CompositionPrincipes actifs
Acalabrutinib (sous forme de maléate d'acalabrutinib monohydraté)
Excipients
Noyau du comprimé:
Mannitol (E 421)
Cellulose microcristalline
Hydroxypropylcellulose faiblement substituée
Fumarate de stéaryle sodique
Pelliculage:
Hypromellose
Copovidone
Dioxyde de titane (E 171)
Macrogol 3350
Triglycérides à chaîne moyenne
Oxyde de fer jaune (E 172)
Oxyde de fer rouge (E 172)
1 comprimé pelliculé contient 0,59 mg de sodium.
Indications/Possibilités d’emploiIndication autorisée pour une durée limitée
Lymphome à cellules du manteau (LCM)
CALQUENCE en association avec la bendamustine et le rituximab (BR) est indiqué dans le traitement de patients adultes atteints d'un LCM non précédemment traité et qui ne sont pas éligibles à une greffe de cellules souches autologue (voir «Mises en garde et précautions» et «Propriétés/Effets»).
En raison de la documentation incomplète au moment de l'examen de la demande, cette indication est autorisée pour une durée limitée (art. 9a de la loi sur les produits thérapeutiques). L'autorisation à durée limitée est impérativement liée à la satisfaction de charges en temps opportun. Une fois ces charges satisfaites, l'autorisation à durée limitée pourra être transformée en autorisation sans charge spécifique.
Indications autorisées sans limitation de durée
Lymphome à cellules du manteau (LCM)
CALQUENCE en monothérapie est indiqué dans le traitement des patients adultes atteints de LCM n'ayant pas obtenu de réponse partielle avec un traitement antérieur ou ayant présenté une progression après un traitement antérieur (voir «Propriétés/Effets»).
Leucémie lymphoïde chronique (LLC)
CALQUENCE, en monothérapie ou en association avec l'obinutuzumab, est indiqué dans le traitement des patients adultes atteints d'une LLC non précédemment traitée âgés d'au moins 65 ans ou présentant des comorbidités (voir «Propriétés/Effets»).
CALQUENCE, en monothérapie, est indiqué dans le traitement des patients adultes atteints d'une LLC ayant reçu au moins un traitement antérieur (voir «Propriétés/Effets»).
CALQUENCE, en association avec le vénétoclax, est indiqué dans le traitement des patients adultes atteints d'une leucémie lymphoïde chronique (LLC) non précédemment traitée (voir «Propriétés/Effets»).
Posologie/Mode d’emploiLe traitement par CALQUENCE doit être initié et supervisé par un médecin expérimenté dans les traitements oncologiques.
Posologie usuelle
LCM
La posologie recommandée de CALQUENCE en monothérapie est de 100 mg (1 comprimé) deux fois par jour. Pour le traitement en association, veuillez vous référer à la posologie recommandée de l'information professionnelle du médicament respectif. Pour de plus amples informations concernant le traitement en association, voir «Propriétés/Effets». Le traitement par CALQUENCE doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
LLC
La posologie recommandée de CALQUENCE dans le traitement de la LLC est de 100 mg (1 comprimé) deux fois par jour en monothérapie ou en association. Veuillez vous référer à la posologie recommandée indiquée dans l'information professionnelle du médicament respectif utilisé pour le traitement en association (pour de plus amples informations concernant les traitements en association, voir «Propriétés/Effets»).
Les doses de CALQUENCE doivent être prises à un intervalle d'environ 12 heures.
Le traitement par CALQUENCE en monothérapie ou en association avec l'obinutuzumab doit être poursuivi jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. Le traitement par CALQUENCE en association avec le vénétoclax doit être poursuivi jusqu'à la progression de la maladie, la survenue d'une toxicité inacceptable ou la fin des 14 cycles de traitement (de 28 jours chacun).
Mode d'administration
Les comprimés CALQUENCE doivent être avalés entiers avec de l'eau, matin et soir toutes les 12 heures, à peu près à la même heure chaque jour. CALQUENCE peut être pris avec ou sans nourriture. Le comprimé ne doit pas être mâché, écrasé, dissout ou divisé.
Oubli d'une dose
Si l'oubli de la dose de CALQUENCE remonte à plus de 3 heures, le patient doit prendre la dose suivante à l'heure habituelle. Le patient ne doit pas prendre de comprimés supplémentaires pour compenser les doses oubliées.
Ajustement de la posologie du fait d'effets indésirables/d'interactions
Ajustements posologiques de CALQUENCE recommandés en monothérapie et en association avec l'obinutuzumab: voir Tableau 1. Les ajustements posologiques de CALQUENCE suite à des effets indésirables chez les patients qui reçoivent CALQUENCE en association avec le vénétoclax sont présentés dans le Tableau 2.
Les ajustements posologiques recommandés suite à des effets indésirables de grade ≥3 chez les patients qui reçoivent CALQUENCE en association avec la bendamustine et le rituximab sont indiqués dans le Tableau 3.
Concernant les ajustements posologiques, veuillez également consulter l'information professionnelle du médicament utilisé en association avec CALQUENCE.
Tableau 1. Ajustements posologiques recommandés en cas d'effets indésirables chez les patients qui reçoivent CALQUENCE en monothérapie et CALQUENCE en association avec l'obinutuzumab*
|
Effet indésirable
|
Survenue de l'effet indésirable
|
Ajustement posologique
(Dose de départ = 100 mg toutes les 12 heures environ)
| |
Thrombopénie de grade 3 associée à une hémorragie,
thrombopénie de grade 4
ou
neutropénie de grade 4 durant plus de 7 jours
Toute autre toxicité de grade 3 ne pouvant être traitée ou toute autre toxicité de grade 4
|
Première et deuxième fois
|
Suspendre CALQUENCE
Une fois la toxicité revenue au grade 1 ou au niveau initial, CALQUENCE peut être repris à 100 mg environ toutes les 12 heures
| |
Troisième fois
|
Suspendre CALQUENCE
Une fois la toxicité revenue au grade 1 ou au niveau initial, CALQUENCE peut être repris à une fréquence d'administration réduite de 100 mg une fois par jour
| |
Quatrième fois
|
Arrêter CALQUENCE
|
*Gradation de la sévérité des effets indésirables établie d'après la version 4.03 des critères CTCAE (Common Terminology Criteria for Adverse Events) du National Cancer Institute (NCI).
Tableau 2. Ajustements posologiques recommandés en cas d'effets indésirables chez les patients qui reçoivent CALQUENCE en association avec le vénétoclax
|
Effet indésirablea
|
Survenue de l'effet indésirable
|
Ajustement posologique de CALQUENCE
| |
Neutropénie de grade 3 ou 4 associée ou non à de la fièvre et/ou à une infection; neutropénie de grade 4 durant plus de 7 jours
|
Première fois
|
Suspendre CALQUENCE et/ou le vénétoclax.b
Une fois la toxicité revenue au grade ≤1 ou au niveau initial, reprendre CALQUENCE et/ou le vénétoclax à la même dose.
| |
Deuxième fois
|
Suspendre CALQUENCE et le vénétoclax.b Une fois la toxicité revenue au grade ≤1 ou au niveau initial, reprendre CALQUENCE à la même dose et réduire le vénétoclax au niveau de dose inférieur.
| |
Survenue ultérieure
|
Suspendre CALQUENCE et le vénétoclax jusqu'à ce que la toxicité soit revenue au grade ≤1 ou au niveau initial.b Le schéma thérapeutique de chaque patient doit se baser sur l'appréciation clinique du médecin traitant, en fonction de l'évaluation bénéfice/risque individuelle du traitement par CALQUENCE en association avec le vénétoclax.
Arrêter CALQUENCE si l'effet indésirable survient une 4e fois.
| |
Thrombopénie de grade 3 ou 4 et/ou hémorragiec
|
Première fois
|
Suspendre CALQUENCE et/ou le vénétoclax. Lorsque l'hémorragie est résolue et la thrombopénie est revenue au grade ≤1 ou au niveau initial, sans aide transfusionnelle pendant 5 jours consécutifs, reprendre CALQUENCE et/ou le vénétoclax à la même dose.
| |
Deuxième fois
|
Suspendre CALQUENCE et le vénétoclax jusqu'à ce que l'hémorragie soit résolue et la thrombopénie soit revenue au grade ≤1 ou au niveau initial.
Reprendre CALQUENCE à la même dose et/ou reprendre le vénétoclax en le réduisant au niveau de dose inférieur.
| |
Survenues ultérieures de thrombopénie sévère
|
Suspendre CALQUENCE et le vénétoclax jusqu'à ce que l'hémorragie soit résolue et la thrombopénie soit revenue au grade ≤1 ou au niveau initial.
Reprendre CALQUENCE à une fréquence d'administration réduite de 100 mg une fois par jour et/ou le vénétoclax en le réduisant au niveau de dose inférieur.
En cas de thrombopénie récurrente sévère malgré la réduction de dose et/ou une hémorragie symptomatique, le schéma thérapeutique doit se baser sur l'appréciation clinique du médecin traitant.
Arrêter CALQUENCE si l'effet indésirable survient une 4e fois.
| |
Syndrome de lyse tumorale (SLT) de grade 3 ou 4, premier épisode et épisodes suivants
|
Premier épisode et épisodes suivants
|
Si un sujet présente des modifications des paramètres biochimiques sanguins suggérant un SLT, la prise du vénétoclax et de CALQUENCE doit être suspendue le lendemain. Si l'épisode est résolu dans les 24 à 48 heures suivant la dernière dose, le traitement peut être repris à la même dose.
Pour les épisodes de SLT cliniques ou les modifications des paramètres biochimiques sanguins dont la résolution demande plus de 48 heures, le vénétoclax doit être repris en le réduisant au niveau de dose inférieur. Lors de la reprise du traitement après une interruption due à un SLT, surveiller la réapparition du SLT et mettre en place la prophylaxie nécessaire.
| |
Autres événements non hématologiques de grade 2d
|
Toute survenue
|
Suspendre CALQUENCE et/ou le vénétoclax, si cela est jugé cliniquement indiqué, jusqu'au retour au grade ≤1. Reprendre CALQUENCE et/ou le vénétoclax à la même dose.
| |
Autres événements non hématologiques de grade 3d
|
Première fois
|
Suspendre CALQUENCE et/ou le vénétoclax jusqu'à ce que la toxicité soit revenue au grade ≤1. Reprendre CALQUENCE et/ou le vénétoclax à la même dose.
| |
Deuxième fois
|
Suspendre CALQUENCE et/ou le vénétoclax jusqu'à ce que la toxicité soit revenue au grade ≤1.
Le schéma thérapeutique de chaque patient doit se baser sur l'appréciation clinique du médecin traitant, en fonction de l'évaluation bénéfice/risque individuel du traitement par CALQUENCE en association avec le vénétoclax.
| |
Autres événements non hématologiques de grade 4d
|
Première fois
|
Suspendre CALQUENCE et/ou le vénétoclax jusqu'à ce que la toxicité soit revenue au grade ≤1. Reprendre CALQUENCE à une fréquence d'administration réduite de 100 mg une fois par jour et/ou le vénétoclax en le réduisant au niveau de dose inférieur.
| |
Deuxième fois
|
Suspendre CALQUENCE et/ou le vénétoclax jusqu'à ce que la toxicité soit revenue au grade ≤1. Le schéma thérapeutique de chaque patient doit se baser sur l'appréciation clinique du médecin traitant, en fonction de l'évaluation bénéfice/risque individuel du traitement par CALQUENCE en association avec le vénétoclax.
|
a Gradation de la sévérité des effets indésirables établie d'après la version 4.03 des critères CTCAE (Common Terminology Criteria for Adverse Events) du National Cancer Institute (NCI).
b Un facteur de croissance peut être administré à la discrétion du médecin.
c Des plaquettes peuvent être administrées à la discrétion du médecin.
d Certains EI non hématologiques apparus sous traitement (p.ex. des événements thromboemboliques veineux) peuvent être gérés et stabilisés cliniquement après une intervention médicale sans toutefois revenir au degré ≤1 selon les définitions CTCAE du NCI. Dans ce cas, si le sujet est cliniquement stable, la reprise du médicament à l'étude sera éventuellement possible, selon l'appréciation clinique du médecin traitant.
Tableau 3. Ajustements posologiques recommandés en cas d'effets indésirables* de grade ≥3 chez les patients qui reçoivent CALQUENCE en association avec la bendamustine et le rituximab
|
Effet indésirable
|
Ajustement posologique de la bendamustine†
|
Ajustement posologique de CALQUENCE
| |
Neutropénie
|
En cas de neutropénie de grade 3 ou de grade 4:
Suspendre la bendamustine.
Une fois la toxicité revenue au grade ≤2 ou au niveau initial, la bendamustine peut être reprise à 70 mg/m2.
Arrêter la bendamustine si une réduction de dose supplémentaire s'avère nécessaire.
|
Suspendre CALQUENCE si la neutropénie de grade 4 dure plus de 7 jours.
Une fois la toxicité revenue au grade ≤2 ou au niveau initial, CALQUENCE peut être repris à la dose initiale (1re survenue de l'effet indésirable) ou à une fréquence d'administration réduite de 100 mg une fois par jour (2e et 3e survenue de l'effet indésirable).
Arrêter CALQUENCE si l'effet indésirable survient une 4e fois.
| |
Thrombopénie
|
En cas de thrombopénie de grade 3 ou de grade 4:
Suspendre la bendamustine.
Une fois la toxicité revenue au grade 2 ou au niveau initial, la bendamustine peut être reprise à 70 mg/m2.
Arrêter la bendamustine si une réduction de dose supplémentaire s'avère nécessaire.
|
Suspendre CALQUENCE en cas de thrombopénie de grade 3 associée à une hémorragie pertinente ou de thrombopénie de grade 4.
Une fois la toxicité revenue au grade ≤2 ou au niveau initial, CALQUENCE peut être repris à la dose initiale (1re survenue de l'effet indésirable) ou à une fréquence d'administration réduite de 100 mg une fois par jour (2e et 3e survenue de l'effet indésirable).
Arrêter CALQUENCE lorsque la thrombopénie associée à une hémorragie pertinente survient comme effet indésirable pour la 3e fois.
Arrêter CALQUENCE si l'effet indésirable survient une 4e fois.
| |
Toute autre toxicité hématologique de grade 4‡ ou toute autre toxicité de grade 3 ne pouvant être traitée
|
Suspendre la bendamustine.
Une fois la toxicité revenue au grade ≤2 ou au niveau initial, la bendamustine peut être reprise à 70 mg/m2.
Arrêter la bendamustine si une réduction de dose supplémentaire s'avère nécessaire.
|
Suspendre CALQUENCE.
Une fois la toxicité revenue au grade ≤2 ou au niveau initial, CALQUENCE peut être repris à la dose initiale (1re survenue de l'effet indésirable) ou à une fréquence d'administration réduite de 100 mg une fois par jour (2e et 3e survenue de l'effet indésirable).
Arrêter CALQUENCE si l'effet indésirable survient une 4e fois.
| |
Toxicité non hématologique de grade 3 ou plus
|
Suspendre la bendamustine.
Une fois la toxicité revenue au grade 1 ou au niveau initial, la bendamustine peut être reprise à 70 mg/m2.
Arrêter la bendamustine si une réduction de dose supplémentaire s'avère nécessaire.
|
Suspendre CALQUENCE.
Une fois la toxicité revenue au grade 2 ou au niveau initial, CALQUENCE peut être repris à la dose initiale (1re survenue de l'effet indésirable) ou à une fréquence d'administration réduite de 100 mg une fois par jour (2e survenue de l'effet indésirable).
Arrêter CALQUENCE si l'effet indésirable survient une 3e fois.
|
*Ordre de gravité des effets indésirables établi d'après la version 4.03 des critères communs de terminologie des Effets Indésirables du National Cancer Institute (NCI CTCAE).
†Les informations concernant les toxicités n'étant pas indiquées dans ce tableau sont disponibles dans l'information professionnelle locale de la bendamustine.
‡La lymphopénie de grade 4 est un résultat attendu du traitement par bendamustine et rituximab. Un ajustement posologique en raison d'une lymphopénie n'est attendu que si le médecin-investigateur estime que cela est cliniquement important, par exemple en cas d'infections récurrentes.
Des informations complémentaires sur le traitement des toxicités sont disponibles dans l'information professionnelle du médicament utilisé en association avec CALQUENCE.
Le Tableau 4 contient des recommandations concernant l'utilisation de CALQUENCE avec des inhibiteurs ou des inducteurs du CYP3A
Tableau 4. Utilisation avec des inhibiteurs ou des inducteurs du CYP3A
|
|
Médicament co-administré
|
Utilisation recommandée de CALQUENCE
| |
Inhibiteurs du CYP3A
|
Inhibiteurs puissants du CYP3A
|
Éviter l'utilisation concomitante.
Si ces inhibiteurs doivent être utilisés à court terme (p.ex. des anti-infectieux sur une durée maximale de 7 jours), suspendre CALQUENCE.
| |
Inhibiteurs modérés du CYP3A
|
Aucun ajustement posologique. Les patients doivent être étroitement surveillés à la recherche d'effets secondaires lorsqu'ils prennent concomitamment des inhibiteurs modérés du CYP3A4.
| |
Inducteurs du CYP3A
|
Inducteurs puissants du CYP3A
|
Éviter l'utilisation concomitante; envisager des principes actifs alternatifs à induction plus faible du CYP3A.
|
Instructions posologiques particulières
Patients présentant des troubles de la fonction rénale
D'après l'équation MDRD (Modification of Diet in Renal Disease Equation - équation utilisée pour modifier le régime alimentaire en cas de maladie rénale), aucun ajustement posologique n'est recommandé chez les patients présentant des troubles légers à modérés de la fonction rénale (DFGe ≥30 ml/min/1,73 m2). La pharmacocinétique et la sécurité de CALQUENCE n'ont pas été étudiées chez les patients atteints de troubles sévères de la fonction rénale (DFGe <29 ml/min/1,73 m2) ou d'insuffisance rénale terminale (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n'est recommandé chez les patients présentant une diminution légère ou modérée de la fonction hépatique (Child-Pugh A, Child-Pugh B ou bilirubine totale comprise entre 1,5 et 3 fois la limite supérieure de la normale (LSN) avec ou sans élévation d'ASAT (aspartate aminotransférase)). Les patients présentant une diminution sévère de la fonction hépatique (Child-Pugh C ou bilirubine totale >3 fois la LSN avec ou sans élévation d'ASAT) ne doivent pas prendre CALQUENCE (voir «Pharmacocinétique»).
Patients âgés (≥65 ans)
Aucun ajustement posologique n'est requis du fait de l'âge (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de CALQUENCE pour les enfants et les adolescents âgés de moins de 18 ans ne sont pas établies.
Contre-indicationsHypersensibilité connue au principe actif ou à l'un des excipients.
Mises en garde et précautionsSeconds cancers primitifs
Chez 17,6% des patients atteints d'un cancer hématologique ayant reçu CALQUENCE en monothérapie (n = 1 478) et chez 12,1% des patients traités par CALQUENCE en association avec d'autres médicaments (n = 1 095), un second cancer primitif a été décrit. Il s'agissait de cancers cutanés et d'autres cancers. Les cancers cutanés non mélanomateux étaient le second cancer primitif le plus fréquent et étaient survenus chez 9,9% (tous grades confondus) des patients traités par CALQUENCE en monothérapie et chez 7,2% des patients traités par CALQUENCE en association avec d'autres médicaments.
Mis à part le cancer cutané non mélanomateux, les seconds cancers primitifs les plus fréquents étaient le cancer de la prostate (1,3% et 0,6%), le carcinome épidermoïde (1,2% et 1,3%) et le mélanome malin (0,9% et 0,5%).
Les patients doivent être surveillés à la recherche de tumeurs secondaires et doivent éviter l'exposition au soleil.
Infections
Des infections sont survenues chez des patients atteints d'un cancer hématologique et traités par CALQUENCE en monothérapie (74,3%, n = 1 478) et par CALQUENCE en association avec d'autres médicaments (66,1%, n = 1 095). Les infections des voies aériennes supérieures (25,8% et 16,5%), les infections à COVID-19 (8,5% et 26,3%) et la pneumonie due à la COVID-19 (2,0% et 10,4%), la pneumonie (15,8% et 9,7%) et la sinusite (11,4% et 6,7%) étaient les infections les plus fréquentes. Des infections (bactériennes, virales ou fongiques) sévères, y compris des événements fatals, sont survenues chez des patients atteints d'un cancer hématologique et traités par CALQUENCE en monothérapie (25,3%) et par CALQUENCE en association avec d'autres médicaments (27,0%). Ces infections se sont principalement produites en l'absence de neutropénie de grade 3 ou 4. Des cas d'infections dues à une réactivation du virus de l'hépatite B (VHB) et du virus varicelle-zona (VZV), d'aspergillose et de leucoencéphalopathie multifocale progressive (LEMP) sont survenus (voir rubrique «Effets indésirables»).
Des cas de réactivation de l'hépatite B ont été rapportés chez des patients traités par CALQUENCE. Le statut vis-à-vis du virus de l'hépatite B (VHB) doit être établi avant d'instaurer le traitement par CALQUENCE. En cas de sérologie positive pour l'hépatite B, il est recommandé de consulter un hépatologue avant le début du traitement et les patients doivent être surveillés et pris en charge afin de prévenir une réactivation de l'hépatite B.
Des cas de leucoencéphalopathie multifocale progressive (LEMP), y compris des cas d'évolution fatale, ont été rapportés après l'utilisation de CALQUENCE dans le contexte d'un traitement immunosuppresseur antérieur ou concomitant. Le médecin doit envisager la LEMP comme diagnostic différentiel en cas d'apparition ou de détérioration de signes ou de symptômes neurologiques, cognitifs ou comportementaux. Si une LEMP est suspectée, les évaluations diagnostiques appropriées doivent être réalisées et le traitement par CALQUENCE doit être suspendu jusqu'à l'exclusion du diagnostic de LEMP. En cas de doute, il convient d'envisager d'adresser le patient à un neurologue et de prendre les mesures diagnostiques appropriées, à savoir notamment une IRM de préférence avec administration d'un produit de contraste, une analyse du liquide céphalo-rachidien à la recherche d'ADN du virus JC et de nouvelles évaluations neurologiques.
Une prophylaxie conformément à la pratique habituelle doit être envisagée chez les patients qui présentent un risque accru d'infections opportunistes. Les patients doivent être surveillés à la recherche de signes et symptômes d'infections et être traités, le cas échéant, de manière appropriée.
Hémorragies
Des événements hémorragiques graves, y compris d'évolution fatale, sont survenus chez des patients atteints d'un cancer hématologique et traités par CALQUENCE en monothérapie (n = 1 478) et par CALQUENCE en association avec d'autres médicaments (n = 1 095). Des hémorragies sévères (événements hémorragiques de grade 3 ou plus, événements graves ou touchant le système nerveux central) ont été observées chez 5,5% et 3,5% des patients et ont eu une issue fatale dans 0,1% et 0,1% des cas. Les hémorragies sévères les plus fréquentes comprenaient les hémorragies gastro-intestinales (1,4 et 0,7%) et les hémorragies gastro-intestinales supérieures (0,5% et 0,1%), les hématuries (0,4% et 0,5%), les hématomes (0,5% et 0,2%), les épistaxis (0,3% et 0%), les hémorragies intracrâniennes (1,1% et 0,5%) et les hématomes sous-duraux (0,3% et 0,2%) et les hémorragies rétiniennes (0,3% et 0,3%). Au total, des événements hémorragiques (tous grades confondus) étaient survenus, notamment des hématomes et des pétéchies, chez 46,1% et 36,1% des patients.
Le mécanisme responsable de la survenue des événements hémorragiques n'a pas encore été élucidé. La warfarine ou d'autres antagonistes de la vitamine K ne doivent pas être administrés en même temps que CALQUENCE.
Les patients recevant un traitement antithrombotique peuvent présenter un risque accru d'hémorragie. La prudence est de rigueur en cas d'utilisation concomitante avec des antithrombotiques. Une surveillance supplémentaire des patients à la recherche de signes et symptômes d'hémorragie doit être mise en place si une utilisation concomitante est médicalement nécessaire.
Il convient d'évaluer le rapport bénéfice/risque d'une suspension de CALQUENCE pendant au moins 3 jours avant et 3 jours après une intervention chirurgicale.
Cytopénies
Des cytopénies sont survenues chez des patients atteints d'un cancer hématologique et traités par CALQUENCE en monothérapie (n = 1 478) et par CALQUENCE en association avec d'autres médicaments (n = 1 095). La fréquence totale des cas de neutropénie était de 19,4% et 44,7%, celle des cas d'anémie de 17,1% et 12,6% et celle des cas de thrombopénie de 11,5% et 14,2%. Chez les patients atteints d'un cancer hématologique et ayant reçu CALQUENCE en monothérapie et CALQUENCE en association avec d'autres médicaments, des cytopénies de grade ≥3 décelées par des examens de laboratoire sont apparues sous traitement, notamment neutropénie (17,5% et 40,5%), anémie (9,5% et 5,6%) et thrombopénie (6,2% et 7,4%).
Il est recommandé de surveiller la numération de la formule sanguine lorsque cela est médicalement indiqué.
Fibrillation auriculaire
Des cas de fibrillation/flutter auriculaire (tous grades confondus) sont survenus chez 7,4% des patients atteints d'un cancer hématologique et ayant reçu CALQUENCE en monothérapie (n = 1 478); des cas de fibrillation/flutter auriculaire de grade 3 ou plus sont survenus chez 2,3% des patients. Des cas de fibrillation/flutter auriculaire (tous grades confondus) sont survenus chez 4,1% des patients traités par CALQUENCE en association avec d'autres médicaments (n = 1 095); des cas de fibrillation/flutter auriculaire de grade 3 ou plus sont survenus chez 1,7% des patients. Les patients doivent être surveillés à la recherche de symptômes (p.ex. palpitations, sensation d'étourdissement, syncope, douleur thoracique, dyspnée) de fibrillation auriculaire ou de flutter auriculaire et un ECG doit être réalisé lorsque cela est nécessaire.
Syndrome de lyse tumorale (SLT)
Un syndrome de lyse tumorale a été rapporté en lien avec l'utilisation d'acalabrutinib. Il y a un risque de syndrome de lyse tumorale chez les patients ayant une charge tumorale élevée. Les patients doivent être étroitement surveillés et des mesures de précaution appropriées doivent être prises.
Hépatotoxicité, y compris atteinte hépatique médicamenteuse (drug-induced liver injury, DILI)
Une hépatotoxicité, y compris des cas graves, engageant le pronostic vital et potentiellement mortels d'atteinte hépatique médicamenteuse (drug-induced liver injury, DILI), est survenue chez des patients ayant été traités par des inhibiteurs de la tyrosine kinase de Bruton, y compris CALQUENCE. Les taux de bilirubine et de transaminases doivent être surveillés avant et pendant le traitement par CALQUENCE. Les patients qui présentent des paramètres de la fonction hépatique anormaux après l'utilisation de CALQUENCE doivent faire l'objet d'une surveillance plus fréquente afin de détecter des anomalies dans les analyses de la fonction hépatique et des signes cliniques d'une hépatotoxicité. En cas de suspicion d'une DILI, le traitement par CALQUENCE doit être interrompu. Si la DILI est confirmée, le traitement par CALQUENCE doit être arrêté.
Populations potentielles à risque non évaluées
Patients atteints d'un lymphome ou d'une leucémie du système nerveux central (SNC), patients atteints d'une leucémie prolymphocytaire avérée ou présentant une suspicion actuelle ou des antécédents connus de transformation de Richter, patients atteints d'une maladie cardiovasculaire pertinente, patients présentant une infection bactérienne, virale ou fongique systémique active non contrôlée ou toute autre infection, y compris hépatite B ou C active, patients présentant des antécédents connus d'infection au VIH, patients souffrant de pneumopathie induite par le médicament, patients ayant des antécédents connus d'accident vasculaire cérébral ou d'hémorragie intracrânienne lors des 6 mois précédant l'administration de la première dose du médicament à l'essai, patients présentant des antécédents connus de diathèse hémorragique, anticoagulation par la warfarine ou par antivitamines K équivalents, un traitement par des inhibiteurs de la pompe à protons ou la nécessité d'administration de stéroïdes avec une exposition systémique quotidienne de >20 mg d'un équivalent de la prednisone constituaient un critère d'exclusion des études cliniques.
Ce médicament contient moins de 1 mmol de sodium (23 mg) par comprimé, c.-à-d. qu'il est essentiellement «sans sodium».
InteractionsPrincipes actifs pouvant augmenter les concentrations plasmatiques d'acalabrutinib
Inhibiteurs du CYP3A
La co-administration avec un inhibiteur puissant du CYP3A (200 mg d'itraconazole une fois par jour pendant 5 jours) a multiplié la Cmax de l'acalabrutinib par 3,9 et l'ASC par 5,1 chez des sujets sains (n = 17).
L'utilisation concomitante avec des inhibiteurs puissants du CYP3A/ de la P-gp doit être évitée. Si les inhibiteurs puissants du CYP3A/de la P-gp (p.ex. kétoconazole, conivaptan, clarithromycine, indinavir, itraconazole, ritonavir, télaprévir, posaconazole, voriconazole) doivent être utilisés à court terme, le traitement avec Calquence doit être interrompu (voir «Posologie/Mode d'emploi»).La co-administration avec des inhibiteurs modérés du CYP3A (400 mg de fluconazole en dose unique ou 200 mg d'isavuconazole en dose répétée pendant 5 jours) chez des sujets sains a augmenté la Cmax et l'ASC de l'acalabrutinib de 1,4 fois à 2 fois tandis que la Cmax et l'ASC du métabolite actif ACP-5862 ont diminué de 0,65 fois à 0,88 fois par rapport à l'acalabrutinib lorsqu'il est administré seul. Aucun ajustement de dose n'est nécessaire en cas d'association avec des inhibiteurs modérés du CYP3A. Surveiller étroitement les patients pour détecter les effets indésirables (voir «Posologie/Mode d'emploi»).
Principes actifs pouvant diminuer les concentrations plasmatiques d'acalabrutinib
Inducteurs du CYP3A
La co-administration avec un inducteur puissant du CYP3A (600 mg de rifampicine une fois par jour pendant 9 jours) a diminué la Cmax de l'acalabrutinib de 68% et l'ASC de 77% chez des sujets sains (n = 24).
Médicaments diminuant l'acidité gastrique
Aucune différence cliniquement significative de la pharmacocinétique de l'acalabrutinib n'a été constatée lorsque le médicament est utilisé concomitamment avec l'inhibiteur de la pompe à protons rabéprazole. Les comprimés d'acalabrutinib peuvent être administrés concomitamment avec des principes actifs diminuant l'acidité gastrique (inhibiteurs de la pompe à protons, antagonistes des récepteurs H2, antiacides).
Effet de l'acalabrutinib et de son métabolite actif, l'ACP-5862, sur le métabolisme d'autres substances
In vitro, l'acalabrutinib est un inhibiteur faible du CYP3A4/5, du CYP2C8 et du CYP2C9, mais n'a toutefois pas d'effet inhibiteur sur le CYP1A2, le CYP2B6, le CYP2C19, le CYP2D6, l'UGT1A1 et l'UGT2B7.
In vitro, l'ACP-5862 est un faible inhibiteur du CYP2C8, du CYP2C9 et du CYP2C19, tandis qu'il n'inhibe pas le CYP1A2, le CYP2B6, le CYP2D6, le CYP3A4/5, l'UGT1A1 et l'UGT2B7.
L'acalabrutinib est un inducteur faible des ARNm du CYP1A2, du CYP2B6 et du CYP3A4; l'ACP-5862 induit faiblement le CYP3A4.
Substrats du CYP3A
D'après les données in vitro, les données cliniques et la modélisation pharmacocinétique basée sur la physiologie (PBPK), aucune interaction avec les substrats du CYP3A4 n'est à prévoir aux concentrations cliniquement pertinentes (voir «Propriétés/Effets»).
Effet de l'acalabrutinib et de son métabolite actif, l'ACP-5862, sur les systèmes de transport des principes actifs
L'acalabrutinib peut augmenter l'exposition aux substrats de la BCRP co-administrés (p.ex. méthotrexate) par inhibition de la BCRP (Breast Cancer Resistance Protein) intestinale.
L'ACP-5862 peut augmenter l'exposition aux substrats de la MATE1 co-administrés (p.ex. metformine) par inhibition de la MATE1.
Interactions avec les protéines de transport
In vitro, l'acalabrutinib et son métabolite actif, l'ACP-5862, sont des substrats de la P-glycoprotéine (P-gp) et de la protéine de résistance du cancer du sein (Breast Cancer Resistance Protein, BCRP). In vitro, l'acalabrutinib n'est pas un substrat des transporteurs d'influx rénaux OAT1, OAT3 et OCT2 ou des transporteurs hépatiques OATP1B1 et OATP1B3. L'ACP-5862 n'est pas un substrat de l'OATP1B1 ou de l'OATP1B3.
L'acalabrutinib et l'ACP-5862 n'ont pas d'effet inhibiteur sur la P-gp, l'OAT1, l'OAT3, l'OCT2, l'OATP1B1, l'OATP1B3 et la MATE2-K aux concentrations cliniquement pertinentes.
Grossesse, allaitementGrossesse
Il n'existe pas suffisamment de données cliniques sur l'utilisation de CALQUENCE chez les femmes enceintes. Selon les résultats d'études sur les animaux, il pourrait y avoir un risque pour le fœtus et des difficultés lors de l'accouchement (dystocie) en cas d'exposition à l'acalabrutinib pendant la grossesse (voir «Données précliniques»).
CALQUENCE ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue.
Les femmes en âge de procréer et les patients dont la partenaire est en âge de procréer doivent utiliser une méthode contraceptive très efficace pendant le traitement par CALQUENCE et pendant au moins 1 semaine après l'administration de la dernière dose. Si la méthode de contraception utilisée est de type hormonal, une méthode barrière doit être utilisée en complément.
Si la patiente tombe enceinte pendant le traitement par CALQUENCE, elle doit être informée du risque potentiel pour le fœtus.
Allaitement
On ignore si l'acalabrutinib et ses métabolites passent dans le lait maternel. Il n'existe pas de données sur l'effet de l'acalabrutinib sur le nourrisson allaité ou sur la production de lait. L'acalabrutinib et son métabolite actif étaient excrétés dans le lait de rates (voir «Données précliniques»). Un risque pour le nourrisson allaité ne peut être exclu. Il est conseillé aux mères de ne pas allaiter pendant le traitement par CALQUENCE et pendant au moins 2 semaines après l'administration de la dernière dose.
Fertilité
Il n'existe pas de données sur l'effet de CALQUENCE sur la fertilité humaine. Dans une étude préclinique conduite sur l'acalabrutinib chez des rats mâles et femelles, aucun effet indésirable n'a été observé sur les paramètres de la fertilité (voir «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesL'effet de CALQUENCE sur l'aptitude à la conduite et l'utilisation de machines n'a pas fait l'objet d'études. Cependant, pendant le traitement par CALQUENCE, des céphalées, de la fatigue, des vertiges, des chutes et des syncopes ayant été rapportés, il doit être conseillé aux patients qui présentent ces symptômes de ne pas conduire et de ne pas utiliser de machines jusqu'à la disparition des symptômes. Les patients doivent être informés de la survenue potentielle de ces effets (voir «Effets indésirables»).
Effets indésirablesLe profil de sécurité global de l'acalabrutinib se base sur les données groupées de 1 478 patients atteints d'un cancer hématologique ayant reçu l'acalabrutinib en monothérapie et sur les données groupées de 1 095 patients qui ont été traités par l'acalabrutinib en association avec l'obinutuzumab (n = 223 patients), la bendamustine et le rituximab (n = 297 patients), le vénétoclax (n = 291 patients) ou le vénétoclax et l'obinutuzumab (n = 284 patients). La durée médiane du traitement par CALQUENCE était de 28,2 mois en monothérapie et de 49,7 mois pour le traitement en association. La durée médiane du traitement par CALQUENCE en monothérapie dans la totalité de la population incluse était de 38,2 mois.
CALQUENCE en monothérapie
Chez 1 478 patients traités par CALQUENCE en monothérapie, les effets indésirables (≥10%) les plus fréquemment signalés, tous grades confondus, étaient les suivants: diminution de l'hémoglobine (47,4%), diminution du nombre absolu de neutrophiles (43,9%), diminution des plaquettes (36,9%), diarrhée (36,7%), céphalées (36,5%), douleurs musculo-squelettiques (31,9%), ecchymose (30,9%), infection des voies aériennes supérieures (25,8%), toux (25,2%), arthralgie (24,0%), fatigue (23,6%), nausées (21,8%), leucopénie (20,8%), éruption (20,3%), contusion (20,2%), neutropénie (19,4%), second cancer primitif (17,6%),anémie (17,1%), sensation d'étourdissement /vertiges (17,9%), hémorragie/hématome (16,3%), pneumonie (15,8%), constipation (15,2%), douleur abdominale (14,5%), vomissements (14,0%), thrombopénie (11,5%), sinusite (11,4%) et hypertension artérielle (11,2%).
Les effets indésirables les plus fréquemment signalés (≥5%) de grade ≥3 étaient les suivants: diminution du nombre absolu de neutrophiles (24,0%), leucopénie (18,2%), neutropénie (17,5%), diminution de l'hémoglobine (10,8%), diminution des plaquettes (9,5%), anémie (9,5%), pneumonie (8,7%), second cancer primitif (6,7%), thrombopénie (6,2%) et second cancer primitif sans cancer cutané non mélanocytaire (5,5%).
Les effets indésirables graves les plus fréquents (≥1%) comprenant également des événements létaux étaient les suivants: infections, dont la pneumonie (8,3%) et le sepsis (2,8%), et les seconds cancers primitifs (7,1%) ainsi que les hémorragies/hématomes (2,8%), la leucopénie (2,2%), la neutropénie (2,2%) et l'anémie (2,7%). Ces infections se sont principalement produites en l'absence de neutropénie de grade 3 ou 4 (voir rubrique «Mises en garde et précautions»).
Des réductions de dose en raison d'effets indésirables ont été rapportées chez 5,9% des patients. Le traitement a été arrêté en raison d'effets indésirables chez 15,8% des patients. Il s'agissait le plus fréquemment de cas de pneumonie (0,8%), de COVID-19 (0,6%), de pneumonie due à la COVID-19 (0,5%) et de thrombopénie (0,4%).
Les effets indésirables survenus chez les patients ayant reçu CALQUENCE en monothérapie sont indiqués dans le Tableau 4.
CALQUENCE en association
Chez les 1 095 patients traités par CALQUENCE en association, les effets indésirables les plus fréquemment signalés (≥10%), tous grades confondus, étaient les suivants: diminution du nombre absolu de neutrophiles (75,1%), diminution des plaquettes (54,2%), diminution de l'hémoglobine (52,8%), leucopénie (46,5%), neutropénie (44,7%), diarrhée (38,3%), céphalées (33,7%), douleurs musculo-squelettiques (32,1%), nausées (27,5%), éruption (24,7%), ecchymose (23,9%), fatigue (22,5%), arthralgie (19,5%), toux (18,0%), contusion (17,4%), infection des voies aériennes supérieures (16,5%), constipation (15,4%), vomissements (15%), thrombopénie (14,2%), sensation d'étourdissement/vertiges (15,3%), hémorragie/hématome (13,6%), anémie (12,6%), second cancer primitif (12,1%), douleur abdominale (11,8%).
Les effets indésirables les plus fréquemment signalés (≥5%) de grade ≥3 étaient les suivants: diminution du nombre absolu de neutrophiles (47,0%), leucopénie (41,9%), neutropénie (40,5%), diminution des plaquettes (12,3%), diminution de l'hémoglobine (7,7%), thrombopénie (7,4%), anémie (5,6%) et pneumonie (5,2%).
Dans les analyses groupées des patients traités par l'acalabrutinib en association avec l'obinutuzumab (n = 223), une fréquence globale plus élevée des effets indésirables suivants a été observée par rapport aux patients traités par l'acalabrutinib en monothérapie (n =1 040): infections (74 vs 66,7%), y compris infections de grade ≥3, (21,5 vs 17,6%), infections des voies aériennes supérieures (31,4 vs 22%) et autres infections très fréquentes, affections musculo-squelettiques et systémiques (58,3 vs 51,6%), principalement arthralgie (26,9 vs 19,1%) et douleurs aux extrémités (13,9 vs 8,9%), fatigue (30,5 vs 21,3%), contusion (27,4 vs 21,7%), sensation d'étourdissement (23,8 vs 13,4%) et chutes (14,8 vs 7,9%). La fréquence globale des effets indésirables ≥ grade 3 (70,4 vs 54,1%) était dans le groupe de traitement en association également plus élevée par rapport au groupe en monothérapie et principalement due à une incidence plus élevée de neutropénie de grade ≥3 (23,8 vs 11,2%). De plus, des taux plus élevés avec une différence de PT ≥10% ont été observés pour la neutropénie (25,1 vs 12,3%), les réactions liées à la perfusion (19,3 vs 0,8%) et l'éruption maculo-papuleuse (17 vs 4,9%).
Les effets indésirables observés lors des études cliniques chez les patients traités par l'acalabrutinib en monothérapie vs traitement associant l'acalabrutinib et d'autres médicamentssont indiqués dans le Tableau 5.
Les effets indésirables sont présentés par classe de systèmes d'organes (SOC) MedDRA. Au sein de chaque classe de systèmes d'organes, les effets indésirables sont présentés par fréquence, les effets indésirables les plus fréquents figurant en premier. De plus, la catégorie de fréquence correspondant à chaque effet indésirable selon la convention CIOMS III est définie de la manière suivante: très fréquents (≥1/10), fréquents (≥1/100 à <1/10); occasionnels (≥1/1 000 à <1/100); rares (≥1/10 000 à <1/1000); très rares (<1/10 000); fréquence inconnue (ne peut être estimée sur la base des données disponibles).
Tableau 5. Effets indésirables* des patients atteints d'un cancer hématologique et traités par l'acalabrutinib en monothérapie (n = 1 095) ou en association avec d'autres médicaments (n = 520)
|
SOC MedDRA
|
Terme MedDRA
|
Descripteur CIOMS/Fréquence globale
tous grades CTCAE [fréquence des événements de grade CTCAE ≥3]†
| |
Monothérapie
|
Traitement en association
| |
Infections et infestations
|
Infection des voies aériennes supérieures
|
Très fréquents (25,8%) [1,2%]
|
Très fréquents (16,5%) [0,6%]
| |
COVID-19
|
Fréquents (7,4%) [2,0%]
|
Très fréquents (22,7%) [6,3%]
| |
Pneumonie due à la COVID-19
|
Fréquents (2,0%) [1,9%]
|
Très fréquents (10,4%) [9%]
| |
Sinusite
|
Très fréquents (11,4%) [0,4%]
|
Fréquents (6,7%) [0,2%]
| |
Pneumonie
|
Très fréquents (15,8%) [8,7%]
|
Fréquents (9,7%) [5,2%]
| |
Infection des voies urinaires
|
Fréquents (9,9%) [1,8%]
|
Très fréquents (9,0%) [0,8%]
| |
Rhinopharyngite
|
Fréquents (8,3%) [0%]
|
Fréquents (5,6%) [0,1%]
| |
Bronchite
|
Fréquents (9,7%) [0,6%]
|
Fréquents (5,8%) [0,3%]
| |
Infections à herpèsvirus1
|
Fréquents (8,9%) [0,9%]
|
Fréquents (7,8%) [0,7%]
| |
Sepsis1
|
Fréquents (3,2%) [3,0%]
|
Fréquents (2,6%) [2,6%]
| |
Infections à Aspergillus1
|
Occasionnels (0,3%) [0,2%]
|
Très rares (0,2%) [0,2%]
| |
Réactivation de l'hépatite B
|
Occasionnels (0,4%) [0,3%]
|
Occasionnels (0,5%) [0,1%]
| |
Tumeurs bénignes, malignes et non précisées6
|
Second cancer primitif2
|
Très fréquents (17,6%) [6,7%]
|
Très fréquents (12,1%) [4,7%]
| |
Second cancer primitif sans cancer cutané non mélanocytaire3
|
Fréquents (9,7%) [5,5%]
|
Fréquents (6,8%) [3,7%]
| |
Cancer cutané non mélanocytaire
|
Fréquents (9,9%) [1,4%]
|
Fréquents (7,2%) [1,2%]
| |
Affections hématologiques et du système lymphatique
|
Neutropénie1
|
Très fréquents (19,4%) [17,5%]
|
Très fréquents (44,7%) [40,5%]
| |
Anémie1
|
Très fréquents (17,1%) [9,5%]
|
Très fréquents (12,6%) [5,6%]
| |
Thrombopénie1
|
Très fréquents (11,5%) [6,2%]
|
Très fréquents (14,2%) [7,4%]
| |
Leucopénie1
|
Très fréquents (20,8%) [18,2%]
|
Très fréquents (46,5%) [41,9%]
| |
Lymphocytose
|
Occasionnels (0,5%) [0,3%]
|
Occasionnels (0,5%) [0,2%]
| |
Diminution du nombre absolu de neutrophiles7
|
Très fréquents (43,9%) [24,0%]
|
Très fréquents (75,1%) [47,0%]
| |
Diminution de l'hémoglobine7
|
Très fréquents (47,4%) [10,8%]
|
Très fréquents (52,8%) [7,7%]
| |
Diminution des plaquettes7
|
Très fréquents (36,9%) [9,5%]
|
Très fréquents (54,2%) [12,3%]
| |
Troubles du métabolisme et de la nutrition
|
Syndrome de lyse tumorale
|
Occasionnels (0,5%) [0,4%]
|
Occasionnels (0,9%) [0,9%]
| |
Affections du système nerveux
|
Céphalées
|
Très fréquents (36,5%) [1,2%]
|
Très fréquents (33,7%) [1,1%]
| |
Sensation d'étourdissement/vertiges1
|
Très fréquents (16,5%) [0,3%]
|
Très fréquents (15,3%) [0,5%]
| |
Affections cardiaques
|
Fibrillation/flutter auriculaires4
|
Fréquents (7,4%) [2,3%]
|
Fréquents (4,1%) [1,7%]
| |
Affections vasculaires
|
Hématomes1
|
Très fréquents (30,9%) [0%]
|
Très fréquents (23,9%) [0,1%]
| |
Contusion
|
Très fréquents (20,2%) [0%]
|
Très fréquents (17,4%) [0%]
| |
Pétéchies
|
Fréquents (8,9%) [0%]
|
Fréquents (5,6%) [0%]
| |
Ecchymoses
|
Fréquents (5,7%) [0%]
|
Fréquents (3,2%) [0,1%]
| |
Hémorragie/hématome1
|
Très fréquents (16,3%) [3,2%]
|
Très fréquents (13,6%) [1,6%]
| |
Hémorragie gastro-intestinale
|
Occasionnels (0,6%) [0,5%]
|
Occasionnels (0,3%) [0,3%]
| |
Hémorragie intracrânienne
|
Occasionnels (0,3%) [0,2%]
|
Occasionnels (0%) [0%]
| |
Épistaxis
|
Fréquents (8,0%) [0,3%]
|
Fréquents (4,6%) [0%]
| |
Hypertension artérielle
|
Très fréquents (11,2%) [4,7%]
|
Fréquents (8,8%) [3,8%]
| |
Affections respiratoires, thoraciques et médiastinales
|
Toux
|
Très fréquents (25,2%) [0,4%]
|
Très fréquents (18,0%) [0,3%]
| |
Affections gastro-intestinales
|
Diarrhée
|
Très fréquents (36,7%) [2,6%]
|
Très fréquents (38,3%) [3,0%]
| |
Nausées
|
Très fréquents (21,8%) [0,8%]
|
Très fréquents (27,5%) [0,5%]
| |
Constipation
|
Très fréquents (15,2%) [0,1%]
|
Très fréquents (15,4%) [0,4%]
| |
Douleur abdominale1
|
Très fréquents (14,5%) [1,2%]
|
Très fréquents (11,8%) [1,5%]
| |
Vomissements
|
Très fréquents (14,0%) [0,7%]
|
Très fréquents (15,0%) [0,5%]
| |
Affections de la peau et du tissu sous-cutané
|
Éruption1
|
Très fréquents (20,3%) [0,9%]
|
Très fréquents (24,7%) [3,4%]
| |
Affections musculo-squelettiques et systémiques
|
Douleurs musculo-squelettiques5
|
Très fréquents (31,9%) [1,8%]
|
Très fréquents (32,1%) [2,0%]
| |
Arthralgie
|
Très fréquents (24,0%) [0,9%]
|
Très fréquents (19,5%) [1,0%]
| |
Troubles généraux et anomalies au site d'administration
|
Fatigue
|
Très fréquents (23,6%) [2,0%]
|
Très fréquents (22,5%) [1,3%]
| |
Asthénie
|
Fréquents (7,0%) [0,9%]
|
Fréquents (6,7%) [0,4%]
| |
*D'après la version 4.03 de la classification NCI CTCAE (Common Terminology Criteria for Adverse Events du National Cancer Institute).
1 Inclut de multiples termes d'effets indésirables.
2 Les seconds cancers primitifs étaient définis par la SMQ Tumeurs malignes (y compris la SMQ Tumeurs hématologiques et la SMQ Tumeurs non hématologiques), par la SMQ Lymphomes malins [terme étroit] et par la SMQ Syndrome myélodysplastique [terme étroit].
3 Les seconds cancers primitifs (excepté le cancer cutané non mélanocytaire) étaient définis par les critères pour les seconds cancers primitifs, excepté les termes préférentiels (PT) sous le terme de haut niveau «Tumeurs cutanées malignes et non précisées (excepté les mélanomes)».
4 Inclut tous les PT avec les fibrillations auriculaires ou le flutter auriculaire.
5 Inclut douleurs dorsales, douleurs osseuses, douleurs thoraciques musculo-squelettiques, douleurs musculo-squelettiques, troubles musculo-squelettiques, syndrome myofascial douloureux, douleurs cervicales, douleurs aux extrémités, myalgie, douleurs du rachis.
6 Inclut des événements survenus au terme de la phase d'annonce dans le cadre des études.
7 Variations des paramètres biologiques dues au traitement.
|
Description d'effets indésirables spécifiques et informations complémentaires
Hépatotoxicité
Une hépatotoxicité a été rapportée, principalement sous forme d'élévation des transaminases, chez des patients qui recevaient Calquence. Des cas d'hépatotoxicité sévères ont également été observés. Un lien de causalité entre Calquence et l'hépatotoxicité/les élévations des transaminases n'a pas été établi.
Syncopes et chutes
Des syncopes et des chutes ont été observées chez des patients traités par Calquence au cours des études cliniques et après la mise sur le marché (voir «Effet sur l'aptitude à la conduite et l'utilisation de machines»).
Patients âgés
Sur les 1 478 participants des études cliniques sur CALQUENCE en monothérapie, 42,2% avaient ≥65 ans et <75 ans et 20,6% avaient ≥75 ans. Chez les patients de 75 ans ou plus, les événements indésirables de grade ≥3 étaient plus fréquents (77,4%) que chez les patients ≥65 ans et <75 ans (68,4%) ou que les patients âgés de moins de 65 ans (59,3%). Des taux plus élevés ont été observés chez les patients de 75 ans ou plus par comparaison avec les deux autres groupes d'âge pour la pneumonie tous grades confondus (19,7%, 15,9% et 13,5%), y compris la pneumonie de grade ≥3 (14,4%, 8,2% et 6,0%).
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageIl n'existe aucun traitement spécifique en cas de surdosage par CALQUENCE et les symptômes du surdosage n'ont pas été établis. En cas de surdosage, les patients doivent être surveillés étroitement à la recherche de signes ou de symptômes d'effets indésirables et un traitement symptomatique approprié doit être mis en place.
Propriétés/EffetsCode ATC
L01EL02
Mécanisme d'action
L'acalabrutinib est un inhibiteur de faible poids moléculaire sélectif de la tyrosine kinase de Bruton (BTK). La BTK est une molécule de signalisation des voies des récepteurs antigéniques des lymphocytes B (BCR) et des récepteurs de cytokines. La signalisation induite par la BTK stimule la survie et la prolifération des lymphocytes B et est essentielle à l'adhésion, au transport et au chimiotactisme cellulaire.
L'acalabrutinib et son métabolite actif (l'ACP-5862) forment une liaison covalente avec un résidu cystéine au niveau du site actif de la BTK, entraînant ainsi l'inactivation irréversible et sélective de la BTK (CI50 ≤5nM) avec des interactions hors cible minimes. Lors d'un criblage de plus de 380 kinases mammaliennes de type sauvage, les seules autres kinases à interagir avec l'acalabrutinib et l'ACP-5862 à des concentrations pertinentes sur le plan clinique ont été les tyrosines kinases BMX et ERBB4, leur interaction étant de 3 à 4 fois moins forte que celle de la BTK.
Des études non cliniques ont montré que l'acalabrutinib inhibait en aval l'activation des protéines de signalisation CD86 et CD69 médiée par la BTK et qu'il exerçait une activité minime sur d'autres cellules immunitaires (lymphocytes T et cellules NK).
Pharmacodynamique
Chez des patients atteints d'hémopathies malignes B et prenant de l'acalabrutinib à raison de 100 mg deux fois par jour, l'occupation médiane à l'état d'équilibre de la BTK de ≥95% dans le sang périphérique s'est maintenue sur 12 heures, entraînant l'inactivation de la BTK pendant l'intégralité de l'intervalle posologique recommandé.
Électrophysiologie cardiaque
Dans le cadre d'une étude spécifique de l'intervalle QT, l'administration de doses uniques d'acalabrutinib de 100 mg et de 400 mg n'a pas induit d'allongement cliniquement pertinent de l'intervalle QT/QTc (soit pas de façon ≥10 ms).
Efficacité clinique
Patients atteints d'une LLC non précédemment traitée – ELEVATE-TN
La sécurité et l'efficacité de CALQUENCE chez des patients atteints de LLC non précédemment traitée ont été évaluées dans le cadre d'une étude de phase III randomisée, multicentrique, en ouvert (ELEVATE-TN), à laquelle 535 patients ont participé. Il devait s'agir d'une LLC CD20+ active/nécessitant un traitement et diagnostiquée selon les critères IWCLL de 2008. En outre, le nombre absolu de neutrophiles et de plaquettes, indépendamment du facteur de croissance ou d'une aide transfusionnelle, devait être de 750 et 50 000 cellules/μl (en cas d'atteinte de la moelle osseuse de 500 et 30 000 cellules/μl). Les patients présentant une atteinte du SNC, une leucémie prolymphocytique ou une transformation de Richter étaient exclus de l'étude. Les patients ont reçu l'association CALQUENCE plus obinutuzumab, CALQUENCE en monothérapie ou l'association obinutuzumab plus chlorambucil. Des patients âgés de 65 ans ou plus ou entre 18 et 65 ans avec des maladies coexistantes (clairance de la créatinine 30-69 ml/min et/ou score CIRS-G >6) ont été inclus dans l'étude ELEVATE-TN. Les patients pouvaient recevoir en complément des antithrombotiques, à l'exception de la warfarine et des antivitamines K équivalents.
Les patients ont été répartis selon un rapport 1:1:1 dans l'un des 3 bras de traitement suivants:
·Association CALQUENCE plus obinutuzumab (CALQUENCE+G): CALQUENCE a été administré à une posologie de 100 mg deux fois par jour à partir du Jour 1 du Cycle 1 jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. L'obinutuzumab a été administré à partir du Jour 1 du Cycle 2 pendant un maximum de 6 cycles de traitement. L'obinutuzumab a été administré à la posologie de 1 000 mg les Jours 1 et 2 (100 mg le Jour 1 et 900 mg le Jour 2), 8 et 15 du Cycle 2 puis à raison de 1 000 mg le Jour 1 des Cycles 3 à 7. Chaque cycle durait 28 jours.
·CALQUENCE en monothérapie: CALQUENCE a été administré à une posologie 100 mg deux fois par jour jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
·Association obinutuzumab plus chlorambucil (GClb): l'obinutuzumab et le chlorambucil ont été administrés pendant un maximum de 6 cycles de traitement. L'obinutuzumab a été administré à la posologie de 1 000 mg les Jours 1 et 2 (100 mg le Jour 1 et 900 mg le Jour 2), 8 et 15 du Cycle 1 puis à raison de 1 000 mg le Jour 1 des Cycles 2 à 6. Le chlorambucil a été administré à la posologie de 0,5 mg/kg les Jours 1 et 15 des Cycles 1 à 6. Chaque cycle durait 28 jours.
Les patients ont été stratifiés selon le statut mutationnel pour la délétion 17p (oui/non), l'indice de performance ECOG (indice selon l'Eastern Cooperative Oncology Group, 0 ou 1 versus 2) et la région géographique (Amérique du Nord et Europe occidentale versus autre). Après une progression confirmée de la maladie, 45 patients randomisés dans le bras GClb ont reçu CALQUENCE en monothérapie dans le cadre d'une phase de traitement croisé.
Les caractéristiques à l'inclusion étaient généralement équilibrées dans les trois bras (association Calquence plus obinutuzumab [n=179], Calquence en monothérapie [n = 179] et association obinutuzumab plus chlorambucil [n = 177]): âge médian 70, 70 et 71 ans; 62%, 62% et 59,9% des participants étaient de sexe masculin; 94,4%, 92,2% et 94,4% avaient un indice de performance ECOG-PS de 0-1; le temps médian écoulé depuis le diagnostic était de 30,5, 24,4 et 30,7 mois; les facteurs cytogénétiques (del17p, del11q, mutation de TP53, IGHV non muté, caryotype complexe) et le stade Rai étaient généralement équilibrés.
Le critère d'évaluation principal était la survie sans progression (SSP) évaluée par un comité de revue indépendant (Independent Review Committee, IRC) d'après les critères IWCLL (International Workshop on Chronic Lymphocytic Leukaemia) de 2008 avec incorporation de la clarification pour la lymphocytose liée au traitement (Cheson 2012). Après un suivi médian de 28,3 mois, la SSP évaluée par l'IRC montrait une réduction statistiquement significative de 90% du risque de progression de la maladie ou de décès pour les patients atteints d'une LLC non précédemment traitée dans le bras de traitement CALQUENCE+G versus le bras de traitement GClb. Au moment de l'analyse, la survie globale médiane avec au total 37 décès n'avait été encore atteinte dans aucun des bras de traitement: 9 (5%) dans le bras de traitement CALQUENCE+G, 11 (6,1%) dans le bras de traitement CALQUENCE en monothérapie et 17 (9,6%) dans le bras GClb. Les données d'efficacité sont présentées dans le Tableau 6.
Tableau 6. Résultats d'efficacité chez les patients atteints d'une LLC (étude ELEVATE-TN)
|
Caractéristique
|
Association CALQUENCE plus obinutuzumab
n = 179
|
CALQUENCE en monothérapie
n = 179
|
Association obinutuzumab plus chlorambucil
n = 177
| |
Survie sans progression*
| |
Nombre d'événements (%)
|
14 (7,8)
|
26 (14,5)
|
93 (52,5)
| |
Médiane (IC à 95%), mois
|
NA
|
NA (34,2; NA)
|
22,6 (20,2; 27,6)
| |
HR† (IC à 95%)
|
0,10 (0,06; 0,17)
|
0,20 (0,13; 0,30)
|
-
| |
Taux de réponse globale*
| |
TRG, n (%)
(IC à 95%)
|
168 (93,9)
(89,3; 96,5)
|
153 (85,5)
(79,6; 89,9)
|
139 (78,5)
(71,9; 83,9)
| |
RC, n (%)
|
23 (12,8)
|
1 (0,6)
|
8 (4,5)
|
IC = intervalle de confiance; HR = hazard ratio; NA = non atteinte; RC = rémission complète (Complete Response); Rci = rémission complète avec récupération incomplète de la numération sanguine; Rpn = rémission partielle nodulaire; RP = rémission partielle (Partial response);
*D'après l'évaluation de l'IRC
†D'après un modèle à risques proportionnels de Cox stratifié
Les résultats de SSP pour le traitement par CALQUENCE avec ou sans obinutuzumab étaient comparables dans les différents sous-groupes, y compris pour les caractéristiques à haut risque (délétion 17p, délétion 11q, mutation de TP53 ou IGHV non muté).
Une transformation de Richter est survenue chez 6 patients (3,4%) dans le bras acalabrutinib en monothérapie (et chez aucun patient dans le bras de traitement en association) pendant la phase de randomisation et chez un patient (2,2%) dans le bras chlorambucil/obinutuzumab pendant la phase croisée.
Patients atteints d'une LLC non précédemment traitée – durée de traitement fixe – AMPLIFY
La sécurité et l'efficacité de CALQUENCE en association avec le vénétoclax dans le traitement de la LLC non précédemment traitée ont été évaluées dans le cadre d'une étude de phase III randomisée, multicentrique, en ouvert (AMPLIFY). Les patients ont reçu l'association CALQUENCE plus vénétoclax ou une immunochimiothérapie au choix du médecin investigateur, à savoir le protocole FCR (fludarabine plus cyclophosphamide plus rituximab) ou BR (bendamustine plus rituximab). L'étude AMPLIFY incluait des patients atteints d'une LLC non précédemment traitée, âgés d'au moins 18 ans et ne présentant ni délétion 17p ni mutation de TP53.
Les patients ayant un score CIRS-G individuel de 4 ou un score CIRS-G > 6, une anémie hémolytique auto-immune non contrôlée, un purpura thrombopénique idiopathique non contrôlé ou des antécédents de leucoencéphalopathie multifocale progressive confirmée, et les patients ayant reçu un vaccin vivant dans les 28 jours précédant la première dose du médicament à l'essai ont été exclus de l'étude. Les patients pouvaient recevoir en complément des antithrombotiques, à l'exception de la warfarine et des antivitamines K équivalents. Des patients ont été inclus dans l'étude lors de la pandémie de COVID-19.
Les patients ont été répartis selon un rapport 1:1 dans l'un des bras de traitement suivants:
·CALQUENCE plus vénétoclax (AV): CALQUENCE 100 mg a été administré deux fois par jour à partir du Jour 1 du Cycle 1 pendant un total de 14 cycles ou jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. À partir du Jour 1 du Cycle 3, les patients ont suivi le schéma de titration de la dose de vénétoclax pendant 5 semaines, en commençant par une dose de 20 mg une fois par jour et en augmentant ensuite chaque semaine à 50 mg, 100 mg, 200 mg et enfin 400 mg. Le vénétoclax a été administré pendant 12 cycles au total. Chaque cycle durait 28 jours.
·Immunochimiothérapie au choix du médecin investigateur (FCR/BR):
·Fludarabine plus cyclophosphamide plus rituximab (FCR): la fludarabine (25 mg/m2) et le cyclophosphamide (250 mg/m2) ont été administrés les Jours 1 à 3 pendant un maximum de 6 cycles. Le rituximab a été administré à une dose de 375 mg/m2 le Jour 1 du Cycle 1 et à une dose de 500 mg/m2 le Jour 1 des Cycles 2 à 6 au maximum. Chaque cycle durait 28 jours.
·Bendamustine plus rituximab (BR): la bendamustine 90 mg/m2 a été administrée les Jours 1 et 2 pendant un maximum de 6 cycles. Le rituximab a été administré à une dose de 375 mg/m2 le Jour 1 du Cycle 1 et à une dose de 500 mg/m2 le Jour 1 des Cycles 2 à 6 au maximum. Chaque cycle durait 28 jours.
Les patients ont été stratifiés selon l'âge (> 65 ans ou ≤65 ans), le statut mutationnel de l'IGHV (muté versus non muté), le stade Rai (risque élevé [≥3] versus risque non élevé) et la région géographique (Amérique du Nord versus Europe de l'Ouest versus autre). Le Tableau 7 récapitule les caractéristiques démographiques à l'inclusion et les caractéristiques de la maladie dans les deux bras de traitement.
Tableau 7. Caractéristiques à l'inclusion (AMPLIFY) des patients atteints d'une LLC non précédemment traitée
|
Caractéristique
|
AV
n = 291
|
FCR/BR
n = 290
| |
Âge, ans; médiane (intervalle)
|
61 (31-84)
|
61 (26-86)
| |
Hommes, %
|
61,2
|
63,1
| |
Caucasiens, %
|
91,1
|
86,9
| |
Indice de performance ECOG 0-1, %
|
90,0
|
90,3
| |
Temps médian écoulé entre le diagnostic et la randomisation (mois)
|
28,5
|
29,6
| |
Tumeur volumineuse avec ganglions lymphatiques ≥5 cm, %
|
38,8
|
42,8
| |
Catégorie cytogénétique/FISH, %
|
|
| |
Délétion 11q
|
17,5
|
15,9
| |
Caryotype complexe (≥3 anomalies)
|
15,5
|
14,5
| |
IGHV non muté, %
|
57,4
|
59,3
| |
Stade Rai, %
|
|
| |
0
|
1,0
|
1,4
| |
I
|
16,2
|
21,4
| |
II
|
35,7
|
33,4
| |
III
|
23,7
|
20,3
| |
IV
|
23,4
|
23,4
|
Le critère d'évaluation principal était la survie sans progression (SSP) dans le bras AV par rapport à l'immunochimiothérapie au choix du médecin investigateur (FCR/BR), évaluée par un comité de revue indépendant (Independent Review Committee, IRC) d'après les critères IWCLL (International Workshop on Chronic Lymphocytic Leukaemia) de 2018. Les autres critères d'évaluation de l'efficacité étaient la maladie résiduelle minimale (MRD) dans le sang périphérique, mesurée par cytométrie en flux (10-4), et la survie globale (SG). La comparaison de la MRD se basait sur le Cycle 9 (AV) ou sur l'échéance à 12 semaines après le début du Cycle 6 (FCR/BR).
Après un suivi médian de 41,3 mois, la SSP évaluée par l'IRC montrait une réduction statistiquement significative de 35% du risque de progression de la maladie ou de décès chez les patients traités par AV par rapport au traitement FCR/BR (hazard ratio 0,65 [IC à 95% 0,49 à 0,87]).
Une MRD négative aux échéances susmentionnées a été atteinte chez 78 (26,8%) des patients traités par AV et chez 148 (51,0%) des patients du bras FCR/BR (risque relatif en faveur du bras FCR/BR: 0,5 [IC à 95% 0,4; 0,7]).
Après un suivi médian de 46,4 mois, le HR de la SG était de 0,42 (IC à 95% [0,25; 0,70]) pour le bras AV par rapport au bras FCR/BR, avec un total de 67 décès, dont 23 (7,9%) dans le bras AV et 44 (15,2%) dans le bras FCR/BR. Comme la procédure d'analyse statistique n'a pas pu être poursuivie en raison du résultat de la MRD, la signification statistique du résultat de SG n'a pas pu être établie.
Le bénéfice de CALQUENCE en association avec le vénétoclax concernant la réduction du risque de SSP était cohérent dans le sous-groupe des patients atteints de LLC et ayant un IGHV non muté.
Patients atteints d'une LLC ayant reçu au moins un traitement antérieur – ASCEND
La sécurité et l'efficacité de CALQUENCE chez des patients atteints de LLC en rechute ou réfractaire ont été évaluées dans le cadre d'une étude de phase III randomisée, multicentrique, en ouvert (ASCEND), à laquelle 310 patients ayant reçu au moins un traitement antérieur ont participé. Un traitement antérieur par un inhibiteur de BCL-2 (p.ex. vénétoclax), un inhibiteur du récepteur des cellules B (BCR) (p.ex. inhibiteurs de la BTK ou de la PI3K) ou une radiothérapie ou un traitement anticorps conjugué à une toxine n'était pas autorisé. Il devait s'agir d'une LLC CD20+ active/nécessitant un traitement et diagnostiquée selon les critères IWCLL de 2008. En outre, le nombre absolu de neutrophiles et de plaquettes, indépendamment du facteur de croissance ou d'une aide transfusionnelle, devait être de 750 et 50 000 cellules/μl (en cas d'atteinte de la moelle osseuse de 500 et 30 000 cellules/μl). Les patients présentant une atteinte du SNC, une leucémie prolymphocytique ou une transformation de Richter étaient exclus de l'étude. Les patients ont reçu soit CALQUENCE en monothérapie, soit, au choix du médecin investigateur, l'association idélalisib plus rituximab ou l'association bendamustine plus rituximab. L'utilisation d'antithrombotiques était autorisée à l'exception de la warfarine et des antivitamines K équivalents.
Les patients ont été répartis selon un rapport 1:1 dans l'un des groupes de traitement suivants:
·CALQUENCE à raison de 100 mg deux fois par jour jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable ou
·au choix du médecin investigateur:
·idélalisib à raison de 150 mg deux fois par jour en association avec ≤8 perfusions de rituximab (375 mg/m2/500 mg/m2) le Jour 1 de chaque cycle de 28 jours pendant un maximum de 6 cycles jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
·bendamustine à raison de 70 mg/m2 (Jours 1 et 2 de chaque cycle de 28 jours) en association avec le rituximab (375 mg/m2/500 mg/m2) le Jour 1 de chaque cycle de 28 jours pendant un maximum de 6 cycles.
Les patients ont été stratifiés selon les facteurs suivants: statut mutationnel pour la délétion 17p (oui/non), indice de performance ECOG (indice 0 ou 1 versus 2) et nombre de traitements antérieurs (1 à 3 versus ≥4). Après une progression confirmée de la maladie, 35 patients, qui avaient été dans un premier temps traités au choix du médecin investigateur par l'association idélalisib plus rituximab ou l'association bendamustine plus rituximab, ont reçu CALQUENCE dans le cadre d'une phase de traitement croisé.
Les caractéristiques à l'inclusion dans les deux bras (CALQUENCE en monothérapie [n = 155] ou le traitement choisi par le médecin investigateur par l'association idélalisib + rituximab ou l'association bendamustine + rituximab [n = 155]) étaient généralement équilibrées: âge médian 68 et 67 ans; 69,7% et 64,5% des participants étaient de sexe masculin; 87,7% et 86,5% avaient un indice de performance ECOG de 0-1; le temps médian écoulé depuis le diagnostic était de 85,3 et 79 mois; le temps médian écoulé entre le dernier traitement précédent de la LLC et la première dose était de 26,4 et 22,7 mois; les facteurs cytogénétiques (del17p, del11q, mutation de TP53, IGHV non muté, caryotype complexe) et le stade Rai étaient généralement équilibrés.
Le critère d'évaluation principal était la SSP évaluée par un IRC d'après les critères IWCLL de 2008 avec incorporation de la clarification pour la lymphocytose liée au traitement (Cheson 2012). Au moment de la première analyse pré-spécifiée avec un suivi médian de 16,1 mois, CALQUENCE indiquait une amélioration cliniquement et statistiquement significative de la SSP évaluée par l'IRC par rapport au bras IR/BR (hazard ratio 0,31 [IC à 95% 0,20 à 0,49] p < 0,0001). Au moment de l'analyse, la survie globale médiane avec au total 33 décès n'avait été encore atteinte dans aucun des bras de traitement: 15 (9,7%) dans le bras CALQUENCE en monothérapie et 18 (11,6%) dans le bras de traitement comportant l'administration du traitement choisi par le médecin investigateur (association idélalisib plus rituximab ou association bendamustine plus rituximab). Dans une analyse ultérieure non pré-spécifiée, après une durée de suivi médiane de 22 mois, la SSP médiane, qui contrairement au critère d'évaluation principal a été évaluée par le médecin investigateur, n'a pas été atteinte dans le bras Calquence et était de 16,8 mois dans le bras IR/BR (hazard ratio 0,27 [IC à 95% 0,18 à 0,40]. Les données de survie globale étaient toujours immatures avec respectivement 21 (13,5%) et 26 (16,8%) événements dans le bras Calquence et le bras comparateur. Les résultats d'efficacité de l'analyse pré-spécifiée sont présentés dans le Tableau 8.
Tableau 8. Résultats d'efficacité chez les patients atteints d'une LLC (étude ASCEND)
|
|
CALQUENCE
Monothérapie
n = 155
|
Traitement choisi par le médecin investigateur par l'association idélalisib + rituximab (n = 119) ou l'association bendamustine + rituximab (n = 36)
n = 155
| |
Survie sans progression*
| |
Nombre d'événements (%)
|
27 (17,4)
|
68 (43,9)
| |
Médiane (IC à 95%), mois
|
NA
|
16,5 (14,0; 17,1)
| |
HR† (IC à 95%)
|
0,31 (0,20; 0,49)
| |
Taux de réponse globale*
| |
TRG, n (%)
(IC à 95%)
|
126 (81,3)
(74,4; 86,6)
|
117 (75,5)
(68,1; 81,6)
| |
RC, n (%)
|
0
|
2 (1,3)
| |
Durée de la réponse* (DR)
| |
Médiane (IC à 95%), mois
|
NA
|
13,6 (11,9; NA)
|
IC = intervalle de confiance; HR = hazard ratio; NA = non atteinte; RC = rémission complète (Complete Response); RP = rémission partielle (Partial Response)
*D'après l'évaluation de l'IRC
†D'après un modèle à risques proportionnels de Cox stratifié
Les résultats de SSP pour le traitement par CALQUENCE étaient comparables dans les différents sous-groupes, y compris pour les caractéristiques à haut risque (délétion 17p, délétion 11q, mutation de TP53 ou IGHV non muté).
Une transformation de Richter est survenue chez 4 patients (2,6%) dans le bras acalabrutinib en monothérapie et chez 3 patients (2,0%) dans le bras idélalisib plus rituximab/bendamustine plus rituximab lors de la phase de randomisation et chez 2 patients (5,7%) dans le bras idélalisib plus rituximab/bendamustine plus rituximab pendant la phase croisée.
Patients atteints de LCM non précédemment traité – ECHO
L'efficacité de CALQUENCE chez des patients atteints d'un LCM non précédemment traité a été évaluée dans le cadre de l'étude ECHO, une étude de phase III multicentrique, randomisée, en double aveugle et contrôlée contre placebo. 598 patients atteints d'un LCM confirmé non précédemment traité et âgés d'au moins 65 ans ont participé à l'étude ECHO. Les patients dont l'objectif de traitement était une réduction tumorale avant une greffe de cellules souches autologue étaient exclus de l'étude. La randomisation des patients a été stratifiée en fonction de la région géographique (Amérique du Nord versus Europe de l'Ouest versus Autres) et du score MIPI (Mantle Cell Lymphoma International Prognostic Index) simplifié (0-3 versus 4-5 versus 6-11). Des patients ont été inclus dans l'étude lors de la pandémie de COVID-19.
Les patients ont été randomisés dans 2 bras selon un rapport 1:1 et ont reçu:
·Bras CALQUENCE plus bendamustine et rituximab (CALQUENCE + BR) – CALQUENCE 100 mg a été administré deux fois par jour en continu à partir du jour 1 du cycle 1. La bendamustine, 90 mg/m2, a été administrée les jours 1 et 2 de chacun des six cycles de 28 jours par voie intraveineuse pendant 30 minutes et le rituximab, 375 mg/m2, a été administré le jour 1 de chacun des six cycles de 28 jours par voie intraveineuse. L'association CALQUENCE + BR a été administrée lors de 6 cycles de traitement au maximum (traitement d'induction).
·Bras placebo plus bendamustine et rituximab (Placebo + BR) – le placebo a été administré deux fois par jour en continu à partir du jour 1 du cycle 1. La bendamustine, 90 mg/m2, a été administrée les jours 1 et 2 de chacun des six cycles de 28 jours par voie intraveineuse pendant 30 minutes et le rituximab, 375 mg/m2, a été administré le jour 1 de chacun des six cycles de 28 jours par voie intraveineuse. L'association placebo + BR a été administrée lors de 6 cycles de traitement au maximum (traitement d'induction).
CALQUENCE ou le placebo ont été administrés en continu jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. Après le traitement d'induction, les patients du bras placebo + BR ayant obtenu une réponse (rémission partielle ou rémission complète) ont reçu une dose d'entretien de rituximab de 375 mg/m2 le jour 1 d'un cycle sur deux, soit 12 doses supplémentaires au maximum jusqu'au cycle 30. Les patients randomisés dans le bras placebo + BR dont la progression de la maladie a été confirmée ont pu recevoir CALQUENCE en monothérapie à une dose de 100 mg deux fois par jour dans le cadre d'une phase de traitement croisé jusqu'à une nouvelle progression de la maladie ou jusqu'à la survenue d'une toxicité inacceptable pour la seconde fois.
L'âge médian était de 71 ans (65-86); 70,7% étaient de sexe masculin; 78,3% étaient blancs; 93,1% avaient un indice de performance ECOG entre 0 et 1. Le score MIPI simplifié était bas (0-3) chez 33,1% des patients, intermédiaire (4-5) chez 42,8% et élevé (6-11) chez 24,1%. Au total, 37,7% des patients avaient un volume tumoral ≥5 cm et 86% présentaient le stade IV de la classification de Ann Arbor. Des variantes agressives du LCM comme les formes blastoïdes et pléomorphes sont apparues chez 7,7% et 5,5% des patients, respectivement. Au total, 47,8% des patients avaient un indice Ki-67 ≥30%. Les caractéristiques initiales étaient similaires dans les deux bras de traitement.
Le critère d'évaluation principal était la survie sans progression (SSP), évaluée par un comité de revue indépendant (Independent Review Committee, IRC) d'après les critères de la classification de Lugano pour le LNH chez les patients atteints de LCM non précédemment traité. Les autres critèresd'évaluation de l'efficacité contrôlés en fonction de la multiplicité étaient le taux de réponse globale (TRG) évalué par l'IRC et la survie globale (SG). Le plan de l'étude sans 2e randomisation pour la phase post-traitement d'induction ne permet pas de se prononcer sur le bénéfice d'un traitement continu avec Calquence jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable.
Pour une durée de suivi médiane de 46,1 mois dans le bras CALQUENCE + BR et de 44,4 mois dans le bras Placebo + BR, l'évaluation de la SSP par l'IRC a révélé une réduction statistiquement significative du risque de progression de la maladie ou de décès chez les patients traités par CALQUENCE + BR de 27% par rapport au bras Placebo + BR.
Des écarts ont cependant été remarqués dans certains sous-groupes, aussi bien pour la SSP que pour la SG. L'écart concernant le sexe est particulièrement notable. Le HR pour la SSP chez les 423 hommes de l'étude était de 0,91 (0,68, 1,21) par rapport à 0,34 (0,19, 0,58) chez les 175 femmes, le HR correspondant pour la SG était de 1,01 (0,74, 1,38) pour les hommes et de 0,52 (0,28, 0,94) pour les femmes.
Le TRG évalué par l'IRC n'a pas montré de différence statistiquement significative entre les associations Calquence + BR et placebo + BR.
Au moment de l'analyse de la SSP, la SG médiane n'avait été atteinte dans aucun des deux bras, avec un total de 203 décès: 97 (32,4%) dans le bras CALQUENCE + BR, 106 (35,5%) dans le bras Placebo + BR. Aucune différence statistiquement significative n'a été mise en évidence entre les deux bras pour la SG: HR (IC à 95%) (stratifié) 0,86 (0,65, 1,13). Les résultats d'efficacité sont présentés dans le Tableau 9. Les courbes de Kaplan-Meier de la SSP sont représentées dans la Figure 1.
Tableau 9. Résultats d'efficacité chez les patients atteints de LCM non précédemment traité dans l'étude ECHO
|
|
CALQUENCE + BR
N = 299
|
Placebo + BR
N = 299
| |
SSP évaluée par l'IRC
| |
Médiane (IC à 95%)
|
66,4 (55,1; n.e.)
|
49,6 (36,0; 64,1)
| |
HR (IC à 95%) (stratifié)*
|
0,73 (0,57; 0,94)
| |
Valeur p‡
|
0,0160
| |
TRG évalué par l'IRC
| |
n RC + RP (%)
|
272 (91,0)
|
263 (88,0)
| |
IC à 95%
|
87,3; 93,8
|
83,9; 91,3
| |
n RC (%)
|
199 (66,6)
|
160 (53,5)
| |
n RP (%)
|
73 (24,4)
|
103 (34,4)
| |
Différence du TRG (vs. bras PBR)
|
3,0%
|
-
| |
Valeur p
|
0,2196
|
-
|
HR = hazard ratio, RC = rémission complète (Complete Response), RP = rémission partielle (Partial Response), n.e. – non évaluable
* Stratifié en fonction des facteurs de stratification de la randomisation: régions géographiques (Amérique du Nord, Europe de l'Ouest, Autres) et du score MIPI simplifié (risque bas [0 à 3], risque intermédiaire [4 à 5], risque élevé [6 à 11]), telles que recueillis via IXRS. Modèle à risques proportionnels de Cox stratifié utilisé pour estimer le hazard ratio (IC à 95%).
‡ Test du log-rank stratifié utilisé pour estimer la valeur p.
Figure 1. Courbes de Kaplan-Meier de la SSP selon l'évaluation par l'IRC chez les patients atteints de LCM non précédemment traité (ECHO)
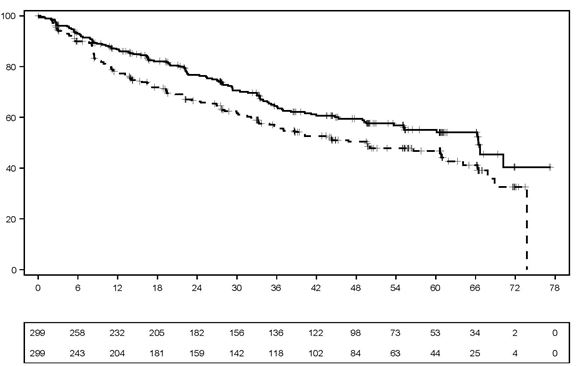
Patients atteints de LCM ayant reçu au moins un traitement antérieur – ACE-LY-004
La sécurité et l'efficacité de CALQUENCE chez des patients atteints de LCM ont été évaluées dans le cadre d'une étude de phase II multicentrique, à un seul bras, en ouvert (ACE-LY-004), à laquelle 124 patients n'ayant pas obtenu de réponse partielle avec un traitement antérieur ou ayant présenté une progression suite à un traitement antérieur ont participé. Tous les patients ont reçu CALQUENCE à raison de 100 mg deux fois par jour par voie orale jusqu'à la progression de la maladie ou la survenue d'une toxicité inacceptable. Aucun patient ayant reçu un traitement antérieur par des inhibiteurs de la BTK, par d'autres inhibiteurs de la voie de signalisation du récepteur des cellules B (inhibiteur de la phosphoinositide 3 kinase [PI3K] ou de la SYK) ou par un inhibiteur de BCL-2 n'a participé à cette étude. Le critère d'évaluation principal était le taux de réponse globale (TRG) évalué par le médecin investigateur d'après les critères de la classification de Lugano pour le lymphome non hodgkinien (LNH). La durée de la réponse (DR) était une mesure de résultat supplémentaire. Les résultats d'efficacité des analyses primaire (12 mois) et finale (54 mois) sont présentés dans le Tableau 10.
Au moment de l'analyse primaire, l'âge médian était de 68 ans (intervalle: 42 à 90), 79,8% étaient de sexe masculin et 74,2% étaient blancs. Au début de l'étude, 92,8% des patients avaient un indice de performance ECOG de 0 ou 1. Le temps médian écoulé depuis le diagnostic était de 46,3 mois et le nombre médian de traitements antérieurs était de 2 (intervalle: 1 à 5), dont 17,7% avec une greffe de cellules souches antérieure. Les protocoles de traitement antérieurs les plus fréquents étaient le schéma thérapeutique à base de CHOP (51,6%) et d'ARA-C (33,9%). Au début de l'étude, 37,1% des patients présentaient au moins une tumeur dont le diamètre le plus important était ≥5 cm; 72,6% présentaient une atteinte ganglionnaire supplémentaire, dont 50,8% une atteinte de la moelle osseuse. Le score MIPI simplifié (qui prend en compte l'âge, le score ECOG ainsi que le taux initial de lactate deshydrogénase et la numération leucocytaire) était intermédiaire chez 43,5% des patients et élevé chez 16,9%.
Tableau 10. Taux de réponse globale et durée de réponse chez les patients atteints de LCM (étude ACE-LY-004) lors de l'analyse à 12 mois et à 54 mois
|
|
Évaluation du médecin investigateur après 12 mois
n = 124
n (%) (IC à 95%*)
|
Évaluation du médecin investigateur après 54 mois
n = 124
n (%) (IC à 95%*)
| |
Taux de réponse globale (TRG)
| |
Taux de réponse globale
|
100 (80,6%) (72,6; 87,2)
|
101 (81,5%) (73,5; 87,9)
| |
Rémission complète
|
49 (39,5%) (30,9; 48,7)
|
59 (47,6%) (38,5; 56,7)
| |
Rémission partielle
|
51 (41,1%) (32,4; 50,3)
|
42 (33,9%) (25,6; 42,9)
| |
Maladie stable
|
11 (8,9%) (4,5; 15,3)
|
10 (8,1%) (3,9; 14,3)
| |
Progression de la maladie
|
10 (8,1%) (3,9; 14,3)
|
10 (8,1%) (3,9; 14,3)
| |
Non évaluable†
|
3 (2,4%) (0,5; 6,9)
|
3 (2,4%) (0,5; 6,9)
| |
Durée de la réponse (DR)
| |
médiane (mois)
|
NA (13,5; NA)
|
28,6 (17,5; 39,1)
| |
IC = intervalle de confiance; NA = non atteinte
*Intervalle de confiance binomial exact à 95%.
†Comprend des sujets sans évaluation suffisante de la maladie après le début de l'étude.
|
PharmacocinétiqueLa pharmacocinétique de l'acalabrutinib et de son métabolite actif, l'ACP-5862, a été étudiée chez des sujets sains et chez des patients atteints d'une hémopathie maligne B. La pharmacocinétique de l'acalabrutinib est proportionnelle à la dose, et l'acalabrutinib ainsi que l'ACP-5862 présentent une pharmacocinétique presque linéaire sur un intervalle de doses de 75 à 250 mg. La modélisation pharmacocinétique de population suggère que la pharmacocinétique de l'acalabrutinib et de l'ACP-5862 est comparable chez les patients atteints de différentes hémopathies malignes B. À la dose recommandée de 100 mg deux fois par jour chez des patients atteints d'hémopathies malignes B (y compris lymphome à cellules du manteau (LCM) et LLC), les moyennes géométriques de l'aire sous la courbe de la concentration plasmatique en fonction du temps quotidienne (ASC24h) et de la concentration plasmatique maximale (Cmax) pour l'acalabrutinib étaient de 1 679 ng•h/ml et 438 ng/ml, respectivement, et pour l'ACP-5862, ces valeurs étaient de 4 166 ng•h/ml et 446 ng/ml, respectivement.
Absorption
Le temps médian nécessaire pour atteindre les concentrations plasmatiques maximales (tmax) était de 0,5 heure (fourchette: 0,2 à 3,0) pour CALQUENCE et 0,75 heure (0,5 à 4,0) pour l'ACP-5862. La biodisponibilité absolue de CALQUENCE était de 25%.
Effet de la nourriture sur l'acalabrutinib
Chez des sujets sains, l'administration d'une dose unique de 100 mg d'acalabrutinib avec un repas riche en lipides et en calories (environ 918 calories, 59 grammes de glucides, 59 grammes de lipides et 39 grammes de protéines) n'a pas affecté l'ASC moyenne par rapport à l'administration à jeun. La Cmax résultante a diminué de 54% et le tmax a été allongé de 1 à 2 heures.
Distribution
La liaison réversible aux protéines plasmatiques humaines était de 97,5% pour l'acalabrutinib et de 98,6% pour l'ACP-5862. In vitro, le ratio moyen sang/plasma était de 0,8 pour l'acalabrutinib et de 0,7 pour l'ACP-5862. Le volume de distribution moyen à l'état d'équilibre (Vee) était d'environ 34 litres.
Métabolisme
In vitro, l'acalabrutinib est principalement métabolisé par les enzymes CYP3A, et dans une moindre mesure par conjugaison au glutathion et hydrolyse de la fonction amide. L'ACP-5862 est le principal métabolite mis en évidence dans le plasma, et la moyenne géométrique de son exposition (ASC) est de 2 à 3 fois plus élevée que celle de l'acalabrutinib. L'ACP-5862 est environ 50% moins puissant que l'acalabrutinib en termes d'inhibition de la BTK.
Élimination
Après une dose unique orale de 100 mg d'acalabrutinib, la moyenne arithmétique de la demi-vie terminale (t1/2) de l'acalabrutinib était de 1,6 heure (fourchette: 0,8 à 9,0 h). La moyenne arithmétique de la t1/2 du métabolite actif, l'ACP-5862, était de 6,9 heures (fourchette: 2,5 à 10,1 h).
La clairance orale (CL/F) moyenne était de 134 l/h pour l'acalabrutinib et de 22 l/h pour l'ACP-5862.
Après l'administration d'une dose unique de 100 mg d'acalabrutinib radiomarqué au [14C] chez des sujets sains, 84% de la dose ont été retrouvés dans les selles et 12% dans les urines, moins de 2% de la dose ayant été excrété sous forme inchangée dans les selles ou l'urine.
Cinétique pour certains groupes de patients
D'après des analyses pharmacocinétiques de population, ni l'âge, le sexe, l'origine ethnique (Caucasiens, Afro-Américains) ni le poids corporel n'ont eu d'effets cliniquement significatifs sur la pharmacocinétique de l'acalabrutinib et de son métabolite actif, l'ACP-5862.
Troubles de la fonction rénale
L'élimination rénale de l'acalabrutinib est minime. Aucune étude pharmacocinétique n'a été conduite chez des patients présentant une diminution de la fonction rénale.
D'après une analyse pharmacocinétique de population, aucune différence cliniquement pertinente n'a été observée au niveau de la pharmacocinétique entre 408 sujets présentant des troubles légers de la fonction rénale (DFGe compris entre 60 et 89 ml/min/1,73 m2, évalué d'après l'équation MDRD), 109 sujets présentant des troubles modérés de la fonction rénale (DFGe compris entre 30 et 59 ml/min/1,73 m2) et 192 sujets présentant une fonction rénale normale (DFGe supérieur ou égal à 90 ml/min/1,73 m2). La pharmacocinétique de l'acalabrutinib n'a pas été caractérisée chez les patients présentant des troubles sévères de la fonction rénale (DFGe <29 ml/min/1,73 m2) ou une insuffisance rénale nécessitant une dialyse. Les patients présentant des taux de créatinine correspondant à plus de 2,5 fois la LSN n'ont pas été inclus dans les études cliniques (voir «Posologie/Mode d'emploi»).
Troubles de la fonction hépatique
L'acalabrutinib est métabolisé dans le foie. Dans des études spécifiques des troubles de la fonction hépatique, l'exposition (ASC) à l'acalabrutinib a été multipliée par respectivement 1,9, 1,5 et 5,3 chez les patients présentant des troubles de la fonction hépatique légers (n = 6) (Child-Pugh A), modérés (n = 6) (Child-Pugh B) et sévères (n = 8) (Child-Pugh C) par rapport aux sujets présentant une fonction hépatique normale (n = 6). Des analyses pharmacocinétiques de population n'ont révélé aucune différence cliniquement pertinente entre les sujets présentant des troubles de la fonction hépatique légers (n = 79) ou modérés (n = 6) (bilirubine totale comprise entre 1,5 et 3 fois la LSN avec ou sans élévation d'ASAT) et les sujets présentant une fonction hépatique normale (n = 613) (taux de bilirubine totale et d'ASAT normaux).
Données précliniquesToxicité en cas d'administration répétée
Dans le cadre d'études de toxicité, l'administration journalière d'acalabrutinib par voie orale pendant une période pouvant aller jusqu'à 6 mois chez le rat et 9 mois chez le chien a été tolérée à des degrés d'exposition qui dépassaient l'exposition thérapeutique humaine à la dose recommandée (1,1 fois l'exposition chez les rats et 8,2 fois l'exposition chez les chiens, selon l'ASC).
Chez le rat, des effets ont été observés au niveau des reins, y compris une dégénérescence tubulaire, sur une ASC correspondant à ≥7 fois l'exposition à la dose recommandée chez l'être humain. Chez le rat, à une exposition correspondant à 4,2 fois l'exposition à la dose recommandée chez l'être humain, les effets au niveau du rein étaient réversibles et la récupération était complète. Chez le rat, à des degrés d'exposition plus élevés (correspondant à ≥6,8 fois l'exposition clinique), les effets n'étaient que partiellement réversibles.
Chez le rat, des anomalies hépatiques, y compris une nécrose hépatocytaire, dose-dépendantes et réversibles ont été observées après des expositions correspondant à ≥4,2 fois l'exposition à la dose recommandée chez l'être humain.
Chez des rats exposés à des doses correspondant à ≥6,8 fois l'exposition à la dose recommandée chez l'être humain, une toxicité cardiaque (hémorragie, inflammation et nécrose myocardique) a été observée. Ces rats sont morts pendant l'étude. La réversibilité des anomalies cardiaques n'a pas pu être évaluée, car ces anomalies ont uniquement été observées à des doses supérieures à la dose maximale tolérée (MTD). À des expositions correspondant à 4,2 fois la dose recommandée chez l'être humain, aucune toxicité cardiaque n'a été observée.
Génotoxicité/Mutagénicité
L'acalabrutinib s'est révélé non mutagène dans un test de mutation réverse sur bactéries, un test d'aberrations chromosomiques in vitro et un test in vivo du micronoyau dans la moelle osseuse chez la souris.
Carcinogénicité
Aucune étude de carcinogénicité n'a été conduite avec l'acalabrutinib.
Toxicité sur la reproduction
Aucun effet sur la fertilité n'a été observé chez des rats mâles et femelles à des expositions correspondant respectivement à 10 ou 9 fois l'exposition chez l'être humain (ASC) à la dose recommandée.
Dans le cadre d'une étude portant à la fois sur la fertilité et le développement embryo-fœtal réalisée chez des rates, l'acalabrutinib a été administré par voie orale à des doses allant jusqu'à 200 mg/kg/jour, de 14 jours avant l'accouplement jusqu'au jour de gestation (JG) 17. Aucun effet n'a été observé sur le développement embryo-fœtal et la survie. À des doses orales quotidiennes de 200 mg/kg chez les rates gravides, l'ASC correspondait à environ 9 fois l'ASC observée chez les patients à la dose recommandée de 100 mg deux fois par jour. La présence de l'acalabrutinib et de son métabolite actif a été confirmée dans le plasma fœtal des rats.
Dans le cadre d'une étude sur le développement embryo-fœtal, réalisée chez des lapines gravides, l'acalabrutinib a été administré par voie orale à des doses quotidiennes allant jusqu'à 200 mg/kg durant l'organogenèse (JG 6 à JG 8). Une diminution du poids des fœtus et un retard d'ossification ont été observés aux niveaux d'exposition ayant produit une toxicité maternelle (doses journalières ≥100 mg/kg) et correspondant à environ 2,4 fois l'exposition atteinte chez l'être humain à la dose recommandée. Dans le cadre d'une étude de reproduction conduite chez le rat, une dystocie (mise bas prolongée/difficile) a été observée à des expositions correspondant à >2,3 fois l'exposition clinique atteinte avec 100 mg deux fois par jour.
L'acalabrutinib et son métabolite actif étaient présents dans le lait de rates allaitantes.
Dans le cadre d'une étude sur le développement prénatal et postnatal chez le rat, l'acalabrutinib a été administré par voie orale à des animaux gravides pendant l'organogenèse, la parturition et la lactation à des doses de 50, 100 et 150 mg/kg/jour. À des doses ≥100 mg/kg/jour, une dystocie (contractions prolongées ou fortes contractions) et une mortalité des jeunes rats ont été observées. À la dose de 100 mg/kg/jour chez les rates gravides, l'ASC correspondait à environ 2 fois l'ASC observée chez les patients ayant reçu environ 100 mg toutes les 12 heures. Dans la génération F1, à des doses de 150 mg/kg/jour, des papilles rénales sous-développées ont été également observées, l'ASC correspondait à environ 5 fois l'ASC des patients ayant reçu environ 100 mg toutes les 12 heures.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine.
Ne pas conserver au-dessus de 30°C.
Conserver hors de portée des enfants.
Numéro d’autorisation68817 (Swissmedic).
PrésentationCALQUENCE 100 mg comprimés pelliculés:
Plaquettes en aluminium/aluminium. Boîtes de 6 x 10 comprimés pelliculés [A].
Titulaire de l’autorisationAstraZeneca AG, 6340 Baar.
Mise à jour de l’informationJuillet 2025.
|