Propriétés/EffetsCode ATC
L04AC21
Mécanisme d'action
Le bimekizumab est un anticorps monoclonal IgG1/κ humanisé qui se lie de manière sélective aux cytokines IL-17A, IL-17F et IL-17AF avec une affinité élevée, bloquant leur interaction avec le complexe récepteur IL-17RA/IL-17RC. Des concentrations élevées d'IL-17A et d'IL-17F ont été impliquées dans la pathogenèse de plusieurs maladies inflammatoires à médiation immunitaire, dont le psoriasis en plaques, le rhumatisme psoriasique et la spondyloarthrite axiale. L'IL-17A et l'IL-17F coopèrent et/ou agisse en synergie avec d'autres cytokines inflammatoires afin d'induire l'inflammation. L'IL-17F est produit en quantité importante par les cellules de l'immunité inné. Cette production peut être indépendante de l'IL-23. Le bimekizumab inhibe les cytokines pro-inflammatoires, ce qui entraîne la normalisation de l'inflammation cutanée et une diminution importante de l'inflammation locale et systémique, et par conséquent une amélioration des signes et symptômes cliniques associés au psoriasis, au rhumatisme psoriasique et à la spondyloarthrite axiale. À partir de modèles in vitro, il a été démontré que le bimekizumab inhibe l'expression du gène lié au psoriasis, la production de cytokines, la migration des cellules inflammatoires et l'ostéogénèse pathologique dans une plus grande mesure que l'inhibition de l'IL 17A seule.
Pharmacodynamique
Aucune étude formelle sur la pharmacodynamique du bimekizumab n'a été menée.
Efficacité clinique
Psoriasis en plaques
La sécurité d'emploi et l'efficacité du bimekizumab ont été évaluées chez 1 480 patients atteints de psoriasis en plaques modéré à sévère dans trois études de phase III multicentriques, randomisées, contrôlées par placebo et/ou par comparateur actif. Les patients étaient âgés d'au moins 18 ans, avaient un score PASI (indice d'étendue et de sévérité du psoriasis [Psoriasis Area and Severity Index]) ≥12 et un score d'évaluation globale des investigateurs (Investigators Global Assessment, IGA) ≥3 sur une échelle de 5 points, une surface corporelle (BSA, Body Surface Area) affectée par le psoriasis (PSO) ≥10 %, et étaient candidats pour un traitement systémique du psoriasis et/ou une photothérapie. L'efficacité et la sécurité d'emploi du bimekizumab ont été évaluées versus placebo et ustékinumab (BE VIVID – PS0009), versus placebo (BE READY – PS0013) et versus adalimumab (BE SURE – PS0008).
L'étude BE VIVID a évalué 567 patients pendant 52 semaines. Les patients y ont été randomisés afin de recevoir du bimekizumab (320 mg toutes les 4 semaines), de l'ustékinumab (en fonction du poids du patient 45 mg ou 90 mg à l'inclusion, à la semaine 4, puis toutes les 12 semaines) ou un placebo pendant une période initiale de 16 semaines, puis du bimekizumab (320 mg toutes les 4 semaines).
L'étude BE READY a évalué 435 patients pendant 56 semaines. Les patients ont été randomisés afin de recevoir du bimekizumab 320 mg toutes les 4 semaines ou un placebo. À la semaine 16, les patients ayant obtenu une réponse PASI 90 sont entrés dans une période randomisée de 40 semaines permettant un retrait. Les patients initialement randomisés pour recevoir du bimekizumab 320 mg toutes les 4 semaines ont été de nouveau randomisés afin de recevoir du bimekizumab 320 mg toutes les 4 semaines, du bimekizumab 320 mg toutes les 8 semaines ou un placebo (c'est-à-dire, retrait du bimekizumab). Les patients initialement randomisés pour recevoir un placebo ont continué à recevoir le placebo s'ils étaient répondeurs PASI 90. Les patients n'ayant pas obtenu une réponse PASI 90 à la semaine 16 sont entrés dans un bras d'échappement en ouvert et ont reçu du bimekizumab 320 mg toutes les 4 semaines pendant 12 semaines. Les patients en rechute (qui n'ont pas obtenu de réponse PASI 75) pendant la période randomisée permettant un retrait sont également entrés dans le bras d'échappement de 12 semaines.
L'étude BE SURE a évalué 478 patients pendant 56 semaines. Les patients ont été randomisés pour recevoir du bimekizumab 320 mg toutes les 4 semaines jusqu'à la semaine 56, du bimekizumab 320 mg toutes les 4 semaines jusqu'à la semaine 16 puis du bimekizumab 320 mg toutes les 8 semaines jusqu'à la semaine 56 ou de l'adalimumab selon la recommandation autorisée jusqu'à la semaine 24 puis du bimekizumab 320 mg toutes les 4 semaines jusqu'à la semaine 56.
Les caractéristiques à l'inclusion étaient homogènes sur l'ensemble des 3 études. La surface corporelle BSA médiane à l'inclusion était de 20 %, le score PASI médian à l'inclusion était de 18 et le score IGA à l'inclusion était sévère chez 33 % des patients. Les scores médians à l'inclusion des éléments douleur, démangeaisons et desquamation du Journal des Symptômes du patient (JSP) étaient compris entre 6 et 7 sur une échelle de 0 à 10 points et le score total médian de l'Indice de qualité de vie en dermatologie (Dermatology Life Quality Index, DLQI) était de 9.
Dans les 3 études, 38 % des patients avaient reçu un traitement biologique antérieur; 23 % avaient reçu au moins un agent anti-IL17 et 13 % avaient reçu au moins un anti-TNF. 22 % étaient naïfs de tout traitement systémique (biologique et non biologique) et 39 % des patients avaient précédemment reçu une photothérapie ou une photochimiothérapie.
L'efficacité du bimekizumab a été évaluée en termes d'impact sur la maladie cutanée globale, au niveau de régions corporelles spécifiques (cuir chevelu, ongles, paumes des mains et plantes des pieds), de symptômes rapportés par les patients et d'impact sur la qualité de vie. Les deux co-critères d'évaluation principaux dans les 3 études étaient la proportion de patients ayant obtenu 1) une réponse PASI 90 et 2) une réponse IGA « blanchi ou quasiment blanchi » (IGA 0/1 avec au moins deux points d'amélioration par rapport à l'inclusion) à la semaine 16. La réponse PASI 100, IGA 0 à la semaine 16 ainsi que la réponse PASI 75 à la semaine 4 étaient les critères d'évaluation secondaires les plus importants dans les 3 études.
Maladie cutanée globale
Le traitement par bimekizumab a entraîné une amélioration significative de tous les paramètres d'activité de la maladie par rapport au placebo, à l'ustékinumab ou à l'adalimumab à la semaine 16. Les principaux résultats d'efficacité sont présentés dans le Tableau 2.
Tableau 2: Résumé des réponses cliniques dans les études BE VIVID, BE READY et BE SURE
|
|
BE VIVID
|
BE READY
|
BE SURE
| |
|
Placebo
(n = 83)
n (%)
|
Bimekizumab 320 mg/4 sem.
(n = 321)
n(%))
|
Ustékinumab
(n = 163)
n (%)
|
Placebo
(n = 86)
n (%)
|
Bimekizumab 320 mg/4 sem.
(n = 349)
n (%)
|
Bimekizumab 320 mg/4 sem.
(n = 319)
n (%)
|
Adalimumab
(n = 159)
n (%)
| |
PASI 100
Semaine 16
|
0 (0,0)
|
188 (58,6)a
|
34 (20,9)
|
1 (1,2)
|
238 (68,2)a
|
194 (60,8)a
|
38 (23,9)
| |
PASI 90
Semaine 16
|
4 (4,8)
|
273 (85,0)a, b
|
81 (49,7)
|
1 (1,2)
|
317 (90,8)a
|
275 (86,2)a
|
75 (47,2)
| |
PASI 75
Semaine 4
Semaine 16
|
2 (2,4)
6 (7,2)
|
247 (76,9)a, b
296 (92,2)
|
25 (15,3)
119 (73,0)
|
1 (1,2)
2 (2,3)
|
265 (75,9)a
333 (95,4)
|
244 (76,5)a
295 (92,5)
|
50 (31,4)
110 (69,2)
| |
IGA 0
Semaine 16
|
0 (0,0)
|
188 (58,6)a
|
36 (22,1)
|
1 (1,2)
|
243 (69,6)a
|
-
|
-
| |
IGA 0/1
Semaine 16
|
4 (4,8)
|
270 (84,1)a, b
|
87 (53,4)
|
1 (1,2)
|
323 (92,6)a
|
272 (85,3)a
|
91 (57,2)
| |
PASI absolu ≤2
Semaine 16
|
3 (3,6)
|
273 (85,0)
|
84 (51,5)
|
1 (1,2)
|
315 (90,3)
|
280 (87,8)
|
86 (54,1)
| |
Douleurs JSP (n)
Semaine 16
|
(n = 54)
9 (16,7)
|
(n = 229)
177 (77,3)a
|
(n = 107)
73 (68,2)
|
(n = 67)
6 (9,0)
|
(n = 255)
201 (78,8)a
|
-
|
-
| |
Démangeaisons JSP (n)
Semaine 16
|
(n = 61)
8 (13,1)
|
(n = 244)
187 (76,6)a
|
(n = 117)
77 (65,8)
|
(n = 72)
4 (5,6)
|
(n = 278)
210 (75,5)a
|
-
|
-
| |
Desquamation JSP (n)
Semaine 16
|
(n = 63)
8 (12,7)
|
(n = 246)
193 (78,5)a
|
(n = 116)
69 (59,5)
|
(n = 70)
4 (5,7)
|
(n = 286)
223 (78,0)a
|
-
|
-
|
Bimekizumab 320 mg /4 sem. = bimekizumab toutes les 4 semaines. L'imputation des non-répondeurs (NRI) a été utilisée.
La réponse IGA 0/1 était définie comme Blanchi (0) ou Quasiment blanchi (1) avec une amélioration d'au moins 2 catégories entre l'inclusion et la semaine 16. La réponse IGA 0 était définie comme Blanchi (0) avec une amélioration d'au moins 2 catégories entre l'inclusion et la semaine 16.
JSP est un Journal des symptômes du patient. La réponse JSP est définie comme une évolution de l'inclusion à la semaine 16 ≥ un seuil défini (1,98, 2,39 et 2,86 respectivement pour les douleurs, les démangeaisons et la desquamation).
a) p < 0,001 versus placebo (BE VIVID et BE READY), versus adalimumab (BE SURE), ajusté pour la multiplicité.
b) p < 0,001 versus ustékinumab (BE VIVID), ajusté pour la multiplicité.
Le traitement par bimekizumab a été associé à une apparition rapide de l'efficacité. Dans l'étude BE VIVID, aux semaines 2 et 4, les taux de réponse PASI 90 étaient significativement plus élevés chez les patients traités par bimekizumab (12,1 % et 43,6 % respectivement) par rapport à ceux traités par placebo (1,2 % et 2,4 % respectivement) et par ustékinumab (1,2 % et 3,1 % respectivement).
Figure 1: Taux de répondeurs PASI 90 au fil du temps dans l'étude BE VIVID
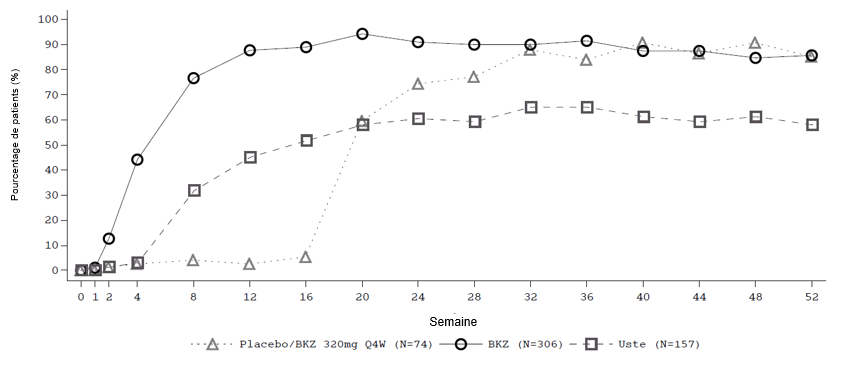
BKZ = Bimekizumab, Usté = Ustékinumab; la NRI est utilisée.
Remarque: Les patients du groupe placebo/BKZ sont passés du placebo au BKZ lors de la période d'entretien à partir de la semaine 16.
Dans l'étude BE VIVID, à la semaine 52, les patients traités par bimekizumab ont obtenu des taux de réponse significativement plus élevés que ceux traités par ustékinumab pour les critères d'évaluation PASI 90 (81,6 % sous bimekizumab vs 55,8 % sous ustékinumab, p < 0,001), de l'IGA 0/1 (77,9 % sous bimekizumab vs 60,7 % sous ustékinumab, p < 0,001) et du PASI 100 (64,2 % sous bimekizumab vs 38,0 % sous ustékinumab).
Dans l'étude BE SURE à la semaine 24, un pourcentage plus élevé de patients traités par bimekizumab ont obtenu des réponses PASI 90 et IGA 0/1 par rapport à l'adalimumab (85,6 % et 86,5 % respectivement vs 51,6 % et 57,9 % respectivement, p < 0,001). Parmi les 65 non-répondeurs à l'adalimumab à la semaine 24 (PASI < 90), 78,5 % ont obtenu une réponse PASI 90 après 16 semaines de traitement par bimekizumab. Chez les patients qui sont passés de l'adalimumab au bimekizumab, le profil de sécurité d'emploi est resté semblable. À la semaine 56, 70,2 % des patients traités par bimekizumab ont obtenu une réponse PASI 100.
Figure 2: Taux de répondeurs PASI 90 au fil du temps dans l'étude BE SURE
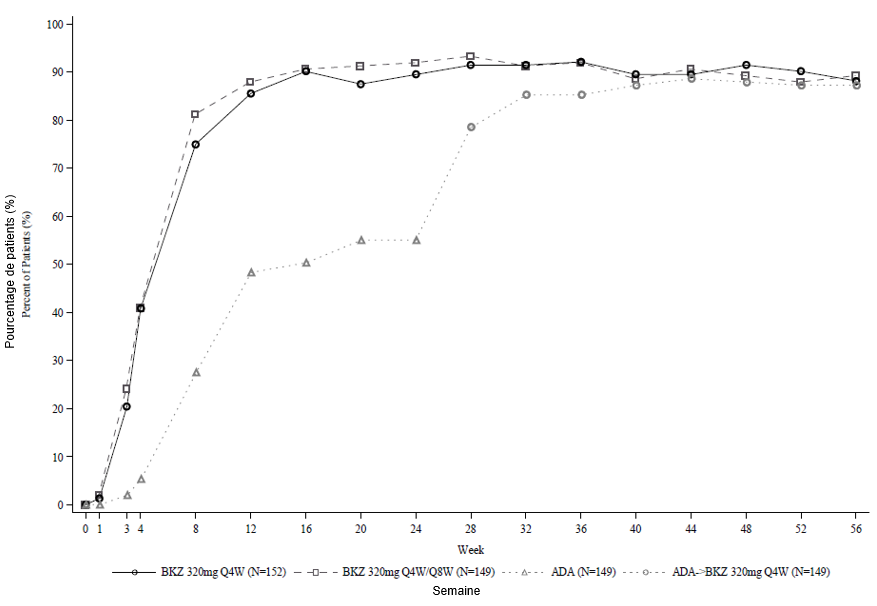
BKZ 320 mg /4 sem. = bimekizumab toutes les 4 semaines; BKZ 320 mg /8 sem. = bimekizumab toutes les 8 semaines; ADA = adalimumab.
Remarque: Ce graphique ne contient que les patients ayant reçu du bimekizumab à la semaine 24 ou ultérieurement. Les patients du groupe BKZ /4 sem./8 sem. sont passés d'une administration /4 sem. à /8 sem. à la semaine 16. Les patients du groupe ADA/BKZ 320 mg /4 sem. sont passés de l'ADA au BKZ /4 sem. à la semaine 24. La NRI est utilisée.
L'efficacité du bimekizumab a été démontrée indépendamment de l'âge, du sexe, de l'origine ethnique, de la durée de la maladie, du poids corporel, de la sévérité PASI à l'inclusion et d'un traitement antérieur par un agent biologique. Le bimekizumab a été efficace chez les patients exposés à un agent biologique antérieur, notamment un anti-TNF/anti-IL-17 ainsi que chez les patients naïfs de traitement systémique. Compte-tenu des analyses PK/PD de population étayées par les données cliniques, les patients avec un poids corporel élevé (≥120 kg) qui n'avaient pas obtenu un blanchiment cutané complet à la semaine 16 ont bénéficié de la poursuite du bimekizumab 320 mg toutes les quatre semaines (/4 sem.) après les 16 premières semaines de traitement.
Dans l'étude BE SURE, les patients ont reçu du bimekizumab 320 mg /4 sem. jusqu'à la semaine 16, puis une administration /4 sem. ou toutes les huit semaines (/8 sem.) jusqu'à la semaine 56, indépendamment du statut de répondeur à la semaine 16. Les patients du groupe ≥120 kg (N = 37) traités par le schéma d'entretien /4 sem. ont présenté une amélioration plus importante du PASI 100 entre la semaine 16 (23,5 %) et la semaine 56 (70,6 %) par rapport à ceux traités par le schéma d'entretien 8 sem. (semaine 16: 45,0 % vs. semaine 56: 60,0 %).
Maintien de la réponse
Tableau 3: Maintien des réponses à la semaine 52 chez les répondeurs à la semaine 16*
|
PASI 100
|
PASI 90
|
IGA 0/1
|
PASI absolu ≤2
| |
BKZ 320 mg /4 sem./4 sem.
(N = 355)
n (%)
|
BKZ 320 mg /4 sem./8 sem.
(N = 182)
n (%)
|
BKZ 320 mg /4 sem./4 sem.
(N = 516)
n (%)
|
BKZ 320 mg /4 sem./8 sem.
(N = 237)
n (%)
|
BKZ 320 mg /4 sem./4 sem.
(N = 511)
n (%)
|
BKZ 320 mg /4 sem./8 sem.
(N = 234)
n (%)
|
BKZ 320 mg /4 sem. /4 sem.
(N = 511)
n (%)
|
BKZ 320 mg /4 sem./8 sem.
(N = 238)
n (%)
| |
295 (83,1)
|
161 (88,5)
|
464 (89,9)
|
214 (90,3)
|
447 (87,5)
|
214 (91,5)
|
460 (90,0)
|
215 (90,3)
|
* Analyse intégrée des études BE VIVID, BE READY et BE SURE. La NRI est utilisée.
BKZ 320 mg /4 sem.: bimekizumab 320 mg toutes les 4 semaines, puis bimekizumab 320 mg toutes les 4 semaines à partir de la semaine 16. BKZ 320 mg /8 sem.: bimekizumab 320 mg toutes les 4 semaines, puis bimekizumab 320 mg toutes les 8 semaines à partir de la semaine 16.
Durabilité de la réponse PASI 90 (après l'arrêt du bimekizumab)
Figure 3: Taux de répondeurs PASI 90 au fil du temps – Période randomisée permettant un retrait de l'étude BE READY
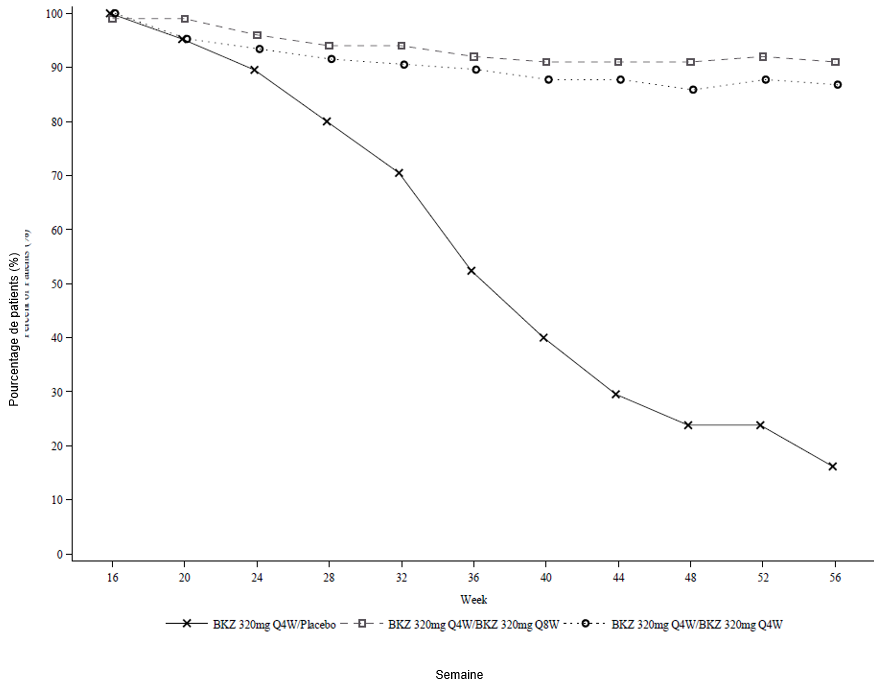
La NRI est utilisée.
Dans l'étude BE READY, pour les répondeurs PASI 90 à la semaine 16 qui ont été à nouveau randomisés afin de recevoir le placebo et de retirer le bimekizumab, le délai médian jusqu'à la rechute, définie comme une perte de réponse PASI 75, était d'environ 28 semaines (32 semaines après la dernière dose de bimekizumab). Parmi ces patients, 88,1 % ont de nouveau obtenu une réponse PASI 90 dans les 12 semaines suivant la reprise du traitement par bimekizumab 320 mg toutes les 4 semaines.
Régions corporelles spécifiques
Au cours des études BE VIVID et BE READY, des améliorations du psoriasis du cuir chevelu, des ongles, de la paume des mains et de la plante des pieds ont été observées chez les patients traités par bimekizumab vs. placebo à la semaine 16 (voir Tableau 4).
Tableau 4: Réponses des régions corporelles spécifiques dans les études BE VIVID et BE READY à la semaine 16
|
|
BE VIVID
|
BE READY
| |
|
Placebo
|
BKZ 320 mg /4 sem.
|
Ustékinumab
|
Placebo
|
BKZ 320 mg /4 sem.
| |
IGA cuir chevelu (n)a
IGA cuir chevelu 0/ 1, n(%)
|
(72)
11 (15,3)
|
(285)
240 (84,2)b
|
(146)
103 (70,5)
|
(74)
5 (6,8)
|
(310)
286 (92,3)b
| |
IGA-pp (n)a
IGA-pp 0/1, n (%)
|
(29)
7 (24,1)
|
(105)
85 (81,0)
|
(47)
39 (83,0)
|
(31)
10 (32,3)
|
(97)
91 (93,8)
| |
mNAPSI 100 (n)a
mNAPSI 100, n(%)
|
(51)
4 (7,8)
|
(194)
57 (29,4)
|
(109)
15 (13,8)
|
(50)
3 (6,0)
|
(210)
73 (34,8)
|
La NRI est utilisée.
a) Comprend uniquement les patients avec une évaluation globale de l'investigateur (IGA) du cuir chevelu de 2 ou plus, une IGA palmo-plantaire de 2 ou plus et un score de l'Indice modifié de gravité du psoriasis unguéal (modified Nail Psoriasis Severity Index, mNAPSI) > 0 à l'inclusion. Les réponses IGA 0/1 pour le cuir chevelu et palmo-plantaire (IGA-pp 0/1) ont été définies comme Blanchi (0) ou Quasiment blanchi (1) avec une amélioration d'au moins 2 catégories par rapport à l'inclusion.
b) p < 0,001 versus placebo, ajusté pour la multiplicité.
Les réponses IGA du cuir chevelu et palmo-plantaire se sont maintenues jusqu'à la semaine 52/56. Le psoriasis unguéal a continué à s'améliorer au-delà de la semaine 16. Dans l'étude BE VIVID, à la semaine 52, une plus grande proportion de patients traités par bimekizumab a obtenu une disparition complète du psoriasis unguéal (mNAPSI 100) que chez les patients traités par ustékinumab (60,3 % contre 40,4 %). Dans l'étude BE READY, à la semaine 56, 67,7 % et 69,8 % des répondeurs PASI 90 à la semaine 16 ont obtenu une disparition complète du psoriasis unguéal sous respectivement bimekizumab 320 mg toutes les 8 semaines et bimekizumab 320 mg toutes les 4 semaines.
Qualité de vie / Résultats rapportés par le patient
Dans les 3 études, à la semaine 16, le psoriasis ne présentait plus aucun impact sur la qualité de vie, mesurée par l'Indice de la qualité de vie en dermatologie (DLQI) chez une plus grande proportion de patients traités par bimekizumab.
Dans l'étude BE READY, le taux de réponse DLQI 0/1 (pas d'impact du psoriasis sur la qualité de vie) à la semaine 16 était de 75,6 % dans le groupe bimekizumab contre 5,8 % dans le groupe placebo.
Dans l'étude BE VIVID, les taux de réponse DLQI 0/1 à la semaine 16 étaient de 67,3 %, 42,3 % et 12,0 % respectivement dans les groupes bimekizumab, ustékinumab- et placebo. Les réponses DLQI 0/1 ont continué à augmenter au-delà de la semaine 16, puis se sont maintenues jusqu'à la semaine 52 (74,8 % chez les patients traités par bimekizumab 320 mg toutes les 4 semaines).
Dans l'étude BE SURE, les taux de réponse DLQI 0/1 à la semaine 16 étaient de 63,0 % et 46,5 % respectivement dans les groupes bimekizumab et adalimumab. Le taux de réponse DLQI 0/1 à la semaine 56 était de 78,9 % et de 74,1 % chez les patients traités par bimekizumab 320 mg toutes les 8 semaines et par bimekizumab 320 mg toutes les 4 semaines, respectivement.
Étude d'extension de phase III en ouvert
Les patients ayant terminé l'une des études pivots de phase III (« études mères ») pouvaient participer à une étude d'extension en ouvert de 144 semaines (PS0014) visant à évaluer la sécurité d'emploi et l'efficacité à long terme du bimekizumab.
344 patients traités par 320 mg de bimekizumab toutes les 8 semaines (BKZ 320 mg 1x/8 sem.) ou toutes les 4 semaines (BKZ 320 mg 1x/4 sem.) au cours de l'étude mère et ayant obtenu un score PASI 90 à la fin de celle-ci, ont reçu le bimekizumab 320 mg 1x/8 sem. pendant toute la durée de l'étude PS0014. Sur le total de ces patients, 293 (85,2 %) ont terminé les 144 semaines de traitement par bimekizumab 320 mg 1x/8 sem. 48 patients (14,0 %) ont arrêté l'étude pendant la période de traitement, dont 21 (6,1 %) en raison d'un événement indésirable et 4 (1,2 %) en raison d'un manque d'efficacité.
Parmi les patients restés dans l'étude, les améliorations obtenues avec le bimekizumab sur les critères d'évaluation de l'efficacité PASI 90 et IGA 0/1 dans les études mères ont été maintenues pendant les 144 semaines supplémentaires du traitement en ouvert.
Période d'extension de la phase IIIb en ouvert
À la semaine 48, les patients pouvaient participer à une période d'extension en ouvert (open label extension, OLE) de 96 semaines et commencer ou continuer le traitement par bimekizumab 320 mg 1x/4 semaines ou 320 mg 1x/8 semaines en fonction de leur statut de répondeur PASI 90 à la semaine 48. Les participants à l'étude qui avaient initialement reçu le bimekizumab 320 mg 1x/4 semaines au cours de la phase OLE étaient passés sous bimekizumab 320 mg 1x/8 semaines à la semaine 72 ou plus tard.
231 patients qui avaient été traités par bimekizumab 320 mg 1x/8 semaines ou bimekizumab 320 mg 1x/4 semaines et qui avaient obtenu un score PASI 90 à la semaine 48 ont reçu du bimekizumab 320 mg 1x/8 semaines pendant toute la période OLE. Parmi ces patients, 31 (13,4 %) ont arrêté l'étude pendant la période OLE, dont 10 (4,3 %) ont arrêté à cause d'un événement indésirable et 1 (0,4 %) en raison d'un manque d'efficacité.
116 patients qui avaient été traités par le secukinumab et qui avaient obtenu un score PASI 90 à la semaine 48 ont reçu du bimekizumab 320 mg 1x/8 semaines pendant toute la période OLE. Parmi ces patients, 16 (13,8 %) ont arrêté l'étude pendant la période OLE, dont 6 (5,2 %) ont arrêté à cause d'un événement indésirable et 1 (0,9 %) en raison d'un manque d'efficacité.
Chez les patients restant dans l'étude, les améliorations obtenues à la semaine 48 avec le bimekizumab ou le secukinumab pour les critères d'évaluation de l'efficacité suivants, les scores PASI 100, PASI 90, PASI 75 et la réponse PASI ≤2, ont été maintenues sous traitement par bimekizumab 320 mg 1x/8 semaines pendant 96 semaines supplémentaires de traitement en ouvert.
Le profil de sécurité du bimekizumab jusqu'à la semaine 144 était cohérent avec le profil de sécurité observé pendant 48 semaines.
Rhumatisme psoriasique (RP)
La sécurité et l'efficacité du bimekizumab ont été évaluées chez 1112 patients adultes (âgés d'au moins 18 ans) atteints de rhumatisme psoriasique (RP) actif dans deux études multicentriques, randomisées, en double aveugle, contrôlées contre placebo (PA0010 – BE OPTIMAL et PA0011 – BE COMPLETE). L'étude BE OPTIMAL comprenait un bras de traitement référence actif (adalimumab) (N = 140).
Pour les deux études, les patients avaient reçu un diagnostic de rhumatisme psoriasique actif depuis au moins 6 mois, basé sur les Critères de classification du rhumatisme psoriasique (Classification Criteria for Psoriatic Arthritis, CASPAR) et présentaient une maladie active avec un nombre d'articulations douloureuses (NAD) ≥3 et un nombre d'articulations gonflées (NAG) ≥3. Les patients avaient reçu un diagnostic de RP depuis, 3,6 ans en médiane dans BE OPTIMAL et 6,8 ans dans BE COMPLETE. Des patients présentant plusieurs sous-types de RP ont été inclus dans ces études, dont l'arthrite symétrique polyarticulaire, l'arthrite asymétrique oligoarticulaire, une atteinte prédominante des articulations interphalangiennes distales, une atteinte prédominante axiale et l'arthrite mutilante. À l'inclusion, 55,9 % des patients présentaient ≥3 % de la surface corporelle (SCo) atteinte par un psoriasis en plaques actif. 10,4 % des patients étaient atteints d'un psoriasis en plaque modéré à sévère et 31,9 % et 12,3 % étaient atteints d'enthésite et de dactylite à l'inclusion, respectivement. Le critère d'évaluation principal dans les deux études était la réponse ACR 50 (American College of Rheumatology) à la semaine 16.
Les principaux critères d'évaluation secondaires à la semaine 16 dans les deux études étaient les suivants: variation par rapport à l'inclusion du Health Assessment Questionnaire - Disability Index (cfB HAQ-DI), réduction du Psoriasis Area and Severity Index de 90 % par rapport à l'inclusion (PASI90), variation par rapport à l'inclusion du score du questionnaire Short Form-36 (Short Form 36-item Health Survey (SF-36) Physical Component Summary [PCS]), réponse à l'activité minimale de la maladie (Minimal Disease Activity [MDA]), ainsi qu'état exempt d'enthésite et de dactylite basé sur les données poolées des deux études. Dans l'étude BE OPTIMAL, la variation par rapport à l'inclusion du score total de Sharp modifié par Van der Heijde (Van der Heijde modified Total Sharp Score, vdHmTSS) était également un critère secondaire important.
L'étude BE OPTIMAL a évalué 852 patients naïfs de toute exposition à un traitement biologique (bDMARDs) pour le traitement du rhumatisme psoriasique ou du psoriasis. Les patients ont été randomisés (selon un rapport de 3:2:1) afin de recevoir bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52 ou le placebo jusqu'à la semaine 16, suivi par bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52 ou ont été affectés à un bras de traitement de référence actif (adalimumab 40 mg toutes les 2 semaines) jusqu'à la semaine 52. Dans cette étude, 78,3 % des patients avaient reçu un traitement antérieur par ≥1 cDMARDs et 21,7 % des patients étaient naïfs de traitement antérieur par cDMARDs. À l'inclusion, 58,2 % des patients recevaient un traitement concomitant par méthotrexate (MTX), 11,3 % recevaient un traitement concomitant par d'autres cDMARDs que le MTX, et 30,5 % ne recevaient aucun cDMARDs.
L'étude BE COMPLETE a évalué 400 patients présentant une réponse inadéquate (manque d'efficacité) ou une intolérance au traitement par 1 ou 2 inhibiteurs du facteur de nécrose tumorale alpha (patients anti-TNFα-IR) pour le rhumatisme psoriasique ou le psoriasis. Les patients ont été randomisés (selon un rapport de 2:1) pour recevoir bimekizumab 160 mg toutes les 4 semaines ou le placebo jusqu'à la semaine 16. À l'inclusion, 42,5 % des patients recevaient un traitement concomitant par MTX, 8,0 % recevaient un traitement concomitant par d'autres cDMARDs que le MTX, et 49,5 % ne recevaient aucun cDMARDs. Dans cette étude, 76,5 % des participants ont présenté une réponse inadéquate à un inhibiteur du TNFα, 11,3% ont présenté une réponse inadéquate à 2 inhibiteurs du TNFα et 12,3 % étaient intolérants aux inhibiteurs du TNFα.
Signes et symptômes
Chez les patients naïfs de bDMARD (BE OPTIMAL) et les patients anti-TNFα-IR (BE COMPLETE), le traitement par bimekizumab s'est traduit par une amélioration significative des signes et symptômes et des mesures de l'activité de la maladie comparativement au placebo à la semaine 16, avec des taux de réponse similaires observés dans les deux populations de patients (voir Tableau 5). Dans l'évaluation selon ACR 50, MDA, PASI 90, la réponse clinique s'est maintenue jusqu'à la semaine 52 dans l'étude BE OPTIMAL.
Tableau 5: Réponse clinique dans les études BE OPTIMAL et BE COMPLETE
|
|
BE OPTIMAL (patients naïfs de bDMARD)
|
BE COMPLETE (anti-TNFα-IR)
| |
|
Placebo
(N = 281)
n (%)
|
BKZ 160 mg toutes les 4 semaines
(N = 431)
n (%)
|
Différence par rapport au placebo (IC à 95 %)(d)
|
Traitement de référence (e) (Adalimumab)
(N = 140)
n (%)
|
Placebo
(N = 133)
n (%)
|
BKZ 160 mg toutes les 4 semaines
(N = 267)
n (%)
|
Différence par rapport au placebo (IC à 95 %)(d)
| |
ACR 50
|
|
|
|
|
|
|
| |
Semaine 16
|
28 (10,0)
|
189 (43,9)*
|
33,9 (27,4; 40,4)
|
64 (45,7)
|
9 (6,8)
|
116 (43,4)*
|
36,7 (27,7; 45,7)
| |
Semaine 24
|
-
|
196 (45,5)
|
|
66 (47,1)
|
|
|
| |
Semaine 52
|
|
235 (54,5)
|
|
70 (50,0)
|
|
|
| |
MDA(a)
|
|
|
|
|
|
|
| |
Semaine 16
|
37 (13,2)
|
194 (45,0)*
|
31,8 (25,2; 38,5)
|
63 (45,0)
|
8 (6,0)
|
118 (44,2)*
|
38,2 (29,2; 47,2)
| |
Semaine 24
|
-
|
209 (48,5)
|
|
67 (47,9)
|
|
|
| |
Semaine 52
|
|
237 (55,0)
|
|
74 (52,9)
|
|
|
| |
Patients avec BSA ≥3 %
|
(N = 140)
|
(N = 217)
|
|
(N = 68)
|
(N = 88)
|
(N = 176)
|
| |
PASI 90
|
|
|
|
|
|
|
| |
Semaine 16
|
4 (2,9)
|
133 (61,3)*
|
58,4 (49,9; 66,9)
|
28 (41,2)
|
6 (6,8)
|
121 (68,8)*
|
61,9 (51,5; 72,4)
| |
Semaine 24
|
-
|
158 (72,8)
|
|
32 (47,1)
|
|
|
| |
Semaine 52
|
|
155 (71,4)
|
|
41 (60,3)
|
|
|
| |
Patients avec LDI > 0 (b)
|
(N = 47)
|
(N = 90)
|
|
|
| |
Résolution totale de dactylite (b)
Semaine 16
|
24 (51,1)
|
68 (75,6)***
|
24,5 (8,4; 40,6)
|
| |
Patients avec LEI > 0 (c)
|
(N = 106)
|
(N = 249)
|
|
| |
Résolution totale d'enthésite (c)
Semaine 16
|
37 (34,9)
|
124 (49,8)**
|
14,9 (3,7; 26,1)
|
|
|
BKZ 160 mg toutes les 4 semaines = bimekizumab 160 mg toutes les 4 semaines. IC = intervalle de confiance, NC = ne peut être déterminé (not calculable)
(a) Un patient était classé comme atteignant l'activité minimale de la maladie (MDA) lorsqu'il répondait à 5 des 7 critères suivants: nombre d'articulations douloureuses ≤1; nombre d'articulations gonflées ≤1; Indice d'activité et de sévérité du psoriasis ≤1 ou surface corporelle ≤3; échelle analogique visuelle (EVA) de la douleur du patient ≤15; EVA de l'activité globale de la maladie du patient ≤20; Questionnaire d'évaluation de la santé- Indice d'invalidité(HAQ-DI) ≤0,5; points d'enthèse douloureux ≤1
(b) Basé sur les données regroupées des études BE OPTIMAL et BE COMPLETE pour les patients présentant un Indice de dactylite de Leeds (LDI) à l'inclusion > 0. La résolution totale de la dactylite correspond à un LDI = 0
(c) Basé sur les données regroupées des études BE OPTIMAL et BE COMPLETE pour les patients présentant un Indice de d'enthésite de Leeds (LEI) à l'inclusion > 0. La résolution totale de l'enthésite correspond à un LEI = 0
(d) Les différences non ajustées sont présentées
(e) Aucune comparaison statistique avec le bimekizumab ou le placebo n'a été effectuée
* p < 0,001 versus placebo ajusté pour la multiplicité.
** p = 0,008 versus placebo ajusté pour la multiplicité.
*** p = 0,002 versus placebo ajusté pour la multiplicité. La NRI est utilisée. Les autres critères d'évaluation à la semaine 16 et tous les critères d'évaluation aux semaines 24 et 52 ne faisaient pas partie de la hiérarchie des tests séquentiels et toutes les comparaisons sont nominales
Les résultats de l'étude BE OPTIMAL ont été similaires pour les patients traités par cDMARD (ACR50 semaine 16: bimekizumab 160 mg toutes les 4 semaines: 43,5 %, placebo: 9,5 %).
Pour les patients de BE OPTIMAL sous bimekizumab, des améliorations par rapport à l'inclusion ont été constatées à la semaine 16 pour chacun des composants ACR, qui se sont poursuivies jusqu'à la semaine 52.
La réponse au traitement par bimekizumab était significativement plus élevée à la semaine 4 pour l'ACR 50 (BE OPTIMAL 17,6 % versus 3,2 %, p nominal < 0,001 et BE COMPLETE 16,1 % versus 1,5 %, p nominal < 0,001) que sous placebo.
Pour les patients traités par bimekizumab ayant obtenu une réponse ACR50 à la semaine 16 dans l'étude BE OPTIMAL, 87,2 % maintenaient cette réponse à la semaine 52.
L'efficacité et la sécurité d'emploi du bimekizumab ont été démontrées indépendamment de l' âge, du sexe, de l'origine ethnique, du poids corporel à l'inclusion, de l'atteinte psoriasique à l'inclusion, de la CRP à l'inclusion, de la durée de la maladie et de l'utilisation antérieure de cDMARD. Dans les deux études, des réponses similaires ont été observées avec bimekizumab que les patients soient sous traitement concomitant par cDMARDs, comprenant du MTX, ou non.
Réponse radiographique
Dans l'étude BE OPTIMAL, l'inhibition de la progression des lésions structurales a été évaluée radiographiquement et exprimée en variation par rapport à l'inclusion, du score total de Sharp modifié par Van der Heijde (vdHmTSS) et ses composants, le score d'érosion (SE) et le score de pincement articulaire (JSN) à la semaine 16 (voir Tableau 6).
Tableau 6: Variation du vdHmTSS dans l'étude BE OPTIMAL à la semaine 16
|
|
Placebo
|
BKZ 160 mg toutes les 4 semaines
|
Différence par rapport au placebo (IC à 95 %) a)
| |
Population présentant une augmentation de la hs-CRP et/ou au moins 1 érosion osseuse par rapport à l'inclusion
|
(N = 227)
|
(N = 361)
|
| |
Variation moyenne par rapport à l'inclusion (SE)
|
0,36 (0,10)
|
0,04 (0,05)*
|
-0,32 (-0,35; -0,30)
| |
Population globale
|
(N = 269)
|
(N = 420)
|
| |
Variation moyenne par rapport à l'inclusion (SE)
|
0,32 (0,09)
|
0,04 (0,04)*
|
-0,26 (-0,29; -0,23)
|
*p = 0,001 par rapport au placebo. Les valeurs p reposent sur une imputation basée sur les références utilisant la différence de moyenne des moindres carrés à l'aide d'un modèle ANCOVA avec le traitement, l'érosion osseuse à l'inclusion et la région comme effets fixes et le score à l'inclusion comme covariables.
Les données récapitulatives de la semaine 16 sont basées sur le premier ensemble de valeurs pour l'analyse primaire.
a)Les différences non ajustées sont affichées
Le bimekizumab a significativement inhibé la progression des dommages articulaires jusqu'à la semaine 16, à la fois dans la population présentant une hs-CRP élevée et/ou au moins une érosion osseuse par rapport à l'inclusion et dans la population globale par rapport au placebo. Bien que l'imputation basée sur les références ait été définie comme une méthode permettant de traiter les données manquantes dans la procédure de test statistique pour comparer le bimekizumab et le placebo, les variations par rapport à l'inclusion ont été observés à la fois dans la population présentant une hs-CRP élevée et/ou au moins une érosion osseuse et dans la population totale à la semaine 16 dans le bras bimekizumab (variation moyenne par rapport à l'inclusion de 0,01 et 0,01, respectivement) et dans le bras adalimumab (variation moyenne par rapport à l'inclusion de -0,05 et 0,03, respectivement) également calculée par imputation multiple standard. L'inhibition de la progression des dommages articulaires a été maintenue à la fois dans la population présentant une hs-CRP élevée et/ou au moins une érosion osseuse par rapport à l'inclusion et dans la population globale jusqu'à la semaine 52 dans le bras bimekizumab (variation moyenne par rapport à l'inclusion 0,10 et 0,10 respectivement) et dans le bras adalimumab (variation moyenne par rapport à l'inclusion -0,17 et 0,12 respectivement).
Le pourcentage observé de patients ne présentant pas de progression radiographique des dommages articulaires (définie comme une variation du mTSS ≤0,5 par rapport à l'inclusion) depuis la randomisation jusqu'à la semaine 52 était de 87,9 % (N = 276/314) pour bimekizumab, de 84,8 % (N = 168/198) pour les participants à l'étude sous placebo et passés au bimekizumab et de 94,1 % (N = 96/102) pour l'adalimumab dans la population présentant une CRP-hs élevée et/ou au moins 1 érosion osseuse. Des taux similaires ont été observés dans la population globale (89,3 % (N = 326/365) pour bimekizumab, 87.3 % (N = 207-237) pour les participants à l'étude sous placebo et passé au bimekizumab, et 94,1% (N = 111/118) pour l'adalimumab).
Fonction physique et autres résultats liés à la santé
A la fois les patients naïfs de bDMARDs (BE OPTIMAL) et ceux antiTNFα-IR (BE COMPLETE) recevant bimekizumab ont montré par rapport à l'inclusion une amélioration significative de la fonction physique comparativement aux patients sous placebo à la semaine 16 (p < 0,001) selon l'évaluation HAQ-DI (variation de la moyenne des moindres carrés par rapport à l'inclusion: - 0,3 versus – 0,1 dans l'étude BE OPTIMAL et – 0,3 versus 0 dans l'étude BE COMPLETE, respectivement). Dans les deux études, une plus grande proportion de patients a obtenu une réduction cliniquement significative par rapport à l'inclusion, d'au moins 0,35 du score HAQ-DI dans le groupe bimekizumab comparé au groupe placebo à la semaine 16.
Les patients traités par bimekizumab ont rapporté une amélioration significative par rapport à l'inclusion de la composante physique du score SF-36 PCS (Short-Form 36 item - Health Survey Physical Component Summary) à la semaine 16 comparativement au placebo (variation de la moyenne des moindres carrés par rapport à l'inclusion: 6,3 versus 1,9, p < 0,001 dans l'étude BE OPTIMAL et 6,2 versus 0,1, p < 0,001 dans l'étude BE COMPLETE).
Dans les deux études, les patients traités par bimekizumab ont rapporté une réduction significative par rapport à l'inclusion de la fatigue, mesurée par le score FACIT-F (Functional Assessment of Chronic llness Therapy -Fatigue) à la semaine 16 comparativement au placebo. Une amélioration significative par rapport à l'inclusion a également été observée pour le score PsAID-12 (Psoriatic Arthritis Impact of Disease-12) dans le groupe traité par bimekizumab comparativement au groupe placebo à la semaine 16.
Les patients présentant une atteinte axiale à l'inclusion, environ 74 % des patients, (définie comme un score BASDAI ≥4 [Bath Ankylosing Spondylitis Disease Activity Index]) ont montré une amélioration plus élevée du score BASDAI par rapport à l'inclusion comparativement au placebo à la semaine 16.
Les améliorations obtenues à la semaine 16 dans toutes les mesures de la fonction physique et autres résultats de santé mentionnées ci-dessus (scores HAQ-DI, SF-36 PCS, FACIT-Fatigue, PsAID-12 et BASDAI) ont été maintenues jusqu'à la semaine 52 dans l'étude BE OPTIMAL.
Dans l'étude BE OPTIMAL, à la semaine 52, 65,5 % des patients traités par bimekizumab ont obtenu une résolution unguéale complète (mNAPSI=0 [Indice de sévérité du psoriasis unguéal modifié] chez les patients présentant un mNAPSI supérieur à 0 à l'inclusion).
Spondyloarthrite axiale (nr-axSpA-nr et SA)
La sécurité et l'efficacité du bimekizumab ont été évaluées chez 586 patients adultes (âgés d'au moins 18 ans) atteints de spondyloarthrite axiale (axSpA) active, dans deux études multicentriques, randomisées, en double aveugle, contrôlées contre placebo, l'une menée sur la spondyloarthrite axiale non radiographique (nr-axSpA) et l'autre menée sur la spondylarthrite ankylosante (SA), également appelée axSpA radiographique. Le critère d'évaluation principal dans les deux études était le pourcentage de patients ayant obtenu une réponse à l'Assessment of SpondyloArthritis International Society (ASAS) 40 à la semaine 16. Des résultats cohérents ont été observés dans les deux populations de patients.
L'étude BE MOBILE 1 (AS0010) a évalué 254 patients atteints de nr-axSpA active. Les patients avaient une axSpA (âge d'apparition des symptômes < 45 ans) répondant aux critères de classification de l'ASAS et avaient une maladie active, telle que définie par un indice BASDAI (Bath Ankylosing Spondylitis Disease Activity Index) ≥4 et une douleur rachidienne ≥4 sur une échelle de notation numérique (ENN) de 0 à 10 (d'après le 2ème Item du BASDAI 2) et aucune preuve d'évolution radiographique au niveau des articulations sacro-iliaques susceptible de répondre aux critères de New York modifiés pour la SA. Les patients présentaient également des signes objectifs d'inflammation, se traduisant par des concentrations élevées de protéine C réactive (CRP) et/ou des signes de sacro-iliite à l'imagerie par résonance magnétique (IRM), ainsi que des antécédents de réponse inadéquate à 2 antiinflammatoires non stéroïdiens (AINS) différents ou une intolérance ou une contre-indication aux AINS. Les patients ont été randomisés (selon un rapport de 1:1) pour recevoir bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52 ou du placebo jusqu'à la semaine 16, suivi par bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52. Lors de la visite d'inclusion, les patients présentaient des symptômes de nr-axSpA en moyenne depuis 9 ans (médiane de 5,5 ans). 10,6 % des patients avaient reçu un traitement antérieur par anti-TNFα.
L'étude BE MOBILE 2 (AS0011) a évalué 332 patients atteints de SA active, documentée par des preuves radiologiques (radiographie) répondant aux critères de New York modifiés pour la SA. Les patients étaient atteints d'une maladie active, telle que définie par un BASDAI ≥4 et une douleur rachidienne ≥4 sur une échelle de notation numérique (ENN) de 0 à 10 (d'après le 2ème Item du BASDAI). Les patients devaient avoir des antécédents de réponse inadéquate à 2 AINS différents, ou une intolérance ou une contre-indication aux AINS. Les patients ont été randomisés (selon un rapport de 2:1) pour recevoir bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52 ou du placebo jusqu'à la semaine 16 suivi par bimekizumab 160 mg toutes les 4 semaines jusqu'à la semaine 52. Lors de la visite d'inclusion, les patients présentaient des symptômes de SA en moyenne depuis 13,5 ans (médiane de 11 ans). 16,3 % des patients avaient reçu un traitement antérieur par anti-TNFα.
Réponse clinique
Le traitement par bimekizumab s'est traduit par une amélioration significative des signes et des symptômes et des mesures de l'activité de la maladie par rapport au placebo à la semaine 16 dans les populations de patients nr-axSpA et SA (voir Tableau 7). Les réponses cliniques ont été maintenues jusqu'à la semaine 52 dans les deux populations de patients, comme montré par tous les critères mesurés présentés dans le Tableau 7.
Tableau 7: Réponse clinique dans les études BE MOBILE 1 et BE MOBILE 2
|
|
BE MOBILE 1 (nr-ax-SpAax)
|
BE MOBILE 2 (SA)
| |
|
Placebo
(N = 126)
n (%)
|
BKZ 160 mg Q4W
(N = 128)
n (%)
|
Différence par rapport au placebo (IC à 95 %)a)
|
Placebo
(N = 111)
n (%)
|
BKZ 160 mg toutes les 4 semaines
(N = 221)
n (%)
|
Différence par rapport au placebo (IC à 95 %)a)
| |
ASAS 40
|
|
|
|
|
|
| |
Semaine 16
Semaine 52
|
27 (21,4)
|
61 (47,7)*
78 (60,9)
|
26,2 (14,9; 37,5)
|
25 (22,5)
|
99 (44,8)*
129 (58,4)
|
22,3 (11,5; 33,0)
| |
ASAS 40 sans prétraitement anti-TNFα
|
|
|
|
|
|
| |
Semaine 16
Semaine 52
|
(N = 109)
25 (22,9)
|
(N = 118)
55 (46,6)
73 (61,9)
|
24,8 (12,4; 37,1)
|
(N = 94)
22 (23,4)
|
(N = 184)
84 (45,7)*
108 (58,7)
|
22,3 (10,5; 34,0)
| |
ASAS 20
|
|
|
|
|
|
| |
Semaine 16
Semaine 52
|
48 (38,1)
|
88 (68,8)*
94 (73,4)
|
30,7 (19,0; 42,3)
|
48 (43,2)
|
146 (66,1)*
158 (71,5)
|
22,8 (11,8; 33,8)
| |
ASDAS – amélioration significative
|
|
|
|
|
|
| |
Semaine 16
Semaine 52
|
9 (7,1)
|
35 (27,3)*
47 (36,7)
|
20,2 (11,2; 29,3)
|
6 (5,4)
|
57 (25,8)*
71 (32,1)
|
20,4 (11,7; 29,1)
| |
BASDAI 50
|
|
|
|
|
|
| |
Semaine 16
Semaine 52
|
27 (21,4)
|
60 (46,9)
69 (53,9)
|
25,3 (14,0; 36,6)
|
29 (26,1)
|
103 (46,6)
119 (53,8)
|
20,5 (9,6; 31,4)
|
BKZ 160 mg Q4W = bimekizumab 160 mg toutes les 4 semaines. ASDAS = Ankylosing Spondylitis Disease Activity Score.
La NRI est utilisée.
a) Les différences non ajustées sont présentées.
* p < 0,001 vs placebo, corrigé pour la multiplicité.
La proportion de patients dans l'étude BE MOBILE 1 atteignant un score ASDAS < 2,1 (combinant ASDAS-inactive disease [maladie inactive; ID] et ASDAS-low disease [faible activité de la maladie; LD] à la semaine 16 était de 46,1 % dans le groupe bimekizumab contre 21,1 % dans le groupe placebo (imputation multiple). A la semaine 52, 61,6 % des patients du groupe bimekizumab ont obtenu un score ASDAS < 2,1, dont 25,2 % étaient en état de maladie inactive (score ASDAS < 1,3).
La proportion de patients dans l'étude BE MOBILE 2 atteignant un score ASDAS < 2,1 (combinant ASDAS-ID et ASDAS-LD) à la semaine 16 était de 44,8 % dans le groupe bimekizumab contre 17,4 % dans le groupe placebo (imputation multiple). A la semaine 52, 57,1 % des patients dans le groupe bimekizumab ont obtenu un score ASDAS < 2,1, dont 23,4 % étaient en état de maladie inactive (score ASDAS < 1,3).
Les quatre composantes de l'ASAS 40 (douleur rachidienne totale, raideur matinale, indice de fonction BASFI [Bath Ankylosing Spondylitis Functional Index] et PGADA [évaluation globale de l'activité de la maladie par le patient]) ont été améliorées avec le traitement par bimekizumab et ont contribué à la réponse ASAS 40 globale à la semaine 16; ces améliorations ont été maintenues jusqu'à la semaine 52 dans les deux populations de patients.
Les améliorations d'autre critères d'efficacité sont présentées dans le Tableau 8.
Tableau 8: Autres citères d'efficacité dans les études BE MOBILE 1 et BE MOBILE 2
|
|
BE MOBILE 1 (SpAax-nr)
|
BE MOBILE 2 (SA)
| |
|
Placebo
(N = 126)
|
BKZ 160 mg toutes les 4 semaines
(N = 128)
|
Placebo
(N = 111)
|
BKZ 160 mg toutes les 4 semaines
(N = 221)
| |
Douleurs rachidiennes nocturnes
|
|
|
|
| |
Inclusion
Variation moyenne entre l'inclusion et la semaine 16
|
6,7
-1,7
|
6,9
-3,6*
|
6,8
-1,9
|
6,6
-3,3*
| |
Variation moyenne entre l'inclusion et la semaine 52
|
|
-4,3
|
|
-4,1
| |
BASDAI
|
|
|
|
| |
Inclusion
Variation moyenne entre l'inclusion et la semaine 16
|
6,7
-1,5
|
6,9
-3,1*
|
6,5
-1,9
|
6,5
-2,9*
| |
Variation moyenne entre l'inclusion et la semaine 52
|
|
-3,9
|
|
-3,6
| |
BASMI
|
|
|
|
| |
Inclusion
Variation moyenne entre l'inclusion et la semaine 16
|
3,0
-0,1
|
2,9
-0,4
|
3,8
-0,2
|
3,9
-0,5**
| |
Variation moyenne entre l'inclusion et la semaine 52
|
|
-0,6
|
|
-0,7
| |
hs-CRP (mg/l)
|
|
|
|
| |
Inclusion (moyenne géométrique)
Ratio entre l'inclusion et la Semaine 16
|
5,0
0,8
|
4,6
0,4
|
6,7
0,9
|
6,5
0,4
| |
Ratio entre l'inclusion et la Semaine 52
|
|
0,4
|
|
0,3
|
BASMI = Bath Ankylosing Spondylitis Metrology Index. Hs-CRP = protéine C-réactive à haute sensibilité
La MI est utilisée.
*p < = 0,001 imputation basée sur les références, versus placebo, corrigée pour la multiplicité. **p < 0,01 imputation basée sur les références, versus placebo, corrigée pour la multiplicité.
Le bimekizumab était associé à une apparition rapide de l'efficacité dans les populations de patients atteints de nr-axSpA et de SA.
Figure 4: Réponse ASAS 40 au fil du temps jusqu'à la semaine 52 dans l'étude BE MOBILE 1 (NRI)
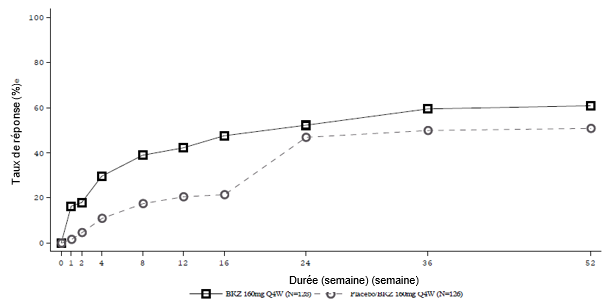
Les patients sous placebo sont passés à 160 mg de bimekizumab toutes les 4 semaines à la semaine 16.
Figure 5: Réponse ASAS 40 au fil du temps jusqu'à la semaine 52 dans l'étude BE MOBILE 2 (NRI)
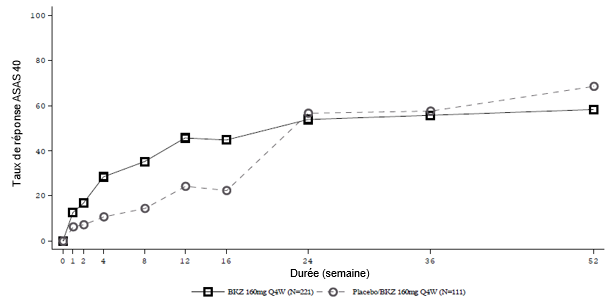
Les patients sous placebo sont passés à 160 mg de bimekizumab toutes les 4 semaines à la semaine 16.
Dans une analyse groupée des études BE MOBILE 1 et BE MOBILE 2, parmi les patients traités par bimekizumab qui ont obtenu une réponse ASAS 40 à la semaine 16, 82,1 % ont maintenu cette réponse à la semaine 52.
L'efficacité du bimekizumab a été démontrée quels que soient l'âge, le sexe, l'origine ethnique, la durée de la maladie, le statut inflammatoire à l'inclusion, le score ASDAS à l'inclusion, et les traitements concomitants par cDMARDs.
A la semaine 16, parmi les patients atteints d'enthésite à l'inclusion, la proportion de patients (NRI) présentant une résolution enthésitique évaluée selon l'indice MASES (Maastricht Ankylosing Spondylitis Enthesitis) était supérieure avec bimekizumab par rapport au placebo (BE MOBILE 1: 51,1 % versus 23,9 % et BE MOBILE 2: 51,5 % versus 32,8 %). La résolution enthésitique avec bimekizumab s'est maintenue jusqu'à la semaine 52 dans les deux études (BE MOBILE 1: 54,3 % et BE MOBILE 2: 50,8 %).
Réduction de l'inflammation
Bimekizumab a réduit l'inflammation évaluée par la CRP-us (voir Tableau 9) et par IRM dans une sous-étude d'imagerie. Les signes d'inflammation ont été évalués par IRM à l'inclusion et à la semaine 16 et exprimés en variation par rapport à l'inclusion du score SPARCC (Spondyloarthritis Research Consortium of Canada) pour les articulations sacro-iliaques et du score ASspiMRI-a (Ankylosing Spondylitis spine Magnetic Resonance Imagine-activity, score modifié de Berlin) pour le rachis. Une réduction des signes d'inflammation a été observée à la fois dans les articulations sacroiliaques et le rachis chez les patients traités par bimekizumab comparativement au placebo (voir Tableau 9). La réduction de l'inflammation mesurée par la CRP-us et par IRM a été maintenue jusqu'à la semaine 52.
Tableau 9: Réduction de l'inflammation selon l'IRM dans les études BE MOBILE 1 et BE MOBILE 2
|
|
BE MOBILE 1 ((nr-axSpA)
|
BE MOBILE 2 (SA)
| |
|
Placebo
|
BKZ 160 mg toutes les 4 semaines
|
Placebo
|
BKZ 160 mg toutes les 4 semaines
| |
Score SPARCC
|
|
|
|
| |
Variation moyenne entre l'inclusion a) et la semaine 16
|
-1,56
(N = 62)
|
-6,15
(N = 78)
|
0,59
(N = 46)
|
-4,51
(N = 81)
| |
Variation moyenne entre l'inclusion a) et la semaine 52
|
|
-7,57
(N = 67)
|
|
-4,67
(N = 78)
| |
Score ASspiMRI (dans la modification de Berlin)
|
|
|
|
| |
Variation moyenne entre l'inclusion a) et la semaine 16
|
0,03
(N = 60)
|
-0,36
(N = 74)
|
-0,34
(N = 46)
|
-2,23
(N = 81)
| |
Variation moyenne entre l'inclusion a) et la semaine 52
|
|
-0,70
(N = 65)
|
|
-2,38
(N = 77)
|
a)Les valeurs de la variation par rapport à l'inclusion sont basées sur les cas observés et ont été déterminées par l'évaluation centrale de l'ensemble de données de la semaine 52.
Fonction physique et autres résultats de santé
Les patients traités par bimekizumab ont montré une amélioration significative par rapport à l'inclusion de la fonction physique évaluée par l'indice BASFI par rapport au placebo (Variation de la moyenne des moindres carrés entre l'inclusion et la semaine 16 dans BE MOBILE 1: -2,4 versus -0,9, p < 0,001 et dans BE MOBILE 2: -2,0 versus -1,0, p < 0,001). Les patients traités par bimekizumab ont rapporté une amélioration significative par rapport à l'inclusion du score SF-36 PCS par rapport aux patients traités par placebo (Variation de la moyenne des moindres carrés entre l'inclusion et la semaine 16 dans BE MOBILE 1: 9,3 versus 5,4, p < 0,001 et dans BE MOBILE 2: 8,5 versus 5,2, p < 0.001).
Les patients traités par bimekizumab ont rapporté une amélioration significative par rapport à l'inclusion en termes de qualité de vie liée à la santé mesurée par le questionnaire ASQoL (AS Quality of Life Questionnaire) par rapport aux patients sous placebo (variation de la moyenne des moindres carrés entre l'inclusion et la semaine 16 dans BE MOBILE 1: -4,9 versus -2,3, p < 0,001 et dans BE MOBILE 2: -4,6 versus -3,0, p < 0,001) ainsi qu'une réduction significative de la fatigue, évaluée par le score FACIT-Fatigue (variation moyenne entre l'inclusion et la semaine 16 dans BE MOBILE 1: 8,5 pour bimekizumab versus 3,9 pour le placebo et dans BE MOBILE 2: 8,4 pour bimekizumab versus 5,0 pour le placebo).
Les améliorations obtenues à la semaine 16 dans toutes les mesures de la fonction physique et autres résultats de santé mentionnées ci-dessus (BASFI, SF-36 PCS, ASQoL et FACIT-Fatigue) ont été maintenues jusqu'à la semaine 52 dans les deux études.
Manifestation extra-articulaire
Dans les données groupées de BE MOBILE 1 (nr-axSpA) et BE MOBILE 2 (SA), à la semaine 16, la proportion de patients développant un événement d'uvéite était inférieure avec bimekizumab (0,6 %) par rapport au placebo (4,6 %). L'incidence d'uvéite est restée faible avec le traitement à long-terme par bimekizumab (1,2/100 patient-années dans les études groupées de phase II/III).
|