisme d'action
Le sémaglutide est un analogue du GLP-1 présentant une homologie de séquence de 94 % avec le GLP-1 humain. Le sémaglutide agit comme un agoniste du récepteur du GLP-1 qui se lie sélectivement au récepteur du GLP-1, la cible du GLP-1 natif, et l'active.
Comparé au GLP-1 natif, le sémaglutide présente une demi-vie prolongée d'environ une semaine, ce qui le rend approprié pour une administration sous-cutanée une fois par semaine. Le principal mécanisme de retard est la liaison à l'albumine qui entraîne une diminution de la clairance rénale et une protection contre la dégradation métabolique. Le sémaglutide est par ailleurs stabilisé contre la dégradation par l'enzyme DDP-4.
Le GLP-1 est une hormone physiologique qui agit dans la régulation de l'appétit et de l'apport calorique; le récepteur du GLP-1 est présent dans différentes zones du cerveau impliquées dans la régulation de l'appétit. Le sémaglutide a des effets directs sur les régions du cerveau impliquées dans la régulation homéostatique de la consommation alimentaire dans l'hypothalamus et le tronc cérébral. Le sémaglutide influence le système de récompense hédonique par des effets directs et indirects sur des régions du cerveau comme le septum, le thalamus et les amygdales.
Des études cliniques montrent que le sémaglutide réduit l'absorption d'énergie, augmente la perception de la satiété, la sensation de réplétion et le contrôle de l'alimentation, atténue la sensation de faim et réduit la fréquence et l'intensité des fringales.
Des études cliniques ont par ailleurs montré que le sémaglutide réduit la glycémie en fonction du glucose en stimulant la sécrétion d'insuline et en réduisant la sécrétion de glucagon lorsque la glycémie est élevée. Le mécanisme de réduction de la glycémie s'accompagne également d'un léger ralentissement de la vidange gastrique au début de la phase postprandiale. Le sémaglutide réduit la sécrétion d'insuline pendant une hypoglycémie, mais ne diminue pas la sécrétion de glucagon.
Les récepteurs GLP-1 sont également exprimés dans le cœur, les vaisseaux sanguins, le système immunitaire et les reins.
Le mécanisme d'action du sémaglutide pour réduire le risque cardiovasculaire est probablement multifactoriel et n'est pas complètement élucidé, en partie par les effets sur les facteurs de risque cardiovasculaire connus (y compris la réduction de la tension artérielle, l'amélioration du profil lipidique et les effets anti-inflammatoires).
Pharmacodynamique
Appétit, apport énergétique et choix alimentaire
Le sémaglutide réduit l'appétit en augmentant les sensations de satiété et de réplétion et en diminuant la faim et la prise alimentaire prospective. Après 20 semaines de traitement, l'apport énergétique d'un repas ad libitum était 35 % plus faible sous Wegovy que sous placebo. Cela s'est accompagné d'un meilleur contrôle de l'alimentation, de moins de fringales (de produits laitiers et d'aliments salés), de moins d'envies de sucré et d'une préférence relativement moins marquée pour les aliments riches en graisses.
Électrophysiologie cardiaque (QTc)
L'effet du sémaglutide sur la repolarisation cardiaque a été étudié dans une étude détaillée de QTc. Le sémaglutide n'a pas prolongé l'intervalle QTc à des doses allant jusqu'à 1.5 mg à l'état d'équilibre.
L'exposition au sémaglutide chez les sujets en surpoids ou obèses traités par Wegovy est comparable à l'exposition étudiée chez les sujets sains dans l'étude QTc sur le sémaglutide.
Efficacité clinique
L'efficacité et la sécurité de Wegovy pour la régulation du poids en association avec un régime hypocalorique et une activité physique accrue ont été évaluées dans quatre études de phase 3a, randomisées, en double aveugle et contrôlées par placebo d'une durée de 68 semaines (STEP 1-4). Au total, 4684 patients (2652 randomisés pour recevoir Wegovy) ont été inclus dans les études. L'efficacité et la sécurité du sémaglutide ont par ailleurs été étudiées dans une étude de phase 3b en double aveugle, randomisée et contrôlée par placebo sur une durée de deux ans (STEP 5) chez 304 patients (152 traités avec du sémaglutide) par rapport au placebo.
Le traitement par Wegovy a permis d'obtenir une perte de poids supérieure, cliniquement pertinente et durable par rapport au placebo chez les patients souffrant d'obésité (IMC ≥30 kg/m2) ou de surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) et d'au moins une maladie concomitante liée au poids.
En outre, dans l'ensemble des essais, une proportion plus élevée de patients a obtenu une perte de poids ≥5 %, ≥10 %, ≥15 % et ≥20 % sous sémaglutide par rapport au placebo.
Le traitement par Wegovy a également montré des améliorations statistiquement significatives du tour de taille et de la pression artérielle systolique par rapport au placebo. Le sémaglutide 2.4 mg a par ailleurs eu un effet globalement favorable sur les lipides plasmatiques et la CRP (marqueurs de l'inflammation) par rapport au placebo (cf. Tableaux 4 et 5).
L'efficacité de la perte de poids s'est avérée indépendante de l'âge, du sexe, de l'appartenance ethnique, des valeurs initiales du poids corporel et de l'IMC, de la présence d'un diabète de type 2 et du degré de la fonction rénale.
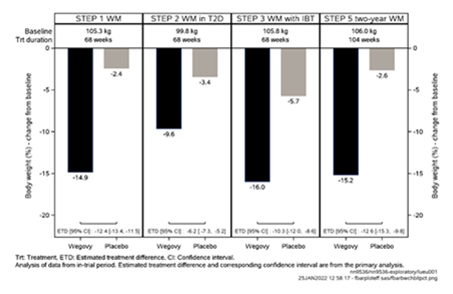
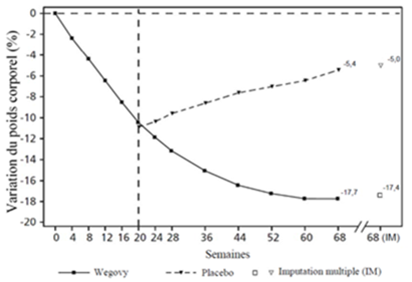
Figure 1 Changement du poids corporel (%) entre le début de l'étude et les semaines 68 et 104
STEP 1: gestion du poids
Dans le cadre d'une étude en double aveugle de 68 semaines, 1 961 patients souffrant d'obésité (IMC ≥30 kg/m2) ou de surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) et présentant au moins une pathologie associée liée au poids ont été randomisés pour recevoir un traitement par Wegovy ou par placebo. Tous les patients ont suivi un régime hypocalorique et augmenté leur activité physique tout au long de l'étude. La majorité des patients avaient au moins une maladie concomitante liée au poids. Il s'agissait notamment du prédiabète (43.7 %), de la dyslipidémie (37.0 %), de l'hypertension (36.0 %), de l'ostéoarthrose du genou ou de la hanche (15.9 %), de l'apnée obstructive du sommeil (11.7 %), de l'asthme/la bronchopneumopathie chronique obstructive (BPCO) (11.6 %), de la maladie du foie (stéatose hépatique non alcoolique (NAFLD) ou stéatohépatite non alcoolique (NASH)) (8.6 %) et le syndrome des ovaires polykystiques (SOPK) (6.6 %).
La perte de poids est apparue tôt et s'est poursuivie pendant l'étude. À la fin du traitement (semaine 68), la perte de poids était supérieure et cliniquement significative par rapport au placebo (cf. Tableau 4 et Figure 2).
À la suite de l'étude de 68 semaines, une prolongation de 52 semaines sans traitement a été effectuée, qui comprenait 327 patients ayant terminé la période d'étude principale avec la dose d'entretien de sémaglutide ou de placebo. Pendant la période sans traitement de la semaine 68 à la semaine 120, le poids corporel moyen a augmenté dans les deux groupes de traitement. Le poids est cependant resté inférieur de 5.6 % à la valeur initiale chez les patients traités par du sémaglutide pendant la période d'étude principale, contre 0.1 % chez les patients du groupe ayant reçu le placebo.
Tableau 4: Résultats d'une étude de 68 semaines comparant Wegovy à un placebo chez des patients souffrant d'obésité ou de surpoids et d'au moins une maladie associée au poids (STEP 1)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
1 306
|
655
| |
Poids corporel
| |
Valeur initiale (kg)
|
105.4
|
105.2
| |
Variation (%) par rapport à la valeur initiale1.2
|
-14.9
|
-2.4
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-12.4
[-13.4; -11.5]*
|
-
| |
Variation (kg) par rapport au placebo
|
-15.3
|
-2.6
| |
Différence (kg) par rapport au placebo1
[IC à 95 %]
|
-12.7
[-13.7; -11.7]
|
-
| |
Patients (%) avec perte de poids ≥5 %3
|
83.5*
|
31.1
| |
Patients (%) avec perte de poids ≥10 %3
|
66.1*
|
12.0
| |
Patients (%) avec perte de poids ≥15 %3
|
47.9*
|
4.8
| |
Patients (%) avec perte de poids ≥
20 %3
|
30.2
|
1.7
| |
Tour de taille (cm)
| |
Valeur initiale
|
114.6
|
114.8
| |
Variation par rapport à la valeur initiale1
|
-13.5
|
-4.1
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-9.4
[-10.3; -8.5]*
|
-
| |
Facteurs cardiométaboliques
| |
Pression artérielle systolique (mmHg)
| |
Valeur initiale
|
126
|
127
| |
Variation par rapport à la valeur initiale1
|
-6.2
|
-1.1
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-5.1
[-6.3; -3.9]*
|
-
| |
Pression artérielle diastolique (mmHg)
| |
Valeur initiale
|
80
|
80
| |
Variation par rapport à la valeur initiale
|
-2.8
|
-0.4
| |
Différence par rapport au placebo
[IC à 95 %]
|
-2.4
[-3.3; -1.6]
|
-
| |
Lipides
| |
Cholestérol total
| |
Valeur initiale (mmol/l)4
|
4.9
|
5.0
| |
Variation (%) par rapport à la valeur initiale1
|
-3.3
|
0.1
| |
Différence relative (%) par rapport au placebo [IC à 95 %]1
|
-3.3
[-4.8; -1.8]
|
-
| |
Cholestérol LDL
| |
Valeur initiale (mmol/l)
|
2.9
|
2.9
| |
Variation (%) par rapport à la valeur initiale1
|
-2.5
|
1.3
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
-3.8
[-5.9; -1.5]
|
-
| |
Cholestérol HDL
| |
Valeur initiale (mmol/l)4
|
1.3
|
1.3
| |
Variation (%) par rapport à la valeur initiale1
|
5.2
|
1.4
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
3.8
[2.2; 5.4]
|
-
| |
Triglycérides
| |
Valeur initiale (mmol/l)4
|
1.4
|
1.4
| |
Variation (%) par rapport à la valeur initiale1
|
-21.9
|
-7.3
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
-15.8
[-18.8; -12.7]
|
-
| |
CRP
| |
Valeur initiale (mg/l)
|
3.9
|
3.9
| |
Variation (%) par rapport à la valeur initiale1
|
-52.6
|
-15.0
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
-44.3
[-49.5; -38.5]
|
-
| |
Statut glycémique
| |
Patients (%) atteints de prédiabète à la valeur initiale
|
43.7
| |
Patients (%) avec statut normoglyclémique à la fin du traitement
|
84.1
|
47.8
| |
*
p < 0.0001 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Au cours de l'étude, le traitement randomisé a été interrompu par 17.1 % et 22.4 % des patients randomisés pour recevoir respectivement Wegovy et un placebo. En supposant que tous les patients randomisés ont continué le traitement et n'ont pas reçu de traitement supplémentaire contre l'obésité, les changements estimés du poids corporel entre la randomisation et la semaine 68, basés sur un modèle mixte de mesures répétées incluant toutes les observations jusqu'au premier arrêt, étaient de -16.9 % pour 2.4 mg de sémaglutide et de -2.4 % pour le placebo.
3 Estimé à l'aide d'un modèle de régression binaire basé sur la même procédure d'imputation que celle utilisée pour l'analyse primaire.
4 Moyenne géométrique.
|
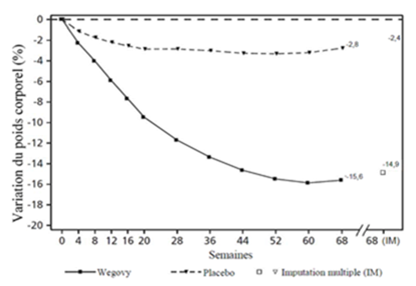
Valeurs observées pour les patients ayant effectué chaque visite prévue et estimations avec imputations multiples (MI) pour les abandons avec examen final («retrieved dropouts»).
Figure 2 STEP 1: variation moyenne du poids corporel (%) de la ligne de base à la semaine 68
STEP 2: Gestion du poids chez les patients atteints de diabète de type 2
Dans une étude en double aveugle de 68 semaines, 1 210 patients souffrant de surpoids ou d'obésité (IMC ≥27 kg/m2) et de diabète de type 2 ont été randomisés pour recevoir Wegovy, le sémaglutide 1 mg une fois par semaine ou un placebo. L'étude a porté sur des patients dont le diabète était insuffisamment contrôlé (HbA1c 7-10 %) et traités soit par un régime et de l'exercice physique seuls, soit par 1 à 3 antidiabétique(s) oral(ux). Tous les patients ont suivi un régime hypocalorique et augmenté leur activité physique tout au long de l'étude. La majorité des patients présentaient au moins deux maladies concomitantes liées au poids. Outre le diabète de type 2, il s'agissait notamment d'hypertension (69.8 %), de dyslipidémie (68.0 %), d'ostéoarthrose du genou ou de la hanche (19.6 %), de maladie du foie (NAFLD ou NASH) (22.6 %), d'apnée obstructive du sommeil (15.1 %), d'asthme/BPCO (8.4 %) et de SOPK (4.1 %).
Le traitement par Wegovy pendant 68 semaines a entraîné une réduction supérieure et cliniquement significative du poids corporel et de l'HbA1c par rapport au placebo (cf. Tableau 5 et Figure 3).
Tableau 5: Résultats d'une étude de 68 semaines comparant Wegovy à un placebo chez des patients souffrant d'obésité ou de surpoids et de diabète de type 2 (STEP 2)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
404
|
403
| |
Poids corporel
| |
Valeur initiale (kg)
|
99.9
|
100.5
| |
Variation (%) par rapport à la valeur initiale1.2
|
-9.6
|
-3.4
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-6.2
[-7.3; -5.2]*
|
-
| |
Variation kg) par rapport à la valeur initiale
|
-9.7
|
-3.5
| |
Différence (kg) par rapport au placebo1
[IC à 95 %]
|
-6.1
[-7.2; -5.0]
|
-
| |
Patients (%) avec perte de poids ≥5 %3
|
67.4*
|
30.2
| |
Patients (%) avec perte de poids ≥10 %3
|
44.5*
|
10.2
| |
Patients (%) avec perte de poids ≥15 %3
|
25.0*
|
4.3
| |
Patients (%) avec perte de poids ≥20 %3
|
12.8
|
2.3
| |
Tour de taille (cm)
| |
Valeur initiale
|
114.5
|
115.5
| |
Variation par rapport à la valeur initiale1
|
-9.4
|
-4.5
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-4.9
[-6.0; -3.8]*
|
-
| |
Facteurs cardiométaboliques
| |
Pression artérielle systolique (mmHg)
| |
Valeur initiale
|
130
|
130
| |
Variation par rapport à la valeur initiale1
|
-3.9
|
-0.5
| |
Différence par rapport au placebo1 [IC à 95 %]
|
-3.4
[-5.6; -1.3]**
|
-
| |
Pression artérielle diastolique (mmHg)
| |
Valeur initiale
|
80
|
80
| |
Variation par rapport à la valeur initiale
|
-1.6
|
-0.9
| |
Différence par rapport au placebo
[IC à 95 %]
|
-0.7
[-2.0; 0.6]
|
-
| |
Lipides
| |
Cholestérol total
| |
Valeur initiale (mmol/l)4
|
4.4
|
4.4
| |
Variation (%) par rapport à la valeur initiale1
|
-1.4
|
-0.5
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
-0.9
[-3.6; 2.0]
|
-
| |
Cholestérol LDL
| |
Valeur initiale (mmol/l)4
|
2.3
|
2.3
| |
Variation (%) par rapport à la valeur initiale1
|
0.5
|
0.1
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
0.4
[-4.0; 4.9]
|
-
| |
Cholestérol HDL
| |
Valeur initiale (mmol/l)4
|
1.2
|
1.1
| |
Variation (%) par rapport à la valeur initiale1
|
6.9
|
4.1
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
2.7
[0.3; 5.1]
|
-
| |
Triglycérides
| |
Valeur initiale (mmol/l)4
|
1.7
|
1.8
| |
Variation (%) par rapport à la valeur initiale1
|
-22.0
|
-9.4
| |
Différence relative (%) par rapport à un placebo1
[IC à 95 %]
|
-13.9
[-19.0; -8.4]
|
-
| |
CRP
| |
Valeur initiale (mg/l)
|
3.5
|
3.4
| |
Variation (%) par rapport à la valeur initiale1
|
-48.9
|
-16.7
| |
Différence relative (%) par rapport au placebo1
[IC à 95 %]
|
-38.7
[-46.5; -29.8]
|
-
| |
Facteurs glycémiques
| |
HbA1c (mmol/mol (%)
| |
Valeur initiale
|
65.3 (8.1)
|
65.3 (8.1)
| |
Variation par rapport à la valeur initiale1.2
|
-17.5 (-1.6)
|
-4.1 (-0.4)
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-13.5
[-15.5; -11.4]
[-1.2 (-1.4; -1.1)]*
|
-
-
| |
Patients (%) qui ont atteint un taux d'HbA1c < 7 %
|
77.4
|
26.0
| |
Patients (%) ayant atteint un taux d'HbA1c ≤6.5 %3
|
65.9
|
15.1
| |
* p < 0.001 (non corrigé bilatéralement) pour la supériorité; ** p < 0.05 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Au cours de l'étude, le traitement randomisé de 11.6 % et 13.9 % des patients randomisés sur Wegovy ou placebo a été interrompu. En supposant que tous les patients randomisés aient continué le traitement et n'aient pas reçu de traitement complémentaire contre l'obésité, les variations estimées du poids corporel depuis la randomisation jusqu'à la semaine 68 sur la base d'un modèle mixte de mesures répétées incluant toutes les observations effectuées jusqu'au premier sevrage étaient de -10.6 % pour 2.4 mg de sémaglutide et de -3.1 % pour le placebo.
3 Estimé à l'aide d'un modèle de régression binaire basé sur la même procédure d'imputation que celle utilisée pour l'analyse primaire.
4 Moyenne géométrique
|
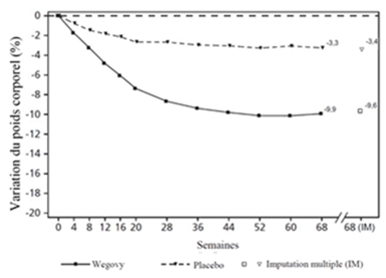
Valeurs observées pour les patients ayant effectué chaque visite prévue et estimations avec imputations multiples (MI) pour les abandons avec examen final («retrieved dropouts»).

HbA1c: hémoglobine A1c
Valeurs observées pour les patients ayant effectué chaque visite prévue et estimations avec imputations multiples (MI) pour les abandons avec examen final («retrieved dropouts») (RD-MI).
Figure 3 STEP 2: Variation moyenne du poids corporel (kg) et du taux d'HbA1c (%) entre le début de l'étude et la semaine 68
STEP 3: Gestion du poids avec une thérapie comportementale intensive
Dans le cadre d'une étude en double aveugle de 68 semaines, 611 patients souffrant d'obésité (IMC ≥30 kg/m2) ou de surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) et d'au moins une pathologie associée liée au poids ont été randomisés pour recevoir un traitement par Wegovy ou par placebo. Pendant l'étude, tous les patients ont suivi une thérapie comportementale intensive (TCI), consistant en un régime très restrictif, une augmentation de l'activité physique et des conseils comportementaux.
La majorité des patients avaient au moins une maladie concomitante liée au poids. Il s'agissait notamment du prédiabète (49.8 %), de l'hypertension (34.7 %), de la dyslipidémie (34.7 %), de l'ostéoarthrose du genou ou de la hanche (18.7 %), de l'asthme/la BPCO (15.1 %), de l'apnée obstructive du sommeil (12.6 %), d'une maladie hépatique (NAFLD ou NASH) (6.1 %) et du SOPK (5.5 %).
Le traitement par Wegovy et IBT pendant 68 semaines a entraîné une réduction du poids corporel supérieure et cliniquement pertinente par rapport au placebo (cf. Tableau 6).
Tableau 6: Résultats d'une étude de 68 semaines comparant Wegovy à un placebo chez des patients souffrant d'obésité ou de surpoids et d'au moins une maladie concomitante liée au poids sous thérapie comportementale intensive (STEP 3)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
407
|
204
| |
Poids corporel
| |
Valeur initiale (kg)
|
106.9
|
103.7
| |
Variation (%) par rapport à la valeur initiale1,2
|
-16.0
|
-5.7
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-10.3
[-12.0; -8.6]*
|
-
| |
Variation (kg) par rapport à la valeur initiale
|
-16.8
|
-6.2
| |
Différence (kg) par rapport au placebo1
[IC à 95 %]
|
-10.6
[-12.5; -8.8]
|
-
| |
Patients (%) avec perte de poids ≥5 %3
|
84.8*
|
47.8
| |
Patients (%) avec perte de poids ≥10 %3
|
73.0*
|
27.1
| |
Patients (%) avec perte de poids ≥15 %3
|
53.5*
|
13.2
| |
Patients (%) avec perte de poids ≥20 %3
|
33.9
|
3.5
| |
Tour de taille (cm)
| |
Valeur initiale
|
113.6
|
111.8
| |
Variation par rapport à la valeur initiale1
|
-14.6
|
-6.3
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-8.3
[-10.1; -6.6]*
|
-
| |
* p < 0.0001 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Au cours de l'étude, le traitement randomisé a été interrompu par 16.7 % et 18.6 % des patients randomisés pour recevoir respectivement Wegovy et un placebo. En supposant que tous les patients randomisés aient conservé le traitement et n'aient pas reçu de traitement complémentaire contre l'obésité, les variations estimées du poids corporel depuis la randomisation jusqu'à la semaine 68 sur la base d'un modèle mixte de mesures répétées incluant toutes les observations effectuées jusqu'au premier sevrage étaient de -17.6 % pour 2.4 mg de sémaglutide et de -5.0 % pour le placebo.
3 Estimé à l'aide d'un modèle de régression binaire basé sur la même procédure d'imputation que celle utilisée pour l'analyse primaire.
|
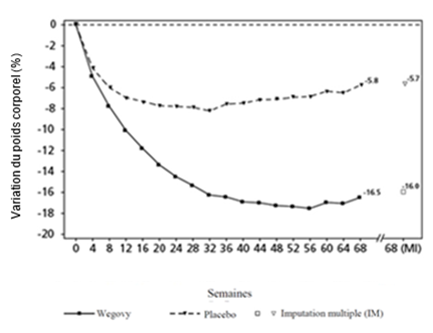
Valeurs observées pour les patients ayant effectué chaque visite prévue et estimations avec imputations multiples (MI) des dropouts
Figure 4 STEP 3: variation moyenne du poids corporel (%) de la ligne de base à la semaine 68
STEP 4: gestion du poids à long terme
Dans le cadre d'une étude en double aveugle de 68 semaines, 902 patients souffrant d'obésité (IMC ≥30 kg/m2) ou de surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) et d'au moins une maladie associée liée au poids ont été inclus. Tous les patients ont suivi un régime hypocalorique et augmenté leur activité physique tout au long de l'étude. De la semaine 0 à la semaine 20 (phase de run-in) tous les patients ont reçu Wegovy. À la semaine 20 (valeur de référence), 803 patients ayant atteint la dose d'entretien de 2.4 mg ont été randomisés pour poursuivre le traitement ou passer au placebo pour les 48 semaines restantes.
La majorité des patients avaient au moins une maladie concomitante liée au poids. Il s'agissait notamment du prédiabète (46.8 %), de l'hypertension (37.1 %), de la dyslipidémie (35.9 %), de l'ostéoarthrose du genou ou de la hanche (13.3 %), de l'apnée obstructive du sommeil (11.7 %), de l'asthme/de la BPCO (11.5 %), des hépatites (NAFLD ou NASH) (7.3 %) et du SOPK (3.9 %).
Les patients ayant atteint la dose d'entretien de 2.4 mg à la semaine 20 (valeur de référence) et ayant continué à recevoir Wegovy pendant 48 semaines (semaines 20 à 68) ont continué à perdre du poids et obtenu une réduction supérieure et cliniquement significative de leur poids corporel par rapport à ceux qui étaient passés au placebo (cf. Tableau 7 et Figure 5). En revanche, chez les patients passés au placebo à la semaine 20 (valeur de référence), le poids corporel a de nouveau augmenté régulièrement entre les semaines 20 et 68. Néanmoins, le poids corporel observé à la semaine 68 est resté inférieur à celui observé au début de la phase de run-in (semaine 0) (cf. Figure 5). Les patients traités par Wegovy de la semaine 0 (initiation) à la semaine 68 (fin du traitement) ont obtenu une variation moyenne du poids corporel de 17.4 %, une perte de poids de ≥5 %, ≥10 % ≥15 % ou ≥20 % ayant été atteinte par 87.8 %, 78.0 %, 62.2 % et 38.6 % de ces patients, respectivement.
Tableau 7: résultats de la période randomisée de 48 semaines (semaine 20 à semaine 68) de l'étude comparant Wegovy à un placebo chez des patients souffrant d'obésité ou de surcharge pondérale et d'au moins une maladie associée liée au poids (STEP 4)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
535
|
268
| |
Poids corporel
| |
Valeur initiale1 (kg)
|
96.5
|
95.4
| |
Variation (%) par rapport à la valeur initiale1,2,3
|
-7.9
|
6.9
| |
Différence (%) par rapport au placebo2
[IC à 95 %]
|
-14.8
[-16.0; -13.5]*
|
-
| |
Variation (kg) par rapport à la valeur initiale1
|
-7.1
|
6.1
| |
Différence (kg) par rapport au placebo2
[IC à 95 %]
|
-13.2
[-14.3; -12.0]
|
-
| |
Tour de taille (cm)
| |
Valeur initiale1
|
105.5
|
104.7
| |
Variation par rapport à la valeur initiale1,2
|
-6.4
|
3.3
| |
Différence par rapport au placebo2
[IC de 95 %]
|
-9.7
[-10.9; -8.5]*
|
-
| |
* p < 0.0001 (non corrigé des bilatéralement) pour la supériorité.
1 Valeur initiale = semaine 20.
2 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
3 Au cours de l'étude, le traitement randomisé a été interrompu par 5.8 % et 11.6 % des patients randomisés pour recevoir respectivement Wegovy et un placebo. En supposant que tous les patients randomisés aient continué le traitement et n'aient pas reçu de traitement complémentaire contre l'obésité, les variations estimées du poids corporel depuis la randomisation jusqu'à la semaine 68 sur la base d'un modèle mixte de mesures répétées incluant toutes les observations effectuées jusqu'au premier sevrage étaient de -8.8 % pour 2.4 mg de sémaglutide et de 6.5 % pour le placebo.
|
Valeurs observées pour les patients ayant effectué chaque visite prévue et estimations avec imputations multiples (MI) pour les abandons avec examen final («retrieved dropouts»).
Figure 5 STEP 4: Valeur moyenne du poids corporel (%) entre la semaine 0 et la semaine 68
STEP 5: Données sur 2 ans
Dans une étude en double aveugle de 104 semaines, 304 patients présentant une obésité (IMC ≥30 kg/m2) ou un surpoids (IMC ≥27 à moins de 30 kg/m2) et au moins une maladie concomitante liée au poids ont été randomisés pour recevoir du sémaglutide ou un placebo. Tous les patients ont reçu un régime hypocalorique avait une activité physique accrue pendant la durée de l'étude.
Au début de l'étude, les patients avaient un IMC moyen de 38.5 kg/m2 et un poids corporel moyen de 106.0 kg.
Le traitement par Wegovy pendant 104 semaines a entraîné une réduction du poids corporel supérieure et cliniquement pertinente par rapport au placebo (cf. Tableau 8 et Figure 6). Le poids corporel moyen a diminué sous Wegovy dès le début de l'étude et jusqu'à la semaine 68, puis a atteint un plateau. Le poids corporel moyen a diminué moins fortement diminué sous placebo et un plateau a été atteint après environ 20 semaines de traitement. Les patients traités par sémaglutide ont atteint une variation moyenne du poids corporel de -15.2 %, avec une perte de poids de 5 % chez 74.7 % des patients, de 10 % chez 59.2 % et de 15 % chez 49.7 %.
Tableau 8: Résultats d'une étude de 104 semaines comparant Wegovy à un placebo chez des patients présentant une obésité ou un surpoids et atteint d'au moins une maladie concomitante liée au poids (STEP 5)
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
152
|
152
| |
Poids corporel
| |
Valeur initiale1 (kg)
|
105.6
|
106.5
| |
Variation (%) par rapport à la valeur initiale1,2
|
-15.2
|
-2.6
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-12.6
[-15.3; -9.8]*
|
-
| |
Variation (kg) par rapport à la valeur initiale
|
-16.1
|
-3.2
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-12.9
[-16.1; -9.8]
|
-
| |
Patients (%) avec perte de poids ≥5 %3
|
74.7*
|
37.3
| |
Tour de taille (cm)
| |
Valeur initiale
|
115.8
|
115.7
| |
Variation (%) par rapport à la valeur initiale1
|
-14.4
|
-5.2
| |
Différence par rapport au placebo1
[IC à 95 %]
|
-9.2
[-12.2; -6.2]*
|
-
| |
* p < 0.0001 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Au cours de l'étude, le traitement randomisé de 13.2 % et 27.0 % des patients randomisés sur sémaglutide ou placebo a été interrompu. En supposant que tous les patients randomisés aient continué le traitement et n'aient pas reçu de traitement complémentaire contre l'obésité, les variations estimées du poids corporel depuis la randomisation jusqu'à la semaine 68 sur la base d'un modèle mixte de mesures répétées incluant toutes les observations effectuées jusqu'au premier sevrage étaient de -16.7 % pour 2.4 mg de sémaglutide et de -0.6 % pour le placebo.
3 Estimé à l'aide d'un modèle de régression binaire basé sur la même procédure d'imputation que celle utilisée pour l'analyse primaire.
|
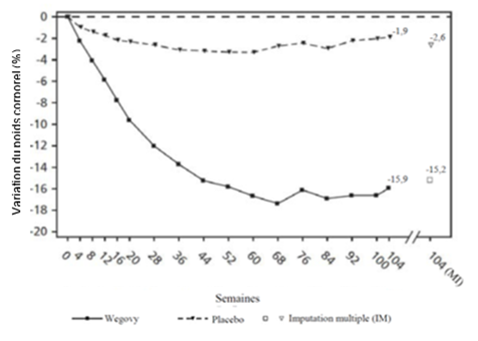
Valeurs observées pour les patients ayant effectué chaque visite planifiée et estimations avec imputations multiples (IM) pour les personnes ayant abandonné l'examen final (« retrieved dropouts »).
Figure 6 STEP 5 - Variation moyenne du poids corporel (%) entre la semaine 0 et la semaine 104
STEP 8: sémaglutide par rapport au liraglutide
Dans une étude de 68 semaines, randomisée, non aveugle, contrôlée par placebo, par paires, 338 patients présentant une obésité (IMC ≥30 kg/m2) ou un surpoids (IMC ≥27 à < 30 kg/m2) et au moins une maladie concomitante liée au poids ont été randomisés pour recevoir Wegovy une fois par semaine, du liraglutide 3 mg une fois par jour ou un placebo. Les groupes Wegovy une fois par semaine et liraglutide 3 mg n'étaient pas aveugles, mais chaque groupe de traitement actif était doublement aveugle par rapport au placebo administré à la même fréquence posologique. Tous les patients ont reçu un régime hypocalorique avec une activité physique accrue pendant la durée de l'étude.
Au début de l'étude, les patients avaient un IMC moyen de 37.5 kg/m2 et un poids corporel moyen de 104.5 kg.
Le poids corporel moyen a diminué sous Wegovy depuis le début de l'étude jusqu'à la semaine 68. Avec le liraglutide, la réduction du poids corporel moyen était plus faible (cf. Tableau 9). 37.4 % des patients traités par sémaglutide ont perdu 20 % de poids contre 7.0 % avec liraglutide.
Tableau 9: STEP 8: Résultats d'une étude de 68 semaines comparant le semaglutide et le liraglutide
|
|
Wegovy
|
Liraglutide 3 mg
| |
Full analysis set (N)
|
126
|
127
| |
Poids corporel
| |
Valeur initiale (kg)
|
102.5
|
103.7
| |
Variation (%) par rapport à la valeur initiale1,2,3
|
-15.8
|
-6.4
| |
Différence (%) par rapport au placebo1
[IC à 95 %]
|
-9.4
[-12.0;-6.8]*
|
-
| |
Variation (kg) par rapport à la valeur initiale
|
-15.3
|
-6.8
| |
Différence (%) par rapport au liraglutide1
[IC à 95 %]
|
-8.5
[-11.2;-5.7]
|
-
| |
* p < 0.005 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Au cours de l'étude, le traitement randomisé de 13.5 % et 27.6 % des patients randomisés pour recevoir du sémaglutide ou du liraglutide a été interrompu. En supposant que tous les patients randomisés aient continué le traitement et n'aient pas reçu de traitement complémentaire contre l'obésité, les variations estimées du poids corporel depuis la randomisation jusqu'à la semaine 68 sur la base d'un modèle mixte de mesures répétées incluant toutes les observations effectuées jusqu'au premier sevrage étaient de -16.7 % pour le sémaglutide et de -6.7 % pour le liraglutide.
3 La variation (%) par rapport à la valeur initiale dans les groupes de placebo regroupés était de -1.9 %.
|
STEP 9: Gestion du poids chez les patients souffrant d'ostéoarthrose du genou – sémaglutide
Dans une étude en double aveugle de 68 semaines, 407 patients (âge moyen 56 ans) souffrant d'obésité et d'ostéoarthrose modérée du genou dans un ou les deux genoux ont été randomisés pour recevoir Wegovy (n=271) ou un placebo (n=136). Au début de l'étude, le poids corporel moyen était de 108.6 kg pour un IMC moyen de 40.3 kg/m². Conformément aux résultats d'études antérieures, une différence cliniquement significative de la perte de poids entre les groupes de traitement a été observée en faveur de Wegovy (différence par rapport au placebo [IC 95 %]: -10.5 % [-12.3, -8.6]). Parallèlement, l'indice Western Ontario and McMaster Universities Osteoarthritis 3.1 (WOMAC), une mesure de la douleur liée à l'ostéoarthrose du genou, s'est amélioré de manière significativement plus importante dans le groupe Wegovy que dans le groupe placebo (différence [IC 95 %]: -14.1 [-20.0, -8.3]), 59 % des patients du groupe Wegovy ayant obtenu une amélioration cliniquement significative, contre 35 % dans le groupe placebo.
Effet sur le taux de graisse corporelle
Une sous-étude de STEP 1 (n=140) a montré, à l'aide de l'absorptiométrie à double rayon X (DEXA), que le traitement par Wegovy entraînait une réduction plus importante de la masse graisseuse que de la masse maigre, ce qui correspond à une amélioration du pourcentage de graisse corporelle après 68 semaines par rapport au placebo. De plus, cette réduction de la masse grasse totale s'est accompagnée d'une réduction de la graisse viscérale.
Résultats cardiovasculaires
SELECT: Étude des résultats cardiovasculaires chez des patients en surpoids ou atteints d'obésité
SELECT était une étude randomisée, en double aveugle, contrôlée par placebo et basée sur des événements, incluant 17 604 patients ayant une maladie cardiovasculaire établie (67.6 % avec un infarctus du myocarde antérieur, 17.8 % avec un accident cérébral vasculaire antérieur et 4.4 % avec une maladie artérielle périphérique (PAD); 8.2 % avec 2 événements cardiovasculaires antérieurs ou plus) et un IMC ≥27 kg/m2. Les patients ayant des antécédents de diabète de type 1 et de type 2 ont été exclus. La durée moyenne de séjour dans l'étude était de 41.8 mois. La population étudiée était composée de 27.7 % de femmes et 72.3 % d'hommes, avec une moyenne d'âge de 61.6 ans, dont 38.2 % de patients ≥65 ans (n=6728) et 7.8 % de patients ≥75 ans (n=1366). L'IMC moyen était de 33.3 kg/m2 et le poids corporel moyen était de 96.7 kg.
Les patients ont été randomisés et ont reçu soit du sémaglutide à une dose maximale à atteindre de 2.4 mg par semaine (n=8803), soit du placebo (n=8801) en complément d'un traitement standard pour la maladie cardiovasculaire antérieure. Au début de l'étude, 92.0 % des patients recevaient des médicaments cardiovasculaires (70.2 % des bêtabloquants, 45.0 % des inhibiteurs de l'enzyme de conversion de l'angiotensine (ECA), 29.5 % des inhibiteurs du récepteur de l'angiotensine et 26.9 % des inhibiteurs du canal calcique), 90.1 % de hypolipidémiants (essentiellement des statines 87.6 %), et 86.2 % de médicaments antiplaquettaires.
La plupart des patients présentaient initialement une maladie cardiovasculaire concomitante, dont 66.4 % avec HbA1c ≥5.7 % et < 6.5 % comme indice de prédiabète, 24.3 % avec insuffisance cardiaque chronique, 81.8 % avec hypertension, 46.8 % avec inflammation (hsCRP ≥2 mg/L) et des patients ayant une insuffisance rénale légère (48.7 %), modérée (10.4 %) ou sévère (0.4 %).
Le critère d'évaluation principal était la période entre la randomisation et la première apparition d'événements cardiovasculaires graves (MACE), définie comme un critère composite de décès cardiovasculaire, d'infarctus du myocarde non fatal ou d'accident vasculaire cérébral non mortel. La supériorité de sémaglutide 2.4 mg sur le placebo a été confirmée avec un Hazard Ratio de 0.80 [0.72; 0.90] [IC à 95 %], ce qui correspond à une réduction relative du risque de 20 % (cf. Figures 7 et 8). Les trois composantes ont contribué à la réduction de MACE (Tableau 10). La réduction du risque cardiovasculaire est apparue largement indépendante de la perte de poids.
L'effet du traitement sur la réduction des risques de MACE était comparable dans les sous-groupes principaux définis par âge, sexe, race, origine ethnique, type de maladie cardiovasculaire antérieure, IMC, normoglycémie/prédiabète et fonction rénale.
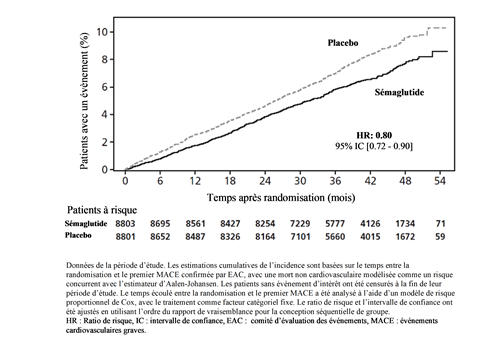
Figure 7 Représentation cumulative de la fonction d'incidence: temps entre la randomisation et le premier MACE
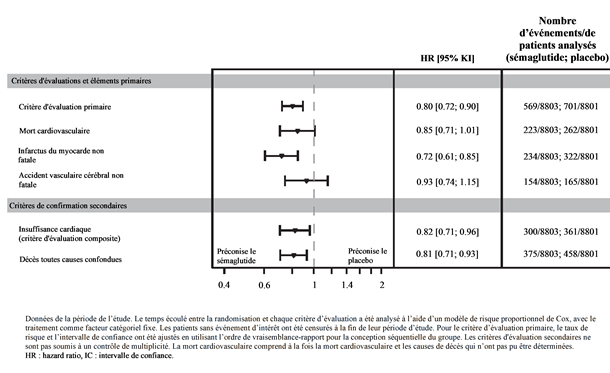
Figure 8 Forest Plot de temps depuis la randomisation jusqu'au premier MACE, composants MACE et critères d'évaluation secondaires
Tableau 10: Temps écoulé entre la randomisation et le premier MACE et ses composants, critère d'évaluation secondaire, mort cardiovasculaire.
|
|
HR [IC de 95 %]
|
Valeur p
(bilatérale)
|
Nombre d'événements/ Patients analysés (Séma 2.4 mg)
|
Nombre d'événements/ Patients analysés (Placebo)
| |
Critère composite primaire d'évaluation (MACE)
|
0.80 [0.72; 0.90]
|
<0.0001
|
569/8803
|
701/8801
| |
Mort cardiovasculaire
|
0.85 [0.71; 1.01]
|
0.0653
|
223/8803
|
262/8801
| |
Infarctus du myocarde non fatal
|
0.72 [0.61; 0.85]
|
|
234/8803
|
322/8801
| |
Accident vasculaire cérébral non fatal
|
0.93 [0.74; 1.15]
|
|
154/8803
|
165/8801
| |
Critères d'évaluation secondaires
| |
Mort cardiovasculaire1
|
0.85 [0.71; 1.01]
|
0.0653
|
223/8803
|
262/8801
| |
Insuffisance cardiaque (critère composite)1
|
0.82 [0.71; 0.96]
|
|
300/8803
|
361/8801
| |
Décès toutes causes confondues1
|
0.81 [0.71; 0.93]
|
|
375/8803
|
458/8801
| |
Sous-groupes:
| |
Maladie cardiovasculaire
| |
Maladie cardiovasculaire: infarctus du myocarde uniquement
|
0.78 [0.68; 0.90]
|
0.4803
|
362/5962
|
455/5944
| |
Maladie cardiovasculaire: accident vasculaire cérébral uniquement
|
0.98 [0.75; 1.27]
|
|
109/578
|
109/1556
| |
Maladie cardiovasculaire: PAD uniquement
|
0.74 [0.36; 1.48]
|
|
13/376
|
19/401
| |
Maladie cardiovasculaire: 2 maladies cardiovasculaires ou plus
|
0.75 [0.55; 1.00]
|
|
76/718
|
100/719
| |
Sexe
| |
Sexe féminin
|
0.84 [0.66; 1.07]
|
0.6324
|
126/2448
|
147/2424
| |
Sexe masculin
|
0.79 [0.70; 0.90]
|
|
443/6355
|
554/6377
|
Remarque: le temps écoulé entre la randomisation et chaque critère a été analysé à l'aide d'un modèle de risque proportionnel de Cox, avec le traitement comme facteur catégoriel fixe. Les sujets sans événement d'intérêt ont été censurés à la fin de leur période d'étude. Pour le critère d'évaluation primaire, l'HR, l'intervalle de confiance et la valeur p ont été ajustés en utilisant l'ordre de vraisemblance-ratio pour les conceptions de séquençage de groupe. Les analyses de sous-groupes pour le critère d'évaluation primaire ont été analysées dans un modèle de risque proportionnel de Cox avec une interaction entre le groupe traité et le sous-groupe pertinent comme facteur fixe. La mort cardiovasculaire comprend à la fois la mort cardiovasculaire et les causes de décès qui n'ont pas pu être déterminées.
1Critère d'évaluation secondaire. Non statistiquement significatif selon la hiérarchie des tests prédéfinie.
HR: Ratio de risque, IC: intervalle de confiance, valeur p: valeur p bilatérale pour le test sans différence. Pour les analyses de sous-groupes, la valeur p fait référence au test d'absence d'effet d'interaction. MACE: événements cardiovasculaires graves.
Tableau 11: SELECT – Évaluation des facteurs de risque cardiovasculaire à la semaine 104
|
|
Sémaglutide
|
Placebo
| |
Full analysis set (N)
|
8803
|
8801
| |
Facteurs cardiométaboliques:
| |
Pression artérielle systolique (mmHg)
| |
Valeur initiale
moyenne (SD)
|
131.0 (15.6)
|
130.9 (15.3)
| |
Variation par rapport à la valeur initiale1
|
-3.82
|
-0.51
| |
Différence par rapport au placebo [IC à 95 %]1
|
-3.1 [-3.75; -2.88]
|
-
| |
Pression artérielle diastolique (mmHg)
| |
Valeur initiale
moyenne (SD)
|
79.4 (10.0)
|
79.2 (9.9)
| |
Variation par rapport à la valeur initiale1
|
-1.02
|
-0.47
| |
Différence par rapport au placebo [IC à 95 %]1
|
-0.55 [-0.83; -0.27]
|
-
| |
Fréquence cardiaque
| |
Valeur initiale
moyenne (SD)
|
68.9 (10.6)
|
68.6 (10.7)
| |
Variation par rapport à la valeur initiale1
|
3.79
|
0.69
| |
Différence par rapport au placebo [IC à 95 %]1
|
3.10 [2.80; 3.39]
|
-
| |
Lipides:
| |
Total Cholestérol
| |
Valeur initiale (mmol/L)
Moyenne géométrique (CV)
|
4.03 (25.82)
|
4.04 (25.41)
| |
Variation (%) par rapport à la valeur initiale1
|
-4.63
|
-1.92
| |
Différence relative (%) par rapport au placebo1
|
-2.77 [-3.37; -2.16]
|
-
| |
LDL Cholestérol
| |
Valeur initiale (mmol/L)
Moyenne géométrique (CV)
|
2.03 (43.70)
|
2.03 (43.56)
| |
Variation (%) par rapport à la valeur initiale1
|
-5.25
|
-3.14
| |
Différence relative (%) par rapport au placebo [IC à 95 %]1
|
-2.18 [-3.22; -1.12]
|
-
| |
HDL Cholestérol
| |
Valeur initiale (mmol/L)
Moyenne géométrique (CV)
|
1.14 (25.52)
|
1.15 (25.02)
| |
Variation (%) par rapport à la valeur initiale1
|
4.86
|
0.59
| |
Différence relative (%) par rapport au placebo [IC à 95 %]1
|
4.24 [3.70; 4.79]
|
-
| |
Triglycérides
| |
Valeur initiale (mmol/L)
Moyenne géométrique (CV)
|
1.56 (51.75)
|
1.57 (50.84)
| |
Variation (%) par rapport à la valeur initiale1
|
-18.34
|
-3.20
| |
Différence relative (%) par rapport au placebo [IC à 95 %]1
|
-15.64 [-16.7; -14.6]
|
-
| |
Facteurs de risque cardiovasculaires liés au poids:
| |
Poids corporel (%)
| |
Valeur initiale
moyenne (SD) (kg)
|
96.53 (17.52)
|
96.82 (17.80)
| |
Variation par rapport à la valeur initiale1
|
-9.39
|
-0.88
| |
Différence (%) par rapport au placebo [IC à 95 %]1
|
-8.51 [-8.75; -8.27]
|
-
|
1Les réponses ont été analysées à l'aide d'une ANCOVA, le traitement étant considéré comme un facteur fixe et la valeur initiale comme une covariable. Avant l'analyse, les données manquantes ont été imputées à plusieurs reprises. Le modèle d'imputation (régression linéaire) a été réalisé séparément pour chaque groupe de traitement et comprenait la valeur initiale sous forme de covariables ainsi que tous les participants ayant des mesures, quel que soit leur statut de traitement à la semaine 104. Le modèle ajusté a été utilisé pour imputer les valeurs des participants sans mesures à la semaine 104. Les estimations moyennes ont été ajustées en fonction de la distribution initiale observée.
STEP TEENS: Régulation du poids chez les patients adolescents
Dans une étude en double aveugle de 68 semaines, 201 adolescents âgés de 12 à 18 ans révolus et présentant une obésité (n=200) ou une surcharge pondérale et présentant au moins une comorbidité liée au poids (n=1) ont été randomisés 2:1 sur sémaglutide ou placebo pendant la puberté. Tous les patients ont reçu un régime hypocalorique et avaient une activité physique accrue pendant la durée de l'étude.
À la fin du traitement (semaine 68), l'amélioration de l'IMC était supérieure avec le sémaglutide et cliniquement significative par rapport au placebo (cf. Tableau 12 et Figure 9). Une plus grande proportion de patients a par ailleurs perdu 5 %, 10 % et 15 % de son poids en prenant du sémaglutide par rapport au placebo (cf. Tableau 12).
Tableau 12: STEP TEENS: Résultats à la semaine 68
|
|
Wegovy
|
Placebo
| |
Full analysis set (N)
|
134
|
67
| |
IMC
| |
Valeur initiale (IMC)
|
37.7
|
35.7
| |
Variation (%) par rapport à la valeur initiale de l'IMC1, 3
|
-16.1
|
0.6
| |
Différence (%) par rapport au placebo1 [IC à 95 %]
|
-16.7 [-20.3; -13.2]*
|
-
| |
Valeur initiale (IMC SDS)
|
3.4
|
3.1
| |
Modification de la valeur initiale en IMC SDS1
|
-1.1
|
-0.1
| |
Différence avec le placebo1 [IC à 95 %]
|
-1.0 [-1.3; -0.8]
|
-
| |
Poids corporel
| |
Valeur initiale (kg)
|
109.9
|
102.6
| |
Variation (%) par rapport à la valeur initiale1
|
-14.7
|
2.8
| |
Différence (%) par rapport au placebo1 [IC à 95 %]
|
-17.4 [-21.1; -13.8]
|
-
| |
Variation (kg) par rapport à la valeur initiale1
|
-15.3
|
2.4
| |
Différence (kg) par rapport au placebo1 [IC à 95 %]
|
-17.7 [-21.8; -13.7]
|
-
| |
Patients (%) avec une perte de masse corporelle ≥5 %4
|
72.5*
|
17.7
| |
Patients (%) avec une perte de masse corporelle ≥10 %4
|
61.8
|
8.1
| |
Patients (%) avec une perte de masse corporelle ≥15 %4
|
53.4
|
4.8
| |
Patients (%) avec une perte de masse corporelle ≥20 %4
|
37.4
|
3.2
| |
Tour de taille (cm)
| |
Valeur initiale
|
111.9
|
107.3
| |
Variation (%) par rapport à la valeur initiale1
|
-12.7
|
-0.6
| |
Différence avec le placebo1 [IC à 95 %]
|
-12.1 [-15.6; -8.7]
|
-
|
* p < 0.005 (non corrigé bilatéralement) pour la supériorité.
1 Estimé à l'aide d'un modèle ANCOVA avec imputation multiple basé sur toutes les données, indépendamment de l'arrêt du traitement randomisé ou du début d'un autre traitement médicamenteux contre l'obésité ou de la chirurgie bariatrique.
2 Les chiffres se réfèrent aux patients ne souffrant pas de diabète de type 2.
3 Au cours de l'étude, le traitement randomisé a été interrompu de manière permanente par 10.4 % des patients ayant reçu 2.4 mg de sémaglutide et 10.4 % de patients randomisés sur placebo. En supposant que tous les patients randomisés ont continué le traitement et n'ont pas reçu de traitement supplémentaire de l'obésité, les modifications de l'IMC depuis la randomisation jusqu'à la semaine 68 étaient de -17.9 % pour 2.4 mg de sémaglutide et -0.6 % pour le placebo, sur la base d'un modèle mixte de mesures répétées, incluant toutes les observations jusqu'au premier arrêt.
4 Estimation à l'aide d'un modèle de régression logistique basé sur la même méthode d'imputation que dans l'analyse primaire.
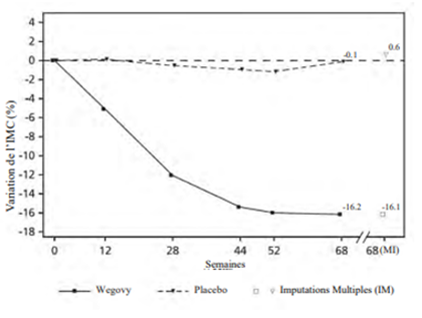
Valeurs observées pour les patients ayant effectué chaque visite planifiée et estimations avec imputations multiples (IM) pour les personnes ayant abandonné l'examen final
Figure 9 STEP TEENS: Variation moyenne de l'IMC (%) de la valeur initiale à la semaine 68
PharmacocinétiqueAbsorption
Sur la base d'une analyse pharmacocinétique de population, la concentration moyenne de sémaglutide à l'état d'équilibre après l'administration s. c. de 2.4 mg de sémaglutide était d'environ 75 nmol/l chez les patients présentant un surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) ou une obésité (IMC ≥30 kg/m2). La concentration maximale a été atteinte environ 24 heures après l'injection. L'exposition au sémaglutide à l'état d'équilibre a augmenté à des doses allant jusqu'à 2.4 mg par semaine. Une exposition similaire a été obtenue lors de l'administration s. c. de sémaglutide dans l'abdomen, la cuisse ou le bras. La biodisponibilité absolue du sémaglutide était de 89 %.
Distribution
Le volume de distribution moyen du sémaglutide après administration s. c. à des patients souffrant de surpoids ou d'obésité était d'environ 12.4 litres. Le sémaglutide est fortement lié à l'albumine plasmatique (> 99 %).
Métabolisme
Le sémaglutide est largement métabolisé par clivage protéolytique du backbone peptidique et par bêta-oxydation séquentielle de la chaîne latérale des acides gras. Le métabolite le plus courant représentait moins de 8 % de l'exposition totale et a été identifié comme étant le sémaglutide tronqué des 13 premiers acides aminés de l'extrémité N-terminale.
Élimination
Les voies d'élimination primaires des matières liées au sémaglutide sont l'urine et les fèces. Environ 3 % de la dose absorbée ont été excrétés dans l'urine sous forme de sémaglutide intact.
La clairance du sémaglutide chez les patients présentant un surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) ou une obésité (IMC ≥30 kg/m2) était d'environ 0.05 l/h. Avec une demi-vie d'élimination d'environ 1 semaine, le sémaglutide sera encore présent dans la circulation sanguine pendant environ 7 semaines après la dernière dose de 2.4 mg.
Cinétique pour certains groupes de patients
Sur la base d'une analyse pharmacocinétique de population, l'âge, le sexe, la race et l'ethnie ainsi que les troubles de la fonction rénale n'ont pas d'influence cliniquement significative sur la pharmacocinétique du sémaglutide.
Poids corporel
Le poids corporel se répercutait sur l'exposition au sémaglutide. Un poids corporel supérieur était associé à une exposition plus faible. La dose hebdomadaire de 2.4 mg de sémaglutide a entraîné une exposition systémique adéquate dans une fourchette de 54.4 à 245.6 kg de poids corporel, évaluée dans les études cliniques pour l'exposition obtenue.
Troubles de la fonction hépatique
Une fonction hépatique réduite n'a pas eu d'influence sur l'exposition au sémaglutide. La pharmacocinétique du sémaglutide a été évaluée dans une étude avec une dose unique de 0.5 mg de sémaglutide chez des patients présentant des degrés divers d'altération de la fonction hépatique (légère, modérée, sévère) par rapport à des patients à la fonction hépatique normale.
Troubles de la fonction rénale
Une limitation de la fonction rénale n'a pas influencé la pharmacocinétique du sémaglutide de façon cliniquement significative. Cela a été démontré à l'aide d'une dose unique de 0.5 mg de sémaglutide chez des patients présentant des degrés divers d'altération de la fonction rénale (légère, modérée, sévère ou dialysée) par rapport à des patients à la fonction rénale normale. Sur la base d'une analyse pharmacocinétique de population, cela a également été démontré à partir des données des études de phase 3a de patients en surpoids (IMC ≥27 kg/m2 à < 30 kg/m2) ou obèses (IMC ≥30 kg/m2) et présentant une restriction légère à modérée de la fonction rénale.
Patients âgés
Selon les données des études de phase 3 menées chez des patients âgés de 18 à 86 ans, l'âge n'a pas eu d'influence sur la pharmacocinétique du sémaglutide.
Enfants et adolescents
Les propriétés pharmacocinétiques du sémaglutide ont été observées dans une étude clinique chez des patients adolescents présentant une obésité ou une surcharge pondérale et au moins une comorbidité liée au poids, âgés de 12 à 18 ans révolus (124 patients, poids corporel compris entre 61.6 et 211.9 kg). L'exposition au sémaglutide chez les adolescents était similaire à celle des adultes obèses ou en surcharge pondérale.
La sécurité et l'efficacité de Wegovy chez les enfants de moins de 12 ans n'ont pas été démontrées.
Polymorphismes génétiques
Le sexe et l'appartenance ethnique n'ont pas eu d'influence sur la pharmacocinétique du sémaglutide.
Données précliniquesSur la base des études conventionnelles de pharmacologie de sécurité, de toxicité à doses répétées ou de génotoxicité, les données précliniques ne révèlent aucun risque particulier pour les humains.
Carcinogénicité
Les tumeurs non létales des cellules C de la thyroïde observées chez les rongeurs sont un effet de classe des agonistes des récepteurs du GLP-1. Dans des études de carcinogénicité de 2 ans chez le rat et la souris, le sémaglutide a provoqué des tumeurs à cellules C de la thyroïde à des expositions cliniquement significatives. Les tumeurs des cellules C chez les rongeurs sont provoquées par un mécanisme non génotoxique, à médiation spécifique par le récepteur GLP-1, auquel les rongeurs sont particulièrement sensibles. La pertinence pour les humains est considérée comme faible, mais ne peut pas être totalement exclue.
Toxicité sur la reproduction
Le sémaglutide n'a pas affecté le comportement de saillie ou la fertilité des rats mâles dans les études de fertilité chez le rat. Un allongement de l'œstrus et une faible diminution du nombre de Corpora lutea (ovulations) ont été observés chez les rates à des doses entraînant une perte de poids maternelle.
Dans les études de développement embryo-fœtal chez le rat, le sémaglutide a provoqué une embryotoxicité à des expositions inférieures aux niveaux cliniquement pertinents. Le sémaglutide a provoqué des réductions significatives du poids corporel de la mère et des diminutions de la survie et de la croissance des embryons. Des malformations squelettiques et viscérales importantes ont été observées chez les fœtus, notamment des effets sur les os longs, les côtes, les vertèbres, la queue, les vaisseaux sanguins et les ventricules cérébraux. Des études sur le mécanisme indiquent que l'embryotoxicité est due à une altération de l'approvisionnement en nutriments de l'embryon via le sac vitellin chez le rat par l'intermédiaire du récepteur du GLP-1. En raison des différences anatomiques et fonctionnelles du sac vitellin entre les espèces et de l'absence d'expression du récepteur du GLP-1 dans le sac vitellin des primates non humains, il est considéré comme peu probable que ce mécanisme soit pertinent pour les humains. Un effet direct du sémaglutide sur le fœtus ne peut cependant être exclu.
Une augmentation des avortements et une légère augmentation de l'incidence des anomalies fœtales à des expositions cliniquement significatives ont été observées dans les études de toxicité sur le développement menées chez le lapin et le macaque crabier. Les résultats coïncident avec une perte de poids maternelle significative allant jusqu'à 16 %. La question se pose de savoir si ces effets sont liés à la diminution de la prise alimentaire maternelle en tant qu'effet direct du GLP-1.
La croissance et le développement postnatal ont été évalués chez des macaques crabiers. Les nouveau-nés étaient légèrement plus petits à la naissance, mais ont rattrapé leur retard pendant l'allaitement.
Le sémaglutide a provoqué un retard de la maturité sexuelle chez des rats juvéniles mâles et femelles. Ces retards n'ont eu aucun effet sur la fécondité et la capacité reproductive des deux sexes, ni sur la capacité des femelles à maintenir une grossesse.
Remarques particulièresIncompatibilités
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé à d'autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Stabilité après ouverture
Wegovy Multi FixDose peut être conservé hors du réfrigérateur jusqu'à 42 jours (6 semaines) à une température inférieure à 30°C ou au réfrigérateur (2-8°C).
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8°C). Tenir à l'écart de l'élément de refroidissement.
Ne pas congeler Wegovy et ne pas l'utiliser si son contenu a été congelé.
Wegovy devrait être protégé d'une chaleur excessive.
Après l'emploi: Jetez Wegovy après l'emploi.
Conserver hors de portée des enfants.
Wegovy FixDose: Conserver le stylo dans son emballage d'origine afin de protéger son contenu de la lumière. Wegovy FixDose peut être conservé non réfrigéré pendant un maximum de 28 jours à une température inférieure à 30°C.
Wegovy Multi FixDose: Laisser le capuchon sur le stylo lorsqu'il n'est pas utilisé afin de protéger le contenu de la lumière.
Remarques concernant la manipulation
Wegovy FixDose: Le stylo contient 1 dose.
Wegovy Multi FixDose: Le stylo contient 4 doses.
Retirer et jeter l'aiguille d'injection après chaque injection. Conserver le stylo sans aiguille.
Wegovy ne doit pas être utilisé s'il n'est pas transparent et incolore.
Le stylo ne doit plus être utilisé si le contenu a été congelé.
Le médicament non utilisé ou les déchets doivent être éliminés conformément aux directives nationales.
Numéro d’autorisation68238, 68798 (Swissmedic)
PrésentationWegovy FixDose
Il existe cinq dosages du stylo prérempli pour Wegovy FixDose:
Wegovy FixDose 0.25 mg solution injectable en stylo prérempli:
4 stylos préremplis (B)
Wegovy FixDose 0.5 mg solution injectable en stylo prérempli:
4 stylos préremplis (B)
Wegovy FixDose 1 mg solution injectable en stylo prérempli:
4 stylos préremplis (B)
Wegovy FixDose 1.7 mg solution injectable en stylo prérempli:
4 stylos préremplis (B)
Wegovy FixDose 2.4 mg solution injectable en stylo prérempli:
4 stylos préremplis (B)
Wegovy Multi FixDose
Il existe cinq dosages du stylo prérempli pour Wegovy Multi FixDose:
Wegovy Multi FixDose 0.25 mg solution injectable en stylo prérempli:
1 stylo prérempli et 4 aiguilles jetables NovoFine Plus (B).
Wegovy Multi FixDose 0.5 mg solution injectable en stylo prérempli:
1 stylo prérempli et 4 aiguilles jetables NovoFine Plus (B).
Wegovy Multi FixDose 1 mg solution injectable en stylo prérempli:
1 stylo prérempli et 4 aiguilles jetables NovoFine Plus (B).
Wegovy Multi FixDose 1.7 mg solution injectable en stylo prérempli:
1 stylo prérempli et 4 aiguilles jetables NovoFine Plus (B).
Wegovy Multi FixDose 2.4 mg solution injectable en stylo prérempli:
1 stylo prérempli et 4 aiguilles jetables NovoFine Plus (B).
Titulaire de l’autorisationNovo Nordisk Pharma AG, Kloten
Domicile: Zürich
Mise à jour de l’informationJanvier 2025
|