CompositionPrincipes actifs
Icatibantum (ut icatibantum acetas).
Excipients
Natrii chloridum, acidum acetas glaciale (E260), natrii hydroxidum (E524), aqua ad injectabilia q.s. ad solutionem pro 3 ml.
1 seringue pré-remplie à 3 ml contient 9,9 mg de sodium.
Indications/Possibilités d’emploiIcatibant Spirig HC est indiqué dans le traitement symptomatique des crises aiguës d'angio-œdème héréditaire (AOH) chez les adultes, les adolescents et les enfants âgés de 2 ans et plus présentant une carence en inhibiteur de la C1 estérase.
Posologie/Mode d’emploiIcatibant Spirig HC doit être administré sous la supervision d'un professionnel de santé.
Posologie
Adultes
La dose recommandée chez les adultes est une injection unique d'Icatibant Spirig HC 30 mg en sous-cutanée.
Dans la majorité des cas, une seule injection d'Icatibant Spirig HC suffit à traiter une crise. En cas de soulagement insuffisant ou de récurrence des symptômes, une deuxième injection d'Icatibant Spirig HC peut être administrée 6 heures plus tard. Si la deuxième injection produit un soulagement insuffisant ou en cas de récurrence des symptômes, une troisième injection d'Icatibant Spirig HC peut être administrée de nouveau 6 heures plus tard. Il convient de ne pas dépasser 3 injections d'Icatibant Spirig HC sur une période de 24 heures.
Lors des essais cliniques, 8 injections d'icatibant par mois ont été administrées au maximum.
Enfants et adolescents
La dose recommandée d'Icatibant Spirig HC déterminée en fonction du poids corporel chez les enfants et adolescents (âgés de 2 à 17 ans) est présentée dans le tableau 1 ci-dessous.
Tableau 1: Posologie chez les enfants et adolescents
|
Poids
|
Dose (volume à injecter)
| |
12 kg à 25 kg
|
10 mg (1,0 ml)
| |
26 kg à 40 kg
|
15 mg (1,5 ml)
| |
41 kg à 50 kg
|
20 mg (2,0 ml)
| |
51 kg à 65 kg
|
25 mg (2,5 ml)
| |
> 65 kg
|
30 mg (3,0 ml)
|
Dans l'étude clinique, il n'a pas été administré plus d'une injection d'icatibant par crise d'AOH.
Aucune posologie ne peut être recommandée chez les enfants âgés de moins de 2 ans ou pesant moins de 12 kg car la sécurité et l'efficacité dans ce groupe de la population pédiatrique n'ont pas été établies.
Patients âgés
Des données limitées sont disponibles sur les patients de plus de 65 ans.
Il a été démontré que les patients âgés présentent une exposition systémique accrue à l'icatibant. L'importance de ceci en terme desécurité d'Icatibant Spirig HC n'est pas connue (cf. Pharmacocinétique).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients souffrant de troubles hépatiques.
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients souffrant de troubles rénaux.
Mode d'administration
La voie d'administration d'Icatibant Spirig HC est la voie sous-cutanée, de préférence dans la région abdominale.
Icatibant Spirig HC solution injectable doit être injecté lentement en raison du volume à administrer.
Chaque seringue d'Icatibant Spirig HC est à usage unique.
Pour des instructions sur l'utilisation, voir l'annexe de cette notice d'information professionnelle ou l'information destinée aux patients comme notice d'emballage.
Administration par une tierce personne/auto-administration
La décision de recourir à l'administration par une tierce personne ou à l'auto-administration ne doit être prise que par un médecin expérimenté dans le diagnostic et le traitement de l'angio-œdème héréditaire (voir Mises en garde et précautions).
Adultes
En cas d'auto-administration ou d'administration par une tierce personne, une formation préalable sur la technique de l'injection sous-cutanée devra avoir été dispensée par un professionnel de santé.
Enfants et adolescents âgés de 2 à 17 ans
Chez les patients n'ayant jamais reçu Icatibant Spirig HC, il convient d'instaurer le premier traitement au sein d'un établissement médical ou sous la supervision d'un médecin. Les tierces personnes ne peuvent administrer Icatibant Spirig HC que s'ils ont été suffisamment formés par la technique d'injection sous-cutanée par les professionnels de la santé, y compris les étapes nécessaires pour aspirer la solution dans une seringue vide. En cas de soulagement insuffisant ou de récurrence des symptômes après administration par une tierce personne, un avis médical doit être obtenu.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients.
Mises en garde et précautionsŒdème laryngé
Les patients souffrant d'œdèmes laryngés doivent être traités dans un établissement médical approprié après injection jusqu'à ce que le médecin estime qu'ils peuvent quitter l'établissement.
Cardiopathie ischémique
En cas de cardiopathie ischémique, une détérioration de la fonction cardiaque et une diminution du débit sanguin coronaire seraient théoriquement provoquées par l'antagonisme du récepteur bradykinine de type 2. Il convient donc d'être prudent lors de l'administration d'Icatibant Spirig HC aux patients présentant une cardiopathie ischémique aiguë ou une angine de poitrine instable (voir Données précliniques).
Accident vasculaire cérébral
Bien que certaines données prouvent l'effet bénéfique du blocage du récepteur B2 immédiatement après un accident vasculaire cérébral, il existe une possibilité théorique que l'icatibant puisse atténuer les effets neuroprotecteurs positifs de phase tardive de la bradykinine. Ainsi, il conviendrait d'être prudent lors de l'administration d'icatibant aux patients dans les semaines suivant un accident vasculaire cérébral.
Administration par une tierce personne/auto-administration
Chez les patients n'ayant jamais reçu Icatibant Spirig HC, il convient d'instaurer le premier traitement au sein d'un établissement médical ou sous la supervision d'un médecin.
En cas de soulagement insuffisant ou de récurrence des symptômes après une auto-administration ou l'administration par une tierce personne, il est recommandé que le patient ou la tierce personne consulte un médecin. Chez les adultes, plusieurs doses suivantes qui peuvent être nécessaires pour traiter une crise doivent être administrées au sein d'un établissement médical (voir Posologie/mode d'emploi). Il n'existe pas de données sur l'administration de doses supplémentaires pour la même crise chez les adolescents ou les enfants.
Les patients souffrant d'œdèmes laryngés doivent toujours consulter un médecin et être gardés sous observation au sein d'un établissement médical, même si l'injection a été effectuée à domicile.
Enfants et adolescents
L'expérience du traitement de plus d'une crise d'AOH par Icatibant Spirig HC chez les enfants et adolescents est limitée.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par seringue pré-remplie, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsAucune interaction pharmacocinétique des médicaments impliquant le CYP450 ne devrait être observée (voir Pharmacocinétique).
La co-administration d'Icatibant Spirig HC avec des inhibiteurs de l'enzyme de conversion (EC) n'a pas été étudiée. Les inhibiteurs de l'enzyme de conversion sont contre-indiqués chez les patients souffrant d'AOH en raison de l'augmentation possible des taux de bradykinine.
Grossesse, allaitementGrossesse
Il n'existe aucune donnée disponible sur l'exposition à l'icatibant des femmes enceintes. Des études menées chez l'animal ont montré des effets sur l'implantation utérine et la mise bas (voir Données précliniques) mais le risque possible chez l'homme n'est pas connu.
Icatibant Spirig HC ne doit être utilisé pendant la grossesse que si le bénéfice escompté justifie le risque pour le fœtus (ex.: pour traiter des œdèmes laryngés susceptibles de mettre en jeu le pronostic vital).
Allaitement
On ne sait pas si l'icatibant est excrété dans le lait maternel humain mais il est recommandé aux femmes allaitantes souhaitant utiliser de l'Icatibant Spirig HC de ne pas allaiter pendant les 12 heures qui suivent l'administration du traitement.
L'icatibant est excrété dans le lait des rates en lactation à des concentrations similaires à celles retrouvées dans le sang maternel.
Fertilité
Dans une étude menée chez 39 hommes et femmes adultes sains ayant reçu 3 doses de 30 mg à intervalle de 6 heures tous les 3 jours pour un total de 9 doses, aucune modification cliniquement significative des taux d'hormones sexuelles de base et après stimulation par la GnRH n'a été observée chez les femmes ou les hommes. L'icatibant n'a pas eu d'effets significatifs sur le taux de progestérone de la phase lutéale et la fonction lutéale ou sur la durée du cycle menstruel chez les femmes, ni sur le nombre, la motilité et la morphologie des spermatozoïdes chez les hommes. Il est peu probable que le schéma posologique utilisé dans cette étude est maintenu en pratique clinique. Des études chez l'animal ont mis en évidence une toxicité sur la reproduction. Icatibant n'a cependant eu aucun effet sur la fertilité (voir Données précliniques).
Effet sur l’aptitude à la conduite et l’utilisation de machinesIcatibant Spirig HC a une influence mineure sur 'l'aptitude à conduire des véhicules et à utiliser des machines. Une fatigue, une léthargie, une fatigabilité, une somnolence et des vertiges ont été rapportés après l'administration d'Icatibant Spirig HC. Ces symptômes peuvent survenir à la suite d'une crise d'AOH. Il convient de recommander aux patients de ne pas conduire ni utiliser de machines s'ils se sentent fatigués ou ressentent des vertiges.
Effets indésirablesDans les études cliniques d'enregistrement, 999 crises d'AOH au total ont été traitées par 30 mg d'icatibant administré par voie sous-cutanée par un professionnel de santé. L'icatibant 30 mg SC a été administré par un professionnel de santé à 129 volontaires sains et 236 patients atteints d'AOH.
La quasi-totalité des sujets ayant reçu de l'icatibant en injection sous-cutanée lors des essais cliniques ont présenté des réactions au niveau du site d'injection (caractérisées par des irritations cutanées, un œdème, une douleur, des démangeaisons, un érythème, une sensation de brûlure). Ces réactions ont été généralement légères à modérées, transitoires et se sont résolues sans intervention.
La fréquence des effets indésirables est définie comme: très fréquent (≥1/10); fréquent (> 1/100, < 1/10); peu fréquent (> 1/1'000, < 1/100); rare (> 1/10'000, < 1/1'000); très rare (< 1/10'000); inconnus (fréquence ne peut pas être estimée sur la base des données disponibles).
Affections du système nerveux
Fréquents: sensation vertigineuse, céphalées
Affections gastro intestinales
Occasionnels: nausées
Affections de la peau et du tissu sous cutané
Fréquents: irritation cutanée, érythème, prurit
Troubles généraux et anomalies au site d'administration
Très fréquents: réactions au site d'injection*
Fréquents: fièvre
Investigations
Fréquents: augmentation des transaminases
* Ecchymose au point d'injection, hématome au site d'injection, brûlure au point d'injection, érythème au point d'injection, hypoesthésie au site d'injection, irritation au point d'injection, engourdissement au site d'injection, œdème au point d'injection, douleur au point d'injection, sensation de pression au site d'injection, prurit au point d'injection, gonflement au point d'injection, urticaire au point d'injection et chaleur au niveau du site d'injection.
Enfants et adolescents
Au total, 32 enfants et adolescents (8 enfants âgés de 2 à 11ans et 24 adolescents âgés de 12 à 17 ans) atteints d'AOH ont été traités par l'icatibant au cours des études cliniques. Trente et un patients ont reçu une dose unique d'icatibant et un patient (un adolescent) a reçu l'icatibant pour deux crises d'AOH (deux doses au total). L'icatibant était administré en injection sous-cutanée à la dose de 0,4 mg/kg de poids corporel, jusqu'à une dose maximale de 30 mg.
Chez la majorité des patients pédiatriques, des réactions au site d'injection sont survenues après une injection sous-cutanée d'icatibant (par exemple, érythème, gonflement, sensation de brûlures, douleur cutanée et démangeaisons / prurit). Celles-ci étaient de généralement sévérité légère à modérées et concordaient avec les réactions rapportées chez les adultes (comme érythème, gonflement, sensation de brûlures, prurit, chaleur et douleur de la peau). Deux patients pédiatriques ont présenté des réactions au site d'injection qui ont été évaluées comme sévères et qui se sont complètement résolues dans les 6 heures. Elles consistaient en érythème, gonflement, sensation de brûlure et sensation de chaleur.
Il n'a pas été observé de modifications cliniquement significatives des taux d'hormones sexuelles lors des études cliniques.
Effets indésirables après commercialisation
Affections de la peau et du tissu sous cutané
Inconnus: urticaire
Immunogénicité
Pendant le traitement en administrations répétées chez les adultes dans les études de phase III contrôlées, une positivité transitoire pour les anticorps anti-icatibant a été observée dans de rares cas. L'efficacité a été maintenue chez tous les patients. Un patient traité par l'icatibant était positif pour les anticorps anti-icatibant avant et après le traitement par l'icatibant. Ce patient a été suivi pendant 5 mois et la recherche d'anticorps anti-icatibant a été négative lors des prélèvements ultérieurs. Aucune réaction d'hypersensibilité ou anaphylactique n'a été rapportée avec Icatibant Spirig HC.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageAucune donnée clinique sur les surdosages n'est disponible.
Une dose de 3,2 mg/kg administrée par voie intraveineuse (environ 8 fois la dose thérapeutique) a provoqué un érythème, des démangeaisons, des bouffées congestives ou une hypotension transitoire chez des sujets sains. Aucune intervention thérapeutique n'a été nécessaire.
Propriétés/EffetsCode ATC
B06AC02
Mécanisme d'action
L'AOH (maladie dominante autosomique) est provoqué par une absence ou un dysfonctionnement de l'inhibiteur de la C1 estérase. Les crises d'AOH s'accompagnent d'une libération accrue de bradykinine, qui constitue le principal médiateur dans le développement des symptômes cliniques.
L'AOH se manifeste par des crises intermittentes d'œdème sous-cutané et/ou sous-muqueux touchant les voies respiratoires supérieures, la peau et le tractus gastro-intestinal. Une crise dure généralement de 2 à 5 jours.
L'icatibant est un antagoniste compétitif sélectif au niveau du récepteur de la bradykinine de type 2 (B2). Il s'agit d'un décapeptide de synthèse ayant une structure similaire à celle de la bradykinine mais comportant 5 acides aminés non protéinogènes. Dans l'AOH, les concentrations accrues de bradykinine constituent le principal médiateur dans le développement des symptômes cliniques.
Pharmacodynamique
Chez les sujets jeunes et sains recevant de l'icatibant à des doses de 0,8 mg/kg sur 4 heures, de 1,5 mg/kg/jour ou de 0,15 mg/kg/jour pendant 3 jours, le développement d'hypotension, une vasodilatation et une tachycardie réflexe induites par la bradykinine ont pu être évitées. Il a été démontré que l'icatibant est un antagoniste compétitif lorsque la dose de bradykinine de départ était multipliée par 4.
Efficacité clinique
Des données d'efficacité ont été obtenues au cours d'une étude initiale ouverte de phase II et de trois études de phase III contrôlées.
Les études cliniques de phase III (FAST-1 et FAST-2) étaient des études randomisées en double aveugle contrôlées, dont le plan expérimental était identique à l'exception du comparateur (une étude contrôlée versus acide tranexamique oral et une étude versus placebo).
Au total, 130 patients ont été randomisés pour recevoir une dose de 30 mg d'icatibant (63 patients) ou le comparateur (acide tranexamique, 38 patients ou placebo, 29 patients). Les épisodes ultérieurs d'AOH ont été traités dans le cadre d'une extension en ouvert. Les patients présentant des symptômes d'angio-œdème laryngé ont reçu un traitement en ouvert par l'icatibant. Dans les études cliniques de phase III, le critère d'efficacité primaire était le délai jusqu'au début du soulagement des symptômes, évalué à l'aide d'une échelle visuelle analogue (EVA). Le tableau 2 présente les résultats d'efficacité de ces études.
L'étude FAST-3 était une étude randomisée, contrôlée versus placebo, en groupes parallèles, menée chez 98 patients adultes (âge médian, 36 ans). Les patients ont été randomisés pour recevoir l'icatibant 30 mg ou le placebo en injection sous-cutanée. Un sous-groupe de patients de cette étude présentait des crises aiguës d'AOH malgré l'administration d'androgènes, d'antifibrinolytiques ou d'inhibiteurs de C1. Le critère d'évaluation principal était le délai jusqu'au début du soulagement des symptômes, évalué par le score composite en 3 items d'une échelle visuelle analogique (EVA-3), consistant en évaluations de l'œdème cutané, de la douleur cutanée et de la douleur abdominale. Le tableau 3 présente les résultats d'efficacité de l'étude FAST-3.
Dans ces études, le délai médian jusqu'au début du soulagement des symptômes a été plus court chez les patients traités par l'icatibant (2,0, 2,5 et 2,0 heures, respectivement) par rapport à l'acide tranexamique (12,0 heures) et au placebo (4,6 et 19,8 heures). L'effet du traitement par icatibant a été confirmé par les critères d'efficacité secondaires.
Dans une analyse intégrée de ces études de phase III contrôlées, le délai jusqu'au début du soulagement des symptômes et le délai jusqu' au début du soulagement du symptôme primaire ont été similaires quels que soient la tranche d'âge, le sexe, les particularités ethniques, le poids et l'utilisation ou non d'androgènes ou d'antifibrinolytiques.
La réponse a également été uniforme lors des crises répétées dans les études de phase III contrôlées. Au total, 237 patients ont été traités par 1'386 doses d'icatibant 30 mg pour 1'278 crises aiguës d'AOH. Pour les 15 premières crises traitées par icatibant (1'114 doses pour 1'030 crises), on a décrit un délai médian jusqu'au début du soulagement des symptômes similaire entre les crises (2,0 à 2,5 heures). 92,4 % de ces crises d'AOH ont été traitées par une dose unique d'icatibant.
Tableau 2: Résultats d'efficacité des études FAST-1 et FAST-2
|
Etude clinique contrôlée d'ICATIBANT versus acide tranexamique ou placebo:résultats d'efficacité
| |
FAST-2
|
FAST-1
| |
|
Icatibant
|
Acide tranexamique
|
|
Icatibant
|
Placebo
| |
Nombre de sujets dans la population en intention de traiter (ITT)
|
36
|
38
|
Nombre de sujets dans la population en intention de traiter (ITT)
|
27
|
29
| |
EVA de départ(mm)
|
63,7
|
61,5
|
EVA de départ(mm)
|
69,3
|
67,7
| |
Modification après 4 heures par rapport aux valeurs de départ
|
-41,6
|
-14,6
|
Modification après 4 heures par rapport aux valeurs de départ
|
-44,8
|
-23,5
| |
Différence entre les traitements (95 % CI, valeur p)
|
-27,8 (-39,4, -16,2) p < 0,001
|
Différence entre les traitements (95 % CI, valeur p)
|
-23,3 (-37,1, -9,4) p = 0,002
| |
Modification après 12 heures par rapport aux valeurs de départ
|
-54,0
|
-30,3
|
Modification après 12 heures par rapport aux valeurs de départ
|
-54,2
|
-42,4
| |
Différence entre les traitements (95% CI, valeur p)
|
-24,1 (-33,6, -14,6) p < 0,001
|
Différence entre les traitements (95% CI, valeur p)
|
-15,2 (-28,6, -1,7) p = 0,028
| |
Temps médian avant début de soulagement des symptômes (heures)
|
|
|
Temps médian avant début de soulagement des symptômes (heures)
|
|
| |
Tous les épisodes(N = 74)
|
2,0
|
12,0
|
Tous les épisodes(N = 56)
|
2,5
|
4,6
| |
Taux de réponse (%, IC) 4 heures après le début du traitement
|
|
|
Taux de réponse (%, IC) 4 heures après le début du traitement
|
|
| |
Tous les épisodes(N = 74)
|
80,0
(63,1, 91,6)
|
30,6
(16,3, 48,1)
|
Tous les épisodes(N = 56)
|
66,7
(46,0, 83,5)
|
46,4
(27,5, 66,1)
| |
Temps médian avant début de soulagement des symptômes: tous les symptômes (h):
|
|
|
Temps médian avant début de soulagement des symptômes: tous les symptômes (h):
|
|
| |
Douleur abdominale
|
1,6
|
3,5
|
Douleur abdominale
|
2,0
|
3,3
| |
Gonflement cutané
|
2,6
|
18,1
|
Gonflement cutané
|
3,1
|
10,2
| |
Douleur cutanée
|
1,5
|
12,0
|
Douleur cutanée
|
1,6
|
9,0
| |
Temps médian avant soulagement quasi complet des symptômes (heures)
|
|
|
Temps médian avant soulagement quasi complet des symptômes (heures)
|
|
| |
Tous les épisodes(N = 74)
|
10,0
|
51,0
|
Tous les épisodes(N = 56)
|
8,5
|
19,4
| |
Temps médian avant régression des symptômes, par le patient (heures)
|
|
|
Temps médian avant régression des symptômes, par le patient (heures)
|
|
| |
Tous les épisodes(N = 74)
|
0,8
|
7,9
|
Tous les épisodes(N = 56)
|
0,8
|
16,9
| |
Temps médian pour une amélioration globale du patient, par le médecin (heures)
|
|
|
Temps médian pour une amélioration globale du patient, par le médecin (heures)
|
|
| |
Tous les épisodes (N = 74)
|
1,5
|
6,9
|
Tous les épisodes (N = 56)
|
1,0
|
5,7
|
Tableau 3: Résultats d'efficacité de l'étude FAST-3
|
Résultats d'efficacité: FAST-3; phase contrôlée – population ITT
|
| |
Critère
|
Statistique
|
Icatibant
|
Placebo
|
Valeur P
| |
|
|
(n = 43)
|
(n = 45)
|
| |
Critère principal
| |
élai jusqu'au début du soulagement des symptômes – Score EVA composite (heures)
|
Médiane
|
2,0
|
19,8
|
<0,001
| |
Autres critères
| |
Délai jusqu'au début du soulagement du symptôme primaire (heures)
|
Médiane
|
1,5
|
18,5
|
<0,001
| |
Variation du score EVA composite 2 heures après le traitement
|
Moyenne
|
-19,74
|
-7,49
|
<0,001
| |
Variation du score composite de symptômes évalués par le patient 2 heures après le traitement
|
Moyenne
|
-0,53
|
-0,22
|
<0,001
| |
Variation du score composite de symptômes évalués par l'investigateur 2 heures après le traitement
|
Moyenne
|
-0,44
|
-0,19
|
<0,001
| |
Délai jusqu' au soulagement quasi-complet des symptômes (heures)
|
Médiane
|
8,0
|
36,0
|
0,012
| |
Délai jusqu' à la régression initiale des symptômes, évaluation par le patient (heures)
|
Médiane
|
0,8
|
3,5
|
<0,001
| |
Délai jusqu' à la régression initiale des symptômes, évaluation visuelle par l'investigateur (heures)
|
Médiane
|
0,8
|
3,4
|
<0,001
|
Au total, 66 patients présentant des crises d'AOH touchant le larynx ont été traités dans ces études cliniques de phase III contrôlées. Les résultats ont été similaires à ceux observés chez les patients ayant présenté des crises d'AOH non laryngé en termes de délai jusqu'au début du soulagement des symptômes.
Sécurité et efficacité en pédiatrie
Une étude en ouvert non randomisée, en un seul bras (HGT-FIR-086), a été menée chez 32 patients au total. À une exception près, tous les patients ont reçu une seule dose sous-cutanée d'icatibant (0,4 mg/kg de poids corporel jusqu'à une dose maximale de 30 mg). La majorité des patients ont été suivis pendant au moins 6 mois. Onze patients étaient au stade prépubertaire et 21 patients étaient au stade pubertaire ou postpubertaire.
La population pertinente pour l'analyse de l'efficacité était composée de 22 patients (11 patients au stade prépubertaire et 11 patients au stade pubertaire/postpubertaire) qui avaient été traités par l'icatibant pour une crise acute d'AOH.
Le critère d'évaluation principal était le délai jusqu'au début du soulagement des symptômes (time to onset of symptom relief, TOSR). La sévérité des symptômes a été évalué par l'investigateur à l'aide d'une échelle prédéfinie d'évaluation des symptômes (0 à 5). Le délai jusqu'au soulagement des symptômes était défini comme la durée (en heures) nécessaire pour observer une amélioration de 20 % des symptômes.
Globalement, le délai médian jusqu'au début du soulagement des symptômes a été de 1,0 heure (intervalle de confiance à 95 %: 1,0; 1,1 heure). Une heure et deux heures après le traitement, un début de soulagement des symptômes a été observé chez 50 % et 90 % respectivement des patients.
Globalement, le délai médian jusqu'aux symptômes minimaux (moment le plus proche après le traitement où tous les symptômes étaient légers ou absents) était de 1,1 heure (intervalle de confiance à 95 %: 1,0; 2,0 heures).
PharmacocinétiqueLa pharmacocinétique de l'icatibant a été caractérisée par des études utilisant à la fois l'administration intraveineuse et sous-cutanée à des volontaires sains et à des patients. Le profil pharmacocinétique de l'icatibant chez les patients souffrant d'AOH était similaire à celui des volontaires sains.
Absorption
Après administration sous-cutanée, la biodisponibilité absolue de l'icatibant est de 97 %. Le temps nécessaire pour atteindre la concentration maximale est d'environ d'environ 30 minutes.
Distribution
Le volume de distribution de l'icatibant (Vss) est d'environ 20-25 litres. La liaison aux protéines plasmatiques est de 44 %.
Métabolisme
L'icatibant est métabolisé de façon importante par les enzymes protéolytiques en métabolites inactifs qui sont principalement excrétés dans les urines.
Des études in vitro ont confirmé que l'icatibant n'est pas dégradé par les voies métaboliques oxydatives et qu'il n'est pas un inhibiteur des principales isoenzymes du cytochrome P450 (CYP) (CYP 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 et 3A4) et qu'il n'est pas un inducteur des CYP 1A2 et 3A4.
Élimination
L'icatibant est principalement éliminé par le métabolisme avec moins de 10 % de la dose excrétée dans les urines sous forme non modifiée. La clairance est d'environ 15 à 20 l/h et est indépendante de la dose. La demi-vie terminale plasmatique est d'environ 1 à 2 heures.
Cinétique pour certains groupes de patients
Patients âgés
Les données suggèrent une diminution, liée à l'âge, de la clairance, qui entraîne une exposition supérieure d'environ 50 à 60 % chez les patients âgés (75-80 ans) par rapport à un patient de 40 ans.
Sexe
Les données semblent indiquer qu'il n'y a pas de différence de la clairance entre les sujets de sexe masculin et féminin après correction pour le poids.
Troubles de la fonction hépatique et rénale
Des données limitées suggèrent que l'exposition à l'icatibant n'est pas influencée par les troubles hépatiques ou rénaux.
Groupe ethnique
Les données sur l'effet des particularités ethniques sont limitées. Les données disponibles n'indiquent pas de différences de la clairance entre les différents groupes ethniques.
Enfants et adolescents
La pharmacocinétique de l'icatibant a été caractérisée chez des patients pédiatriques atteints d'AOH dans l'étude HGT-FIR-086 (voir rubrique Pharmacodynamique, efficacité clinique). Après administration d'une dose unique par voie sous-cutanée (0,4 mg/kg jusqu'à un maximum de 30 mg), le temps jusqu'à la concentration maximale est d'environ 30 minutes et la demi-vie terminale est d'environ 2 heures. Il n'a pas été observé de différences de l'exposition à l'icatibant chez les patients présentant ou non une crise d'AOH. Le modèle pharmacocinétique de population utilisant à la fois les données chez les adultes et chez les enfants et adolescents a montré que la clairance de l'icatibant est corrélée au poids corporel, des valeurs de clairance plus faibles étant observées pour les poids plus faibles dans la population pédiatrique atteinte d'AOH. Sur la base de la modélisation de la posologie en fonction du poids, l'exposition prédite à l'icatibant dans la population pédiatrique (voir rubrique Posologie/mode d'emploi) est plus faible que celle observée dans les études menées chez des patients adultes atteints d'AOH.
Données précliniquesPharmacologie de sécurité
L'icatibant n'a provoqué aucune modification de la conduction cardiaque in vitro (canal hERG) ou in vivo chez plusieurs modèles animaux (régulation du rythme ventriculaire, effort physique et ligature coronaire les chiens par rapport aux contrôles sains) chez lesquels aucune modification hémodynamique associée n'a été observée. Il a été démontré que l'icatibant aggrave l'ischémie cardiaque induite chez plusieurs modèles animaux, mais il n'a pas été prouvé qu'il ait un effet délétère systématique dans les cas d'ischémie aiguë.
Toxicité en cas d'administration répétée
Des études à doses répétées menées pendant une période maximum de 6 mois chez le rat et de 9 mois chez le chien ont montré une diminution dose-dépendante du taux d'hormones sexuelles circulantes, ainsi qu'un retard réversible de la maturation sexuelle dû à l'administration répétée d'icatibant chez ces deux espèces.
L'exposition journalière maximale définie par l'aire sous la courbe (ASC) à la dose sans effet nocif observé (NOAEL) lors de l'étude de 9 mois menée chez le chien était 2,3 fois l'ASC chez l'adulte après une dose sous-cutanée de 30 mg. Une concentration NOAEL n'était pas mesurable dans l'étude chez le rat, toutefois, tous les résultats issus de cette étude ont montré un effet soit totalement, soit partiellement réversible chez les rats traités. Une hypertrophie surrénalienne a été observée chez le rat à toutes les doses testées. Une réversibilité de l'hypertrophie surrénalienne a été constatée une fois que le traitement par icatibant a été interrompu. La pertinence clinique de l'exploration des glandes surrénales n'est pas connue.
Toxicité sur la reproduction
L'icatibant n'a eu aucun effet sur la fertilité des souris mâles (dose maximale: 80,8 mg/kg/jour) et des rats mâles (dose maximale: 10 mg/kg/jour).
L'icatibant n'était pas tératogène lorsqu'il était administré par injection sous-cutanée pendant le développement embryonnaire et fœtal précoce chez le rat (dose maximale de 25 mg/kg/jour) et chez le lapin (dose maximale de 10 mg/kg/jour). L'icatibant est un antagoniste puissant de la bradykinine et, par conséquent, à des doses élevées, le traitement peut avoir des effets sur le processus d'implantation utérine et sur la stabilité utérine ultérieure en début de gestation. Ces effets sur l'utérus se manifestent également plus tard au cours de la gestation lorsque l'icatibant présente un effet tocolytique entraînant le retard de la mise bas chez le rat, avec une souffrance fœtale accrue et une mort périnatale lors de l'administration d'une forte dose (10 mg/kg/jour). Aucun effet n'a été constaté dans le développement post-natal des rats nouveau-nés.
Une atrophie des testicules et des épididymes a été observée dans l'étude pivot de toxicité juvénile au cours de laquelle des rats sexuellement immatures ont été traités à la dose de 3 mg/kg par jour pendant 7 semaines. Les anomalies microscopiques observées étaient partiellement réversibles. Des effets similaires de l'icatibant sur les tissus reproducteurs ont été observés chez des rats et des chiens sexuellement matures. Ces anomalies tissulaires étaient compatibles avec les effets observés sur les gonadotrophines et semblent être réversibles pendant la période sans traitement ultérieure.
Carcinogénicité
Pour évaluer le potentiel carcinogène de l'icatibant, des études de 2 ans ont été réalisées sur les souris et les rats. Aucune évidence de carcinogénicité n'a été observée chez les souris et chez les rats recevant de l'icatibant par voie sous-cutanée à des doses jusqu'à 15 mg/kg/jour (2 fois par semaine) et 6 mg/kg/jour (quotidiennement), respectivement (approximativement 3x et 2x supérieures à la dose journalière humaine maximale recommandée [3 doses à 30 mg par jour] sur la base de l'AUC).
Mutagénicité
Lors d'une batterie classique d'analyses in vitro et in vivo, l'icatibant n'a montré aucun signe de génotoxicité.
Autres données
Dans une étude de recherche de dose en administration sous-cutanée d'une durée de 2 semaines chez le rat juvénile, la dose maximale tolérée a été établie à 25 mg/kg/jour
Remarques particulièresChaque seringue pré-remplie de 3 ml contient de l'acétate d'icatibant équivalent à 30 mg d'icatibant.
La solution doit être un liquide transparent et incolore et sans particules. Si elle est colorée ou contient des grumeaux, des flocons ou des particules, il ne faut pas l'utiliser.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
A conserver à une température ne dépassant pas 30°C. Ne pas congeler.
Conserver dans l'emballage d'origine.
A conserver hors de portée des enfants.
Remarques concernant la manipulation
La solution doit être transparente et incolore et sans particules.
Si elle est colorée ou contient des grumeaux, des flocons ou des particules, il ne faut pas l'utiliser.
Usage chez les enfants et adolescents
La dose appropriée à administrer est déterminée en fonction du poids corporel (voir Posologie/mode d'emploi).
Si la dose requise est inférieure à 30 mg (3 ml), les accessoires ci-dessous sont on outre nécessaires pour prélever et administrer la dose appropriée:
·adaptateur (raccord Luer-lock femelle proximal et/ou distal);
·seringue graduée de 3 ml (recommandée).
La seringue préremplie d'icatibant et tous les autres composants sont à usage unique.
Tout produit non utilisé ou déchet doit être éliminé conformément.
Toutes les aiguilles et seringues doivent être éliminées dans un collecteur d'aiguilles.
Numéro d’autorisation68263 (Swissmedic)
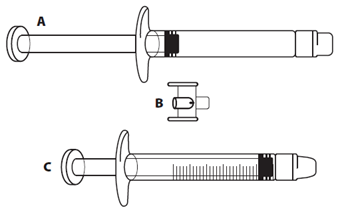
PrésentationSeringue pré-remplie (3-ml seringue pré-remplie, verre de type I) avec bouchon de piston (avec polymère fluorocarbone enduit de bromobutyle).
L'emballage avec:
1 seringue pré-remplie à 3 ml
1 aiguille (25 G; 16 mm)
[B]
Titulaire de l’autorisationSpirig HealthCare SA, 4622 Egerkingen.
Mise à jour de l’informationDécembre 2019
Appendice 1
Instructions étape par étape pour l'injection
1) Informations générales
·Nettoyez le plan de travail (la surface) utilisé avant de commencer.
·Lavez-vous les mains à l'eau et au savon.
·Ouvrez la plaquette en tirant sur le film protecteur.
·Retirez la seringue préremplie d'Icatibant Spirig HC de la plaquette.
·Retirez le capuchon situé à l'extrémité de la seringue préremplie en dévissant celui-ci.
·Une fois le capuchon retiré, posez la seringue préremplie sur une surface plane. Evitez de toucher la surface avec l'embout de la seringue.
2a) Préparation de la seringue
pour les enfants et adolescents (2 à 17 ans)
pesant 65 kg ou moins:
Informations importantes destinées aux professionnels de santé et aux tierce personnes:
Si la dose est inférieure à 3 ml (30 mg), le matériel suivant est nécessaire pour extraire et administrer la dose appropriée:
a) Seringue préremplie d'Icatibant Spirig HC (contenant la solution d'icatibant)
b) Raccord (adaptateur Luer-Lock)
c) Seringue graduée de 3 ml
Le volume à injecter en ml doit être prélevé dans une seringue graduée de 3 ml vide (voir le tableau 1).
Tableau 1: Posologie chez les enfants et adolescents
|
Poids
|
Volume à injecter (dose)
| |
12 kg à 25 kg
|
1,0 ml
| |
26 kg à 40 kg
|
1,5 ml
| |
41 kg à 50 kg
|
2,0 ml
| |
51 kg à 65 kg
|
2,5 ml
|
Chez les patients pesant plus de 65 kg, utiliser le contenu total de la seringue préremplie d'Icatibant Spirig HC (3 ml).
Demandez à votre médecin, à votre pharmacien ou à votre professionnel de la santé si vous n'êtes pas sûr du nombre de ml de solution injectable à extraire.
1.Retirer les capuchons à chaque extrémité du raccord.
Évitez de toucher les extrémités du raccord et l'embout de la seringue pour prévenir une contamination.
2.Vissez le raccord sur la seringue préremplie d'Icatibant Spirig HC.
3.Fixez la seringue graduée de 3 ml à l'autre extrémité du raccord en veillant à ce que les deux connexions soient bien verrouillées.
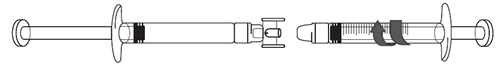
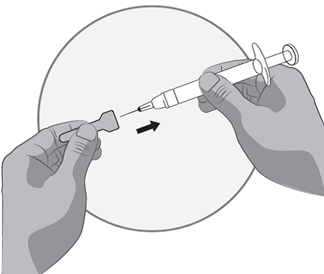
Transfert de la solution d'icatibant dans la seringue graduée:
1.Pour commencer à transférer la solution d'icatibant, appuyez sur le piston de la seringue préremplie (à l'extrême gauche sur l'illustration ci-dessous).
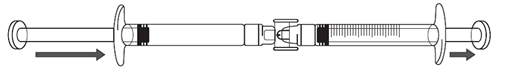
2.Si la solution d'icatibant ne commence pas à passer dans la seringue graduée, tirez légèrement sur le piston de la seringue graduée jusqu'à ce que la solution d'icatibant commence à s'écouler dans la seringue graduée (voir l'illustration ci-dessous).
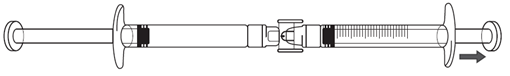
3.Continuez à appuyer sur le piston de la seringue préremplie jusqu'à ce que le volume d'injection en ml (dose) nécessaire soit transféré dans la seringue graduée. Voir le tableau 1 au-dessus pour les informations sur la posologie et volume d'injection.
S'il y a de l'air dans la seringue graduée:
·Retournez les seringues connectées de façon à ce que la seringue préremplie soit en haut (voir l'illustration ci-dessous).
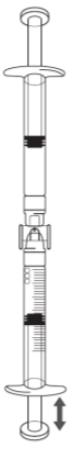
·Appuyez sur le piston de la seringue graduée afin que tout l'air repasse dans la seringue préremplie (il peut être nécessaire de répéter cette étape plusieurs fois).
·Retirez le volume d'injection de solution d'icatibant nécessaire.
4.Retirez la seringue préremplie et le raccord de la seringue graduée.
5.Éliminez la seringue préremplieet le raccord dans un conteneur pour objets tranchants.
2b) Préparation de la seringue (seringue préremplie pour les adultes, seringue graduée pour les adolescents et les enfants) et l'aiguille d'injection:
Tous les patients (adultes, adolescents et enfants)
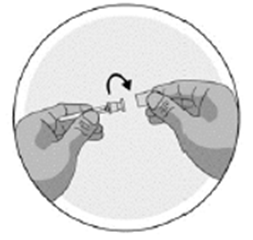
·Retirez de la plaquette l'étui contenant l'aiguille. Ne retirez pas l'aiguille de l'étui protecteur de l'aiguille.
·Tournez le couvercle du capuchon d'aiguille pour briser le sceau (l'aiguille doit toujours être dans le capuchon d'aiguille).
·Munissez-vous de la seringue (seringue préremplie pour les adultes, seringue graduée pour les adolescents et les enfants jusqu'à 65 kg) et maintenez-la fermement. Fixez soigneusement l'aiguille qui est toujours dans l'étui protecteur de l'aiguille, à la seringue contenant la solution incolore.
·Introduisez la seringue dans l'étui protecteur de l'aiguille et vissez la seringue à l'aiguille. L'aiguille avec l'étui protecteur est maintenant fixée à la seringue.
·Maintenez la seringe par le corps de la seringue et retirez l'aiguille de son étui protecteur en tirant sur le corps de la seringue. Ne tirez pas sur le piston.
·La seringue est à présent prête pour l'injection.
3) Comment préparer le point d'injection
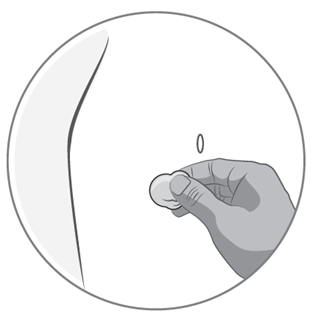
·Choisissez la partie du corps à préparer pour l'injection. L'injection doit être pratiquée dans un pli de la peau sur le côté gauche ou droit de votre ventre à environ 5-10 cm au-dessous de votre nombril. Cette zone doit se trouver à au moins 5 cm de toute cicatrice éventuelle. Ne pas choisir une zone tuméfiée (gonflée), présentant des ecchymoses (bleus) ou douloureuse.
·Nettoyez le point d'injection à l'aide d'un coton imbibé d'alcool et laissez sécher.
4) Injection de la solution
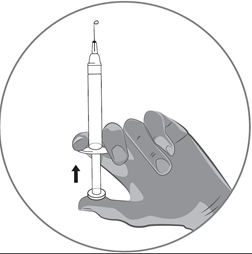
·Maintenez la seringue verticalement avec les deux doigts d'une main, le pouce étant positionné sur le piston.
·Vérifiez l'absence de bulles d'air dans la seringue en appuyant sur le piston jusqu'à l'apparition d'une toute première goutte de produit à l'extrémité de la seringue.
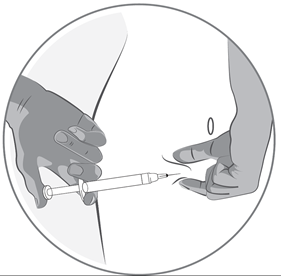
·Maintenez la seringue selon un angle compris entre 45 et 90 degrés par rapport à la surface de la peau, l'aiguille étant dirigée vers la peau.
·Tandis que vous maintenez la seringue d'une main, utilisez votre autre main pour former un pli de peau entre le pouce et l'index à l'endroit que vous avez désinfecté.
·Tout en maintenant le pli de peau, mettez la seringue en contact avec la peau et introduisez l'aiguille rapidement dans le pli de peau.
·Appuyez lentement sur le piston tout en gardant la position initiale de votre main jusqu'à ce que l'intégralité du liquide soit injectée dans la peau et que plus aucun liquide ne reste dans la seringue.
·Il convient d'appuyer lentement sur le piston; une injection doit prendre environ 30 secondes.
·Relâchez le pli de peau et retirez doucement l'aiguille.
5) Elimination du kit d'injection
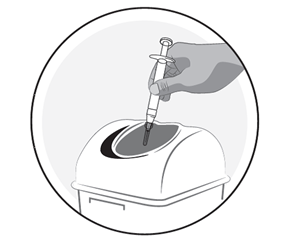
Jetez la seringue, l'aiguille et l'étui protecteur de l'aiguille dans un conteneur pour objets tranchants pour éviter les blessures.
|