CompositionPrincipes actifs
Nitisinonum
Excipients
Capsule dures:
Contenu de la capsule dure: Amylum pregelificatum
Enveloppe de la capsule dure 5mg: Brilliant Blue FCF (E133), Erythrosinum (E127), Titanii dioxidum (E171), Sunset Yellow FCF (E110, 0.008 mg), Gelatinum.
Enveloppe de la capsule dure 10mg: Indigocarmine (E132), Titanii dioxidum (E171), Aqua, Gelatinum.
Enveloppe de la capsule dure 20mg: Titanii dioxidum (E171), Gelatinum.
Suspension:
Hypromellosum (E464), Glycerolum (E422) (500 mg/ml), Polysorbatum 80 (E433), Natrii benzoas (E211) (1 mg/ml), Acidum tartaricum (E334), Aromatica (Fraises) cum Propylenglycolum (E1520), Aqua purificata.
1 ml suspension contient de 0.1595 mg Natrium.
Indications/Possibilités d’emploiNitisinon NOBEL est utilisé pour le traitement de patients adultes et pédiatriques atteints de tyrosinémie héréditaire de type 1 (HT-1), en association avec les mesures diététiques appropriées comprenant également une restriction alimentaire de tyrosine et de phénylalanine.
Posologie/Mode d’emploiLe traitement par la nitisinone doit être instauré et suivi par un médecin expérimenté dans la prise en charge des patients/patientes avec HT-1. Le traitement de tous les génotypes de la maladie doit être instauré dès que possible pour prolonger la survie et éviter les complications telles qu’une insuffisance hépatique, un cancer du foie ou une maladie rénale. Le traitement par la nitisinone doit être associé à un régime alimentaire à faible teneur en phénylalanine et en tyrosine; celui-ci sera suivi en contrôlant régulièrement les taux plasmatiques en acides aminés (se référer aux rubriques «Mises en garde et précautions» et «Effets indésirables»).
Il est en outre recommandé que notamment les patients pédiatriques soient conseillés et accompagnés en matière de nutrition par un personnel qualifié dûment formé et expérimenté.
Dosage
La dose quotidienne initiale recommandée chez l’adulte et l’enfant est de 1 mg/kg de poids corporel à administrer par voie orale. La dose de nitisinone doit être adaptée à chaque patient. Il est recommandé d’administrer la dose une fois par jour. Toutefois, les données concernant les patients/patientes ayant un poids corporel < 20 kg étant limitées, il est recommandé de fractionner la dose quotidienne totale en deux administrations par jour chez cette population de patients/patientes.
Ajustement de la posologie / titration
Dans le cadre de la surveillance régulière, il convient de surveiller la concentration urinaire de succinylacétone, les valeurs des tests fonctionnels hépatiques ainsi que les concentrations en alpha-fœtoprotéine (se référer à la rubrique «Mises en garde et précautions»). Si la succinylacétone est encore détectable dans les urines un mois après l’instauration du traitement par la nitisinone, la dose de nitisinone devra être augmentée jusqu’à 1,5 mg/kg de poids corporel/jour. Il est possible qu’une dose de 2 mg/kg de poids corporel/jour soit nécessaire, en fonction de l’évaluation de tous les paramètres biochimiques. Cette dose doit être considérée comme la dose maximale pour tous les patients/patientes.
En cas de réponse biochimique satisfaisante, la dose doit être ajustée uniquement en fonction du gain de poids corporel. Toutefois, en plus des tests cités ci-dessus, pendant l’instauration du traitement, après le passage d’une administration biquotidienne à une administration quotidienne unique ou lors d’une détérioration, il s’avèrera parfois nécessaire de suivre plus attentivement tous les paramètres biochimiques disponibles (notamment la concentration plasmatique de succinylacétone, la concentration urinaire de 5-aminolévulinate (ALA) et l’activité de la porphobilinogène (PBG)-synthase érythrocytaire).
Instructions posologiques particulières
Il n’existe aucune recommandation de dose spécifique pour les personnes âgées ou les patients présentant une affection rénale ou hépatique.
Enfants et adolescents
La recommandation de dose en mg/kg de poids corporel est identique pour les enfants et les adultes. Toutefois, les données concernant les patients ayant un poids corporel < 20 kg étant limitées, il est recommandé de fractionner la dose quotidienne totale en deux administrations par jour chez cette population de patients.
Mode d’administration
Capsule dures:
Après l’ouverture de la capsule dure, son contenu peut être dispersé dans une petite quantité d’eau ou d’aliments juste avant la prise. Nitisinone NOBEL est également disponible en suspension buvable à 4 mg/ml pour les enfants et les adolescents ayant des difficultés à avaler les capsules dures. Si le traitement par nitisinone est instauré avec de la nourriture, il est recommandé de le poursuivre dans les mêmes conditions, se référer à la rubrique «Interactions».
Suspension:
La suspension est administrée dans la bouche du patient, sans dilution, avec une seringue pour administration orale. Des seringues pour administration orale de 1 ml, 3 ml et 5 ml sont comprises dans la boîte pour mesurer la dose en mL conformément à la posologie prescrite. Elles sont respectivement graduées tous les 0.05 ml (seringue de 1 ml) et 0.1 ml (seringue de 3 ml et 5 ml). Les tableaux ci-dessous indiquent la conversion des doses (mg/ml) pour les trois tailles de seringue pour administration orale.
|
Dose de Nitisinone NOBEL
| |
Seringue pour administration
orale de 1 ml (graduée tous les
0.05 ml)
|
Seringue pour administration
orale de 3 ml (graduée tous les
0.1 ml)
|
Seringue pour administration
orale de 5 ml (graduée tous les
0.1 ml)
| |
mg
|
ml
|
mg
|
ml
|
mg
|
ml
| |
1.00
|
0.25
|
4.5
|
1.1
|
13.0
|
3.2
| |
2.00
|
0.50
|
5.0
|
1.3
|
14.0
|
3.6
| |
3.00
|
0.75
|
5.5
|
1.4
|
15.0
|
3.8
| |
4.00
|
1.00
|
6.0
|
1.5
|
16.0
|
4.0
| |
|
|
6.5
|
1.6
|
17.0
|
4.2
| |
|
|
7.0
|
1.8
|
18.0
|
4.6
| |
|
|
7.5
|
1.9
|
19.0
|
4.8
| |
|
|
8.0
|
2.0
|
20.0
|
5.0
| |
|
|
8.5
|
2.1
|
|
| |
|
|
9.0
|
2.3
|
|
| |
|
|
9.5
|
2.4
|
|
| |
|
|
10.0
|
2.5
|
|
| |
|
|
10.5
|
2.6
|
|
| |
|
|
11.0
|
2.8
|
|
| |
|
|
11.5
|
2.9
|
|
| |
|
|
12.0
|
3.0
|
|
|
Informations importantes concernant les consignes d’utilisation :
Une remise en suspension est nécessaire avant chaque utilisation, par une agitation vigoureuse. Avant agitation, le médicament peut avoir l’aspect d’un agglomérat solide avec un surnageant légèrement opalescent. La dose doit être prélevée et administrée tout de suite après la remise en suspension. Il est important de bien respecter les consignes indiquées dans la rubrique «Remarques particulières» pour la préparation et l’administration de la dose afin de garantir l’exactitude de la dose administrée. Il est recommandé que le médecin ou le professionnel de santé conseille le patient/la patiente ou le soignant sur l’utilisation des seringues pour administration orale pour garantir l’administration du volume correct et s’assurer que la prescription est donnée en ml. Il est recommandé de prendre la suspension buvable au cours des repas, voir rubrique «Interactions».
Précautions à prendre avant/pendant de manipuler ou d’administrer le médicament
Aucun(e) aiguille, tubulure pour perfusion ou autre dispositif d’administration parentérale ne doit être relié(e) à la seringue pour administration orale.
Nitisinone NOBEL est à administrer par voie orale uniquement.
Contre-indicationsHypersensibilité à la substance active ou à l’un des excipients mentionnés sous la rubrique «Composition».
Les femmes recevant de la nitisinone ne doivent pas allaiter (voir rubriques «Grossesse, Allaitement» et «Données précliniques»).
Mises en garde et précautionsAugmentation de la concentration de tyrosine plasmatique, troubles oculaires, retard de développement et plaques hyperkératosiques
La nitisinone est un inhibiteur de la 4-hydroxyphényl-pyruvate-dioxygénase, une enzyme du métabo-lisme de la tyrosine. Un traitement par nitisinone peut donc entraîner une augmentation de la concentration plasmatique de tyrosine chez les patients atteints d'HT-1. Il faut donc veiller à ce que le patient/ la patiente maintienne sa réduction simultanée de l'apport alimentaire en tyrosine et en phénylalanine pendant le traitement par nitisinone.
Il n'est pas recommandé d'ajuster la concentration plasmatique de tyrosine en réduisant la dose de nitisinone, car cela peut entraîner une détérioration de l'état de santé.
Le taux plasmatique de tyrosine doit être maintenu en dessous de 500 μmol/l.
Une restriction insuffisante de l'absorption de tyrosine et de phénylalanine peut entraîner une augmentation des concentrations plasmatiques de tyrosine; des valeurs supérieures à 500 µmol/l peuvent avoir les effets suivants:
-Des troubles oculaires/affections oculaires tels que des ulcères cornéens, des opacités cornéennes, des kératites, des conjonctivites, des douleurs oculaires et une photophobie ont été rapportés chez des patients traités par la nitisinone (voir la rubrique "Effets indésirables"). Dans une étude clinique portant sur une population non-HT-1 sans restrictions alimentaires et avec des taux plasmatiques de tyrosine supérieurs à 500 µmol/l, des kératopathies symptomatiques et asymptomatiques ont été observées. Pour cette raison, un examen ophtalmologique de base, y compris un examen à la lampe à fente, doit être effectué avant le traitement par Nitisinon NOBEL. Cet examen doit être répété à intervalles réguliers. Les patients/patientes qui développent une photophobie, des douleurs oculaires ou des signes d'inflammation tels que rougeur, gonflement ou brûlure des yeux, ou dont le taux de tyrosine plasmatique dépasse 500 µmol/l pendant le traitement, doivent subir un nouvel examen à la lampe à fente et une mesure immédiate du taux de tyrosine plasmatique.
-Différents degrés de déficience mentale et de retard de développement. On ne sait pas dans quelle mesure les cas observés sont imputables à la maladie elle-même, au traitement par la nitisinone ou à d'autres facteurs. Les patients/patientes traités par Nitisinon NOBEL qui présentent un changement brutal de l'état neurologique doivent subir un examen clinique en laboratoire, y compris les taux de tyrosine dans le plasma.
-Plaques hyperkératosiques douloureuses sur la plante des pieds et la paume des mains.
Chez les patients qui doivent suivre des mesures diététiques et qui sont traités par Nitisinon NOBEL et qui développent en conséquence des valeurs plasmatiques de tyrosine élevées, l'apport alimentaire en tyrosine et en phénylalanine doit être surveillé.
Leucopénie et thrombocytopénie sévère
Dans les essais cliniques, les patients traités par Nitision et soumis à des mesures diététiques ont développé une leucopénie transitoire (3%), une thrombocytopénie (3%) ou les deux (1,5%) (voir la rubrique "Effets indésirables"). Aucun patient n'a développé d'infection ou d'hémorragie suite aux épisodes de leucopénie - ou de thrombocytopénie. Une surveillance des plaquettes et des globules blancs est indiquée pendant le traitement par nitisinone.
Surveillance de la fonction hépatique
Les fonctions hépatiques doivent être suivies régulièrement par les tests fonctionnels et par l’imagerie hépatique appropriée. Il est également recommandé de vérifier la concentration sérique en alpha-fœtoprotéine. Une augmentation de la concentration sérique en alpha-foetoprotéine peut indiquer que le traitement est inadapté. Les patients présentant une augmentation en alpha-foetoprotéine ou des nodules hépatiques doivent toujours faire l'objet d'explorations complémentaires pour écarter la possibilité d'une tumeur hépatique maligne.
Des visites de surveillance doivent être réalisées tous les 6 mois; des intervalles plus rapprochés sont recommandés en cas d’événements indésirables.
Utilisation concomitante avec d’autres médicaments
La nitisinone est un inhibiteur modéré du CYP 2C9. Le traitement par la nitisinone peut donc entraîner une augmentation des concentrations plasmatiques des médicaments coadministrés qui sont métabolisés principalement via le CYP 2C9. En cas de traitement concomitant par la nitisinone et des médicaments à marge thérapeutique étroite métabolisés via le CYP 2C9, tels que la warfarine et la phénytoïne, les patients/patientes doivent faire l’objet d’une étroite surveillance. Un ajustement de la dose de ces médicaments coadministrés pourra être nécessaire (voir rubrique «Interactions»).
Autres composants dont l’effet est connu:
Glycérol
1 ml de suspension Nitisinon NOBEL contient 500 mg de glycérol. Une dose de 20 ml ou plus de suspension buvable (10 g de glycérol) peut provoquer des maux de tête, des maux d'estomac et la diarrhée.
Sodium
Ce médicament (suspension Nitisinon NOBEL) contient moins de 1 mmol de sodium (23 mg) par dose journalière maximale, c'est-à-dire qu'il est essentiellement «sans sodium».
Sodium benzoate
1 ml de suspension Nitisinon NOBEL contient 1 mg de benzoate de sodium. L’augmentation de la bilirubine après sa dissociation de l’albumine, due à l’acide benzoïque et à ses sels, peut exacerber un ictère chez les nouveau-nés prématurés et nés à terme présentant déjà un ictère et entraîner un ictère nucléaire (dépôts de bilirubine non conjuguée dans le tissu cérébral). Il est donc très important de réaliser une surveillance étroite des taux plasmatiques de bilirubine chez le nouveau-né. Les taux de bilirubine doivent être mesurés avant le début du traitement: en cas d’élévation significative des taux plasmatiques de bilirubine, en particulier chez les patients prématurés présentant des facteurs de risque tels qu’une acidose et une albuminémie faible, un traitement à l’aide d’une portion correctement pesée d’une capsule dure de Nitisinone NOBEL doit être envisagé en lieu et place de la suspension buvable jusqu’au retour à la normale des taux plasmatiques de bilirubine non conjuguée.
Colorants azoïques
La capsule dure de 5 mg contient le colorant azoïque Sunset Yellow FCF (E110). Le Sunset Yellow FCF (E110) peut provoquer des réactions allergiques.
InteractionsInteractions pharmacocinétiques
Influence d’autres substances sur la pharmacocinétique de la nitisinone
La nitisinone est métabolisée in vitro par l’isoenzyme CYP 3A4 et il peut donc être nécessaire d’ajuster la dose quand la nitisinone est coadministrée avec des inhibiteurs ou des inducteurs de cette enzyme.
Influence de la nitisinone sur la pharmacocinétique d’autres substances
D’après les données issues d’une étude d’interaction clinique effectuée avec 80 mg de nitisinone à l’état d’équilibre, la nitisinone est un inhibiteur modéré du CYP 2C9 (augmentation de l’ASC du tolbutamide d’un facteur 2.3). Le traitement par la nitisinone peut donc entraîner une augmentation des concentrations plasmatiques des médicaments coadministrés qui sont métabolisés principalement via le CYP 2C9 (voir rubrique «Mises en garde et précautions»). La nitisinone est un faible inducteur du CYP 2E1 (diminution de 30 % de l’ASC de la chlorzoxazone) et un faible inhibiteur de l’OAT1 et de l’OAT3 (augmentation de l’ASC du furosémide d’un facteur 1.7), mais la nitisinone n’inhibe pas le CYP 2D6 (voir rubrique «Pharmacocinétique»).
Capsules dures
Aucune étude formelle portant sur des interactions avec l’alimentation n’a été réalisée avec les capsules dures de Nitisione NOBEL. Toutefois, la nitisinone a été coadministrée avec l’alimentation durant les études d’efficacité et de sécurité. De ce fait, si le traitement par la nitisinone avec les capsules dures de Nitisinone NOBEL est instauré avec l’alimentation, il est recommandé de le poursuivre dans les mêmes conditions, se référer à la rubrique «Posologie/Mode d’emploi».
Suspension
L’alimentation n’a pas d’influence sur la biodisponibilité de la nitisinone sous forme de suspension buvable, mais la prise du médicament avec l’alimentation réduit la vitesse d’absorption et donne donc lieu à moins de fluctuations des concentrations sériques pendant l’intervalle de prise. Par conséquent, il est recommandé de prendre la suspension buvable au cours des repas ; voir rubrique «Posologie/Mode d’emploi».
Grossesse, AllaitementGrossesse
Il n'existe pas de données pertinentes concernant l'utilisation de la nitisinone chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique «Données précliniques»). Le risque potentiel chez l’homme n'est pas connu. Nitisinone NOBEL ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la femme ne justifie le traitement avec la nitisinone.
Allaitement
On ne sait pas si la nitisinone est excrétée dans le lait maternel. Les études chez l’animal ont mis en évidence des effets indésirables post-nataux lors de l’exposition à la nitisinone via le lait maternel. En conséquence, les mères recevant de la nitisinone ne doivent pas allaiter puisqu’un risque pour le nourrisson ne peut être exclu (voir rubriques «Contre-indications» et «Données précliniques»).
Fertilité
Il n’existe aucune donnée démontrant que la nitisinone a un effet sur la fertilité.
Effet sur l’aptitude à la conduite et l’utilisation de machinesNitisinone a une influence mineure sur l’aptitude à conduire des véhicules et à utiliser des machines. Les réactions indésirables touchant les yeux (voir la rubrique «Effets indésirables») peuvent altérer la vision. Si la vision est altérée, le patient ne doit pas conduire de véhicules ni utiliser de machines jusqu’à ce que l’effet ait disparu.
Effets indésirablesRésumé du profil de sécurité
En raison de son mécanisme d'action, tous les patients traités par la nitisinone présentent des augmentations de la concentration de tyrosine.
Les effets indésirables qui peuvent survenir fréquemment pendant le traitement par la nitisinone concernent l'œil (tels que conjonctivite, opacité de la cornée, kératite, photophobie et douleur oculaire) et la formule sanguine (thrombocytopénie, leucopénie, granulocytopénie). Une dermatite exfoliative a été occasionnellement observée.
Les patients atteints de HT-1 présentent un risque accru de crises porphyriques, de néoplasmes hépatiques et d'insuffisance hépatique nécessitant une transplantation hépatique. Ces complications de l'HT-1 ont été observées chez des patients traités par nitisinone pendant une durée médiane de 22 mois au cours de l'étude clinique (transplantation hépatique 13%, insuffisance hépatique 7%, néoplasmes hépatiques malins 5%, néoplasmes hépatiques bénins 3%, porphyrie 1%).
Liste des effets indésirables
Les données proviennent d'études cliniques menées sur des patients et de l'utilisation du HT-1 après sa mise sur le marché.
Les effets indésirables sont rangés par classe de système d’organes de la classification MedDRA et par fréquence selon la convention suivante:
«très fréquents» (≥1/10),
«fréquents» (≥1/100 à <1/10),
«occasionnels» (≥1/1000 à <1/100),
«rares» (≥1/10 000 à <1/1000),
«très rares» (<1/10 000).
«Fréquence inconnue» (ne peut être estimée sur la base des données disponibles)
Affections hématologiques et du système lymphatique
Fréquents: Thrombocytopénie, leucopénie, granulocytopénie
Occasionnels: Leucocytose
Affections oculaires
Fréquents: Conjonctivite, opacité cornéenne, kératite, photophobie, douleur oculaire
Occasionnels: Blépharite
Affections de la peau et du tissu sous-cutané
Occasionnels: Dermatite exfoliatrice, rash érythémateux, prurit, rash
Investigations
Très fréquents: Taux de tyrosine élevés
Population pédiatrique
Le profil de sécurité est principalement basé sur la population pédiatrique puisque le traitement par nitisinone doit être instauré dès que le diagnostic d’une tyrosinémie héréditaire de type 1 (HT-1) est établi. Sur la base des études cliniques et des données après commercialisation, aucun élément n'indique que le profil de sécurité est différent selon les sous-groupes de la population pédiatrique ou par rapport au profil de sécurité observé chez les patients adultes.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageUne ingestion accidentelle de nitisinone par un sujet suivant un régime alimentaire normal sans restriction en tyrosine et en phénylalanine conduit à une augmentation des taux en tyrosine. Des taux élevés en tyrosine ont été associés à une toxicité oculaire, cutanée et du système nerveux. Un apport restreint en tyrosine et en phénylalanine dans le régime alimentaire devrait limiter la toxicité associée à ce type de tyrosinémie. Aucune information concernant un traitement spécifique en cas de surdosage n’est disponible.
Propriétés/EffetsCode ATC
A16AX04
Mécanisme d’action
La nitisinone est un inhibiteur compétitif de la 4-hydroxyphénylpyruvate dioxygénase, la deuxième étape du métabolisme de la tyrosine. En inhibant le catabolisme de la tyrosine chez les patients/les patientes atteints d’HT-1, la nitisinone empêche l’accumulation de métabolites nocifs en aval de la 4hydroxyphénylpyruvate dioxygénase.
Dans l’HT-1, l’anomalie biochimique est une carence en fumarylacétoacétate-hydrolase, qui est la dernière enzyme de la voie catabolique de la tyrosine. La nitisinone empêche l’accumulation des produits intermédiaires toxiques, le maleylacétoacétate et le fumarylacétoacétate. Ces produits intermédiaires sont par ailleurs réduits en deux métabolites toxiques, la succinylacétone et le succinylacétoacétate. La succinylacétone inhibe la voie de synthèse des porphyrines, ce qui conduit à une accumulation de 5-aminolévulinate.
Pharmacodynamique
Chez les patients/patientes atteints d’HT-1, le traitement par la nitisinone normalise le métabolisme des porphyrines avec une activité normale de la porphobilinogène (PBG)-synthase érythrocytaire et un taux urinaire normal de 5-aminolévulinate, une excrétion urinaire de succinylacétone réduite, un taux plasmatique de tyrosine accru et une excrétion urinaire d’acides phénoliques accrue. Les données disponibles lors d’une étude clinique indiquent que la concentration urinaire de succinylacétone est redevenue normale chez plus de 90 % des patients au cours de la première semaine de traitement. La succinylacétone n’est détectable ni dans les urines ni dans le plasma lorsque la dose de nitisinone est correctement ajustée.
Efficacité clinique
Une étude non aveugle et non contrôlée a été menée avec n=250 patients/patientes. Dans l'étude, la fréquence d'utilisation était de deux fois par jour.
L'étude a montré une probabilité de survie de 93% à 2, 4 et 6 ans si le traitement a commencé à l'âge de 6 mois ou moins. Si le patient était âgé de plus de 6 mois, la probabilité de survie était de 96% (à 2 ans) et de 95% à 4 et 6 ans ; la probabilité moyenne de survie était de 94%.
Un contrôle historique, dans lequel les probabilités de survie à 1 an et à 2 ans ont été mesurées en fonction du moment de l'apparition des premiers symptômes (< 2 mois, 2 - 6 mois et > 6 mois), a montré que plus les premiers symptômes ont été diagnostiqués tard, plus la probabilité de survie était élevée (> 6 mois : 96%) (van Spronsen et al., 1994). Si les symptômes apparaissaient au cours des deux premiers mois de vie, la probabilité de survie était de 38% à 1 an et de 29% à 2 ans. Par contre, la probabilité de survie était de 74% (à 1 et 2 ans) si les symptômes apparaissaient entre le deuxième et le sixième mois de vie.
Il a été mis en évidence que le traitement par la nitisinone (n = 250) réduit le risque de survenue d’un hépatome lorsqu’il est comparé aux données historiques avec régime alimentaire seul. Il a également été observé que l’instauration précoce du traitement réduisait encore plus ce risque de survenue d’un hépatome.
Si le traitement par nitisinone a débuté à l'âge de 24 mois ou moins, la probabilité de non-apparition d'un CHC (carcinome hépatocellulaire) était de 99% à 2, 4 et 6 ans. Si le patient avait plus de 24 mois, cette probabilité était de 92% après 2 ans, 82% après 4 ans et 75% après 6 ans.
Lors d’une enquête internationale sur les patients avec HT-1 dont le traitement consistait en un régime alimentaire seul, il a été observé qu’un hépatome avait été diagnostiqué chez 18 % de l’ensemble des patients âgés de 2 ans et plus.
Une étude visant à évaluer la pharmacocinétique, l’efficacité et la sécurité d’une administration quotidienne unique comparée à une administration biquotidienne a été réalisée chez 19 patients avec HT-1. Aucune différence cliniquement significative n’a été notée au niveau des effets indésirables ou des autres évaluations de la sécurité entre l’administration biquotidienne et l’administration quotidienne unique. Aucun patient n’a présenté de taux détectables de succinylacétone (SA) à la fin de la période de traitement avec administration quotidienne unique. L’étude indique qu’une administration quotidienne unique est sûre et efficace dans tous les groupes d’âge de patients. Les données concernant les patients ayant un poids corporel < 20 kg sont toutefois limitées.
PharmacocinétiqueAbsorption
Aucune étude d'absorption n'a été réalisée à ce jour.
Distribution
Aucune étude de distribution n'a été réalisée à ce jour.
Métabolisme
Aucune étude de métabolisme n'a été réalisée à ce jour.
Élimination
Aucune étude d'éliminiation n'a été réalisée à ce jour.
Après administration d’une dose unique de capsules de nitisinone (1 mg/kg de poids corporel) chez 10 hommes volontaires sains, la demi-vie terminale (médiane) de la nitisinone dans le plasma était de 54 heures (allant de 39 à 86 heures).
Après administration d’une dose unique de comprimés de nitisinone (10 mg) à 23 volontaires sains, la demi-vie finale (médiane) de la nitisinone dans le plasma était de 59 heures (allant de 41 à 74 heures).
Une analyse pharmacocinétique sur la population a été effectuée une population regroupant 207 patients avec HT-1. Il a été montré que la clairance était de 0.0956 l/kg de poids corporel /jour et que la demi-vie était de 52.1 heures.
Des études in vitro utilisant des microsomes hépatiques humains et des enzymes P450 ADNcexprimées ont montré que le métabolisme ayant pour médiateur l’enzyme CYP 3A4 était limité. D’après les données issues d’une étude d’interaction clinique effectuée avec 80 mg de nitisinone à l’état d’équilibre, la nitisinone a entraîné une augmentation d’un facteur 2.3 de l’ASC∞ du tolbutamide, un substrat du CYP 2C9, ce qui indique une inhibition modérée du CYP 2C9. La nitisinone a entraîné une diminution d’environ 30 % de l’ASC∞ de la chlorzoxazone, ce qui indique une faible induction du CYP 2E1. La nitisinone n’inhibe pas le CYP 2D6 puisque l’ASC∞ du métoprolol n’a pas été affectée par l’administration de la nitisinone. L’ASC∞ du furosémide a augmenté d’un facteur 1.7, ce qui indique une faible inhibition des OAT1/OAT3 (voir rubriques «Mises en garde et précautions» et «Interactions»).
D’après les études in vitro, il n’est pas attendu que la nitisinone inhibe le métabolisme ayant pour médiateur les isoenzymes CYP 1A2, 2C19 ou 3A4, ni qu’elle induise les CYP 1A2, 2B6 ou 3A4/5. La nitisinone ne devrait pas inhiber le transport ayant pour médiateur la P-gp, la BCRP ou l’OCT2. Aux concentrations plasmatiques atteintes dans la pratique clinique, la nitisinone ne devrait pas inhiber le transport ayant pour médiateur l’OATP1B1 et l’OATP1B3.
Données précliniquesPharmacologie de sécurité
La nitisinone s’est avérée avoir un effet toxique sur l’embryon et le fœtus de souris et de lapin à des doses cliniquement pertinentes. Chez le lapin, la nitisinone a induit un effet dose-dépendant sur la survenue des malformations (hernie ombilicale et gastroschisis), ceci à partir d’une dose 2.5 fois plus forte que la dose maximale recommandée chez l’homme (2 mg/kg/jour).
Génotoxicité
Aucun effet mutagène n’a été observé ; par contre, une faible activité clastogène a été observée dans les études in vitro. Il n’y a eu aucun signe de génotoxicité in vivo (test du micronoyau chez la souris et test de synthèse de l’ADN non programmée du foie chez la souris).
Carcinogénicité
La nitisinone ne s’est pas révélée carcinogène au cours d’une étude de carcinogénicité de 26 semaines chez les souris transgéniques (TgrasH2).
Toxicité sur la reproduction
Une étude sur le développement pré- et postnatal chez la souris a montré une réduction statistiquement significative des taux de survie et de croissance de la progéniture pendant la période de sevrage à des doses de 125 et 25 fois la dose thérapeutique maximale recommandée pour l'homme, avec une tendance à un effet négatif sur la survie de la progéniture à partir d'une dose de 5 mg/kg/jour. Chez le rat, l'exposition par le lait a entraîné une diminution du poids moyen de la progéniture et des lésions de la cornée.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Incompatibilités
Non pertinent.
Remarques particulières concernant le stockage
Casules dures
Conserver au réfrigérateur (2-8°C), dans l’emballage d’origine et hors de la portée des enfants.
Le médicament peut être conservé une fois pendant 3 mois à température ambiante (15 - 25 °C) ; ensuite, le médicament doit être éliminé.
Suspension
Conserver au réfrigérateur (2 - 8 °C), bien fermé, dans l'emballage d'origine et hors de portée des enfants.
Ne pas congeler. Conserver en position verticale.
Après ouverture, le médicament peut être conservé une fois pendant 2 mois à température ambiante (15-25 °C), après quoi il doit être éliminé.
Remarques concernant la manipulation
Suspension
Une remise en suspension est nécessaire avant chaque utilisation, par une agitation vigoureuse. Avant agitation, le médicament peut avoir l’aspect d’un agglomérat solide avec un surnageant légèrement opalescent. La dose doit être prélevée et administrée tout de suite après la remise en suspension. Il est important de bien respecter les consignes indiquées ci-dessous pour la préparation et l’administration de la dose afin de garantir l’exactitude de la dose administrée. Trois seringues pour administration orale (1 ml, 3 ml et 5 ml) sont fournies pour mesurer la dose prescrite avec précision. Il est recommandé que le medecin ou le professionnel de santé conseille le patient/la patiente ou le soignant sur l’utilisation des seringues pour administration orale afin de garantir l’administration du volume correct.
Comment préparer un flacon neuf de médicament pour la première utilisation:
Avant de prendre la première dose, le flacon doit être secoué vigoureusement car lors d’une conservation longue les particules forment un agglomérat solide au fond du flacon.
Figure A.
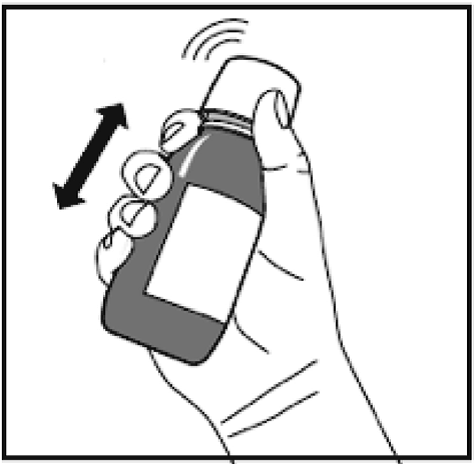
Figure B.
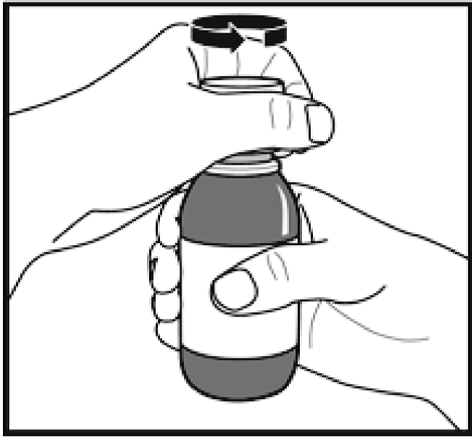
Figure C

1.Le flacon doit être sorti du réfrigérateur et la date de sortie du réfrigérateur doit être notée sur l’étiquette du flacon.
2.Le flacon doit être secoué vigoureusement pendant au moins 20 secondes jusqu’à dispersion complète de l’agglomérat solide au fond du flacon (Figure A).
3.Le bouchon à vis de sécurité enfant doit être retiré en le poussant fermement vers le bas et en tournant dans le sens inverse des aiguilles d’une montre (Figure B).
4.Le flacon ouvert doit être placé en position verticale sur une table et l’adaptateur en plastique doit être fermement enfoncé sur le goulot du flacon aussi loin que possible (Figure C). Le flacon doit être bien refermé avec le bouchon à vis de sécurité enfant.
Pour les administrations suivantes, voir les instructions ci-dessous, dans «Comment préparer une dose de médicament».
Comment préparer une dose de médicament
Figure D.
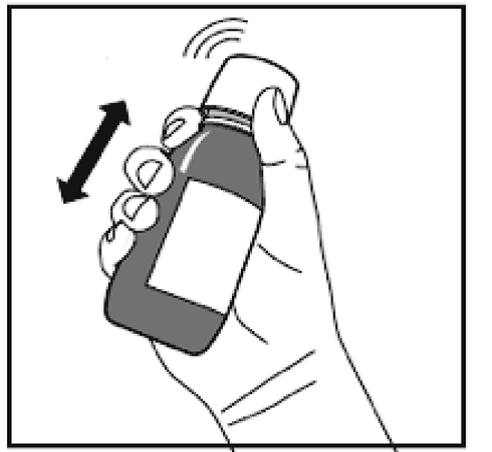
Figure E.
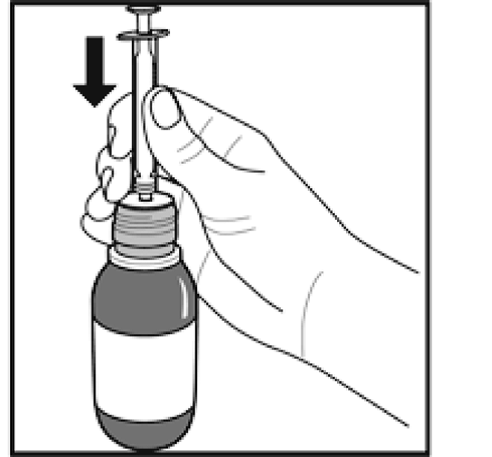
Figure F.
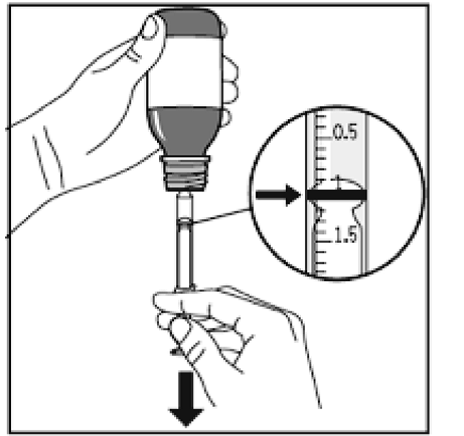
1.Le flacon doit être secoué vigoureusement pendant au moins 5 secondes (Figure D).
2.Immédiatement après, le flacon doit être ouvert en retirant le bouchon à vis de sécurité enfant.
3.Le piston doit être poussé à fond dans la seringue pour administration orale.
4.Le flacon doit être conservé en position verticale et la seringue pour administration orale doit être insérée dans le trou de l’adaptateur, en haut du flacon (Figure E).
5.Le flacon doit être retourné la tête en bas avec précaution en laissant la seringue pour administration orale en place (Figure F).
6.Afin de prélever la dose prescrite (ml), le piston doit être tiré doucement vers le bas jusqu’à ce que le bord supérieur de l’anneau noir soit exactement au niveau de la ligne indiquant la dose (Figure F). Si des bulles d’air sont présentes dans la seringue pour administration orale remplie, le piston doit être repoussé vers le haut jusqu’à ce que les bulles soient chassées. Le piston doit ensuite être tiré à nouveau vers le bas jusqu’à ce que le bord supérieur de l’anneau noir soit exactement au niveau de la ligne indiquant la dose.
7.Le flacon doit être remis à l’endroit et la seringue pour administration orale doit être retirée en la faisant tourner légèrement pour la sortir du flacon.
8.La dose doit être administrée immédiatement dans la bouche (sans dilution) pour éviter une agglutination dans la seringue pour administration orale. La seringue pour administration orale doit être vidée lentement pour permettre d’avaler ; un jet rapide du médicament peut provoquer une fausse route.
9.Le bouchon à vis de sécurité enfant doit être replacé et bien fermé tout de suite après utilisation. L’adaptateur du flacon ne doit pas être retiré.
10.Le flacon peut être conservé à une température ambiante (15 - 25 °C) ou au réfrigérateur (2 - 8 °C).
Nettoyage
La seringue pour administration orale doit être nettoyée immédiatement à l’eau. Le corps et le piston doivent être séparés et rincés tous les deux à l’eau. Les éléments doivent être secoués pour éliminer l’excès d’eau et la seringue doit être mise à sécher telle quelle avant d’être remontée pour une prochaine utilisation.
Numéro d’autorisation68331, 68332 (Swissmedic)
PrésentationCapsule dures
Nitisinone NOBEL 5mg: 60 capsules dures par flacon* [B]
Nitisinone NOBEL 10mg: 60 capsules dures par flacon* [B]
Nitisinone NOBEL 20mg: 60 capsules dures par flacon* [B]
* Flacon en HDPE avec bouchon inviolable en LDPE.
Chaque emballage contient 1 flacon.
Suspension
Nitisinone NOBEL suspension 4mg/ml: emballages de 90ml par flacon [B]
Flacon en verre brun de 100 ml (type III) avec bouchon à vis HDPE de sécurité pour les enfants et scellé (protection contre la première ouverture). Chaque flacon contient 90 ml de suspension buvable. Chaque emballage contient un flacon, un adaptateur pour flacon en LDPE et 3 seringues pour préparations buvables en polypropylène (PP) (1 ml, 3 ml et 5 ml).
Titulaire de l’autorisationNOBEL Pharma Schweiz AG, Risch
Mise à jour de l’informationMédicament de comparaison étranger: Octobre 2020
Avec ajout d’informations pertinentes pour la sécurité par Swissmedic: Juin 2022
|