CompositionPrincipes actifs
Difélikéfaline (sous forme d’acétate de difélikéfaline).
Excipients
Acide acétique (pour ajustement du pH), acétate de sodium trihydraté (pour ajustement du pH), chlorure de sodium, eau pour préparations injectables. Contient au total env. 3,3 mg de sodium par ml.
Indications/Possibilités d’emploiKapruvia est indiqué dans le traitement du prurit modéré à sévère associé à la maladie rénale chronique chez les patients adultes sous hémodialyse.
Posologie/Mode d’emploiPosologie usuelle
La dose recommandée de difélikéfaline est de 0,5 microgramme/kg de poids sec (c.-à-d. le poids cible après dialyse) et est administrée trois fois par semaine par injection intraveineuse en bolus. Calculer le volume correspondant à la dose requise à prélever comme suit : 0,01 x poids sec (kg) conformément à l’ordonnance médicale, arrondi au dixième le plus proche (0,1 ml). Pour les patients dont le poids sec est supérieur ou égal à 195 kg, la dose recommandée est de 100 microgrammes (2 ml). Les doses recommandées sont mentionnées dans le tableau ci-après :
|
Catégorie de poids
(poids sec en kg)
|
Dose*
(ml)
| |
40-44
|
0,4
| |
45-54
|
0,5
| |
55-64
|
0,6
| |
65-74
|
0,7
| |
75-84
|
0,8
| |
85-94
|
0,9
| |
95-104
|
1
| |
105-114
|
1,1
| |
115-124
|
1,2
| |
125-134
|
1,3
| |
135-144
|
1,4
| |
145-154
|
1,5
| |
155-164
|
1,6
| |
165-174
|
1,7
| |
175-184
|
1,8
| |
185-194
|
1,9
| |
> 195
|
2
|
* Lorsqu’une dose de plus de 1 ml est requise, plus d’un flacon doit être utilisé.
Doses manquées
En cas d’omission d’un traitement d’hémodialyse programmé régulièrement, Kapruvia devrait être administré à la dose habituelle lors du prochain traitement d’hémodialyse.
Traitement supplémentaire
Lorsqu’une 4ème séance d’hémodialyse est réalisée durant la même semaine, Kapruvia devrait être administré après l’hémodialyse à la dose recommandée. Le nombre de 4 doses par semaine ne devrait pas être dépassé, même si plus de 4 séances d’hémodialyse sont réalisées en une semaine. Il est peu probable qu’une 4ème dose de Kapruvia entraîne une accumulation de difélikéfaline comportant un risque pour la sécurité du patient, car la majeure partie de la difélikéfaline résiduelle du traitement précédent est éliminée par l’hémodialyse. Cependant, la sécurité et l’efficacité d’une 4ème dose n’ont pas été entièrement établies en raison de données insuffisantes.
Patients avec un traitement par hémodialyse incomplet
En cas de séances d’hémodialyse durant moins d’une heure, l’administration de difélikéfaline devrait être suspendue jusqu’à la prochaine séance d’hémodialyse.
Après l’administration de difélikéfaline chez les patients sous hémodialyse, jusqu’à 70 % sont éliminés de l’organisme avant la prochaine séance d’hémodialyse. Les concentrations plasmatiques de difélikéfaline restantes jusqu’à la prochaine hémodialyse sont réduites de près de 40-50 % après une heure d’hémodialyse.
Patients âgés
Les recommandations posologiques pour les patients âgés sont les mêmes que pour les patients adultes.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n’est nécessaire chez les patients présentant des troubles légers à modérés de la fonction hépatique (voir rubrique « Pharmacocinétique »). L’administration de difélikéfaline chez les patients souffrant de troubles sévères de la fonction hépatique n’a pas encore été étudiée et son utilisation dans cette population de patients n’est donc pas recommandée.
Enfants et adolescents
La sécurité et l’efficacité de Kapruvia chez les enfants et les adolescents âgés de 0 à 17 ans n’ont pas encore été démontrées. Aucune donnée n’est disponible.
Mode d’administration
Kapruvia ne devrait pas être dilué ou mélangé avec d’autres médicaments.
Kapruvia est administré 3 fois par semaine par injection intraveineuse en bolus dans la ligne veineuse du circuit de dialyse à la fin du traitement d’hémodialyse, pendant ou après le rinçage.
En cas d’administration après le rinçage, au moins 10 ml de solution de chlorure de sodium à 9 mg/ml (0,9 %) devraient être administrés comme volume de rinçage après l’injection de Kapruvia. Lorsque la dose est administrée pendant le rinçage, aucune solution de chlorure de sodium supplémentaire à 9 mg/ml (0,9%) n’est nécessaire pour rincer la ligne.
Contre-indicationsHypersensibilité au principe actif ou à l’un des autres composants (voir « Composition »).
Mises en garde et précautionsHyperkaliémie
L’hyperkaliémie est fréquente chez les patients atteints de maladie rénale chronique sous hémodialyse. Dans le cadre d’études cliniques contrôlées par placebo, une incidence numériquement plus élevée d’événements indésirables d’hyperkaliémie a été rapportée chez les patients traités par la difélikéfaline (4,7 % ; 20/424 patients) par rapport au placebo (3,5 % ; 15/424 patients). Aucun lien de causalité n’a pu être déterminé. Un suivi régulier des taux de potassium est recommandé.
Insuffisance cardiaque et fibrillation auriculaire
La difélikéfaline n’a pas été étudiée chez les patients souffrant d’insuffisance cardiaque de classe IV de la New York Heart Association. Un léger déséquilibre numérique des événements d’insuffisance cardiaque et de fibrillation auriculaire a été observé chez les patients sous difélikéfaline par rapport au placebo lors des études pivots, en particulier chez les patients ayant des antécédents de fibrillation auriculaire qui ont arrêté ou suspendu leur traitement pour la fibrillation auriculaire. Aucun lien de causalité n’a pu être déterminé.
Patients présentant une altération de la barrière hémato-encéphalique
La difélikéfaline est un agoniste des récepteurs opioïdes kappa avec un passage restreint dans le système nerveux central (SNC). L’intégrité de la barrière hémato-encéphalique (BHE) est importante pour réduire l’absorption de la difélikéfaline dans le SNC (voir « Propriétés/Effets »). Les patients souffrant de troubles cliniquement significatifs de la BHE (p. ex. dans le cas de tumeurs cérébrales malignes primitives, de métastases du SNC ou d’autres maladies inflammatoires, de sclérose en plaques active, de maladie d’Alzheimer avancée) présentent un risque de pénétration de la difélikéfaline dans le SNC. Kapruvia devrait être prescrit avec prudence chez ces patients, en tenant compte du rapport bénéfice/risque individuel, et en surveillant les éventuels effets sur le SNC.
Vertiges et somnolence
Les patients traités par la difélikéfaline peuvent souffrir de vertiges et de somnolence. Ces symptômes peuvent s’atténuer au fil du temps avec la poursuite du traitement (voir « Effets indésirables »). L’utilisation concomitante d’antihistaminiques sédatifs, d’analgésiques opioïdes ou d’autres dépresseurs du SNC peut augmenter la probabilité de ces effets indésirables et devrait faire l’objet de prudence pendant le traitement par la difélikéfaline (voir « Interactions »).
Par rapport au placebo, l’incidence de la somnolence était supérieure chez les patients traités par la difélikéfaline et âgés de 65 ans et plus (7 %), que chez ceux traités par la difélikéfaline et âgés de moins de 65 ans (2,8 %).
Ce médicament contient moins de 1 mmol (23 mg) de sodium par flacon, c.-à-d. qu’il est essentiellement « sans sodium ».
InteractionsAucune étude clinique n’a été menée sur les interactions.
La difélikéfaline n’est ni un substrat de CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4, ni un inhibiteur de CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4/5 et présente un potentiel minime voire nul d’induction du CYP1A2, CYP2B6 ou CYP3A humain. Elle n’est pas non plus un inhibiteur des enzymes de glucuronidation (UGT1A3, UGT1A9 ou UGT2B7).
En outre, la difélikéfaline n’est pas un inhibiteur de BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2, OAT1, OAT3, OATP1A2, OATP1B1, OATP1B3, OCT1, OCT2, OCT3, glycoprotéine P, PEPT1 ou PEPT2 ni un substrat d’ASBT, BCRP, BSEP, LAT1, MATE1, MATE2-K, MRP2, OAT1, OAT2, OAT3, OATP1A2, OATP1B1, OATP1B3, OATP2B1, OCT1, OCT2, OCT3, OCTN1, OCTN2, OSTαβ, glycoprotéine P, PEPT1 ou PEPT2.
Par conséquent, les interactions de la difélikéfaline avec d’autres médicaments sont peu probables.
L’utilisation concomitante de médicaments tels que les antihistaminiques sédatifs, les analgésiques opioïdes ou d’autres dépresseurs du SNC (par exemple, la clonidine, l’ondansétron, la gabapentine, la prégabaline, le zolpidem, l’alprazolam, la sertraline, la trazodone) peut augmenter le risque de vertiges et de somnolence (voir « Mises en garde et précautions » et « Effets indésirables »).
Grossesse, allaitementGrossesse
Il n’existe pas de données ou il existe des données limitées sur l’utilisation de la difélikéfaline chez les femmes enceintes. Les études expérimentales chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects concernant la toxicité pour la reproduction (voir « Données précliniques »).
Par mesure de précaution, Kapruvia ne devrait pas être utilisé pendant la grossesse.
Allaitement
On ignore si la difélikéfaline est excrétée dans le lait maternel. Les études expérimentales chez l’animal indiquent une excrétion de la difélikéfaline dans le lait maternel. Un risque pour le nouveau-né/nourrisson ne peut pas être exclu.
La décision d’une interruption de l’allaitement ou d’une interruption/abstention du traitement par Kapruvia devrait être prise, en prenant en compte le bénéfice de l’allaitement pour l’enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Il n’existe aucune donnée disponible sur les effets de Kapruvia sur la fertilité humaine. Les études portant sur la difélikéfaline chez le rat n’ont pas démontré d’altération de la capacité d’accouplement ou de la fertilité (voir « Données précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesKapruvia a une légère influence sur l’aptitude à la conduite et sur l’utilisation de machines.
Une somnolence et/ou des vertiges ont été rapportés chez des patients recevant de la difélikéfaline (voir « Effets indésirables »). Les patients devraient être mis en garde à propos de la conduite de véhicules ou de l’utilisation de machines jusqu’à ce que l’effet de la difélikéfaline sur l’aptitude à la conduite et l’utilisation de machines soit connu. La somnolence est survenue au cours des 3 premières semaines de traitement et a eu tendance à s’atténuer avec la poursuite du traitement. Des vertiges sont survenus au cours des 9 premières semaines de traitement et étaient généralement transitoires.
Effets indésirablesRésumé du profil de sécurité
Dans des essais cliniques de phase 3 contrôlés par placebo et non contrôlés, environ 6,6 % des patients ont présenté au moins un effet indésirable au cours du traitement par la difélikéfaline. Les effets secondaires les plus fréquents étaient la somnolence (1,1 %), les vertiges (0,9 %), la paresthésie (y compris l’hypoesthésie, la paresthésie buccale et l’hypoesthésie buccale) (1,1 %), les maux de tête (0,6 %), les nausées (0,7 %), les vomissements (0,7 %), la diarrhée (0,2 %) et les modifications de l’état mental (y compris un état confusionnel) (0,3 %). La plupart des événements étaient de gravité légère ou modérée, n’ont eu aucune conséquence délétère et se sont résolus avec la poursuite du traitement. Aucun événement n’était grave et l’incidence des événements ayant conduit à l’arrêt du traitement était de ≤ 0,5 % pour chacun des effets indésirables susmentionnés.
Liste des effets indésirables
Les effets indésirables sont listés par classe de système d’organes de la classification MedDRA et par fréquence selon la convention suivante :
« très fréquents » (≥1/10),
« fréquents » (≥1/100 à <1/10),
« occasionnels » (≥1/1000 à <1/100),
« rares » (≥1/10 000 à <1/1000),
« très rares » (<1/10 000).
Les effets indésirables constatés lors de l’utilisation de Kapruvia dans des études cliniques à la dose cible de 0,5 microgramme/kg chez ces patients (n = 1 306) sont présentés dans le tableau 1.
Pour chaque catégorie de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Tableau 1 : Effets indésirables survenus en lien avec le traitement par Kapruvia chez des patients sous hémodialyse
|
Classe de système d’organes MedDRA
|
Fréquent
|
Occasionnel
| |
Affections du système nerveux
|
Somnolence, paresthésie
|
Vertiges ; maux de tête
| |
Affections gastro-intestinales
|
|
Vomissements; nausées; diarrhée
| |
Affections psychiatriques
|
|
Modifications de l’état mental1
|
1 Les modifications de l’état mental comprenaient les termes privilégiés MedDRA « état confusionnel » et « modifications de l’état mental ».
Description d’effets indésirables spécifiques
Somnolence
La somnolence en tant qu’événement indésirable lié au traitement a été signalée chez 2,2 % des participants à l’étude, randomisés pour recevoir la difélikéfaline. La grande majorité de ces événements étaient légers ou modérés. Chez 0,3 % des patients, la somnolence a entraîné l’interruption du traitement par la difélikéfaline. La somnolence a été signalée comme événement indésirable grave chez < 0,1 % des patients traités par la difélikéfaline. Chez 1,1 % des patients, un lien de causalité a été rapporté entre la somnolence et le traitement par la difélikéfaline. La somnolence est survenue au cours des 3 premières semaines de traitement et a eu tendance à s’atténuer avec la poursuite du traitement.
Le risque de somnolence peut augmenter lorsque la difélikéfaline est utilisée en concomitance avec d’autres médicaments (voir « Mises en garde et précautions »).
Vertiges
Des vertiges en tant qu’événement indésirable lié au traitement ont été signalés chez 7,9 % des patients, randomisés pour recevoir la difélikéfaline. La grande majorité de ces événements étaient légers ou modérés. Chez 0,5 % des patients, les vertiges ont entraîné l’interruption du traitement par la difélikéfaline. Des vertiges en tant qu’événement indésirable grave ont été signalés chez 0,5 % des patients traités par la difélikéfaline. Chez 0,9 % des patients, un lien de causalité a été rapporté entre les vertiges et le traitement par la difélikéfaline. Les vertiges sont survenus au cours des 9 premières semaines de traitement et étaient généralement transitoires.
Le risque de vertiges peut augmenter lorsque la difélikéfaline est utilisée en concomitance avec d’autres médicaments (voir « Mises en garde et précautions »).
Modifications de l’état mental
Des modifications de l’état mental (y compris un état confusionnel) en tant qu’événement indésirable lié au traitement ont été signalées chez 4,4 % des patients, randomisés pour recevoir la difélikéfaline.
La majorité de ces événements étaient légers ou modérés. Chez moins de 0,2 % des patients, les modifications de l’état mental ont entraîné l’interruption du traitement par la difélikéfaline.
Des modifications de l’état mental ont été signalées comme un événement indésirable grave chez 2,2 % des patients traités par la difélikéfaline. Chez 0,3 % des patients, un lien de causalité a été rapporté entre les modifications de l’état mental et le traitement par la difélikéfaline.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les essais cliniques, des doses uniques de difélikéfaline jusqu’à 12 fois la dose clinique de 0,5 microgramme/kg et des doses multiples de difélikéfaline jusqu’à 5 fois la dose clinique de 0,5 microgramme/kg ont été administrées à des patients sous hémodialyse. Une augmentation dose-dépendante des effets indésirables, notamment vertiges, somnolence, modifications de l’état mental, paresthésie, fatigue, hypertension et vomissements, a été observée.
En cas de surdosage, les soins médicaux appropriés doivent être assurés en fonction de l’état clinique du patient. Une hémodialyse de 4 heures à l’aide d’un dialyseur à haut débit a éliminé environ 70 à 80 % de la difélikéfaline du plasma, et la difélikéfaline était indétectable dans le plasma au terme de deux cycles de dialyse.
Propriétés/EffetsATC-Code
Code ATC : V03AX04
Mécanisme d’action
La difélikéfaline est un agoniste sélectif des récepteurs opioïdes kappa, à faible pénétration dans le SNC.
Les récepteurs opioïdes sont connus pour moduler les signaux de démangeaison et l’inflammation, l’activation des récepteurs opioïdes kappa réduisant les démangeaisons et produisant des effets immunomodulateurs.
Pharmacodynamique
Le rapport exposition/effet de la difélikéfaline et l’évolution dans le temps de la réponse pharmacodynamique ne sont pas connus.
Influence sur l’électrocardiogramme
À une posologie correspondant à 6 fois la dose recommandée, la difélikéfaline ne produit pas d’allongement cliniquement significatif de l’intervalle QTc.
Efficacité clinique
Études contrôlées par placebo
Dans deux études cliniques pivots de phase III de conception similaire, en double aveugle, randomisées et contrôlées par placebo (KALM 1 et KALM 2), des patients atteints de maladie rénale chronique sous hémodialyse et présentant un prurit modéré à sévère ont reçu soit un placebo, soit 0,5 microgramme/kg de difélikéfaline par voie intraveineuse 3 fois par semaine après la session d’hémodialyse, pendant 12 semaines. Un maximum de 4 doses était autorisé chez les patients nécessitant une dialyse supplémentaire dans une même semaine. Cette phase de traitement en double aveugle a été suivie d’une phase de prolongation ouverte de 52 semaines avec traitement actif uniquement. Le critère d’évaluation principal des deux études était le pourcentage de patients qui, après 12 semaines, atteignaient une réduction d’au moins 3 points sur l’échelle Worst Itching Numerical Rating Scale (les valeurs WI-NRS se situent entre 0 et 10, les valeurs plus élevées représentant une plus forte intensité de la démangeaison) par rapport à la valeur initiale. Les critères d’évaluation secondaires les plus importants, qui étaient cohérents dans les deux études, étaient le pourcentage de patients présentant une amélioration du score WI-NRS d’au moins 4 points après 12 semaines, et les changements de la sévérité des démangeaisons et de la qualité de vie (QdV) liée aux démangeaisons, mesurés par le Skinindex-10 total et l’échelle de démangeaison 5-D. Les critères d’inclusion les plus importants étaient la maladie rénale chronique avec hémodialyse trois fois par semaine depuis au moins 3 mois, un prurit modéré à sévère (WI-NRS >4 à l’inclusion) et une hémodialyse appropriée. Les principaux critères d’exclusion étaient le prurit d’origine autre que la maladie rénale chronique ou des complications associées, les démangeaisons sur la paume des mains et les démangeaisons survenant uniquement lors des séances d’hémodialyse.
Les 2 études combinées ont recruté 851 patients. L’âge moyen était de 59 ans, 33,1 % étaient âgés de 65 ans ou plus, 60 % des patients étaient des hommes. Les caractéristiques de la maladie au début de l’étude, comme l’administration de médicaments pour soulager le prurit, le délai depuis l’établissement du diagnostic de la maladie rénale chronique et la durée du prurit, étaient comparables dans les bras sous traitement actif et sous placebo. La valeur moyenne du WI-NRS à l’inclusion était de 7,18 dans les deux bras; la valeur moyenne du score WI-NRS à l’inclusion était de 7,13 (intervalle de 4,2 à 10) dans le groupe difélikéfaline et de 7,13 (intervalle de 4,1 à 10) dans le groupe placebo. Au total, 38 % des patients avaient déjà utilisé des médicaments pour traiter le prurit auparavant. Dans toutes les études, la difélikéfaline a significativement amélioré la sévérité des démangeaisons et la qualité de vie liée aux démangeaisons sur 12 semaines, comme le montre le tableau 2.
Tableau 2 : Résumé des critères d’évaluation principaux et secondaires les plus importants des études KALM-1 et KALM-2 et de la base de données groupée après la semaine 12
|
Critère d’évaluation à la
fin de la semaine 12
|
KALM-1 (n = 378)
|
KALM-2 (n = 473)
| |
Difélikéfaline
(n = 189)
|
Placebo
(n = 189)
|
Difélikéfaline
(n = 237)
|
Placebo
(n = 236)
| |
Critère d’évaluation principal
|
|
|
|
| |
WI-NRS
|
|
|
|
| |
Patients avec une amélioration de ≥ 3 points (%)
|
51,0 %
(p < 0,001)
|
27,6 %
|
54,0 %
(p = 0,02)
|
42,2 %
| |
Critère d’évaluation secondaire
|
|
|
|
| |
WI-NRS
Patients avec une amélioration de ≥ 4 points (%)
|
38,9 %
(p < 0,001)
|
18,0 %
|
41,2 %
(p = 0,01)
|
28,4 %
| |
Skindex-10
Changement par rapport à l’inclusion
[Points]
|
-17,2
(p < 0,001)
|
-12,0
|
-16,6
(p = 0,171)
|
-14,8
| |
Prurit 5-D
Changement par rapport à l’inclusion
[Points]
|
-5,0
(p < 0,001)
|
-3,7
|
-4,9
(p = 0,002)
|
-3,8
|
La figure 1 illustre le pourcentage moyen de patients des études KALM-1 et KALM-2 présentant une amélioration du score WI-NRS de ≥ 3 points par rapport à la valeur initiale par semaine d’étude. Sur la base des rapports de cotes, des améliorations statistiquement significatives en faveur du groupe difélikefaline ont été observées à la semaine 3 dans l’étude KALM-1 et à la semaine 2 dans l’étude KALM-2, se poursuivant chaque semaine suivante jusqu’à la semaine 12 dans les deux études.
Figure 1 : Pourcentage de patients avec une amélioration de ≥ 3 points du score WI-NRS par semaine dans les études KALM-1 et KALM-2 (population ITT)
KALM-1
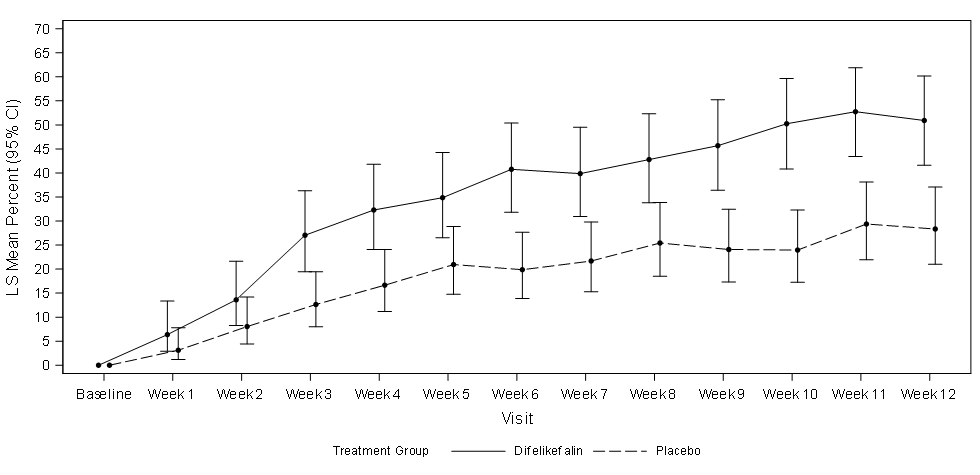
IC = intervalle de confiance ; ITT = Intention To Treat (intention de traiter) ; MC = moindres carrés ; WI-NRS = Worst Itching-Numerical Rating Scale (échelle d’évaluation numérique des pires démangeaisons)
KALM-2
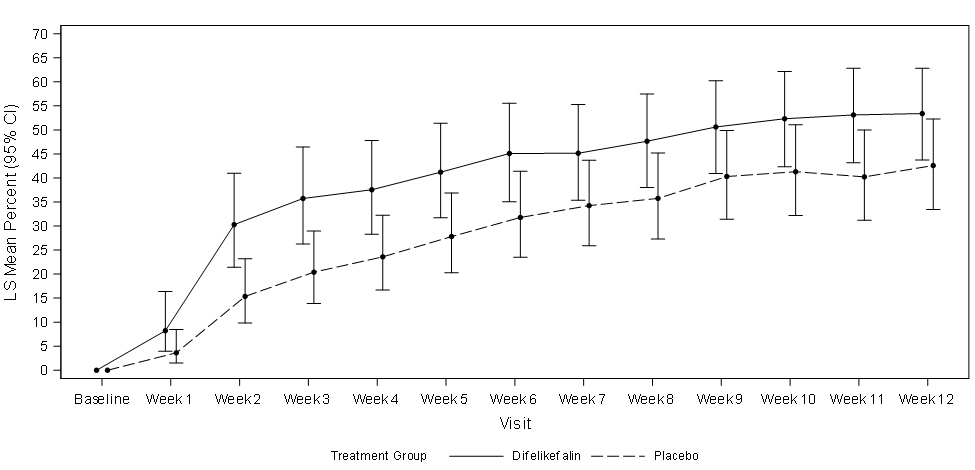
IC = intervalle de confiance ; ITT = Intention To Treat (intention de traiter) ; MC = moindres carrés ; WI-NRS = Worst Itching-Numerical Rating Scale (échelle d’évaluation numérique des pires démangeaisons)
Études de prolongation en phase ouverte
Chez les patients qui sont passés du placebo à la difélikefaline à la fin de la période de traitement en double aveugle, une amélioration sur l’échelle des démangeaisons 5-D a été observée après une période de traitement de 4 semaines, avec une moyenne des moindres carrés (écart-type) de la modification par rapport à l’inclusion semblable à celle des patients qui ont reçu de la difélikefaline depuis le début de l’essai : -6,0 (0,22) contre -5,7 (0,23). L’amélioration sur l’échelle des démangeaisons 5-D a été maintenue dans les deux groupes de traitement pendant la période de traitement de 52 semaines.
PharmacocinétiqueLes propriétés pharmacocinétiques de la difélikéfaline administrée en intraveineuse ont été étudiées chez 319 sujets sains et 115 patients atteints de maladie rénale chronique, dont 91 sous hémodialyse. Le profil pharmacocinétique chez les patients souffrant de troubles légers de la fonction rénale était comparable à celui des participants sains. Cependant, chez les patients atteints d’insuffisance rénale sévère, la clairance corporelle totale de la difélikéfaline était réduite et les concentrations plasmatiques sont restées relativement constantes jusqu’à l’élimination du principe actif pendant la dialyse.
Absorption
Non pertinent.
Distribution
La liaison aux protéines plasmatiques de la difélikéfaline est faible à modérée. Elle se situe entre 24 et 32 % et n’est pas affectée par les troubles de la fonction rénale. Le volume moyen de distribution à l’état d’équilibre varie de 145 à 189 ml/kg chez les sujets sains et de 214 à 301 ml/kg chez les patients sous hémodialyse présentant un prurit modéré à sévère.
Metabolisme
La difélikéfaline n’est pas un substrat du CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4.
Elimination
Chez les sujets sains, la principale voie d’élimination de la difélikéfaline est rénale, environ 81 % de la dose étant excrétée dans les urines et 11 % dans les selles. La clairance totale moyenne varie entre 54 et 71 ml/h/kg et la demi-vie moyenne entre 2 et 3 heures. En revanche, l’élimination chez les patients sous hémodialyse souffrant d’insuffisance rénale s’est effectuée principalement par les selles, dans lesquelles environ 59 % de la dose a été détecté en moyenne ; environ 19 % a été récupéré dans le dialysat et quelque 11 % dans les urines. En comparaison avec les participants présentant une fonction rénale normale, la clairance totale moyenne était réduite et la demi-vie multipliée par 10, avec des valeurs allant de 5,3 à 7,5 ml/h/kg et de 23 à 31 heures, respectivement. Après administration du principe actif radiomarqué, le composé parent représentait >99 % de la radioactivité en circulation. L’hémodialyse réduit la concentration de difélikéfaline de 70 à 80 %. Après 2 cycles de dialyse, la difélikéfaline était indétectable dans le plasma.
Linéarité/non-linéarité
Chez les patients sous hémodialyse et souffrant de maladie rénale chronique, la pharmacocinétique de la difélikéfaline dans la plage de dose intraveineuse unique de 1 à 3 microgrammes/kg (2 à 6 fois la dose recommandée) et dans la plage de doses multiples de 0,5 à 2,5 microgrammes/kg (1 à 5 fois la dose recommandée) est linéaire et proportionnelle à la dose. L’état d’équilibre a été atteint après la deuxième dose administrée, et le taux d’accumulation moyen atteignait 1,6.
Cinétique pour certains groupes de patients
Il n’existe actuellement aucune preuve que des facteurs tels que l’âge (25 à 80 ans), le sexe, l’origine ethnique ou une insuffisance hépatique légère à modérée affectent la pharmacocinétique de la difélikéfaline.
Rapport entre pharmacocinétique et pharmacodynamique
Dans le groupe cible des patients sous hémodialyse souffrant d’insuffisance rénale, aucun rapport dose/effet évident n’a été observé concernant l’effet antiprurigineux. Chez les personnes dont la fonction rénale est intacte, le risque d’aquarèse est dose-dépendant.
Données précliniquesLes données non cliniques issues des études conventionnelles sur la pharmacologie de sécurité, la toxicité en administration répétée, la génotoxicité, la carcinogénicité et le potentiel d’abus et de dépendance n’ont pas révélé de risque particulier pour l’homme.
Toxicité sur la reproduction et le développement
Chez le rat, la fertilité mâle et femelle, le développement embryonnaire précoce et le développement prénatal et postnatal n’ont pas été affectés à des expositions correspondant à au moins 775 fois l’aire sous la courbe (ASC) chez l’être humain. Chez le lapin, le développement prénatal n’a pas non plus été affecté malgré une toxicité maternelle manifeste à 30 fois l’ASC chez l’être humain.
Chez le rat, la difélikéfaline traverse le placenta et pénètre dans le lait maternel.
Remarques particulièresIncompatibilités
En l’absence d’études de compatibilité, Kapruvia ne doit pas être mélangé à d’autres médicaments.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Conserver à 15-30°C.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
La solution injectable Kapruvia en flacon est prête à l’emploi. Kapruvia ne doit pas être mélangé à d’autres médicaments ni dilué.
La solution injectable stérile en flacon ne contient aucun conservateur et est réservée à une injection unique à un patient. Seules les solutions limpides, incolores et exemptes de particules visibles doivent être injectées.
Tout médicament non utilisé ou déchet doit être éliminé conformément aux exigences nationales.
Numéro d’autorisation68653
PrésentationBoîte de 3 flacons (actuellement indisponible sur le marché) et de 12 flacons, contenant chacun 1 ml de solution injectable. (B).
Titulaire de l’autorisationVifor Fresenius Medical Care Renal Pharma Ltd., Saint-Gall
Mise à jour de l’informationMai 2024
|