CompositionPrincipes actifs
Fingolimod sous forme de chlorhydrate de fingolimod.
Excipients
Gélules de 0.25 mg:
Carmellose calcique, fumarate de stéaryle sodique (équivalent à 0.030 mg de sodium)
Enveloppe des gélules: gélatine, dioxyde de titane (E171), oxyde de fer jaune (E172)
Encre d'impression: gommes laques, oxyde de fer noir (E172), alcool butylique, eau purifiée, propylèneglycol, éthanol anhydre, alcool isopropylique, solution concentrée d’ammoniaque.
Gélules de 0.5 mg:
Carmellose calcique, fumarate de stéaryle sodique (équivalent à 0.030 mg de sodium)
Enveloppe des gélules: gélatine, dioxyde de titane (E171), tartrazine (E102) (0.028 mg), jaune orangé S (E110) (0.003 mg)
Encre d'impression: gommes laques, oxyde de fer noir (E172), alcool butylique, eau purifiée, propylèneglycol, éthanol anhydre, alcool isopropylique, solution concentrée d’ammoniaque.
Indications/Possibilités d’emploiFingolimod Devatis est indiqué dans le traitement des adultes, adolescents et enfants à partir de 10 ans atteints de sclérose en plaques (SEP) récurrente-rémittente (évoluant par poussées et rémissions) pour réduire la fréquence des poussées et ralentir la progression du handicap.
Posologie/Mode d’emploiGroupes de patients généraux
Posologie
La dose recommandée de Fingolimod Devatis chez les adultes est d'une gélule de 0.5 mg une fois par jour par voie orale. La dose peut être prise avec ou sans nourriture.
Chez les enfants et adolescents (à partir de 10 ans), la dose recommandée dépend du poids corporel:
·enfants et adolescents avec un poids corporel allant jusqu'à 40 kg: une gélule de 0.25 mg une fois par jour par voie orale;
·enfants et adolescents avec un poids corporel de plus de 40 kg: une gélule de 0.5 mg une fois par jour par voie orale.
Les enfants et adolescents qui ont reçu des gélules de 0.25 mg au début du traitement doivent passer à des gélules de 0.5 mg lorsqu'ils atteignent un poids corporel stable de plus de 40 kg.
En cas d'omission d'une dose, le traitement doit être poursuivi avec la dose suivante selon le plan prédéfini.
Pour des recommandations sur le passage à Fingolimod Devatis après des traitements antérieurs par d'autres agents modifiant la maladie et d'autres immunosuppresseurs, voir «Mises en garde et précautions: Traitement antérieur par des immunosuppresseurs ou des immunomodulateurs». La durée d'action de ces médicaments doit être prise en considération pour éviter des effets immunosuppresseurs cumulatifs (voir «Mises en garde et précautions: Traitement antérieur par des immunosuppresseurs ou des immunomodulateurs»).
Avant le début du traitement
Évaluation ophtalmologique
Un examen ophtalmologique du fond d'œil, y compris de la macula, doit être exécuté (voir «Mises en garde et précautions»).
Évaluation dermatologique
Un examen dermatologique doit être exécuté. Les lésions cutanées suspectes doivent être clarifiées dans les plus brefs délais (voir «Mises en garde et précautions»).
Instauration du traitement
Un ECG doit être effectué chez tous les patients, avant le début du traitement et à la fin de la période de surveillance de six heures. Le pouls et la pression artérielle doivent être mesurés toutes les heures chez tous les patients pendant au moins les 6 premières heures qui suivent la prise de la première dose, afin de déceler une bradycardie ou des troubles de la conduction auriculoventriculaire. Il faut s'assurer de disposer des moyens appropriés pour le traitement des urgences cardiologiques. Un enregistrement de l'ECG en continu et en temps réel est conseillé au cours des six premières heures qui suivent la première prise de Fingolimod Devatis.
Lors du passage de la dose journalière de 0.25 mg à 0.5 mg, il est recommandé de surveiller l'administration de la première dose augmentée comme après la prise de la première dose au début du traitement.
Après toute interruption de traitement, une surveillance de la dose, comme après l'instauration du traitement, est recommandée(voir «Mises en garde et précautions»).
Une surveillance cardiaque se prolongeant au-delà des six premières heures qui suivent le début du traitement est requise chez certains patients (voir «Surveillance après la première prise de Fingolimod Devatis – Résumé sous forme de tableau» dans la présente rubrique et «Mises en garde et précautions»). En outre, il incombe au médecin traitant de décider de la nécessité d'une surveillance des paramètres vitaux/de l'ECG lors des administrations suivantes (voir «Mises en garde et précautions»).
Le tableau ci-dessous résume les mesures de surveillance cardiaque après la prise de la première dose de Fingolimod Devatis (voir aussi «Mises en garde et précautions»).
Tableau 1 Surveillance après la première prise de Fingolimod Devatis – Résumé sous forme de tableau
|
Chez tous les patients
| |
Une surveillance de 6 heures quant à l‘apparition de symptômes de bradycardie ainsi que de troubles de la conduction auriculoventriculaire devrait comprendre les mesures suivantes:
·Un contrôle toutes les heures de la pression artérielle et de la fréquence cardiaque
·Un ECG avant l‘instauration du traitement et après la période de surveillance de 6 heures
·La possibilité de mettre en œuvre un traitement cardiologique d‘urgence
·Il est recommandé de procéder à un monitorage en continu de l‘ECG (en temps réel).
| |
Chez les patients avec apparition d‘anomalies au cours des 6 premières heures après la dose initiale
| |
En cas d‘apparition de bradyarythmies symptomatiques,
|
la surveillance du patient sera poursuivie après la phase de surveillance de 6 heures jusqu‘à la disparition complète des symptômes.
| |
Lorsque la fréquence cardiaque atteint sa valeur la plus basse 6 heures après la dose initiale,
|
il convient de poursuivre le monitorage cardiaque jusqu‘au rétablissement de la fréquence cardiaque, ceci cependant durant 2 heures au moins.
| |
En présence de l‘un des résultats suivants 6 heures après la dose initiale:
·fréquence cardiaque inférieure à 45 battements/minute
·bloc AV de 2e degré nouvellement apparu et persistant ou tout bloc AV de degré supérieur
·intervalle QTc ≥500 ms
En présence à tout moment du résultat suivant à l‘ECG pendant la phase de surveillance après la dose initiale:
·bloc AV de 3e degré nouvellement apparu
|
il est indiqué de prolonger le monitorage cardiaque au minimum durant la nuit.
| |
En cas de symptômes de bradyarythmie nécessitant un traitement médicamenteux lors de la première prise, il convient de surveiller le patient durant la nuit au sein d‘une unité médicale. La stratégie de surveillance initiale sera appliquée lors de la deuxième prise.
| |
Chez les patients avec affections cardiaques préexistantes
| |
Pour certains groupes de patients, un traitement par Fingolimod Devatis ne doit être envisagé que si les bénéfices attendus dépassent les risques potentiels.
| |
Chez les patients prédisposés avec:
·cardiopathie ischémique connue (y compris angine de poitrine)
·insuffisance cardiaque congestive
·maladie cérébrovasculaire
·hypertension artérielle non contrôlée
·syndrome d'apnées du sommeil sévères non traitées
De même, chez les patients avec les antécédents suivants:
·infarctus du myocarde
·arrêt cardiaque
·syncopes récurrentes
·bradycardie symptomatique
|
il convient, avant d'instaurer le traitement:
·de consulter un cardiologue
·et de déterminer le monitorage cardiaque approprié (au moins durant la nuit)
| |
Chez les patients recevant un traitement à effet bradycardisant
| |
Chez les patients sous
·bêtabloquants
·bloqueurs des canaux calciques (ralentissant la fréquence cardiaque, tels que vérapamil ou diltiazem)
·d'autres substances susceptibles de ralentir la fréquence cardiaque (p.ex. ivabradine, digoxine, inhibiteurs de l'acétylcholinestérase, pilocarpine)
|
il convient, avant d'instaurer le traitement:
·de consulter un cardiologue
afin d'évaluer la possibilité du passage à un médicament sans effet bradycardisant respectivement sans effet ralentisseur sur la conduction AV.
ou
·si le changement de traitement est impossible, il convient de procéder à un monitorage cardiaque approprié (y compris un monitorage continu de l'ECG) au moins durant la nuit.
| |
Chez les patients avec allongement de l'intervalle QT
| |
Chez les patients avec:
·allongement significatif de l'intervalle QTc avant le début du traitement (QTc > 470 ms chez les femmes ou > 450 ms chez les hommes)
·facteurs de risque supplémentaires d'allongement de l'intervalle QT (comme une hypokaliémie, une hypomagnésémie ou un syndrome du QT long congénital)
|
il convient, avant d'instaurer le traitement:
·de consulter un cardiologue
et
·de déterminer le monitorage cardiaque approprié (y compris un monitorage continu de l'ECG au moins durant la nuit au sein d'une unité médicale).
|
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Il n'est pas nécessaire d'adapter la posologie de Fingolimod Devatis chez les patients atteints d'insuffisance hépatique légère (classe A du score de Child-Pugh), mais le traitement sera appliqué avec prudence (voir «Mises en garde et précautions, fonction hépatique» et «Pharmacocinétique»). Fingolimod Devatis ne doit pas être administré en cas d'insuffisance hépatique modérée (classe B du score de Child-Pugh) ou sévère (classe C du score de Child-Pugh) (voir «Contreindications»).
Patients présentant des troubles de la fonction rénale
Il n'existe aucune donnée clinique concernant l'efficacité et la sécurité chez les patients atteints d'insuffisance rénale.
Patients âgés
Les données cliniques concernant les patients atteints de sclérose en plaques âgés de plus de 55 ans sont très limitées.
Enfants et adolescents
La sécurité et l'efficacité de fingolimod pour les enfants âgés de moins de 10 ans n'ont pas encore été étudiées. Fingolimod Devatis ne doit pas être utilisé chez les enfants de moins de 10 ans. Dans la classe d'âge ≥10 et ≤12 ans, seules des données limitées sont disponibles (voir «Efficacité clinique»).
Groupe ethnique
Il n'est pas nécessaire d'adapter la posologie de Fingolimod Devatis en fonction de l'appartenance ethnique (voir «Pharmacocinétique»).
Sexe
Il n'est pas nécessaire d'adapter la posologie de Fingolimod Devatis en fonction du sexe (voir «Pharmacocinétique»).
Contre-indications·Patients victimes d'un infarctus du myocarde, d'une angine de poitrine instable, d'un accident vasculaire cérébral/AIT, d'une insuffisance cardiaque décompensée (nécessitant une hospitalisation) ou d'une insuffisance cardiaque de classe NYHA III/IV au cours des six derniers mois.
·Patients souffrant de troubles du rythme cardiaque sévères nécessitant un traitement par des antiarythmiques de classe Ia et III (voir «Mises en garde et précautions», «Interactions»).
·Patients avec un bloc AV du 2e degré de type Mobitz II ou un bloc AV de 3e degré ou une maladie du sinus (sick-sinus-syndrome), pour autant qu'ils ne soient pas équipés d'un pacemaker.
·Patients avec un intervalle QTc dès 500 ms au début du traitement (voir «Mises en garde et précautions»).
·Patients souffrant d'un syndrome d'immunodéficience.
·Patients présentant un risque élevé d'infections opportunistes, y compris ceux qui suivent actuellement un traitement immunosuppresseur ou dont la protection immunitaire est affaiblie.
·Patients souffrant d'infections graves actives ou d'infections chroniques actives d'origine bactérienne, mycosique ou virale (p.ex. hépatite, tuberculose).
·Patients atteints de maladies malignes actives, à l'exception de ceux présentant un carcinome basocellulaire.
·Patients souffrant d'insuffisance hépatique/de cirrhose du foie modérée et sévère (correspondant à la classe B et C du score de Child-Pugh).
·Patients présentant un œdème maculaire.
·Fingolimod Devatis est contre-indiqué chez les patientes en âge de procréer sans contraception suffisante ainsi que pendant la grossesse et l'allaitement.
·Hypersensibilité connue au fingolimod ou à l'un des excipients.
Mises en garde et précautionsBradyarythmie
L'instauration d'un traitement par Fingolimod Devatis induit un ralentissement passager de la fréquence cardiaque et peut de plus être associée à un ralentissement de la conduction AV (voir «Effets indésirables» et «Pharmacodynamique»). La fréquence cardiaque commence à diminuer dans l'heure qui suit la première dose; la valeur minimale étant atteinte dans les 6 premières heures voire chez certains patients dans les 24 heures. C'est la raison pour laquelle tous les patients doivent être maintenus sous surveillance pendant au moins les 6 premières heures suivant la prise de la première dose de Fingolimod Devatis, afin de détecter tout symptôme de bradycardie. Par la suite, la fréquence cardiaque retrouve son niveau initial en l'espace d'un mois de traitement continu (voir «Pharmacodynamique», paragraphe «Fréquence cardiaque et rythme cardiaque»). Chez les patients sous 0.5 mg de fingolimod, la fréquence cardiaque ralentit d'environ 8 battements par minute (/min). Des fréquences cardiaques inférieures à 40/min (chez les adultes) et 50/min (chez les enfants et les adolescents) n'ont été que rarement constatées (voir «Effets indésirables»). Les patients présentant une bradycardie étaient pour la plupart asymptomatiques. Quelques patients ont cependant développé des symptômes légers tels qu'une hypotension, des vertiges, une fatigue, des palpitations et des douleurs dans la poitrine, symptômes disparaissant habituellement durant les 24 premières heures de traitement. En cas de besoin, la bradycardie pourra être traitée par de l'atropine ou de l'isoprénaline parentérales.
L'instauration d'un traitement par fingolimod a été associée à un ralentissement de la conduction auriculoventriculaire (AV), en général sous forme de bloc AV du premier degré (allongement de l'intervalle PR à l'ECG). Moins de 0.2% des patients adultes traités par 0.5 mg de fingolimod ont développé un bloc auriculoventriculaire du second degré, le plus souvent de type Mobitz I (Wenckebach). Les troubles de la conduction étaient en règle générale transitoires, asymptomatiques, ne nécessitaient pas de traitement, et disparaissaient au cours des 24 premières heures du traitement. Depuis l'introduction sur le marché de fingolimod, des cas isolés de bloc AV complet ayant régressé spontanément, ont été rapportés (voir «Effets indésirables» et «Pharmacodynamique»).
Mesures de surveillance cardiaque après la première prise (voir aussi Résumé sous forme de tableau dans la rubrique «Posologie/Mode d'emploi»)
Il convient de procéder chez tous les patients à un ECG avant la première dose, ainsi qu'à la fin de la période d'observation de 6 heures. Au début d'un traitement par Fingolimod Devatis, des mesures de la fréquence cardiaque et de la tension artérielle doivent être effectuées chez tous les patients, et ce toutes les heures, afin de détecter tout symptôme de bradycardie. Il est par ailleurs recommandé de procéder à un enregistrement en continu de l'ECG (en temps réel) durant les six premières heures.
En cas d'apparition de bradyarythmies symptomatiques après la première dose, des mesures adaptées sont indiquées et le patient ou la patiente devra être surveillé(e) au-delà de la phase d'observation de six heures, jusqu'à la disparition complète des symptômes.
Si un patient ou une patiente requiert un traitement médicamenteux pendant la période d'observation après la première dose, il ou elle devra rester en observation au sein d'une unité médicalisée au cours de la nuit suivante et on appliquera la même stratégie de surveillance après la prise de la seconde dose de Fingolimod Devatis qu'après la première.
Si la fréquence cardiaque atteint son niveau le plus bas à la fin de la surveillance de six heures suivant la première dose (ce qui laisse entendre que l'effet pharmacodynamique maximal au niveau cardiaque n'a pas encore été obtenu), il convient de prolonger la surveillance jusqu'à la récupération de la fréquence cardiaque, mais au moins durant deux heures.
Lors du passage de la dose journalière de 0.25 mg à 0.5 mg chez les enfants et les adolescents, il convient de prendre les mêmes précautions qu'après la première prise.
Une poursuite du monitorage cardiaque, au moins durant la nuit qui suit, est d'autre part indiquée lorsque l'un des critères suivants est vérifié:
·nouveau bloc AV de grade 3 à tout moment de la phase de surveillance après la mise en route du traitement
·présence 6 heures après le début du traitement:
·d'une fréquence cardiaque < 45 battements par minute chez les adultes, < 55 battements par minute chez les adolescents et les enfants à partir de 12 ans ou < 60 battements par minute chez les enfants à partir de 10 ou 11 ans, de la fréquence la plus basse depuis le début du monitorage cardiaque, de sorte que l'effet pharmacodynamique maximal n'a pas encore été obtenu,
·d'un nouveau bloc AV persistant de grade 2 ou d'un bloc de grade supérieur,
·d'un intervalle QTc ≥500 ms.
Un traitement par Fingolimod Devatis ne sera envisagé dans certains groupes de patients que si le bénéfice attendu est supérieur aux risques potentiels. Les patients présentant une maladie ischémique connue (y compris angine de poitrine), des antécédents d'infarctus du myocarde, une insuffisance cardiaque congestive et une maladie cérébrovasculaire sont susceptibles de mal tolérer une éventuelle bradycardie. Si un traitement par Fingolimod Devatis est envisagé, il convient de demander l'avis d'un cardiologue avant son instauration, afin de déterminer le monitorage cardiaque approprié (durant au moins une nuit) (voir «Interactions»).
Compte tenu du risque de troubles graves du rythme cardiaque, Fingolimod Devatis ne doit pas être utilisé chez les patients présentant un bloc sino-auriculaire, en particulier avec des antécédents de bradycardie symptomatique ou de syncopes récurrentes.
Dans la mesure où une bradycardie importante peut être mal tolérée par les patients ayant un antécédent d'arrêt cardiaque, d'hypertension non maîtrisée ou d'apnée du sommeil grave non traitée, Fingolimod Devatis ne doit pas être utilisé chez ces patients.
Les expériences avec fingolimod sont limitées chez les patients traités par des bêtabloquants, des inhibiteurs des canaux calciques ralentissant la fréquence cardiaque (p.ex. vérapamil ou diltiazem) ou d'autres substances bradycardisantes (p.ex. ivabradine, digoxine, inhibiteurs de l'acétylcholinestérase, pilocarpine). L'introduction d'un traitement par Fingolimod Devatis étant également associée à un ralentissement de la fréquence cardiaque (voir «Bradyarythmie»), l'administration concomitante de ces substances pendant l'instauration du traitement par Fingolimod Devatis peut donner lieu à une bradycardie sévère et à un bloc cardiaque. En raison de l'effet additif potentiel sur la fréquence cardiaque, on renoncera d'une manière générale à un traitement par Fingolimod Devatis chez les patients recevant simultanément de telles substances. Si un traitement par Fingolimod Devatis est envisagé, il convient de consulter au préalable un cardiologue afin d'évaluer la possibilité du passage à une substance sans effet bradycardisant respectivement sans effet ralentisseur sur la conduction AV, et pour déterminer les mesures de surveillance appropriées lors de l'instauration du traitement. Les patients chez qui un tel changement de traitement n'est pas envisageable doivent faire l'objet d'un monitorage en continu de l'ECG au moins pendant la nuit suivante (voir «Interactions»).
L'effet sur la fréquence cardiaque et la conduction auriculoventriculaire peut se répéter lors de la reprise du traitement par Fingolimod Devatis, en fonction de la durée de l'interruption du traitement et de la durée du traitement par Fingolimod Devatis préalablement effectué.
Des mesures de précaution identiques à celles requises lors de la première prise sont recommandées après une interruption de traitement de:
·un ou plusieurs jours au cours des deux premières semaines du traitement,
·plus de sept jours au cours des troisième et quatrième semaines du traitement,
·plus de deux semaines après le premier mois du traitement.
·Si la durée d'interruption du traitement est inférieure à celles indiquées ci-dessus, le traitement doit être poursuivi comme prévu avec la dose suivante.
Allongement du QT
Des allongements de l'intervalle QT ont été observés chez certains patients sous fingolimod (dans certains cas, cet allongement du QTcF était de 30 à 60 ms, pas d'allongements du QTcF supérieurs à 60 ms, ni de valeurs individuelles de plus de 500 ms). Les essais cliniques n'ont pas porté sur des patients à risque d'allongement du QTc. La signification clinique de ces résultats est incertaine.
Dans la mesure où l'introduction d'un traitement par Fingolimod Devatis est associée à un ralentissement de la fréquence cardiaque et un allongement de l'intervalle QT, Fingolimod Devatis est contre-indiqué chez les patients avec un intervalle QTc supérieur ou égal à 500 ms avant l'instauration du traitement (voir «Contreindications»).
Dans les groupes de patients suivants, l'utilisation de Fingolimod Devatis doit être évitée dans la mesure du possible. Si toutefois un traitement par Fingolimod Devatis est envisagé, il convient de consulter en premier lieu un cardiologue pour déterminer le monitorage cardiaque approprié (y compris une surveillance par ECG en continu au sein d'une unité médicalisée au moins au cours de la nuit suivante):
·patients avec un allongement significatif du QTc (QTc > 470 ms chez les femmes adultes, QTc > 460 ms chez les filles, QTc > 450 ms chez les garçons et les hommes adultes) avant le début du traitement,
·patients avec des facteurs de risque supplémentaires d'allongement de l'intervalle QT (p.ex. hypokaliémie, hypomagnésémie ou syndrome du QT long congénital) (voir «Pharmacodynamique» et «Interactions»).
Chez les patients avec un intervalle QTc ≥500 ms à la fin de la période de surveillance de six heures post-dose initiale, il est indiqué de prolonger la phase de monitorage cardiaque au moins durant la nuit suivante (voir «Posologie/Mode d'emploi»).
Fingolimod n'a pas été testé chez des patients souffrant d'arythmies nécessitant un traitement par des antiarythmiques de classe Ia (p.ex. quinidine, procaïnamide) ou de classe III (p.ex. amiodarone, sotalol). Les antiarythmiques de classe Ia et de classe III ont été associés à des cas de torsades de pointes entre autres chez les patients bradycardes. Comme le début d'un traitement par Fingolimod Devatis va de pair avec un ralentissement de la fréquence cardiaque, Fingolimod Devatis ne doit pas être administré simultanément avec ce type de médicaments (voir «Contreindications»).
Infections
Un effet pharmacodynamique essentiel de Fingolimod Devatis consiste en une diminution dose-dépendante du nombre de lymphocytes périphériques, à un niveau de 20–30% de la valeur à initiale, en conséquence d'une séquestration réversible des lymphocytes dans les tissus lymphoïdes (voir «Pharmacocinétique»).
En raison des effets de Fingolimod Devatis sur le système immunitaire (voir «Pharmacocinétique»), le risque d'infection (infections opportunistes incluses) peut augmenter (voir «Effets indésirables»).
Avant le début d'un traitement par Fingolimod Devatis, un hémogramme complet récent (à savoir, dans les 6 mois ou après l'arrêt d'un traitement antérieur) doit être disponible.
De plus, il est recommandé qu'un hémogramme complet, notamment un hémogramme différentiel, soit effectué au mois 3 ainsi que régulièrement ensuite – au moins chaque année – pendant le traitement ainsi qu'à titre de contrôle lors de signes d'infection. En cas d'un nombre total des lymphocytes < 0.1 × 109/l, le traitement doit être interrompu jusqu'à amélioration. En cas d'un nombre total des lymphocytes < 0.2 × 109/l, des contrôles étroits de l'hémogramme différentiel doivent être effectués au moins tous les 3 mois.
Chez les patients présentant des infections actives graves ou des infections chroniques actives, un traitement par Fingolimod Devatis ne devrait pas être initié (voir «Contreindications») ou la guérison de l'infection doit être attendue avant d'initier le traitement.
Des mesures diagnostiques et thérapeutiques appropriées immédiates s'imposent chez les patients développant des signes d'infection en cours de traitement, notamment en cas de suspicion d'infection par des virus du groupe herpès (notamment herpès simplex (VHS) et varicelle-zona (VZV)) (voir «Effets indésirables»). L'élimination du fingolimod pouvant durer jusqu'à deux mois après l'arrêt du traitement, il convient de surveiller les patients pendant cette période afin de guetter les signes d'une infection (voir paragraphe cidessous: «Arrêt du traitement»). En raison du risque d'effets cumulatifs sur le système immunitaire, la comédication simultanée avec des traitements antinéoplasiques, immunosuppresseurs ou immunomodulateurs est proscrite. Les décisions spécifiques quant à la posologie et la durée du traitement avec des corticostéroïdes dépendent de l'état clinique. Lors des études cliniques de phase III, l'administration d'un traitement court par corticostéroïdes en concomitance avec le fingolimod (jusqu'à 5 jours selon le protocole d'étude) n'a entraîné aucune augmentation de la fréquence globale des infections en comparaison au placebo. En se basant sur ces données, des traitements courts avec des corticostéroïdes (jusqu'à 5 jours) peuvent être envisagés en concomitance avec Fingolimod Devatis (voir «Effets indésirables» et «Interactions»).
Les patients recevant Fingolimod Devatis doivent être instruits afin qu'ils signalent tout symptôme d'infection à leur médecin. Si un patient développe une infection grave, l'interruption du traitement par Fingolimod Devatis doit être envisagée et les risques et bénéfices du traitement doivent être évalués avant la reprise de celui-ci.
Infection par les virus du groupe herpès
Dans les études cliniques contrôlées contre placebo, 9% des patients adultes sous fingolimod ont développé une infection herpétique, contre 7% des patients sous placebo. Depuis l'autorisation de mise sur le marché, des cas parfois graves, menaçant le pronostic vital de méningite/encéphalite due au virus varicelle-zona (VZV) et au virus herpès simplex (VHS) ont été rapportés, qui sont apparus pendant le traitement par fingolimod. Lors de l'apparition d'infections potentiellement fatales par les virus du groupe herpès, comme une encéphalite/méningite ou une défaillance multi-organique, suite à une infection disséminée, le traitement par Fingolimod Devatis doit être interrompu et le diagnostic et les traitements appropriés doivent être immédiatement mis en place.
Le statut immunitaire des patients vis-à-vis de la varicelle doit être déterminé avant le début du traitement par Fingolimod Devatis. Il est recommandé de rechercher les anticorps contre le virus varicelle-zona avant l'instauration d'un traitement par Fingolimod Devatis chez les patients sans antécédents de varicelle médicalement confirmée et chez les patients qui n'ont pas suivi un cycle complet de vaccination contre la varicelle. Une vaccination complète contre la varicelle est conseillée avant tout traitement par Fingolimod Devatis chez les patients dont la sérologie est négative (voir «Effets indésirables»). Le traitement par Fingolimod Devatis ne débutera qu'un mois après la vaccination, afin de garantir l'entière efficacité du vaccin.
De plus, depuis l'autorisation de mise sur le marché, des cas de sarcome de Kaposi, déclenchés par une infection avec le virus herpès humain de type 8 (VHH-8) ont été observés. Les patients présentant des symptômes ou des signes d'un sarcome de Kaposi doivent être diagnostiqués et traités en temps utile.
Leucoencéphalopathie multifocale progressive
Depuis l'introduction sur le marché, des cas de leucoencéphalopathie multifocale progressive (LEMP) ont été documentés (voir «Effets indésirables»). La LEMP est une infection opportuniste causée par le virus JC, qui peut être fatale ou entraîner une incapacité grave.
La LEMP ne peut survenir qu'en présence d'une infection par le virus JC. Si un test de détection du virus JC est réalisé, il faut tenir compte du fait que l'influence de la lymphopénie sur la précision du test de détection des anticorps anti-JCV n'a pas été étudiée chez les patients traités par fingolimod.
Il faut aussi noter qu'un test de détection des anticorps anti-JCV négatif n'exclut pas la possibilité d'une infection ultérieure par le virus JC.
Au début du traitement par fingolimod, la réalisation d'une IRM (habituellement de moins de 3 mois) est recommandée à titre de référence. Lors des examens IRM de routine (en accord avec les recommandations nationales et locales), les médecins doivent prêter attention aux lésions pouvant évoquer une LEMP. La réalisation d'IRM doit être envisagée dans le cadre d'une surveillance étroite de patients présentant un risque élevé de LEMP.
Les médecins doivent prêter attention aux symptômes tels que des troubles de la parole et de la marche, des changements de personnalité ou des résultats IRM évocateurs d'une LEMP.
En cas de suspicion de LEMP, une IRM doit être immédiatement réalisée à des fins diagnostiques et le traitement par fingolimod doit être arrêté jusqu'à ce qu'une LEMP puisse être exclue. En cas de confirmation d'une LEMP, le traitement par Fingolimod Devatis doit être définitivement arrêté.
Les résultats d'une IRM indiquant une LEMP peuvent déjà être visibles avant l'apparition de signes cliniques ou de symptômes. Des cas de LEMP ayant été diagnostiqués sur la base des résultats d'une IRM et de la détection d'ADN de JVC dans le liquide cérébrospinal en absence de signes cliniques ou de symptômes spécifiques de la LEMP ont été signalés chez des patients ayant été traités par des médicaments contre la sclérose en plaques qui sont associés à un risque de LEMP, dont Fingolimod Devatis fait partie.
Des cas de LEMP sont survenus sans traitement préalable par le natalizumab après environ 2 à 3 ans de traitement. Le risque estimé semble augmenter au cours du temps suite à une exposition cumulée, mais la relation exacte avec la durée du traitement est inconnue. En outre, des cas de LEMP ont été rapportés chez des patients préalablement traités par natalizumab (le natalizumab est associé à un risque accru de LEMP).
Le taux d'incidence de LEMP semble être plus élevé au Japon, sans que la raison en soit connue à ce jour.
Un syndrome inflammatoire de reconstitution immunitaire (en anglais Immune reconstitution inflammatory syndrome, IRIS) a été rapporté chez des patients traités avec des modulateurs du récepteur de S1P, dont le fingolimod, chez lesquels une LEMP est survenue et qui ont ensuite arrêté le traitement. L'IRIS se manifeste par une dégradation de l'état clinique du patient pouvant survenir rapidement; il peut entraîner des complications neurologiques graves ou mener au décès et il s'accompagne souvent de modifications caractéristiques à l'IRM. L'apparition d'un IRIS chez des patients atteints d'une LEMP a eu lieu le plus souvent quelques mois après l'arrêt du modulateur du récepteur de S1P. Il convient de surveiller l'apparition d'un IRIS et de traiter l'inflammation qui l'accompagne de manière appropriée.
Infections cryptococciques
Depuis la mise sur le marché, des cas d'infections cryptococciques, y compris de méningite à cryptococcose, ont été signalés (voir «Effets indésirables»). La plupart des cas sont survenus après environ 2 à 3 ans de traitement. La relation exacte avec la durée du traitement est cependant inconnue. La méningite à cryptococcus peut avoir une issue fatale. Les patients présentant des symptômes et des signes correspondant à une méningite à cryptococcus (maux de tête accompagnés d'une raideur de la nuque, sensibilité à la lumière, nausées et/ou confusion) doivent par conséquent rapidement faire l'objet d'une évaluation. Si une méningite à cryptococcus est diagnostiquée, un traitement approprié doit être instauré.
Infections par le papillomavirus humain
Des infections par le papillomavirus humain (HPV), y compris un papillome, une dysplasie, des verrues et des maladies cancéreuses liées au HPV ont été signalées après la mise sur le marché chez des patients traités par fingolimod (voir «Effets indésirables»). En raison des propriétés immunosuppressives du fingolimod, une vaccination contre le HPV doit être prise en considération en tenant compte des recommandations de vaccination avant le début du traitement par Fingolimod Devatis. Un dépistage du cancer incluant un test Pap est recommandé conformément aux normes en matière de soins.
Vaccins
L'efficacité des vaccinations peut être réduite pendant la prise de Fingolimod Devatis et jusqu'à deux mois après l'arrêt du traitement (voir paragraphe cidessous: «Arrêt du traitement»). L'utilisation des vaccins vivants atténués est à éviter au cours des deux premiers mois suivant l'arrêt du traitement par Fingolimod Devatis.
Pour les enfants et adolescents, veuillez vous reporter également au paragraphe «Enfants et adolescents».
Œdème maculaire
Un œdème maculaire avec ou sans symptômes visuels a été rapporté chez 0.5% des patients traités par 0.5 mg de fingolimod (voir «Effets indésirables»); il apparaissait principalement dans les 3–4 premiers mois du traitement. Un examen ophtalmologique pour évaluation du fond d'œil, macula comprise, doit être exécuté avant le début du traitement et dans les 3 à 4 mois après initiation du traitement par Fingolimod Devatis. Un examen de la vue doit avoir lieu tous les 6 mois chez le neurologue traitant. Si les patients se plaignent de troubles visuels à un moment quelconque du traitement par Fingolimod Devatis, il faut procéder à un examen du fond d'œil, y compris de la macula. Les patients diabétiques ou ceux dont l'anamnèse révèle une uvéite, ainsi que les patients dont l'anamnèse révèle un œdème maculaire doivent être régulièrement suivis sur le plan ophtalmologique pendant le traitement par Fingolimod Devatis (voir «Contreindications»).
Fonction hépatique
On a constaté une élévation des enzymes hépatiques, en particulier du taux d'alanine aminotransférase (ALAT), mais également de la gammaglutamyltransférase (γGT) et de l'aspartate aminotransférase (ASAT), sous traitement par fingolimod. Des études cliniques avec des patients adultes souffrant de SEP ont ainsi mis en évidence chez 8.0% des patients (placebo 1.9%) une augmentation de l'ALAT supérieure à 3 fois la limite supérieure de la normale (LSN). Dans les études cliniques avec le fingolimod, une augmentation supérieure à 5 fois la LSN a été observée chez 1.8% des patients (placebo 0.9%). Dans ces cas-là, le traitement a été interrompu. Une nouvelle exposition au fingolimod a entrainé une nouvelle hausse des transaminases chez certains patients, ce qui démontre la relation de causalité entre les perturbations hépatiques et la prise de fingolimod. De même, depuis l'autorisation de mise sur le marché, des atteintes hépatiques cliniquement significatives ont été observées chez des patients traités par fingolimod. Ainsi, des cas d'insuffisance hépatique aiguë ont été rapportés, pour lesquels une greffe s'est révélée nécessaire (voir «Effets indésirables»). Des signes d'une atteinte hépatique, notamment une nette élévation des transaminases sériques et une élévation de la bilirubine totale, sont apparus dans certains cas, déjà 10 jours après la première prise, mais ont également été signalés après une utilisation prolongée.
Avant l'initiation d'un traitement par Fingolimod Devatis, il convient de disposer d'une détermination des taux de transaminases et de bilirubine, ne datant pas de plus de 6 mois. Pendant le traitement et jusqu'à deux mois après l'arrêt de Fingolimod Devatis, des contrôles réguliers doivent être réalisés, même en l'absence de signes cliniques d'atteintes hépatiques. Après le début du traitement par Fingolimod Devatis, des contrôles des paramètres hépatiques doivent être effectués après 1, 3, 6, 9 et 12 mois; par la suite, les déterminations des paramètres hépatiques doivent être réalisées périodiquement, même en l'absence de symptômes cliniques, jusqu'à 2 mois après la fin du traitement.
En cas d'élévation significative des transaminases hépatiques sans symptôme clinique associé, de plus de 3 fois mais de moins de 5 fois la limite supérieure de la normale (LSN) et SANS élévation concomitante de la bilirubine, des contrôles des paramètres hépatiques plus fréquents, y compris de la bilirubine sérique et de la phosphatase alcaline (PA) sont recommandés afin de détecter toute poursuite de l'augmentation. Il convient également de clarifier en parallèle les autres causes d'atteinte hépatique. Lors d'une élévation des transaminases d'au moins 5 fois la LSN ou d'une élévation de 3 fois la LSN AVEC élévation concomitante de la bilirubine, le traitement par Fingolimod Devatis doit d'abord être interrompu et des analyses biologiques étroites des paramètres hépatiques doivent être réalisées.
Après normalisation des paramètres hépatiques, le traitement ne doit être repris qu'après une analyse approfondie du profil bénéfice-risque, par exemple lorsqu'une autre étiologie plausible pour les signes et symptômes d'atteinte hépatique a pu être mise en évidence.
Par ailleurs, les patients doivent être surveillés cliniquement quant aux signes et symptômes d'une atteinte hépatique. Chez les patients présentant des symptômes suggérant une atteinte hépatique, comme par exemple des nausées, vomissements, douleurs abdominales, douleurs abdominales droites, fatigue d'apparition récente ou s'aggravant, troubles de la concentration, anorexie ou jaunisse et/ou urines foncées inexpliqués, il convient de procéder immédiatement à une détermination des enzymes hépatiques et de la bilirubine. Le traitement par Fingolimod Devatis doit être interrompu en cas de mise en évidence d'une atteinte hépatique significative et ne peut être repris qu'après confirmation d'une autre étiologie plausible de l'atteinte hépatique. Bien que l'on ne dispose d'aucune donnée établissant que les patients avec atteinte hépatique préexistante présentent un risque accru d'élévation des paramètres de la fonction hépatique avec la prise de fingolimod, la prudence s'impose chez les patients avec anamnèse de maladie hépatique grave. Les paramètres hépatiques doivent être déterminés de manière périodique, même en l'absence de symptômes cliniques, et ce jusqu'à 2 mois après la fin du traitement. La prise supplémentaire de médicaments/substances potentiellement hépatotoxiques (dont les boissons alcoolisées) doit être évitée. Les patients atteints de cirrhose hépatique et d'insuffisance hépatique (classes B et C du score de Child-Pugh) ne doivent pas être traités par Fingolimod Devatis. Les patients atteints d'une hépatite B aiguë ou chronique active ne doivent pas être traités par Fingolimod Devatis, en raison du risque d'exacerbation de l'affection hépatique virale (voir «Contreindications»).
Tension artérielle
Dans des études cliniques sur la SEP, les patients qui étaient traités par du fingolimod à 0.5 mg, ont montré une augmentation moyenne de la tension artérielle systolique d'environ 3 mm Hg et de la tension artérielle diastolique d'environ 1 mm Hg, laquelle a été objectivée la première fois environ 1 mois après le début du traitement et qui a persisté pendant le traitement. La tension artérielle doit être contrôlée régulièrement pendant le traitement par Fingolimod Devatis.
Syndrome d'encéphalopathie postérieure réversible
Lors des études cliniques et dans le cadre de la surveillance post-commercialisation, de rares cas de syndrome d'encéphalopathie postérieure réversible (SEPR) ont été rapportés en relation avec la posologie à 0.5 mg chez les adultes (voir «Effets indésirables»). Les symptômes rapportés comprenaient l'apparition soudaine de violents maux de tête, de nausées, de vomissements, une modification de l'état mental, des troubles de la vision et des crises convulsives. Les symptômes de la SEPR sont généralement réversibles, mais peuvent évoluer vers un accident ischémique cérébral ou une hémorragie cérébrale. Un diagnostic et un traitement tardifs peuvent entraîner des complications neurologiques chroniques. En cas de suspicion de SEPR, Fingolimod Devatis ne doit plus être administré.
Fonction pulmonaire
Une réduction dose-dépendante du VEMS et des valeurs de la DLCO (capacité de diffusion) a été observée dès le premier mois suivant le début du traitement par fingolimod, ces valeurs restant stables par la suite. Après 24 mois de traitement, la réduction du VEMS attendu par rapport aux valeurs initiales a été de 2.7% pour le fingolimod 0.5 mg et de 1.2% pour le placebo. Pour la DLCO, les réductions par rapport aux valeurs initiales ont été de 3.3% pour le fingolimod 0.5 mg et de 2.7% pour le placebo après 24 mois de traitement. Les modifications du VEMS semblent réversibles à l'arrêt du traitement. On ne dispose que de données limitées sur la réversibilité de la modification de la DLCO à l'arrêt du traitement. Dans des études cliniques contrôlées, menées chez des patients atteints de SEP, une dyspnée est survenue chez 5% des patients traités par 0.5 mg de fingolimod et chez 4% des patients du groupe placebo. Certains patients ont interrompu le traitement par fingolimod lors des études d'extension (non contrôlées), en raison d'une dyspnée inexpliquée. Fingolimod n'a pas été évalué chez des patients atteints de SEP présentant une diminution de la fonction pulmonaire. En cas de survenue de symptômes évoquant un trouble pneumologique, un examen spécialisé (comprenant spirométrie et mesure de la DLCO) doit être réalisé.
Tumeurs malignes cutanées
Des carcinomes basocellulaires (CBC) et autres néoplasies cutanées telles que le mélanome malin, le cancer épidermoïde, le sarcome de Kaposi et le carcinome à cellules de Merkel ont été signalés chez des patients ayant été traités par fingolimod (voir «Effets indésirables»).
Chez tous les patients, notamment ceux présentant un risque élevé de néoplasies cutanées malignes, mais aussi ceux ne présentant pas de risque élevé de néoplasies cutanées malignes, des examens dermatologiques réguliers devront être effectués avant l'instauration d'un traitement par Fingolimod Devatis, puis au cours du traitement. Toute lésion cutanée suspecte doit être clarifiée sans délai.
En raison du risque potentiel de néoplasies cutanées malignes, les patients qui sont traités par Fingolimod Devatis doivent être mis en garde contre une exposition à la lumière du soleil sans protection. Ces patients ne doivent pas être traités par une photothérapie avec des rayons UVB ou une photochimiothérapie (PUVA-thérapie) concomitante.
Lymphomes
Les patients recevant des immunosuppresseurs ont en général un risque accru de développer un lymphome ou d'autres néoplasies malignes. Des cas de lymphome ont été signalés dans des études cliniques et après la mise sur le marché. Les cas signalés étaient de nature hétérogène; il s'agissait principalement de lymphomes non hodgkiniens, y compris des lymphomes à cellules B et à cellules T. Des cas de lymphomes à cellules T cutanés (mycosis fongoïde) ont également été observés (voir «Effets indésirables»).
Modification du nombre de lymphocytes
En raison de son mécanisme d'action, l'administration de 0.5 mg de Fingolimod Devatis provoque une réduction réversible du nombre de lymphocytes de 70% par rapport à la valeur à l'état d'équilibre. Des contrôles réguliers de la formule sanguine doivent être réalisés.
Traitement antérieur par des immunosuppresseurs ou des immunomodulateurs
Il n'existe aucune étude clinique qui évalue la sécurité et l'efficacité de fingolimod administré après un traitement antérieur par du tériflunomide, par du fumarate de diméthyle ou par de l'alemtuzumab.
Lors du passage d'autres traitements modifiant la maladie à Fingolimod Devatis, il convient de tenir compte de la demi-vie d'élimination et du mode d'action de cet autre traitement pour éviter un effet immunitaire cumulatif et simultanément, diminuer le risque d'une réactivation de la maladie. Avant l'initiation du traitement par Fingolimod Devatis, un hémogramme complet récent (à savoir, après l'arrêt du traitement antérieur) doit être disponible afin de s'assurer que tous les effets immunitaires éventuels (à savoir, cytopénie) ont disparus.
Interféron bêta, acétate de glatiramère ou fumarate de diméthyle
On peut normalement commencer avec Fingolimod Devatis directement après l'arrêt de l'interféron bêta, de l'acétate de glatiramère ou du fumarate de diméthyle.
Natalizumab ou tériflunomide
Du fait de la longue demi-vie d'élimination du natalizumab ou du tériflunomide, la prudence s'impose lors du passage de l'un de ces traitements à Fingolimod Devatis compte tenu des effets immunitaires cumulatifs possibles. Il est recommandé de fixer le début du traitement par Fingolimod Devatis après une évaluation approfondie au cas par cas.
L'élimination du natalizumab dure usuellement jusqu'à 2 à 3 mois après l'arrêt.
L'élimination du tériflunomide du plasma est également lente. Sans un procédé accéléré d'élimination, l'élimination du tériflunomide du plasma peut durer de plusieurs mois à 2 ans. Un procédé accéléré d'élimination est décrit dans l'information professionnelle du tériflunomide.
Alemtuzumab
Sur la base des propriétés et de la durée de l'effet immunosuppresseur de l'alemtuzumab, qui sont décrites dans l'information professionnelle, la mise en place d'un traitement par Fingolimod Devatis après alemtuzumab n'est pas recommandée à moins que les besoins d'un traitement par Fingolimod Devatis dépassent clairement les risques pour le patient particulier.
Retour de l'activité de la maladie (Rebound) après l'arrêt de Fingolimod Devatis
Après la commercialisation, des cas d'exacerbation grave de la maladie avec des événements de poussées évoluant en partie de manière fulminante survenant après l'arrêt de fingolimod ont été rapportés. Ceci a généralement été observé dans les 12 semaines suivant l'arrêt de fingolimod, mais des cas ont également été rapportés jusqu'à 24 semaines après l'arrêt de fingolimod et au-delà de ce délai. La prudence est donc de mise lorsque le traitement par Fingolimod Devatis est arrêté (voir «Arrêt du traitement»). Après l'arrêt de Fingolimod Devatis, les patients doivent faire l'objet d'une surveillance pour déceler les signes et symptômes révélateurs d'une activité renforcée de la maladie. Si besoin, un traitement correspondant doit être initié.
Surveillez les patients atteints de LEMP après l'arrêt de Fingolimod Devatis afin de repérer l'apparition d'un syndrome inflammatoire de reconstitution immunitaire (LEMP-IRIS) (voir Mise en garde «Leuco-encéphalopathie multifocale progressive»).
Lésions pseudo-tumorales
Après la mise sur le marché, des cas rares de lésions pseudo-tumorales en rapport avec des poussées de sclérose en plaques ont été signalés. En cas de poussées graves, un IRM doit être effectué afin d'exclure des lésions pseudo-tumorales. L'arrêt du traitement par Fingolimod Devatis doit être examiné au cas par cas par le médecin en tenant compte des bénéfices et des risques pour le patient concerné.
Arrêt du traitement
Lors de l'arrêt du traitement par Fingolimod Devatis, il faut tenir compte du fait que le fingolimod reste dans le sang jusqu'à deux mois après la dernière dose avec des effets pharmacodynamiques, tels qu'une diminution du nombre de lymphocytes. Ce taux se normalise généralement dans les 1–2 mois après la fin du traitement (voir «Pharmacocinétique»). La surveillance des infections doit être poursuivie pendant 2 mois et les patients doivent également faire part des signes d'infection pendant cette période. L'initiation d'autres traitements pendant cette période implique une exposition simultanée au fingolimod. L'utilisation d'immunosuppresseurs juste après l'arrêt de Fingolimod Devatis peut entraîner des effets cumulatifs sur le système immunitaire. La prudence est donc de mise. (Voir également «Retour de l'activité de la maladie (Rebound) après l'arrêt de Fingolimod Devatis»). De même, le contrôle des transaminases et de la bilirubine doit être poursuivi pendant au moins 2 mois après l'arrêt de Fingolimod Devatis.
Enfants et adolescents (à partir de 10 ans)
Le profil de sécurité chez les enfants et les adolescents est comparable à celui des adultes, c'est pourquoi les mises en garde et précautions pour les adultes sont également applicables aux enfants et aux adolescents. En cas de prescription de Fingolimod Devatis à des enfants et adolescents, il faut en particulier veiller à ce qui suit:
·Lors de l'administration de la première dose, des précautions doivent être prises (voir «Bradyarythmie»). Les mêmes précautions que lors de la première prise sont également recommandées lorsque la dose journalière des patients passe de 0.25 mg à 0.5 mg.
·Lors de l'étude pédiatrique contrôlée D2311, des convulsions, des états anxieux, des humeurs dépressives et des dépressions ont été plus fréquemment rapportés chez les patients traités par le fingolimod que chez les patients traités par l'interféron bêta-1a. La prudence est donc particulièrement de mise dans ce sous-groupe de patients (voir «Enfants et adolescents» dans «Effets indésirables»).
·Chez les enfants et adolescents sous fingolimod, de légères augmentations isolées du taux de bilirubine ont été constatées.
·Chez les enfants et les adolescents, il est recommandé de ne commencer le traitement par Fingolimod Devatis qu'après que toutes les vaccinations prévues conformément aux directives en vigueur en matière de vaccination ont été effectuées.
·Il n'y a que des données très limitées concernant l'utilisation chez les enfants âgés de 10 à 12 ans, les enfants ayant un poids inférieur à 40 kg ou les enfants se trouvant dans le stade de Tanner < 2 (voir «Effets indésirables» et «Efficacité clinique»). En raison des résultats très limités provenant de l'étude clinique, la prudence est particulièrement de mise dans ces sous-groupes d'enfants et d'adolescents.
·Il n'existe pas de donnés de sécurité à long terme pour les enfants et les adolescents.
Enfants et adolescents (de moins de 10 ans)
La sécurité et l'efficacité n'ont pas été étudiées chez les patients de moins de 10 ans. Fingolimod Devatis ne doit donc pas être utilisé chez les enfants de moins de 10 ans.
Grossesse, femmes en âge de procréer, risque pour le fœtus et contraception
Fingolimod Devatis est contre-indiqué pendant la grossesse, chez les femmes en âge de procréer sans contraception suffisante et pendant l'allaitement (voir «Contreindications»). En raison du risque élevé potentiel pour le fœtus, les femmes en âge de procréer doivent effectuer un test de grossesse avant le début du traitement par Fingolimod Devatis, et ce test doit être négatif. Une consultation médicale concernant le risque d'effets nocifs sur le fœtus en rapport avec le traitement doit avoir lieu. Pendant le traitement par Fingolimod Devatis, les femmes ne doivent pas tomber enceintes. Une méthode contraceptive efficace doit être utilisée pendant le traitement et au cours des 2 mois suivant la fin du traitement. (Voir «Contreindications», «Grossesse, Allaitement», ainsi que la section cidessus: «Retour de l'activité de la maladie (Rebound) après l'arrêt de Fingolimod Devatis»).
Fingolimod Devatis 0.5 mg contient des colorants azoïques tartrazine (E102) et jaune orangé S (E110), qui peuvent provoquer des réactions allergiques.
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule , c.-à-d. qu’il est essentiellement « sans sodium ».
InteractionsInteractions pharmacocinétiques
Le fingolimod est métabolisé en premier lieu par le cytochrome P450-4F2 (CYP4F2) et éventuellement par d'autres isoenzymes du CYP4F. Des études in vitro sur des hépatocytes ont montré que le CYP3A4 peut contribuer au métabolisme du fingolimod, si le CYP3A4 est fortement stimulé.
Potentiel du fingolimod et du phosphate de fingolimod à inhiber le métabolisme de médicaments administrés simultanément
Les études d'inhibition in vitro avec des microsomes hépatiques humains en pools et certaines sondes métaboliques (substrats) ont démontré que le fingolimod et le phosphate de fingolimod n'inhibaient qu'à peine, voire pas du tout, l'activité des enzymes CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5, ou CYP4A9/11 (fingolimod uniquement)). Il est donc improbable que le fingolimod et le phosphate de fingolimod réduisent la clairance des substances métabolisées en premier lieu par les principales isoenzymes du CYP.
Potentiel du fingolimod et du phosphate de fingolimod à stimuler son propre métabolisme et/ou le métabolisme de médicaments administrés simultanément
Le potentiel du fingolimod à stimuler les ARN messagers (ARNm) des CYP3A4, CYP1A2, CYP4F2 et ABCB1 (P-gp) humains et l'activité des CYP3A, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 et CYP4F2 a été étudié dans les hépatocytes primaires humains. Le fingolimod n'induit aucune stimulation d'ARNm ou de l'activité des différents enzymes du CYP450 et de l’ABCB1 par rapport au véhicule contrôle. Aussi aucune induction clinique significative des enzymes testées du CYP450 ou de l'ABCB1 (P-gp) n'est attendue lors de l'utilisation du fingolimod à concentration thérapeutique. Les essais in vitro n'ont pas montré d'induction du CYP par le phosphate de fingolimod.
Potentiel du fingolimod et du phosphate de fingolimod à inhiber le transport actif de médicaments administrés simultanément
Sur la base des données in vitro, le fingolimod comme le phosphate de fingolimod ne devraient exercer aucune action inhibitrice sur la prise de médicaments utilisés simultanément et/ou sur les principes actifs biologiques transportés par les transporteurs d'anions organiques (OATP1B1, OATP1B3) ou les cotransporteurs du taurocholate de sodium (NTCP). En conséquence, on en conclut qu'à concentration thérapeutique aucune inhibition du flux des médicaments utilisés simultanément et/ou des principes actifs biologiques n'a lieu, qu'ils soient transportés par BCRP (Breast Cancer Resistant-Protein), BSEP (pompe d'exportation des sels biliaires) ou MRP2 (Multidrug Resistance-Associated-Protein 2) ou exportés par la glycoprotéine P (P-gp).
Contraceptifs oraux
L'administration concomitante de 0.5 mg/jour de fingolimod et de contraceptifs oraux (éthinylestradiol et lévonorgestrel) n'a pas entraîné de modification de l'exposition aux contraceptifs oraux. L'exposition au fingolimod et au phosphate de fingolimod a correspondu aux valeurs mesurées lors des études antérieures. Aucune étude d'interactions n'a été effectuée avec des contraceptifs oraux contenant d'autres progestatifs; mais il n'est pas attendu d'effet du fingolimod sur l'exposition à ces substances.
Cyclosporine
Lors de l'utilisation simultanée de cyclosporine à l'état d'équilibre, la pharmacocinétique d'une dose unique de fingolimod n'a pas été modifiée. Inversement, lors de l'administration d'une dose ou de plusieurs doses (28 jours) de fingolimod, aucune modification de la pharmacocinétique de la cyclosporine à l'état d'équilibre n'est apparue. Ces données indiquent que le fingolimod ne réduit pas et n'augmente pas la clairance des médicaments principalement éliminés par le CYP3A4 et qu'il est improbable que l'inhibition du CYP3A4 réduise la clairance du fingolimod. La forte inhibition des molécules de transport P-gp, MRP2 et OATP1B1 OATP-C n'influence pas l'élimination du fingolimod.
Kétoconazole
L'utilisation simultanée de kétoconazole 200 mg deux fois par jour à l'état d'équilibre et d'une dose unique de 5 mg de fingolimod a provoqué l'élévation de la valeur d'AUC du fingolimod et du phosphate de fingolimod (augmentation de 1.7 fois) du fait de l'inhibition du CYP4F2.
Isoprotérénol, atropine, aténolol et diltiazem
Lors de l'utilisation simultanée d'une dose unique de fingolimod, de phosphate de fingolimod et d'isoprotérénol ou d'atropine, aucune modification n'a été constatée. En conséquence, la pharmacocinétique des doses uniques de fingolimod et de phosphate de fingolimod demeure inchangée, de même que la pharmacocinétique à l'état d'équilibre de l'aténolol et du diltiazem lors de l'utilisation simultanée de ces deux médicaments et du fingolimod.
Carbamazépine
L'utilisation simultanée de carbamazépine 600 mg deux fois par jour à l'état d'équilibre et d'une dose unique de 2 mg de fingolimod a eu un faible impact sur la valeur d'AUC du fingolimod et du phosphate de fingolimod (une diminution d'environ 40% pour chacun), ce qui indique que l'utilisation simultanée de carbamazépine peut réduire l'efficacité du fingolimod.
D'autres inducteurs enzymatiques puissants du CYP3A4, comme la rifampicine, le phénobarbital, la phénytoïne, l'oxcarbazépine, l'éfavirenz et le millepertuis, peuvent réduire la valeur de l'AUC du fingolimod et de ses métabolites au moins dans une mesure comparable. En conséquence, en cas d'utilisation simultanée, ils peuvent nuire à l'efficacité du fingolimod.
Tests de laboratoire
Le fingolimod abaisse le nombre de lymphocytes dans le sang par redistribution dans les organes lymphatiques secondaires; par conséquent, les valeurs de lymphocytes circulants en périphérie ne peuvent pas être utilisées pour évaluer le statut des sous-groupes lymphocytaires chez les patients traités par Fingolimod Devatis.
Les tests de laboratoire pour lesquels des cellules mononucléées circulantes sont nécessaires, nécessitent une plus grande quantité de sang en raison du nombre abaissé de lymphocytes circulants.
Interactions pharmacodynamiques
Il ne faut pas administrer simultanément des traitements antinéoplasiques, immunomodulateurs ou immunosuppresseurs (y compris les corticostéroïdes), en raison du risque d'effets additifs sur le système immunitaire (voir «Mises en garde et précautions»). Les décisions spécifiques quant à la posologie et la durée du traitement concomitant avec corticostéroïdes doivent s'appuyer sur une évaluation clinique. Lors des études cliniques de phase III, l'administration d'un traitement court par corticostéroïdes en concomitance avec le fingolimod (jusqu'à 5 jours selon le protocole d'étude) n'a entraîné aucune augmentation de la fréquence globale des infections en comparaison au placebo (voir «Mises en garde et précautions» et «Effets indésirables»).
La prudence est recommandée lors d'une substitution thérapeutique chez un patient recevant un traitement qui exerce des effets prolongés sur le système immunitaire, comme le natalizumab, le tériflunomide ou la mitoxantrone, et qui passe à Fingolimod Devatis (voir «Mises en garde et précautions, Traitement antérieur par des immunosuppresseurs ou des immunomodulateurs»).
En cas d'association du fingolimod et de l'aténolol, un ralentissement supplémentaire de 15% de la fréquence cardiaque est observé après le début du traitement par le fingolimod. Cet effet n'est pas observé avec le diltiazem.
En raison des effets additifs potentiels sur la fréquence cardiaque, il convient de renoncer à un traitement par Fingolimod Devatis chez les patients recevant des bêtabloquants, des inhibiteurs des canaux calciques ralentissant la fréquence cardiaque (p.ex. vérapamil ou diltiazem) ou d'autres substances bradycardisantes (p.ex. ivabradine, digoxine, inhibiteurs de l'acétylcholinestérase, pilocarpine). Si un traitement par Fingolimod Devatis est envisagé, il convient de consulter un cardiologue au préalable pour évaluer la possibilité du passage à une substance sans effet bradycardisant. Les patients chez qui un tel changement de traitement n'est pas envisageable doivent bénéficier d'une surveillance par ECG cardiaque en continu au moins pendant la nuit suivante (voir «Mises en garde et précautions» et «Posologie/Mode d'emploi»). Fingolimod Devatis est contre-indiqué en cas de prise d'antiarythmiques de classe Ia ou III (voir «Contreindications»).
L'efficacité des vaccins peut être diminuée pendant et jusqu'à deux mois après le traitement par Fingolimod Devatis. L'utilisation de vaccins vivants atténués peut comporter un risque infectieux; elle est donc aussi à éviter au cours du traitement par Fingolimod Devatis et jusqu'à 2 mois après l'arrêt du traitement par Fingolimod Devatis (voir «Effets indésirables» et «Mises en garde et précautions»).
Effet de Fingolimod Devatis sur d'autres médicaments
Analyse en pharmacocinétique de populations des interactions médicamenteuses potentielles
Une étude de pharmacocinétique de populations menée chez les patients atteints de sclérose en plaques n'a révélé aucune influence fondamentale de la fluoxétine et de la paroxétine (puissants inhibiteurs du CYP2D6) sur la concentration de fingolimod ou de phosphate de fingolimod. Lors de la prise de carbamazépine, la concentration de phosphate de fingolimod est réduite de moins de 30%. De plus, les substances suivantes très fréquemment prescrites n'avaient aucun effet clinique significatif (≤20%) sur les concentrations de fingolimod ou de phosphate de fingolimod: baclofène, gabapentine, oxybutynine, amantadine, modafinil, amitriptyline, prégabaline, corticostéroïdes et contraceptifs oraux.
Grossesse, AllaitementFemmes en âge de procréer/contraception pour les femmes
Le fingolimod est contre-indiqué chez les femmes en âge de procréer sans contraception suffisante (voir «Contreindications»). Chez les femmes en âge de procréer, un résultat négatif du test de grossesse doit donc être disponible avant le début d'un traitement par Fingolimod Devatis. Les femmes doivent être averties de la possibilité d'un risque grave pour l'enfant à naître et de la nécessité d'une contraception efficace pendant le traitement et au cours des 2 mois suivant la fin du traitement par Fingolimod Devatis. Étant donné qu'il faut environ 2 mois après l'arrêt du traitement jusqu'à l'élimination du médicament de l'organisme (voir «Mises en garde et précautions»), le potentiel de risque pour le fœtus peut perdurer. La contraception doit donc être poursuivie pendant cette période (voir «Contreindications»).
Si le traitement par Fingolimod Devatis est interrompu à cause d'une grossesse avérée ou prévue, voir les sections «Mises en garde et précautions» ainsi que les sous-sections «Retour de l'activité de la maladie (Rebound) après l'arrêt de Fingolimod Devatis» et «Arrêt du traitement».
Grossesse
Pendant le traitement, les femmes ne doivent pas tomber enceinte et un moyen de contraception efficace doit être utilisé (voir «Contreindications»). Si une femme tombe enceinte pendant la prise de Fingolimod Devatis, Fingolimod Devatis ne doit plus être pris. Une toxicité sur la reproduction est apparue dans les études expérimentales chez l'animal, y compris une déficience et des défauts d'organes du fœtus, c.-à-d. une persistance du tronc artériel et une communication interventriculaire (voir «Données précliniques»). De plus, on sait que le récepteur sur lequel le fingolimod agit (récepteur de la sphingosine-1-phosphate), participe à la formation des vaisseaux pendant l'embryogenèse. À l'heure actuelle, on ignore si des malformations cardiovasculaires apparaissent chez l'être humain.
Il n'existe pas d'études appropriées et bien contrôlées portant sur le fingolimod chez la femme enceinte.
Les données disponibles pour l'utilisation chez l'humain (données après la mise sur le marché et informations issues du registre des grossesses) indiquent que l'utilisation de fingolimod est liée à une augmentation de la prévalence d'anomalies congénitales graves par rapport à la population globale. Pendant le traitement, les femmes ne doivent pas tomber enceintes et l'utilisation d'une méthode contraceptive efficace est recommandée. Si une femme tombe enceinte pendant la prise de Fingolimod Devatis, le traitement par Fingolimod Devatis doit être arrêté. Les patientes doivent être averties des effets nocifs sur le fœtus et un examen de suivi médical doit être effectué (p.ex. échographie). La possibilité d'une exacerbation grave de la maladie doit également être prise en compte lorsque les patientes arrêtent le traitement par Fingolimod Devatis en raison d'une grossesse ou d'une grossesse prévue. Les patientes concernées doivent demander conseil au médecin traitant concernant les possibilités de traitements alternatifs (voir «Mises en garde et précautions»).
Les données issues des registres des grossesses au Canada, dans les pays de l'UE et d'Amérique du Sud montrent jusqu'à présent que le risque de malformations congénitales dans la population atteinte de SEP est semblable à celui observé dans la population générale. Selon les données d'un registre des grossesses aux USA, le risque de fausses couches et d'enfants mort-nés paraît comparable dans la population atteinte de SEP et dans la population générale.
Dans plus de 600 cas prospectifs de grossesses avec des naissances vivantes, des mort-nés ou des avortements en raison d'une anomalie fœtale suite à une exposition maternelle au fingolimod pendant la grossesse qui ont été signalés après la mise sur le marché, la proportion de malformations congénitales graves était de 5%. La prévalence de malformations congénitales graves observée dans la population générale est comprise entre 2 et 4%. Le schéma des malformations rapportées en rapport avec fingolimod est comparable à celui de la population globale. Les malformations les plus fréquentes sont:
·des affections cardiaques congénitales telles que des anomalies du septum auriculaire et ventriculaire, la tétralogie de Fallot;
·des anomalies rénales;
·des anomalies de l'appareil locomoteur.
Il n'existe aucune indication d'une accumulation d'anomalies de naissance spécifiques avec Fingolimod Devatis.
Capacité de contraction et déroulement de la naissance
On ne dispose d'aucune donnée sur les effets du fingolimod sur la capacité de contraction et le déroulement de la naissance.
Allaitement
Le fingolimod est contre-indiqué pendant l'allaitement (voir «Contreindications»). Le fingolimod passe dans le lait des animaux traités pendant la lactation. Il n'existe aucune donnée sur les effets de fingolimod sur les enfants allaités ou sur la production de lait. Étant donné que de nombreuses substances actives passent dans le lait maternel et en raison de la possibilité de réactions médicamenteuses indésirables graves pour l'enfant allaité, consécutives à l'exposition au fingolimod, les femmes traitées par Fingolimod Devatis ne doivent pas allaiter.
Test de grossesse
Chez les femmes en âge de procréer, une éventuelle grossesse doit être vérifiée avant le début du traitement.
Fertilité
Les études chez l'animal n'indiquent pas que le fingolimod est associé à un risque accru de baisse de la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesFingolimod Devatis n'a aucune influence ou une influence négligeable sur l'aptitude à la conduite et l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
La population qui a servi à caractériser le profil de sécurité de fingolimod provenait de deux études cliniques de phase III contrôlées contre placebo et d'une étude clinique de phase III contrôlée par principe actif avec des patients adultes atteints de sclérose en plaques évoluant par poussées et rémissions. Elle comptait au total 2431 patients adultes traités par fingolimod (dose de 0.5 ou 1.25 mg). L'étude D2301 (FREEDOMS) était une étude clinique d'une durée de 2 ans contrôlée contre placebo portant sur 854 patients adultes atteints de sclérose en plaques, qui recevaient du fingolimod (groupe placebo: 418 patients adultes). L'étude D2309 (FREEDOMS II) était une étude clinique sur 2 ans, contrôlée contre placebo, incluant 728 patients adultes présentant une sclérose en plaques et traités par du fingolimod (placebo: 355). Sur la base des données agrégées issues de ces deux études, les réactions indésirables les plus graves observées après le début du traitement à la dose thérapeutique recommandée de 0.5 mg étaient: infections, œdème maculaire et blocs auriculoventriculaires transitoires. Les réactions indésirables les plus fréquentes (incidence ≥10%) lors de la prise de 0.5 mg étaient: céphalées, élévation des enzymes hépatiques, diarrhées, toux, grippe, sinusite et douleurs dorsales. L'événement indésirable le plus fréquent, apparu lors de la prise de 0.5 mg de fingolimod avec une incidence de 1%, et qui a mené à l'interruption du traitement était l'élévation du taux d'ALAT (2.2%).
Les effets indésirables de l'étude D2302 (TRANSFORMS), une étude contrôlée par principe actif, d'une durée de 1 an, dans laquelle 849 patients adultes atteints de sclérose en plaques ont été traités par du fingolimod ou par de l'interféron bêta-1a, la molécule servant à la comparaison, étaient globalement similaires à ceux des études contrôlées contre placebo, si l'on tient compte des durées différentes des deux études.
Fréquences des effets indésirables issues des données agrégées des deux études contrôlées contre placebo FREEDOMS et FREEDOMS II
Les effets indésirables sont répertoriés par classe de système d'organes de la classification MedDRA. Les fréquences sont définies de la manière suivante: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100); rares (≥1/10 000 à < 1/1000); très rares (< 1/10 000). Dans chaque groupe de fréquence, les effets indésirables sont présentés selon un ordre de gravité décroissante.
Infections et infestations
Très fréquents: infections virales grippales (11%, placebo: 8%), sinusite (11%, placebo: 8%).
Fréquents: bronchite, zona, pityriasis versicolor.
Occasionnels: pneumonie.
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Fréquents: basaliome (ou carcinome basocellulaire).
Occasionnels: mélanome malin.
Rares: lymphome, cancer épidermoïde.
Très rares: sarcome de Kaposi.
Affections hématologiques et du système lymphatique
Fréquents: leucopénie, lymphopénie.
Occasionnels: thrombopénie.
Affections psychiatriques
Fréquents: dépressions.
Occasionnels: humeur dépressive.
Affections du système nerveux
Très fréquents: céphalées (25%, placebo: 23%).
Fréquents: vertige, migraine.
Occasionnels: convulsions.
Rares: syndrome d'encéphalopathie postérieure réversible (SEPR)*.
Affections oculaires
Fréquents: vision trouble.
Occasionnels: œdème maculaire.
Affections cardiaques
Fréquents: bradycardie, blocs auriculoventriculaires.
Affections vasculaires
Fréquents: hypertension.
Affections respiratoires, thoraciques et médiastinales
Très fréquents: toux (12%, placebo: 11%).
Fréquents: dyspnée.
Affections gastro-intestinales
Très fréquents: diarrhée (13%, placebo: 10%).
Affections hépatobiliaires
Très fréquents: élévation des enzymes hépatiques (augmentation de l'ALAT, de la GGT et de l'ASAT) (15%, placebo: 4%).
Affections de la peau et du tissu sous-cutané
Fréquents: eczéma, prurit.
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents: dorsalgie (10%, placebo: 9%).
Troubles généraux et anomalies au site d'administration
Fréquents: asthénie.
Investigations
Fréquents: élévation des triglycérides sanguins.
* Non rapporté dans les études FREEDOMS, FREEDOMS II et TRANSFORMS. La catégorie de fréquence se base sur l'incidence observée sur les quelques 10 000 patients sous fingolimod dans toutes les études cliniques.
Effets indésirables après commercialisation
Infections et infestations
Leucoencéphalopathie multifocale progressive (LEMP).
Infections cryptococciques (y compris méningite à cryptococcus).
Infections, notamment méningite/encéphalite due à un virus du groupe herpès (en particulier virus varicelle-zona (VZV) et virus herpès simplex (VHS)).
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Carcinome à cellules de Merkel.
Affections du système immunitaire
Réactions d'hypersensibilité, p.ex. éruption cutanée, urticaire et angio-œdème au début du traitement, anémie hémolytique auto-immune.
Affections du système nerveux
Exacerbation grave de la maladie après l'arrêt de Fingolimod Devatis (voir «Mises en garde et précautions»).
Affections gastro-intestinales
Nausées.
Affections hépatobiliaires
Atteinte hépatique jusqu'à insuffisance hépatique nécessitant une greffe.
Affections musculo-squelettiques et du tissu conjonctif
Myalgie, arthralgie.
Investigations
Perte de poids.
Description de certains effets indésirables
Infections
Dans les études cliniques sur la sclérose en plaques (SEP), la fréquence totale des infections (65.1%) sous traitement par 0.5 mg de fingolimod était semblable à celle rapportée sous placebo. Les bronchites, zonas et pneumonies étaient toutefois plus fréquents chez les patients traités par fingolimod. Les infections graves se sont déclarées dans le groupe traité avec 0.5 mg de fingolimod selon une fréquence de 1.6% et dans le groupe sous placebo selon une fréquence de 1.4%.
Lors des études cliniques de phase III, l'administration d'un traitement court par corticostéroïdes en concomitance avec le fingolimod (jusqu'à 5 jours selon le protocole d'étude) n'a entraîné aucune augmentation de la fréquence globale des infections en comparaison au placebo (voir «Mises en garde et précautions» et «Interactions»).
Des infections par le papillomavirus humain (HPV), y compris des papillomes, des dysplasies, des verrues et des maladies cancéreuses liées au HPV ont été signalées pendant le traitement par fingolimod après la mise sur le marché (voir «Mises en garde et précautions»).
Depuis le lancement sur le marché, des cas d'infections avec des agents pathogènes opportunistes avec issue parfois mortelle ont été rapportés. Outre les infections virales (p.ex. LEMP par le virus JC, méningite/encéphalite par les virus du groupe herpès (entre autres, virus herpès simplex (VHS) et virus varicelle-zona (VZV), sarcome de Kaposi par le virus herpès humain de type 8 (VHH-8)), des infections mycosiques (p.ex. méningite/encéphalite à cryptococcus) ou bactériennes (p.ex. mycobactéries atypiques), ont été signalées (voir «Mises en garde et précautions»).
Œdème maculaire
Dans les études cliniques, des œdèmes maculaires sont apparus chez 0.5% des patients traités par la dose recommandée de 0.5 mg de fingolimod et chez 1.1% des patients recevant la dose supérieure de 1.25 mg.
Dans les études cliniques portant sur la sclérose en plaques, la plupart des cas sont apparus dans les 3–4 premiers mois de traitement. Quelques patients se sont présentés avec une vision trouble ou une diminution de l'acuité visuelle, d'autres par contre étaient asymptomatiques et le diagnostic fut posé au cours d'examens ophtalmologiques de routine. L'œdème maculaire s'est en général amélioré ou a disparu spontanément à l'arrêt du médicament. Le risque de récidive lors de la reprise du traitement n'a pas été examiné.
L'incidence de l'œdème maculaire est accrue chez les patients atteints de SEP présentant une anamnèse d'uvéite (environ 20% lors d'uvéite à l'anamnèse contre 0.6% sans uvéite à l'anamnèse).
Bradyarythmie
L'introduction d'un traitement par Fingolimod Devatis induit un ralentissement passager de la fréquence cardiaque et peut en outre s'accompagner d'un ralentissement de la conduction auriculoventriculaire (voir «Posologie/Mode d'emploi», «Mises en garde et précautions» et «Pharmacodynamique»).
Dans les études cliniques sur la sclérose en plaques, le ralentissement maximal moyen de la fréquence cardiaque a atteint une valeur maximale dans les 6 heures suivant la prise de la première dose, ce qui correspondait à un ralentissement moyen de 8 battements par minute pour une dose de 0.5 mg de fingolimod. Après la deuxième dose, un nouveau ralentissement de moindre importance peut apparaître. Une fréquence cardiaque inférieure à 40 battements/minute n'a été que rarement constatée chez les patients traités par 0.5 mg de fingolimod. À dose constante, la fréquence cardiaque retrouve sa valeur de départ dans le premier mois.
Dans les études cliniques, un bloc auriculoventriculaire du premier degré (allongement de l'intervalle PR sur l'électrocardiogramme) est apparu après l'instauration du traitement chez 4.7% des patients qui recevaient 0.5 mg de fingolimod, chez 2.8% des patients qui recevaient de l'interféron bêta-1a par voie intramusculaire et chez 1.6% des patients du groupe placebo. Un bloc AV du deuxième degré a été constaté chez moins de 0.2% des patients traités par 0.5 mg de fingolimod. Depuis la commercialisation de fingolimod, des cas isolés de blocs AV complets transitoires ayant régressé spontanément ont été signalés au cours de la phase d'observation de six heures après la prise de fingolimod. Les anomalies de conduction rapportées dans les études cliniques et dans le cadre du suivi post-marketing étaient généralement transitoires, asymptomatiques et disparaissaient dans les 24 heures suivant le début du traitement. Chez la plupart des patients, aucune intervention médicale n'a été nécessaire, mais un patient ayant reçu la dose de 0.5 mg dans l'étude clinique a été traité par de l'isoprénaline en raison d'un bloc auriculoventriculaire du deuxième degré de type Mobitz I asymptomatique.
Depuis la commercialisation de fingolimod, des cas isolés d'événements retardés (dans les 24 heures) après la première dose, y compris d'asystolie passagère et de décès de cause indéterminée, ont été observés. Les traitements concomitants et/ou les antécédents médicaux ont rendu difficile l'analyse concluante de ces cas. La relation de causalité entre ces événements et fingolimod reste par conséquent indéterminée.
Tension artérielle
Dans les études cliniques sur la sclérose en plaques, la prise de 0.5 mg de fingolimod a été associée à une légère élévation de la tension artérielle moyenne d'1 mmHg en moyenne, constatée dans les 2 mois suivant le début du traitement et persistante lors de la poursuite du traitement. Une hypertension a été observée chez 6.5% des patients traités par 0.5 mg de fingolimod et chez 3.3% des patients du groupe placebo.
Fonction hépatique
Chez les patients présentant une sclérose en plaques et traités par fingolimod, des augmentations des enzymes hépatiques, principalement de l'alanine aminotransférase (ALAT), ont été constatées. Dans les études cliniques sur la sclérose en plaques, respectivement 8.0% et 1.8% des patients traités par 0.5 mg de fingolimod ont développé une élévation asymptomatique des taux sériques de l'ALAT d'au moins 3 fois et d'au moins 5 fois la limite supérieure de la normale (LSN). Dans la plupart des cas, ces élévations ont été observées dans les 6 à 9 mois après le début du traitement par fingolimod. Après l'arrêt de fingolimod le taux sérique de l'ALAT a rejoint les valeurs normales en l'espace d'environ 2 mois. Chez un petit nombre de patients, c.-à-d. 10 patients recevant 1.25 mg de fingolimod et 2 patients recevant 0.5 mg de fingolimod, dont les transaminases hépatiques étaient élevées à ≥5 × LSN et qui poursuivaient le traitement par fingolimod, le retour à des valeurs normales a duré environ 5 mois.
De même, après la mise sur le marché, des atteintes hépatiques cliniquement significatives ont été observées chez les patients traités par fingolimod (voir «Mises en garde et précautions»). Les signes d'une atteinte hépatique, notamment une nette élévation des transaminases sériques et une élévation de la bilirubine totale, sont apparus dans certains cas déjà 10 jours après la première dose, mais également après une utilisation plus longue. Des cas d'insuffisance hépatique aiguë nécessitant une greffe de foie ont également été rapportés.
Organes respiratoires
Une réduction dose-dépendante du VEMS et des valeurs de la DLCO (capacité de diffusion) ont été observées au cours du traitement par fingolimod (voir «Mises en garde et précautions» et «Pharmacocinétique»).
Convulsions
Des cas de convulsions, incluant un état de mal épileptique, au cours du traitement par fingolimod ont été rapportés, aussi bien dans les études cliniques que dans le cadre de rapports d'expériences pratiques après l'autorisation de mise sur le marché. On ignore si ces évènements étaient liés seulement aux symptômes de la sclérose en plaques, à fingolimod ou bien à une association des deux.
Événements vasculaires
Au cours des études cliniques de phase III, de rares cas d'artériopathie oblitérante des membres inférieurs sont survenus chez des patients traités par des doses élevées de fingolimod (1.25 ou 5.0 mg). Par ailleurs, de rares cas d'accidents vasculaires cérébraux ischémiques ou hémorragiques ont été observés sous 0.5 mg de fingolimod au cours des études cliniques et dans le cadre de la surveillance postcommercialisation; aucun lien de causalité n'a cependant été établi.
Néoplasies cutanées
Dans les données combinées des deux études cliniques contrôlées par placebo, des carcinomes basocellulaires sont survenus chez 14 participants sur 783 (1.8%) sous fingolimod 0.5 mg et chez 5 participants sur 773 (0.6%) sous placebo. Par ailleurs, d'autres cas de néoplasies cutanées malignes (p.ex. mélanomes, sarcome de Kaposi) sont survenus chez des patients qui avaient pris fingolimod au cours d'études cliniques et après la mise sur le marché. Des examens dermatologiques réguliers doivent être effectués chez les patients à risque de néoplasies cutanées malignes avant le début du traitement par fingolimod, puis régulièrement par la suite.
Lymphome
Des cas de lymphomes ont été observés lors des études cliniques et après la mise sur le marché (y compris un cas mortel de lymphome à cellules B positif au virus d'Epstein-Barr (VEB)). Les cas rapportés étaient hétérogènes et contenaient principalement des lymphomes non hodgkiniens et des lymphomes à cellules B et à cellules T. Dans les études cliniques, l'incidence des lymphomes à cellules B et T était plus élevée qu'attendu dans la population générale. Des cas de lymphomes T cutanés (mycosis fongoïde) ont été observés.
Enfants et adolescents (à partir de 10 ans)
Dans une étude pédiatrique contrôlée, le profil de sécurité chez les enfants et les adolescents (âgés de 10 à 18 ans) qui ont reçu 0.25 mg ou 0.5 mg de fingolimod une fois par jour était en grande partie similaire à celui des patients adultes.
Des convulsions sont toutefois apparues au cours de l'étude pédiatrique chez 5.6% des patients traités par le fingolimod et chez 0.9% des patients traités par l'interféron bêta-1a.
On sait que des dépressions et des états anxieux surviennent souvent chez les patients souffrant de sclérose en plaques. Chez les patients pédiatriques qui ont reçu du fingolimod, des dépressions et états anxieux ont également été rapportés.
De légères augmentations isolées du taux de bilirubine ont été constatées chez les patients pédiatriques sous fingolimod.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes doses uniques jusqu'à 80 fois la dose recommandée (0.5 mg) ont été étudiées chez des volontaires sains. Sous 40 mg, 5 des 6 sujets d'expériences ont décrit une légère sensation d'oppression ou des douleurs dans la poitrine, qui étaient la traduction clinique d'une réactivité des petites voies respiratoires.
Le fingolimod peut provoquer une bradycardie et des ralentissements de la conduction AV. En règle générale, la baisse de la fréquence cardiaque commence dès la première heure qui suit la dose initiale et atteint une valeur maximale après environ 6 heures. Par la suite, la fréquence cardiaque retrouve son niveau initial en l'espace d'un mois de traitement continu (voir «Mises en garde et précautions»). Des cas de ralentissement de la conduction auriculoventriculaire ont été signalés et on dispose également de rapports de cas isolés de blocs AV complets transitoires à résolution spontanée (voir «Mises en garde et précautions» et «Effets indésirables»).
Signes et symptômes
En cas de surdosage de Fingolimod Devatis, il est important de surveiller aussi les symptômes d'une bradycardie/ arythmie. Si le surdosage est survenu au début du traitement, il est important de surveiller les patients au moins durant les six premières heures au moyen d'un ECG en continu (en temps réel) et d'effectuer des mesures de la fréquence cardiaque et de la tension artérielle toutes les heures, les mêmes mesures que lors de la surveillance après la dose initiale étant indiquées (voir «Tableau 1: Surveillance après la première prise de Fingolimod Devatis», rubrique «Posologie/Mode d'emploi» et «Mises en garde et précautions»).
Traitement
Une élimination significative du fingolimod de l'organisme n'est possible ni par dialyse ni par échange plasmatique.
Propriétés/EffetsCode ATC
L04AE01
Groupe pharmacothérapeutique: modulateurs des récepteurs de la spingosine-1-phosphate (S1P)
Mécanisme d’action
Le fingolimod est un modulateur du récepteur de la sphingosine-1-phosphate. Le principe actif est métabolisé par la sphingosine kinase en phosphate de fingolimod, son métabolite actif. Le phosphate de fingolimod se lie, dans des domaines de concentrations nanomolaires inférieurs, aux récepteurs 1, 3 et 4 de la sphingosine-1-phosphate (S1P) sur les lymphocytes et après un passage aisé de la barrière hémato-encéphalique, aux récepteurs S1P 1, 3 et 5 des cellules nerveuses dans le système nerveux central (SNC). En agissant comme antagoniste fonctionnel du S1PR sur les lymphocytes, le phosphate de fingolimod bloque la capacité des lymphocytes à quitter les ganglions lymphatiques de sorte qu'une redistribution et non pas une déplétion de lymphocytes ait lieu. Cette redistribution réduit l'infiltration par les lymphocytes pathogènes, y compris par les cellules proinflammatoires Th17, du SNC, dans lequel ils participent à l'inflammation des nerfs et à l'atteinte des tissus nerveux. Des expériences menées sur les animaux et des expérimentations in vitro ont démontré que les effets avantageux du fingolimod dans la sclérose en plaques pourraient aussi être attribués à une interaction avec les récepteurs S1P sur les cellules nerveuses. Chez les humains et les animaux, le fingolimod pénètre le SNC et l'on a démontré une réduction de l'astrogliose, de la démyélinisation et de la dégénérescence neuronale. En outre, le fingolimod élève les niveaux de BDNF (facteur neurotrophique issu du cerveau) dans le cortex, l'hippocampe et le corps strié du cerveau: il participe ainsi à la survie neuronale et améliore les fonctions motrices.
Pharmacodynamique
Système immunitaire
Effets sur le nombre de cellules immunitaires dans le sang. Dans les 4–6 heures suivant la première prise de 0.5 mg de fingolimod, le nombre de lymphocytes s'abaisse à un niveau équivalent à 75% de la valeur initiale. Si le traitement quotidien est poursuivi, le taux de lymphocytes s'abaisse encore pendant une période de deux semaines et atteint finalement sa valeur minimale d'environ 500 cellules/µl ou environ 30% de la valeur initiale. Dixhuit pourcent des patients ont au moins une fois atteint une valeur inférieure à 200 cellules/µl. Lors d'une administration quotidienne continue, les valeurs de lymphocytes restent basses. La plupart des lymphocytes T et B passent régulièrement par les organes lymphoïdes, c'est donc sur ces cellules que le fingolimod exerce ses effets les plus importants. Environ 15–20% des lymphocytes T ont un phénotype de cellules à mémoire-effectrices et jouent ainsi un rôle important dans la surveillance immunitaire périphérique. Ce sous-groupe de lymphocytes ne passant pas par les organes lymphoïdes, il n'est pas influencé par le fingolimod. Au cours des quelques jours qui suivent l'arrêt du fingolimod, on assiste à une augmentation du nombre de lymphocytes périphériques, qui généralement se normalise en un ou deux mois. Une administration continue de fingolimod conduit à une légère diminution du nombre de neutrophiles à environ 80% de la valeur initiale. Le fingolimod n'exerce aucune influence sur les monocytes.
Fréquence cardiaque et rythme cardiaque
Au début du traitement, le fingolimod provoque un ralentissement passager de la fréquence cardiaque et de la conduction auriculoventriculaire (voir «Effets indésirables»). Le ralentissement de la fréquence cardiaque est le plus manifeste 4–5 heures après la prise, le médicament déployant 70% de son effet chronotrope négatif le premier jour. La fréquence cardiaque rejoint le plus souvent les valeurs normales en l'espace d'un mois de traitement continu.
Le traitement par le fingolimod n'exerce aucune influence sur les réactions autonomes du muscle cardiaque, ni sur les variations diurnes de la fréquence cardiaque ou les réactions aux activités physiques.
Au début du traitement par le fingolimod, on assiste à une augmentation des extrasystoles d'origine atriale, mais non à l'augmentation du taux de fibrillation atriale/flutter atrial ou d'arythmies ventriculaires ou de battements ventriculaires ectopiques. Le traitement par le fingolimod n'entraîne aucune diminution du débit cardiaque.
Le ralentissement de la fréquence cardiaque provoqué par le fingolimod peut être combattu avec de l'atropine, de l'isoprénaline ou du salmétérol.
Potentiel d'allongement de l'intervalle QT
Dans une étude approfondie sur l'intervalle QT avec des doses de 1.25 ou 2.5 mg de fingolimod à l'état d'équilibre, alors que l'effet chronotrope négatif du fingolimod était encore présent, le traitement par le fingolimod a provoqué un allongement du QTcI avec une limite supérieure de l'IC à 90% de ≤13.0 ms. Il n'y avait pas de relation dose-effet ou exposition-effet entre le fingolimod et un allongement du QTcI. De même il n'y avait aucun signal cohérent en faveur d'une incidence plus élevée d'échappement du QTcI, ni absolue ni sous forme de modification par rapport à la valeur initiale, en relation avec un traitement par le fingolimod. Dans l'étude 1, des allongements du QTcF compris entre 30 et 60 ms sont cependant survenus chez 6.6% (placebo: 2.4%) des patients lors de la première administration de 0.5 mg de fingolimod et chez 13.9% (placebo: 6.7%) des patients lors d'administrations ultérieures. La pertinence clinique est inconnue.
Système respiratoire
Le traitement par une dose unique ou par des doses répétées de 0.5 mg ou 1.25 mg de fingolimod pendant 2 semaines n'est pas associé à une augmentation détectable de la résistance respiratoire mesurée au moyen du VEMS ou du DEF25–75. Des doses uniques de ≥5 mg (10 fois la dose recommandée) ont par contre provoqué une élévation dosedépendante de la résistance respiratoire. Le traitement par des doses répétées de 0.5, 1.25 ou 5 mg de fingolimod n'a provoqué aucune modification de l'apport en oxygène, de la saturation en oxygène à l'effort et aucune élévation de la sensibilité des voies aériennes à la méthacholine. Les patients traités par le fingolimod ont développé une sensibilité normale aux bêta-agonistes inhalés.
Efficacité clinique
L'efficacité de fingolimod a été mise en évidence dans deux études utilisant une dose unique quotidienne de 0.5 mg et 1.25 mg de fingolimod chez les patients adultes atteints d'une forme récurrente-rémittente de la sclérose en plaques. Les participants aux deux études devaient avoir eu au moins 2 poussées cliniques dans les 2 ans précédant la randomisation, ou au moins 1 poussée clinique dans l'année précédant la randomisation et un score EDSS (Expanded Disability Status Scale) situé entre 0 et 5.5. Une troisième étude avec le même type de patients a été menée après l'inscription de fingolimod.
L'efficacité et la sécurité de la dose unique quotidienne de 0.25 mg et 0.5 mg de fingolimod (choix de la dose en fonction du poids corporel et des mesures de l'exposition) ont été évaluées chez les enfants et les adolescents âgés de 10 à 18 ans atteints d'une forme récurrente-rémittente de la sclérose en plaques.
Étude D2301 (FREEDOMS)
L'étude D2301 (FREEDOMS) était une étude de phase III de 2 ans, randomisée, en double aveugle et contrôlée par placebo chez des patients atteints de sclérose en plaques évoluant par poussées, qui n'avaient pas reçu d'interféron-bêta ou d'acétate de glatiramère pendant au moins 3 mois avant le début de l'étude, et n'avaient pas reçu de natalizumab pendant au moins 6 mois avant le début de l'étude.
L'âge médian était de 37 ans, la durée médiane de la maladie de 6.7 ans et le score EDSS initial médian était de 2.0. Les patients ont reçu après randomisation sur une période allant jusqu'à 24 mois, 0.5 mg de fingolimod (n = 425) ou 1.25 mg de fingolimod (n = 429) ou un placebo (n = 418). La durée moyenne de traitement avec la dose de 0.5 mg a été de 717 jours, de 715 jours avec la dose de 1.25 mg et la durée moyenne de prise de placebo de 718.5 jours.
Le critère d'évaluation principal était le taux annuel de poussées.
Le taux annualisé de poussées (ARR = annualized relapse rate) était significativement plus bas du point de vue statistique chez les patients traités par fingolimod que dans le groupe placebo. Le critère d'évaluation secondaire le plus important était le temps écoulé jusqu'à une progression du handicap persistante pendant 3 mois. La progression était mesurée à l'aide d'une élévation du score EDSS par rapport à la valeur de départ d'au moins 1 point (chez les patients dont la valeur EDSS de départ était de 5.5, au moyen d'une élévation d'au moins 0.5 point), qui persistait pendant au moins 3 mois. L'intervalle jusqu'à l'apparition d'une progression du handicap persistante sur 3 mois était significativement allongé du point de vue statistique lors du traitement par fingolimod en comparaison au traitement par placebo. À aucun moment une différence statistiquement significative entre la dose de 0.5 mg et la dose de 1.25 mg n'a été notée.
Les résultats de cette étude figurent dans le tableau 2 et la figure 1.
Tableau 2 Résultats cliniques et résultats de l'IRM dans l'étude FREEDOMS
|
|
Fingolimod 0.5 mg
|
Placebo
| |
Critères d'évaluation cliniques
|
N=425
|
N=418
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.18
(p<0.001*)
|
0.40
| |
Réduction relative (%)
|
54
|
| |
Proportion de patients toujours sans poussées au 24e mois (%)
|
70.4
(p<0.001*)
|
45.6
| |
Risque de progression du handicap
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.70 (0.52, 0.96)
(p=0.024*)
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 6 mois)
|
0.63 (0.44, 0.90)
(p=0.012*)
|
| |
Critères d'évaluation IRM
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=370
|
n=339
| |
Médiane (moyenne) du nombre pendant 24 mois
|
0.0 (2.5)
(p<0.001*)
|
5.0 (9.8)
| |
Nombre de lésions rehaussées au Gd
|
n=369 (mois 24)
|
n=332 (mois 24)
| |
Médiane (moyenne) du nombre au
| |
6e mois
|
0.0 (0.2)
|
0.0 (1.3)
| |
12e mois
|
0.0 (0.2)
|
0.0 (1.1)
| |
24e mois
|
0.0 (0.2)
(p<0.001* à
toutes les dates)
|
0.0 (1.1)
| |
Pourcentage de modification du volume total des lésions en T2
|
n=368
|
n=339
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-1.7 (10.6)
(p<0.001*)
|
8.6 (33.8)
| |
Modification du volume des lésions hypointenses en T1
|
n=346
|
n=305
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
0.0 (8.8)
(p=0.012*)
|
1.6 (50.7)
| |
Pourcentage de modification du volume cérébral
|
n=357
|
n=331
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-0.7 (-0.8)
(p<0.001*)
|
-1.0 (-1.3)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Montre une signification statistique par rapport au placebo avec un seuil de signification bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS
Figure 1 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée jusqu'au 24e mois étude FREEDOMS (population ITT)
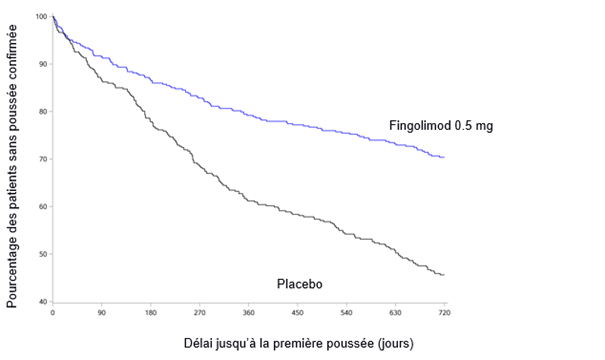
Les patients qui ont participé à l'étude FREEDOMS (D2301) se sont vus offrir la possibilité de participer à une étude de prolongation en aveugle D2301E1. Pour cette étude, 920 patients de l'étude principale traités par le fingolimod ont été sélectionnés (n = 331 ont poursuivi le traitement à la dose de 0.5 mg, 289 ont poursuivi le traitement à la dose de 1.25 mg, 155 sont passés du placebo à la dose de 0.5 mg et 145 sont passés du placebo à la dose de 1.25 mg). Au bout de 12 mois (mois 36), 856 patients (93%) étaient encore présents. On disposait de données de suivi pour un total de 811 patients (88.2%), sur au moins 18 mois pour la phase de prolongation.
Au mois 24 de l'étude de prolongation, le taux annualisé de poussées (Annualized Relapse Rate, ARR) pour les patients qui faisaient partie du groupe sous placébo dans l'étude principale et qui sont passés à la dose de 0.5 mg de fingolimod avait diminué de 55% (ARR 0.45, IC à 95% 0.32 à 0.62, p < 0.001). Chez les patients qui étaient déjà traités avec 0.5 mg de fingolimod dans l'étude principale, l'ARR est resté bas pendant toute la phase de prolongation (ARR = 0.10).
Entre les mois 24 et 36, le taux annualisé de poussées (ARR) a été de 0.17 pour les patients sous 0.5 mg de fingolimod dans l'étude principale et qui continuaient de recevoir 0.5 mg (0.21 dans l'étude principale). Chez les patients qui sont passés du placebo à 0.5 mg de fingolimod, l'ARR s'élevait à 0.22 (0.42 dans l'étude principale).
Étude D2309 (FREEDOMS II)
Dans l'étude de phase III de 2 ans répliquée, randomisée, en double aveugle et contrôlée contre placebo incluant 1083 patients présentant une sclérose en plaques récurrente-rémittente, des résultats comparables à ceux de l'étude D2301 ont été obtenus. Cette étude a été réalisée après l'autorisation de mise sur le marché du fingolimod.
L'âge médian était de 40.5 ans, la durée médiane de la maladie de 8.9 ans et le score EDSS initial médian de 2.5. Les résultats de cette étude sont présentés dans le tableau 3 et la figure 2.
Tableau 3 Résultats cliniques et résultats de l'IRM dans l'étude FREEDOMS II
|
|
0.5 mg Fingolimod
|
Placebo
| |
Critères d'évaluation cliniques
|
N=358
|
N=355
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.21
(p<0.001*)
|
0.40
| |
Réduction relative (en %)
|
48
|
| |
Proportion de patients toujours sans poussées au 24e mois (%)
|
71.5
(p<0.001*)
|
52.7
| |
Risque de progression du handicap†
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.83 (0.61, 1.12)
(p=0.227)
|
| |
Hazard Ratio (IC à 95%) (confirmé à 6 mois)
|
0.72 (0.48, 1.07)
(p=0.113)
|
| |
Critères d'évaluation IRM
| |
Pourcentage de modification du volume cérébral (en %)
|
n=266
|
n=249
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-0.7 (-0.9)
(p<0.001*)
|
-1.0 (-1.3)
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=264
|
n=251
| |
Médiane (moyenne) du nombre pendant 24 mois
|
0.0 (2.3)
(p<0.001*)
|
4.0 (8.9)
| |
Nombre de lésions rehaussées au Gd
|
n=269 (mois 24)
|
n=256 (mois 24)
| |
Médiane (moyenne) du nombre au
| |
6e mois
|
0.0 (0.2)
|
0.0 (1.1)
| |
12e mois
|
0.0 (0.2)
|
0.0 (1.3)
| |
24e mois
|
0.0 (0.4)
(p<0.001* à
toutes les dates)
|
0.0 (1.2)
| |
Pourcentage de modification du volume total des lésions en T2
|
n=262
|
n=247
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-7.1 (13.7)
(p<0.001*)
|
0.8 (25.1)
| |
Modification du volume des lésions hypodenses en T1
|
n=225
|
n=209
| |
Médiane (moyenne) de la modification pendant 24 mois en %
|
-9.9 (12.6)
(p=0.372)
|
-8.5 (26.4)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Statistiquement significatif vs. placebo avec un niveau bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS.
† D'autres analyses ont montré que les résultats pour la population totale n'étaient pas statistiquement significatifs, du fait de progressions faussement positives dans le sous-groupe des patients avec un score EDSS initial = 0 (n = 62, 8.7% de la population de l'étude). Chez les patients affichant un score EDSS > 0 (n = 651; 91.3% de la population de l'étude), on a observé, pour une dose de 0.5 mg de fingolimod, une diminution cliniquement notable et statistiquement significative par rapport au placebo (HR = 0.70; IC (0.50, 0.98); p = 0.040), ce qui concorde avec l'étude FREEDOMS.
Figure 2 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée, jusqu'au 24e mois – étude FREEDOMS II (population ITT)
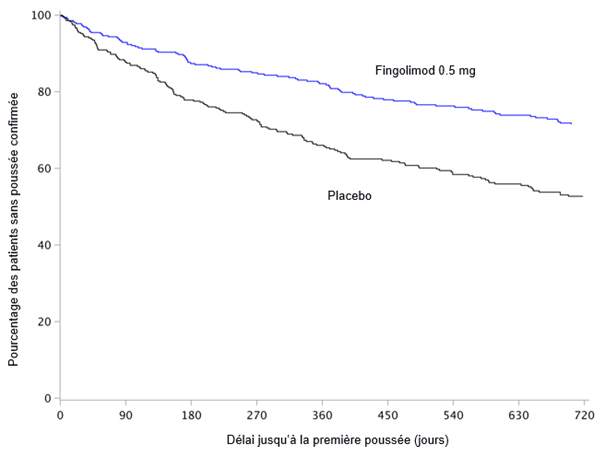
Aucune différence statistiquement significative n'a été constatée entre les doses de 0.5 mg et de 1.25 mg pour aucun des critères d'évaluation.
Étude D2302 (TRANSFORMS)
L'étude D2302 (TRANSFORMS) est une étude de phase III, randomisée, en double aveugle, doubledummy (ou double placebo), contrôlée par traitement actif (interféron bêta-1a, 30 microgrammes en intramusculaire, une fois par semaine) et menée sur un an chez les patients présentant une SEP évoluant par poussées et rémissions, qui n'avaient pas reçu de natalizumab pendant les 6 mois précédant le début de l'étude. Un traitement antérieur par interféron bêta ou par acétate de glatiramère était autorisé jusqu'au moment de la randomisation.
L'âge médian était de 36 ans, la durée médiane de la maladie était de 5.9 ans et le score EDSS initial médian était de 2.0. Après randomisation, les patients ont reçu, pendant une période allant jusqu'à 12 mois, 0.5 mg de fingolimod (n = 431) ou 1.25 mg de fingolimod (n = 426) ou 30 microgrammes d'interféron bêta-1a par voie intramusculaire une fois par semaine (n = 435). La valeur médiane pour la durée du traitement avec le médicament d'étude était de 365 jours (0.5 mg de fingolimod), 354 jours (1.25 mg de fingolimod) et 361 jours (interféron bêta-1a).
Les résultats de cette étude figurent dans le tableau 4 et la figure 3.
Tableau 4 Résultats cliniques et résultats IRM de l'étude TRANSFORMS
|
|
Fingolimod 0.5 mg
|
Interféron bêta-1a,
30 μg
| |
Critères d'évaluation cliniques
|
N=429
|
N=431
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.16
(p<0.001*)
|
0.33
| |
Réduction relative (%)
|
52
|
| |
Proportion de patients toujours sans poussées jusqu'au 12e mois (%)
|
82.5
(p<0.001*)
|
70.1
| |
Risque de progression du handicap
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.71 (0.42, 1.21)
(p=0.209)
|
| |
Critères d'évaluation IRM
| |
Nombre de lésions en T2, nouvelles ou récemment agrandies
|
n=380
|
n=365
| |
Médiane (moyenne) du nombre pendant 12 mois
|
0.0 (1.7)
(p=0.004*)
|
1.0 (2.6)
| |
Nombre de lésions rehaussées au Gd
|
n=374
|
n=354
| |
Médiane (moyenne) du nombre après 12 mois
|
0.0 (0.2)
(p<0.001*)
|
0.0 (0.5)
| |
Pourcentage de modification du volume cérébral
|
n=368
|
n=359
| |
Médiane (moyenne) de la modification pendant 12 mois en %
|
-0.2 (-0.3)
(p<0.001*)
|
-0.4 (-0.5)
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées dans la population Intent-to-Treat (ITT). Les données évaluables ont été utilisées pour les analyses de l'IRM.
* Montre une signification statistique par rapport à l'interféron bêta-1a avec un seuil de signification bilatéral de 0.05.
Détermination des valeurs de p: analyse de la somme totale des ARR par régression négative binomiale, ajustée au traitement, au pays (données regroupées), au nombre de poussées dans les 2 années précédentes et à la valeur initiale du score EDSS.
Figure 3 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée jusqu'au 12e mois étude TRANSFORMS (population ITT)
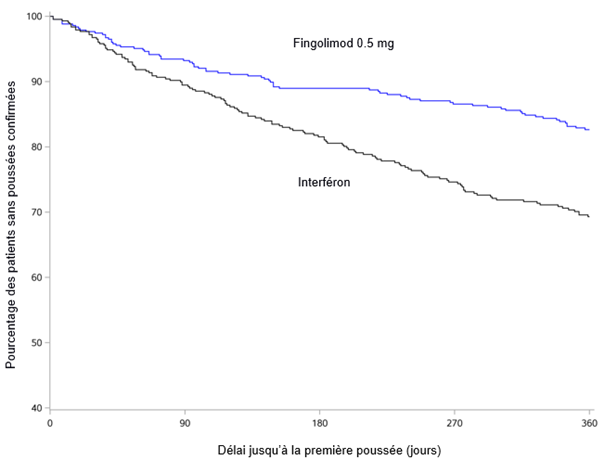
Pour aucun des critères d'évaluation des différences statistiquement significatives n'ont été constatées entre les doses à 0.5 mg et à 1.25 mg.
Les patients qui ont participé à l'étude TRANSFORMS (D2302) se sont vus offrir la possibilité de participer à une étude de prolongation en aveugle (D2302E1). Au total 1030 patients de l'étude principale traités par le fingolimod ont été sélectionnés (n = 357 ont poursuivi le traitement à la dose de 0.5 mg, 330 ont poursuivi le traitement à la dose de 1.25 mg, 167 sont passés de l'interféron bêta-1a à la dose de 0.5 mg et 176 sont passés de l'interféron bêta-1a à la dose de 1.25 mg). On disposait de données de suivi pour un total de 882 de ces patients (85.9%) sur au moins 12 mois pour la phase de prolongation.
Au mois 12 de l'étude de prolongation, chez les patients qui faisaient partie du groupe sous interféron bêta-1a i.m. dans l'étude principale et qui sont passés à la dose de 0.5 mg de fingolimod, l'ARR avait relativement diminué de 30% (ARR 0.70, p = 0.06). Chez les patients qui étaient déjà traités par 0.5 mg de fingolimod dans l'étude principale, l'ARR est resté bas pendant toute l'étude principale et la phase de prolongation (ARR = 0.18 jusqu'au mois 24).
Entre les mois 12 et 24, l'ARR a été de 0.20 (0.19 dans l'étude principale) pour les patients sous 0.5 mg de fingolimod dans l'étude principale et qui continuaient de recevoir 0.5 mg. Chez les patients qui sont passés de l'interféron bêta-1a à 0.5 mg de fingolimod, l'ARR s'élevait à 0.33 (0.48 dans l'étude principale).
Dans l'ensemble, les résultats des études D2301 (FREEDOMS) et D2302 (TRANSFORMS) ont montré une réduction homogène du taux annualisé de poussées par rapport à la préparation utilisée pour la comparaison dans les sous-groupes définis en fonction du sexe, de l'âge, du traitement antérieur de la SEP, de l'activité de la maladie ou du degré de handicap initial.
Étude D2311 (PARADIGMS) chez les enfants et les adolescents à partir de 10 ans
L'étude D2311 (PARADIGMS) est une étude à groupes parallèles en double aveugle, randomisée, contrôlée par substance active et multicentrique, d'une durée flexible allant jusqu'à 24 mois pour l'évaluation de l'efficacité et de la sécurité du fingolimod (n = 107) en comparaison avec l'interféron bêta-1a (n = 107) chez les enfants et les adolescents atteints de SEP récurrente-rémittente âgés de 10 à moins de 18 ans. Un traitement préalable avec de l'interféron bêta, du fumarate de diméthyle ou de l'acétate de glatiramère était autorisé jusqu'au moment de la randomisation. Les patients présentant au moins une poussée clinique l'année précédente ou au moins 2 poussées cliniques dans les 2 ans précédant la randomisation ou la détection de ≥1 lésion rehaussée au Gd en IRM dans les 6 mois précédant la randomisation et avec un score EDSS situé entre 0 et 5.5 ont été inclus. Des examens neurologiques ont été réalisés lors de la sélection ainsi que par la suite tous les 3 mois et lors d'une poussée présumée. Les évaluations des résultats de l'IRM ont eu lieu lors de la sélection et par la suite tous les 6 mois pendant toute la durée de l'étude. Le critère d'évaluation principal était le taux annuel de poussées.
L'âge médian était de 16 ans, la durée médiane de la maladie depuis l'apparition du premier symptôme de 1.5 ans et le score EDSS initial médian était de 1.5. Les patients ont été randomisés et ont reçu du fingolimod ou de l'interféron bêta-1a, qui a été administré par voie intramusculaire une fois par semaine pour une durée allant jusqu'à 24 mois. Dans la classe d'âge ≥10 et ≤12 ans (n = 13 sous fingolimod), ainsi que dans la catégorie de poids ≤40 kg (n = 9 sous fingolimod), peu de patients ont été inclus, de sorte que dans ces groupes de patients seules des données limitées sont disponibles pour évaluer l'efficacité et la sécurité. La durée médiane du traitement par le médicament à l'étude était de 634 jours sous fingolimod et de 547 jours sous interféron bêta-1a.
Le critère d'évaluation principal, le taux annuel de poussées (Annualized Relapse Rate, ARR), était significativement plus faible d'un point de vue statistique chez les patients traités par le fingolimod en comparaison avec ceux qui ont reçu de l'interféron bêta-1a (réduction relative de l'ARR de 81.9%). Le critère d'évaluation secondaire le plus important, le nombre annualisé de nouvelles lésions en T2 ou de lésions en T2 agrandies jusqu'au mois 24, était en outre significativement plus faible d'un point de vue statistique chez les patients traités par le fingolimod en comparaison avec ceux qui ont reçu de l'interféron bêta-1a, de même que le nombre de lésions en T1 rehaussées au Gd par examen jusqu'au mois 24. En outre, le taux annualisé d'atrophie cérébrale entre l'inclusion et le mois 24 était réduit de façon statistiquement significative par le fingolimod. Une analyse post-hoc supplémentaire a montré que l'intervalle jusqu'à l'apparition d'une progression du handicap persistante sur 3 mois était allongé de façon statistiquement significative par le fingolimod en comparaison avec l'interféron bêta-1a.
Les résultats de cette étude sont présentés dans le tableau 5 et les figures 4 et 5.
Tableau 5 Résultats cliniques et résultats de l'IRM dans l'étude PARADIGMS
|
|
Fingolimod
0.25 mg ou 0.5 mg
|
Interféron bêta-1a
30 µg
| |
Critères d'évaluation cliniques
|
N=107
|
N=107#
| |
Taux annualisé de poussées (critère d'évaluation principal)
|
0.122
(p<0.001*)
|
0.675
| |
Réduction relative (%)
|
81.9
|
| |
Proportion de patients toujours sans poussées
jusqu'au 24e mois (%)
|
85.7
(p<0.001*)
|
38.8
| |
Risque de progression du handicap
| |
Hazard Ratio (IC à 95%)
(confirmé à 3 mois)
|
0.23 (0.08, 0.66)
(p=0.007*)
|
| |
Hazard Ratio (IC à 95%)
(confirmé à 6 mois)
|
0.20 (0.04; 0.93)
(p=0.040**)
|
| |
Critères d'évaluation IRM
| |
Nombre annualisé de lésions en T2, nouvelles ou récemment agrandies
|
n=106
|
n=102
| |
Moyenne ajustée
|
4.393
(p<0.001*)
|
9.269
| |
Réduction relative (%)
|
52.6
|
| |
Nombre de lésions en T1 rehaussées au Gd par examen jusqu'au mois 24
|
n=106
|
n=101
| |
Moyenne ajustée
|
0.436
(p<0.001*)
|
1.282
| |
Réduction relative (%)
|
66.0
|
| |
Taux annualisé d'atrophie cérébrale entre l'inclusion et le mois 24
|
n=96
|
n=89
| |
Moyenne des moindres carrés
|
-0.48
(p=0.014*)
|
-0.80
|
Toutes les analyses des critères d'évaluation cliniques ont été effectuées sur la population totale d'analyse. Les données évaluables ont été utilisées pour les analyses de l'IRM.
# Un patient randomisé ayant reçu 30 µg d'interféron bêta-1a en injection intramusculaire une fois par semaine était incapable d'avaler la dose supplémentaire nécessaire de placebo dans le cadre de la procédure doubledummy et a été retiré de l'étude. Le patient a été exclu de la population totale d'analyse et de la population de sécurité.
* Montre une signification statistique par rapport à l'interféron bêta-1a IM, avec un seuil de signification bilatéral de 0.05.
** Analyse post-hoc, modèle à risques proportionnels de Cox. p = 0.180 dans le test du log-rank.
Détermination des valeurs de p: ARR agrégé: par régression binomiale négative, ajustée au traitement, au pays, au stade pubertaire (le facteur de stratification dans le système de dialogue vocal interactif, SVI) et au nombre de poussées durant les 2 années précédentes (offset: durée dans l'étude); Pourcentage des patients sans poussée: sur la base d'une estimation de Kaplan-Meier; Risque de progression du handicap: sur la base du modèle à risques proportionnels de Cox, ajusté au traitement, au pays, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de poussées dans les 2 années précédentes; Nombre annualisé de lésions en T2, nouvelles ou récemment agrandies: par régression binomiale négative, ajustée au traitement, à la région, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de lésions en T2 initial (offset: durée dans l'étude); Nombre de lésions rehaussées au Gd par examen: par régression binomiale négative avec le nombre cumulé de lésions en T1 rehaussées au Gd pour tous les examens IRM programmés réalisés durant l'étude depuis l'initiation comme variable réponse, ajustée au traitement, au pays, au stade pubertaire (le facteur de stratification dans le SVI) et au nombre de lésions en T1 rehaussées au Gd initial (offset: nombre d'examens IRM); Taux annualisé d'atrophie cérébrale: au moyen d'une analyse ANCOVA, ajusté au traitement, à la région, au stade pubertaire (le facteur de stratification dans le SVI) et au volume cérébral total initial
Figure 4 Courbes de Kaplan-Meier du délai jusqu'à la première poussée confirmée, jusqu'au 24e mois – étude PARADIGMS- (population totale d'analyse)
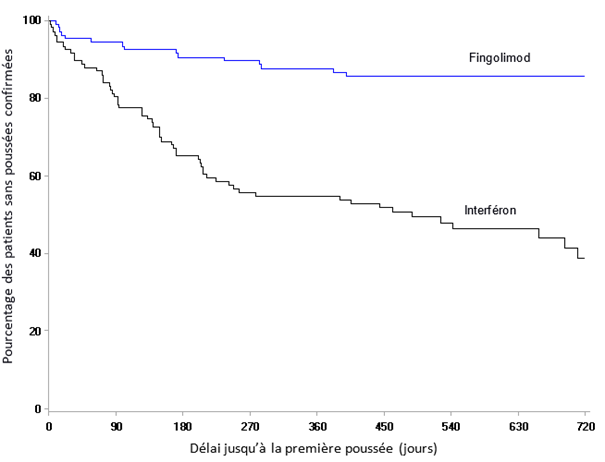
Figure 5 Graphique de Kaplan-Meier du délai jusqu'à la constitution d'une progression confirmée du handicap persistante sur 6 mois étude PARADIGMS- (population totale d'analyse)
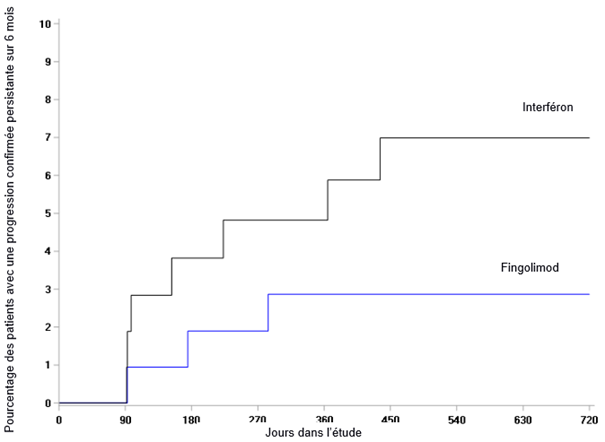
PharmacocinétiqueAbsorption
Le fingolimod est absorbé lentement (tmax de 12–16 heures) et en grande quantité (≥85%, d'après la quantité de radioactivité excrétée dans l'urine et la quantité de métabolites excrétée dans les selles, extrapolée à l'infini). La biodisponibilité orale absolue apparente est élevée (93%).
La prise de nourriture ne modifie ni la Cmax ni la valeur d'exposition (AUC) du fingolimod. Fingolimod Devatis peut donc être pris indépendamment des repas (voir «Posologie/Mode d'emploi»).
Les concentrations sanguines à l'état d'équilibre sont atteintes en 1 à 2 mois lors d'une administration quotidienne unique et sont environ 10 fois plus élevée qu'après la dose initiale.
Distribution
Le fingolimod est distribué en grande quantité dans les érythrocytes, la fraction contenue dans les cellules sanguines est de 86%. La proportion de phosphate de fingolimod lié aux cellules sanguines est < 17%. Le fingolimod et le phosphate de fingolimod sont principalement liés aux protéines (> 99.7%). La liaison aux protéines du fingolimod et du phosphate de fingolimod n'est pas modifiée en cas d'insuffisance rénale ou hépatique.
Le fingolimod est distribué en grande quantité dans les tissus; le volume de distribution est d'environ 1200 ± 260 l. Une étude avec quatre volontaires sains qui ont reçu, par voie intraveineuse, une dose unique de fingolimod marqué à l'iode radioactif, a montré que le fingolimod passe dans le cerveau. Dans une étude portant sur 13 patients masculins atteints de sclérose en plaques et qui recevaient 0.5 mg de fingolimod par jour à l'état d'équilibre, la quantité de fingolimod (et de phosphate de fingolimod) dans l'éjaculat était plus de 10 000 fois inférieure à la dose administrée (0.5 mg).
Métabolisme
Chez l'homme, il existe trois voies principales de biotransformation du fingolimod: par phosphorylation réversible stéréosélective en énantiomère (S) du phosphate de fingolimod, pharmacologiquement actif; par biotransformation oxydative, principalement catalysée par le CYP4F2 et éventuellement par d'autres isoenzymes du CYP4F, et ensuite dégradation par bêta-oxydation en métabolites inactifs et finalement par formation d'analogues de céramides apolaires du fingolimod pharmacologiquement inactifs.
Après une prise orale unique de fingolimod [14C], le fingolimod lui-même (23.3%), le phosphate de fingolimod (10.3%) et les métabolites inactifs (acide carbonique M3 (8.3%), céramide M29 (8.9%) et céramide M30 (7.3%)), sont les composants principaux issus du fingolimod dans le sang, comme déterminé selon leur contribution à l'AUC jusqu'à 816 heures après l'administration de la dose par rapport à la contribution de tous les composés marqués radioactivement.
Élimination
La clairance sanguine du fingolimod est de 6.3 ± 2.3 l/h, et la demi-vie d'élimination apparente terminale moyenne (t1/2) est de 6–9 jours. En phase terminale, la concentration sanguine du phosphate de fingolimod diminue parallèlement à celle du fingolimod, ce qui résulte en des demi-vies similaires des deux substances.
Après une dose orale, environ 81% de la dose administrée est lentement excrétée dans l'urine sous forme de métabolites inactifs. Le fingolimod et le phosphate de fingolimod ne sont pas excrétés dans l'urine sous forme intacte; ils constituent cependant les composés les plus importants dans les selles, les quantités représentant chaque fois moins de 2.5% de la dose. Après 34 jours, la part récupérée de la dose administrée s'élève à 89%.
Linéarité/non-linéarité
Après plusieurs administrations d'une dose unique quotidienne de fingolimod 0.5 mg ou 1.25 mg, les concentrations de fingolimod et de phosphate de fingolimod croissent apparemment de manière proportionnelle à la dose.
Après plusieurs administrations quotidiennes uniques d'une dose de fingolimod de 0.25 mg ou de 0.5 mg, la concentration de phosphate de fingolimod augmente visiblement de manière proportionnelle à la dose chez l'enfant et l'adolescent.
Cinétique pour certains groupes de patients
Groupe ethnique
Les effets de l'appartenance ethnique sur la pharmacocinétique du fingolimod et du phosphate de fingolimod ne sont pas cliniquement significatifs.
Sexe
Le sexe n'a aucune influence sur la pharmacocinétique du fingolimod et du phosphate de fingolimod.
Troubles de la fonction hépatique
Chez les patients présentant une insuffisance hépatique légère, modérée et sévère (Child-Pugh classe A, B et C), la pharmacocinétique d'une dose unique de fingolimod (1 ou 5 mg) était non modifiée en ce qui concerne la Cmax du fingolimod, cependant, l'AUC s'élevait de respectivement 12%, 44% et 103%. La demi-vie apparente d'élimination reste inchangée lors d'insuffisance hépatique légère; par contre elle se prolonge de 49–50% en cas d'insuffisance hépatique modérée et sévère. Chez les patients souffrant d'insuffisance hépatique sévère (Child-Pugh classe C), la Cmax du phosphate de fingolimod a diminué de 22% et la valeur de l'AUC a augmenté de 38%. La pharmacocinétique du phosphate de fingolimod n'a pas été évaluée chez les patients souffrant d'insuffisance hépatique légère ou modérée.
Troubles de la fonction rénale
En cas d'insuffisance rénale grave, la Cmax et l'AUC du fingolimod augmentent de 32% et 43% et la Cmax et l'AUC du phosphate de fingolimod de 25% et 14%. La demi-vie apparente d'élimination des deux analytes reste inchangée. Chez les patients présentant une insuffisance rénale, aucune adaptation de la posologie de Fingolimod Devatis n'est nécessaire.
Patients âgés
Le mécanisme d'élimination et les résultats des études de pharmacocinétiques de population suggèrent qu'aucune adaptation posologique ne serait nécessaire chez les patients âgés. Cependant, les expériences cliniques chez les patients de plus de 55 ans sont limitées.
Enfants et adolescents
La concentration en phosphate de fingolimod à l'état d'équilibre est plus faible chez les enfants et les adolescents que chez les adultes.
La sécurité et l'efficacité de fingolimod n'ont pas été étudiées chez les enfants et les adolescents de moins de 10 ans.
Données précliniquesLe profil de sécurité préclinique du fingolimod a été étudié chez des souris, des rats, des chiens et des singes. Les principaux organes cibles étaient le système lymphoïde (lymphopénie et atrophie lymphoïde), les poumons (augmentation du poids, hypertrophie du muscle lisse au niveau de la transition broncho-alvéolaire) et le cœur (effet chronotrope négatif, élévation de la tension artérielle, modifications périvasculaires et dégénérescence du muscle cardiaque) chez plusieurs espèces; les vaisseaux cardiaques (vasculopathie) uniquement chez les rats, l'hypophyse, la première partie de l'estomac, le foie, les surrénales, le tractus gastro-intestinal, et le système nerveux uniquement à hautes doses (fréquemment dans un contexte de tableau de toxicité généralisée) chez plusieurs espèces.
Mutagénicité et carcinogénicité
Le fingolimod ne s'est pas avéré mutagène in vitro, ni dans le test d'Ames ni sur une lignée cellulaire du lymphome murin L5178Y. Dans les cellules pulmonaires V79 du hamster chinois, on n'a pas mesuré d'effet clastogène in vitro. Dans les cellules V79, le fingolimod a induit des aberrations chromosomiques numériques (polyploïdie) à des concentrations supérieures ou égales à 3.7 µg/ml. Dans le test du micronoyau chez la souris et le rat, le fingolimod n'a pas eu d'effet clastogène in vivo.
Dans un essai biologique de 2 ans chez le rat avec des doses orales de fingolimod allant jusqu'à la dose maximale tolérée de 2.5 mg/kg, ce qui, prenant comme point de départ la valeur d'AUC de l'exposition systémique de l'être humain lors d'une prise de 0.5 mg, représente une limite environ 50 fois supérieure, aucune indication de carcinogénicité n'a été rapportée. Dans une étude portant sur 2 ans chez les souris, on a constaté à des doses partant de 0.25 mg/kg, ce qui représente en prenant comme point de départ la valeur AUC de l'exposition systémique de l'être humain lors de l'administration quotidienne d'une dose de 0.5 mg, une limite environ 6 fois supérieure, une élévation de l'incidence des lymphomes malins.
Toxicité sur la reproduction
Jusqu'aux doses les plus élevées testées (10 mg/kg), le fingolimod n'avait (en prenant comme point de départ la valeur d'AUC de l'exposition systémique chez l'être humain à des doses de 0.5 mg, cette valeur correspondant à environ 150 fois la limite supérieure) aucun effet sur la mobilité/le nombre des spermatozoïdes ni sur la fertilité du rat mâle ou femelle.
Le fingolimod était tératogène lors de l'administration d'une dose de 0.1 mg/kg ou supérieure chez le rat. Parmi les malformations viscérales les plus fréquentes chez le fœtus, on comptait la persistance du tronc artériel et une communication interventriculaire. À des doses de 1 mg/kg ou supérieures, une augmentation de la fréquence de la perte des fœtus après implantation a été observée. À des doses de 3 mg/kg on a constaté une baisse du nombre de fœtus viables. Le fingolimod n'était pas tératogène chez le lapin, alors qu'à des doses de 1.5 mg/kg et supérieures, on observait une augmentation de la mortalité embryo-fœtale, ainsi qu'à 5 mg/kg une baisse du nombre de fœtus viables et un retard de croissance fœtale.
Chez le rat, à des doses ne provoquant aucune toxicité chez les mères, la survie des descendants F1 diminue dans les phases post-natales précoces. Le traitement par le fingolimod n'avait pourtant aucune influence sur le poids corporel, le développement, le comportement et la fertilité des descendants F1.
Le fingolimod a été décelé dans le lait des animaux traités lors de la lactation. Chez les lapines gravides, le fingolimod et ses métabolites ont traversé la barrière placentaire.
Études menées sur les jeunes animaux
Les résultats issus de deux études de toxicité chez les jeunes rats ont montré un effet mineur sur la densité minérale osseuse et sur la réaction neurocomportementale, ainsi qu'un léger retard de la maturité sexuelle et une faible diminution de la réponse immunitaire à des stimulations répétées avec l'hémocyanine de patelle (KLH). Ces effets n'ont pas été considérés comme des évènements indésirables. Globalement, les effets du fingolimod chez les jeunes animaux étaient comparables à ceux observés chez les rats adultes recevant des doses comparables, à l'exception de l'absence d'une hypertrophie de la musculature lisse dans les poumons des rats juvéniles. Les valeurs de DSENO (dose sans effet nocif observé) chez les jeunes animaux ont été principalement déterminées par des effets non spécifiques sur le poids corporel ou la consommation de nourriture plutôt que par une toxicité manifeste.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C.
Tenir hors de portée des enfants.
Numéro d’autorisation68406 (Swissmedic)
PrésentationGélules à 0.25 mg: 7 et 28 [B].
Gélules à 0.5 mg: 7, 28, 84 et 98 [B].
Titulaire de l’autorisationDevatis AG, 6330 Cham.
Mise à jour de l’informationDécembre 2024
|