CompositionPrincipes actifs
Ocrelizumabum (produit par génie génétique à l'aide de cellules CHO [Chinese Hamster Ovary]).
Excipients
Forme pharmaceutique intraveineuse (i.v.)
Natrii acetas trihydricus 21,4 mg (correspond à 3,6 mg de sodium par flacon), acidum aceticum glaciale, α,α-trehalosum dihydricum, polysorbatum 20 (produit à partir de maïs génétiquement modifié), aqua ad iniectabile.
1 ml de solution à diluer contient 0,36 mg de sodium.
Un flacon (10 ml) contient 3,6 mg de sodium.
Forme pharmaceutique sous-cutanée (s.c.)
Hyaluronidasum humanum (rHuPH20), natrii acetas trihydricus 50,1 mg (correspond à 8,4 mg de sodium par flacon), acidum aceticum glaciale, α,α-trehalosum dihydricum, polysorbatum 20 (produit à partir de maïs génétiquement modifié), L-methioninum, aqua ad iniectabile.
1 ml de solution contient 0,37 mg de sodium.
Un flacon (23 ml) contient 8,4 mg de sodium.
Indications/Possibilités d’emploiOcrevus est indiqué dans le traitement des patients adultes atteints de formes actives de sclérose en plaques (SEP) récurrente.
Ocrevus est indiqué dans le traitement des patients adultes atteints de sclérose en plaques primaire progressive (SEP-PP) pour ralentir la progression de la maladie et réduire l'aggravation de la vitesse de marche.
Posologie/Mode d’emploiLe traitement par Ocrevus i.v. ou Ocrevus s.c. doit être instauré et surveillé par un neurologue expérimenté dans le traitement de patients atteints de SEP.
Il est important de vérifier l'étiquetage du produit pour s'assurer que la forme pharmaceutique (Ocrevus i.v. ou Ocrevus s.c.) correspond bien à la prescription et est administrée par la voie correcte au patient.
Les patients ont la possibilité de commencer le traitement par Ocrevus i.v. ou s.c.. Les patients recevant actuellement Ocrevus i.v. peuvent poursuivre le traitement par Ocrevus i.v. ou Ocrevus s.c.
Ocrevus i.v.
Ocrevus i.v. n'est pas destiné à l'administration sous-cutanée.
Les perfusions d'Ocrevus i.v. ne doivent être administrées que sous la surveillance directe et étroite d'un membre expérimenté du personnel médical.
Un traitement médical approprié, y compris un équipement de réanimation complet ainsi que des médicaments, dont entre autres de l'épinéphrine (adrénaline), des antihistaminiques et des glucocorticoïdes, doivent être disponibles pour une utilisation immédiate en cas de survenue d'effets indésirables sévères, comme par exemple des réactions sévères liées à la perfusion ou des réactions d'hypersensibilité.
Chez les patients développant des symptômes pulmonaires sévères comme un bronchospasme ou des exacerbations d'asthme, la perfusion doit être immédiatement et définitivement interrompue. Après avoir mis en œuvre un traitement symptomatique, le patient doit être surveillé jusqu'à la disparition complète des symptômes pulmonaires car une aggravation peut survenir après une amélioration initiale des symptômes cliniques.
Pendant les perfusions d'Ocrevus i.v., la survenue d'une hypotension en tant que symptôme d'une réaction liée à la perfusion est possible. Il convient donc d'envisager l'interruption d'un traitement antihypertenseur 12 heures avant et pendant chaque perfusion d'Ocrevus i.v. Ocrevus i.v. est administré par perfusion intraveineuse (i.v.) au moyen d'une voie de perfusion séparée. Ocrevus i.v. ne doit pas être administré en injection i.v. rapide ou bolus et ne doit pas non plus être perfusé sans avoir été dilué.
Il faut utiliser une solution isotonique de chlorure de sodium (à 0,9 %) comme milieu de perfusion. Dans le cas où la perfusion i.v. ne peut pas être terminée le jour même, le liquide restant dans la poche de perfusion doit être éliminé (voir «Remarques concernant le stockage» et «Remarques concernant la manipulation et l'élimination»).
Observez tous les patients pendant au moins une heure après la fin de la perfusion (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»).
Prémédication pour diminuer les éventuelles réactions liées à la perfusion
Avant chaque perfusion d'Ocrevus i.v., les deux prémédications suivantes doivent être administrées afin de diminuer la fréquence et la sévérité des réactions liées à la perfusion (voir «Mises en garde et précautions»):
·100 mg de méthylprednisolone i.v. (ou équivalent) environ 30 minutes avant chaque perfusion d'Ocrevus i.v.;
·un antihistaminique environ 30 à 60 minutes avant chaque perfusion d'Ocrevus i.v.
De plus, une prémédication avec un antipyrétique (p.ex. paracétamol) peut également être envisagée environ 30 à 60 minutes avant chaque perfusion d'Ocrevus i.v.
Administration d'Ocrevus i.v.
Instauration du traitement
La dose initiale (dose 1) de 600 mg est administrée en deux perfusions i.v. distinctes, c'est-à-dire sous forme de deux perfusions de 300 mg chacune, ayant lieu à intervalle de deux semaines.
Traitement d'entretien
Les doses d'Ocrevus i.v. suivantes sont administrées par perfusion i.v. tous les 6 mois sous forme de doses uniques de 600 mg (voir tableau 1).
Si les patients n'ont pas développé de grave réaction liée à la perfusion (infusion-related reaction, IRR) lors de perfusions précédentes d'Ocrevus i.v., une perfusion plus rapide (2 heures) peut être réalisée pour les doses suivantes (voir tableau 1, Option 2).
Un délai minimum de 5 mois doit être respecté entre l'administration des différentes doses d'Ocrevus.
Tableau 1: Doses et schéma thérapeutique d'Ocrevus i.v.
|
|
Dose d'Ocrevus i.v. à administrer*
|
Remarques concernant la perfusion
| |
Instauration du traitement
(600 mg)
répartie en 2 perfusions
|
Perfusion 1
|
300 mg
dans 250 ml
|
·Commencer la perfusion avec 30 ml/h.
·La vitesse peut ensuite être augmentée toutes les 30 minutes de 30 ml/h, jusqu'à un maximum de 180 ml/h.
·Chaque perfusion devrait être administrée en l'espace d'environ 2,5 h.
| |
Perfusion 2
(2 semaines plus tard)
|
300 mg
dans 250 ml
| |
Traitement d'entretien**
(600 mg)
Perfusion unique
une fois tous les 6 mois
|
Option 1
Perfusion d'une durée de 3,5 heures environ
|
600 mg
dans 500 ml
|
·Commencer la perfusion avec 40 ml/h.
·La vitesse peut ensuite être augmentée toutes les 30 minutes de 40 ml/h, jusqu'à un maximum de 200 ml/h.
·Chaque perfusion devrait être administrée en l'espace d'environ 3,5 h.
| |
OU
| |
Option 2
Perfusion d'une durée de 2 heures environ
|
600 mg
dans 500 ml
|
·Commencer la perfusion avec 100 ml/h pendant les 15 premières minutes.
·Augmenter la vitesse à 200 ml/h pendant les 15 minutes suivantes.
·Augmenter la vitesse à 250 ml/h pendant les 30 minutes suivantes.
·Augmenter la vitesse à 300 ml/h pendant les 60 minutes restantes.
·Chaque perfusion doit être administrée pendant 2 h environ.
|
* Les solutions d'Ocrevus destinées à la perfusion i.v. sont constituées par dilution du médicament dans une poche de perfusion contenant du chlorure de sodium à 0,9 %, pour obtenir une concentration finale du médicament d'environ 1,2 mg/ml.
** La première perfusion unique suivant la dose initiale doit être administrée 6 mois après la perfusion 1 de la dose initiale.
Afin d'assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot d'Ocrevus.
Adaptations posologiques au cours du traitement
Une adaptation des doses d'Ocrevus i.v. n'a pas été étudiée et n'est pas recommandée lorsqu'il n'y a pas de signe d'intolérance.
Ajustement de la posologie du fait d'effets indésirables/d'interactions
Réactions liées à la perfusion
Le traitement par Ocrevus i.v. est associé à des réactions liées à la perfusion pouvant être en relation avec la libération de cytokines et/ou d'autres médiateurs chimiques. De façon générale, les directives suivantes concernant les adaptations posologiques doivent être suivies en cas de réactions liées à la perfusion. Vous trouverez de plus amples informations sur les réactions liées à la perfusion sous «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection».
Réactions mettant en jeu le pronostic vital liées à la perfusion
Si des signes de réactions liées à la perfusion mettant en jeu le pronostic vital ou pouvant entraîner un handicap surviennent, p.ex. une hypersensibilité aiguë ou un syndrome de détresse respiratoire aiguë, la perfusion d'Ocrevus i.v. doit être immédiatement interrompue. Le patient doit recevoir un traitement de soutien approprié. Le traitement par Ocrevus i.v. doit être arrêté de manière définitive chez ces patients et ne peut être repris.
Réactions sévères liées à la perfusion
La perfusion doit immédiatement être interrompue et le patient doit recevoir un traitement symptomatique si une réaction liée à la perfusion sévère ou un ensemble de symptômes regroupant rougeurs cutanées, fièvre et douleurs à la gorge venait à se manifester. La perfusion ne peut être reprise qu'après la disparition de tous les symptômes. La perfusion doit être reprise à un débit correspondant à la moitié du débit auquel le début de la réaction liée à la perfusion a été constaté.
Réactions légères à modérées liées à la perfusion
Le débit de perfusion correspondant à celui utilisé lorsque le début de la réaction liée à la perfusion a été constaté doit être réduit de moitié lorsque le patient souffre d'une réaction liée à la perfusion légère à modérée (p.ex. céphalée). Ce débit réduit doit être maintenu pendant au moins 30 minutes. Il peut ensuite à nouveau être augmenté au débit de perfusion initialement prévu pour le patient, dans la mesure où il est toléré.
Ocrevus s.c.
La première administration doit impérativement être effectuée sous observation clinique avec une prise en charge médicale appropriée, afin de maîtriser les réactions sévères, telles que les réactions liées à l'injection, les réactions d'hypersensibilité et/ou les réactions anaphylactiques (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»).
Prémédication contre les réactions liées à l'injection
Les deux prémédications suivantes doivent être administrées peu de temps avant chaque injection d'Ocrevus s.c., afin de réduire le risque de réactions locales et systémiques liées à l'injection (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»):
·20 mg de dexaméthasone (ou équivalent) par voie orale
·un antihistaminique par voie orale (p.ex. desloratadine ou équivalent).
De plus, l'administration d'un antipyrétique (p.ex. paracétamol) peut également être envisagée peu de temps avant chaque administration d'Ocrevus s.c.
Administration d'Ocrevus s.c.
Ocrevus s.c. n'est pas destiné à l'administration intraveineuse et doit toujours être utilisé sous forme d'injection sous-cutanée par un membre expérimenté du personnel médical (voir «Remarques concernant la manipulation» et «Élimination»). Avant l'utilisation, sortir Ocrevus s.c. du réfrigérateur et laisser la solution revenir à température ambiante.
Lors de l'administration de la dose initiale, une surveillance incluant l'accès à une assistance médicale appropriée pour le traitement des réactions sévères liées à l'injection est recommandée pendant au moins une heure après l'injection. Pour les doses suivantes, la nécessité et la durée d'une surveillance après l'injection sont laissées à la discrétion du médecin traitant (voir «Mises en garde et précautions»).
La dose de 23 ml (920 mg) d'Ocrevus solution s.c. est administrée par voie sous-cutanée dans l'abdomen pendant environ 10 minutes, si possible à l'aide d'un set de perfusion sous-cutanée (p.ex. aiguille à ailettes/papillon). Tout volume résiduel éventuellement présent dans le set de perfusion s.c. ne doit PAS être administré au patient.
L'injection doit être effectuée dans l'abdomen, à l'exception d'une zone de 5 cm autour du nombril. Les injections d'Ocrevus s.c. ne doivent pas être réalisées au niveau des zones où la peau est rouge, sensible ou indurée, ou de zones présentant un hématome, des grains de beauté ou des cicatrices.
Dose et schéma thérapeutique
Ocrevus s.c. est administré en injection sous-cutanée de 920 mg tous les 6 mois.
Il n'est pas nécessaire de répartir la dose initiale ou les doses suivantes en injections séparées.
Un intervalle minimal de 5 mois doit être maintenu entre l'administration de chaque dose d'Ocrevus.
Ocrevus i.v. et s.c.
Patients présentant des troubles de la fonction hépatique
La sécurité et l'efficacité d'Ocrevus n'ont pas été examinées de façon formelle chez les patients atteints de troubles de la fonction hépatique. Les patients atteints d'insuffisance hépatique légère ont été inclus dans les études cliniques. Il n'existe aucune expérience chez les patients atteints d'insuffisance hépatique modérée ou sévère. Ocrevus est un anticorps monoclonal et est éliminé par catabolisme (plutôt que par élimination hépatique). C'est pourquoi une adaptation de la dose n'est vraisemblablement pas nécessaire chez les patients atteints de troubles de la fonction hépatique (voir «Pharmacocinétique, Cinétique pour certains groupes de patients, Troubles de la fonction hépatique»).
Patients présentant des troubles de la fonction rénale
La sécurité et l'efficacité d'Ocrevus n'ont pas été examinées de façon formelle chez les patients atteints de troubles de la fonction rénale. Les patients atteints d'insuffisance rénale légère ont été inclus dans les études cliniques. Il n'existe aucune expérience chez les patients atteints d'insuffisance rénale modérée ou sévère. Ocrevus est un anticorps monoclonal et est éliminé par catabolisme (plutôt que par élimination rénale). C'est pourquoi une adaptation de la dose n'est vraisemblablement pas nécessaire chez les patients atteints de troubles de la fonction rénale (voir «Pharmacocinétique, Cinétique pour certains groupes de patients, Troubles de la fonction rénale»).
Patients âgés
La sécurité et l'efficacité d'Ocrevus n'ont pas été démontrées chez les patients de > 55 ans.
Enfants et adolescents
La sécurité et l'efficacité d'Ocrevus n'ont pas été évaluées chez les enfants et les adolescents (< 18 ans).
Prise retardée
En cas d'omission d'une perfusion d'Ocrevus prévue, il faut la rattraper le plus rapidement possible; il ne faut pas attendre la date prévue d'administration de la dose suivante. L'intervalle thérapeutique entre les différentes doses d'Ocrevus doit être respecté. L'intervalle thérapeutique de 6 mois (au minimum 5 mois) entre l'administration des doses d'Ocrevus doit être respecté.
Contre-indicationsHypersensibilité à l'ocrélizumab ou à l'un des excipients
Patients atteints d'insuffisance cardiaque sévère (stade NYHA IV)
Patients avec une immunodépression sévère, y compris les patients qui suivent actuellement un traitement immunosuppresseur (à l'exception des traitements symptomatiques avec des corticostéroïdes contre les récidives) ou dont le système immunitaire est affaibli par les traitements précédents (voir «Mises en garde et précautions, Traitement immunosuppresseur avant, pendant ou après l'administration d'Ocrevus»)
Présence d'une infection active (voir «Mises en garde et précautions»)
Affection active maligne existante, à l'exception des patients avec un carcinome basocellulaire
Début du traitement durant la grossesse.
Mises en garde et précautionsLes professionnels de la santé doivent s'assurer avant chaque perfusion que le patient a bien lu et compris l'information concernant la sécurité.
Réactions liées à la perfusion (IRR) et réactions liées à l'injection (IR)
Les IRR sont associées à l'administration d'Ocrevus i.v. et les IR à l'administration d'Ocrevus s.c.. Les IRR et les IR peuvent être liées à la libération de cytokines et/ou d'autres médiateurs chimiques. Les médecins doivent informer de manière exhaustive les patients que des IRR et des IR peuvent survenir pendant ou dans les 24 heures qui suivent l'administration du traitement.
Une réaction d'hypersensibilité peut également survenir (réaction allergique aiguë au médicament). Sur le plan clinique, les IRR et les IR ne peuvent pas être distinguées des réactions aiguës d'hypersensibilité de type 1 (médiées par les IgE) (voir «Mises en garde et précautions, Réactions d'hypersensibilité»).
En ce qui concerne la prémédication visant à diminuer la fréquence et la sévérité des IRR et le risque d'IR, voir «Posologie/Mode d'emploi».
Réactions liées à la perfusion lors de l'administration d'Ocrevus i.v.
Les symptômes des réactions liées à la perfusion peuvent survenir au cours de chaque perfusion, mais elles se produisent le plus souvent au cours de la première perfusion (voir «Effets indésirables»). Ces réactions peuvent se manifester sous la forme de prurit, d'éruption cutanée, d'urticaire, d'érythème, d'irritation de la gorge, de douleurs oro-pharyngées, de détresse respiratoire, d'œdème de la gorge ou du larynx, de bouffées de chaleur, d'hypotension, de fièvre, de fatigue, de céphalées, de vertiges, de nausées, de tachycardie et d'anaphylaxie. Lors de l'administration d'ocrélizumab par voie intraveineuse, des réactions graves dues à des IRR ont été rapportées, dont certaines ont nécessité une hospitalisation. Les patients sous traitement par Ocrevus doivent être surveillés pendant au moins une heure après la fin de la perfusion afin de détecter la survenue possible de tout symptôme suspect de réaction liée à la perfusion.
Mesures à prendre en cas de réactions liées à la perfusion lors de l'administration d'Ocrevus i.v.
Pour les mesures à prendre chez les patients avec des réactions liées à la perfusion sévères mettant en jeu le pronostic vital, ou légères à modérées, voir «Posologie/Mode d'emploi, Adaptations posologiques».
Chez les patients présentant des symptômes pulmonaires sévères comme un bronchospasme ou des exacerbations d'asthme, la perfusion doit être immédiatement et définitivement interrompue. Après avoir mis en œuvre le traitement symptomatique, le patient doit être surveillé jusqu'à la disparition complète des symptômes pulmonaires car une aggravation peut survenir après une amélioration initiale.
La survenue d'une hypotension en tant que symptôme d'une réaction liée à la perfusion est possible au cours de chaque perfusion d'Ocrevus. Il convient donc d'envisager l'interruption d'un traitement antihypertenseur 12 heures avant et pendant chaque perfusion d'Ocrevus. Les patients présentant une insuffisance cardiaque congestive dans l'anamnèse (NYHA III & IV) n'ont pas été examinés (voir «Contre-indications»).
Réactions liées à l'injection lors de l'administration d'Ocrevus s.c.
Les symptômes d'IR peuvent survenir pendant ou dans les 24 heures suivant une injection. Des symptômes d'IR ont été plus fréquemment rapportés lors de la première injection. Les IR peuvent être des réactions locales ou systémiques. Les symptômes fréquents des IR locales au site d'injection comprennent érythème, douleurs, gonflement et prurit. Les symptômes fréquents des IR systémiques comprennent céphalées et nausées (voir «Effets indésirables»). Les patients doivent recevoir une prémédication peu avant l'injection pour réduire le risque d'IR (voir «Posologie/Mode d'emploi»).
Les patients traités par la dose initiale d'Ocrevus s.c. doivent rester en observation pendant au moins une heure après l'injection afin de détecter tout signe d'IR sévère. Lors de l'administration de la dose initiale du médicament, un équipement approprié doit être disponible pour la prise en charge d'IR sévères, de réactions d'hypersensibilité sévères et/ou de réactions anaphylactiques sévères.
Pour les doses suivantes, la nécessité d'une surveillance après l'injection est laissée à la discrétion du médecin traitant. Un traitement symptomatique est recommandé en cas de survenue d'IR.
En cas de signes d'IR engageant le pronostic vital, l'administration d'Ocrevus s.c. doit être immédiatement arrêtée et des mesures thérapeutiques de soutien mises en place. Ocrevus doit être définitivement arrêté chez ces patients.
Si un patient présente une IR sévère, l'injection doit être immédiatement interrompue et un traitement symptomatique mis en place. L'injection ne sera reprise qu'après la résolution de tous les symptômes.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité peuvent se produire (réaction allergique aiguë au médicament, médiée par les IgE). Sur le plan des symptômes, il peut être difficile de distinguer une réaction d'hypersensibilité d'une réaction liée à la perfusion ou d'une réaction liée à l'injection. Une réaction d'hypersensibilité peut survenir au cours de chaque administration, mais elle n'a en principe pas lieu au cours de la première administration. En cas de survenue, au cours d'une administration ultérieure, de symptômes plus graves que ceux apparus jusqu'alors ou en cas d'apparition de nouveaux symptômes sévères, il faut immédiatement envisager une réaction d'hypersensibilité potentielle. L'administration et le traitement doivent être immédiatement et définitivement arrêtés si une réaction d'hypersensibilité est suspectée pendant l'administration. Les patients présentant une hypersensibilité connue à l'ocrélizumab, médiée par les IgE, ou à l'un des excipients ne doivent pas recevoir ce traitement (voir «Contre-indications»).
Infections
Ocrevus ne doit pas être administré chez les patients présentant des infections actives sévères (telles que tuberculose, sepsis et infections opportunistes) ou une réponse immunitaire fortement limitée (p.ex. lorsque le nombre de cellules CD4 ou CD8 est considérablement réduit). Chez les patients présentant une infection active, il convient de reporter l'administration de la perfusion d'Ocrevus jusqu'à la guérison de l'infection (voir «Contre-indications»).
Vous trouverez de plus amples informations sur les facteurs de risque d'infections sévères en lien avec des maladies autres que la SEP dans la section «Effets indésirables» (infections sévères dans les études cliniques portant sur des maladies auto-immunes autres que la SEP).
Des infections graves peuvent survenir dans le cadre du traitement par Ocrevus, y compris des cas dont l'évolution est fatale (surtout lors de pneumonies) (voir «Effets indésirables»). La fréquence des infections avec issue fatale rapportées sous traitement par Ocrevus est de l'ordre de la fréquence des infections avec issue fatale observées chez les patients recevant un placebo dans d'autres études portant sur la SEP.
Chez les patients qui rapportent des signes ou symptômes d'infection à la suite d'un traitement par Ocrevus, la cause des symptômes doit être rapidement déterminée et les patients doivent être traités de façon appropriée. Avant un traitement ultérieur, il convient d'examiner à nouveau les patients en ce qui concerne un risque potentiel d'infection.
Leucoencéphalopathie multifocale progressive (LEMP)
Des infections par le virus JC ayant mené à une LEMP ont été observées chez des patients traités par des anticorps anti-CD20, y compris Ocrevus, et présentant majoritairement des facteurs de risque (p.ex. population de patients, polymédication avec immunosuppresseurs, utilisation antérieure d'autres DMT (traitements modificateurs de la maladie) ou faible taux de lymphocytes).
Le risque de LEMP ne pouvant être exclu sous Ocrevus, les médecins doivent être attentifs aux signes précoces et aux symptômes d'une LEMP, qui peuvent inclure tout type de signes et symptômes neurologiques d'apparition nouvelle ou s'aggravant et peuvent ressembler aux symptômes d'une poussée de SEP.
La LEMP est une infection opportuniste induite par le virus JC, susceptible de conduire à de sévères handicaps, voire au décès. La LEMP ne peut survenir qu'en présence d'une infection par le JCV. Il convient de signaler qu'un test d'anticorps anti-JCV négatif n'exclut pas la possibilité d'une infection subséquente par le JCV. Une LEMP résiste souvent à tous les traitements, conduisant alors au décès. Les symptômes de la LEMP sont extrêmement variables, persistent durant des jours, voire des semaines, et peuvent inclure une faiblesse progressive d'un côté du corps ou des troubles de la coordination des membres, des troubles de l'équilibre, des troubles de la vue et des altérations de la pensée, de la mémoire et de l'orientation, pouvant aboutir à un état de confusion et à des troubles de la personnalité.
En cas de suspicion de LEMP, il faut interrompre le traitement par Ocrevus. En cas de suspicion de LEMP, il convient de procéder à un bilan par IRM (de préférence avec produit de contraste) et de le comparer à une IRM effectuée avant le traitement (ne datant idéalement pas de plus de trois mois), ainsi qu'à un examen du liquide céphalo-rachidien de confirmation, avec détermination de l'ADN du virus JC et examens neurologiques répétés.
En cas de confirmation d'une LEMP, il faut définitivement arrêter le traitement.
Colite à médiation immunitaire
Des cas de colite à médiation immunitaire, pouvant se présenter sous une forme sévère et aiguë de colite, ont été rapportés chez des patients ayant reçu Ocrevus après la mise sur le marché. Certains cas de colite étaient graves et ont nécessité une hospitalisation, et chez quelques patients, une intervention chirurgicale a été nécessaire. L'administration de corticostéroïdes systémiques a été nécessaire chez un grand nombre de ces patients. Le délai écoulé entre l'instauration du traitement et la survenue des symptômes a varié dans ces cas de quelques semaines à plusieurs années. Pendant le traitement par Ocrevus, il convient de surveiller les patients pour détecter la survenue d'une colite à médiation immunitaire et d'examiner ceux-ci rapidement en cas d'apparition de signes et de symptômes évoquant une colite à médiation immunitaire, comme une diarrhée nouvelle ou persistante ou d'autres signes et symptômes gastro-intestinaux.
Réactivation d'une hépatite B
Dans le cadre de la surveillance post-commercialisation, des cas de réactivation du virus de l'hépatite B (HBV) ont été observés lors du traitement par Ocrevus. Chez des patients traités par des anticorps anti-CD20, des hépatites fulminantes, des insuffisances hépatiques et des décès ont été rapportés à la suite d'une réactivation du HBV.
Un dépistage du virus de l'hépatite B (HBV) conforme aux dispositions locales en vigueur doit être réalisé chez tous les patients avant l'instauration d'un traitement par Ocrevus. Les patients présentant une infection active par le HBV (autrement dit une infection active confirmée par la présence de l'antigène HBsAg et d'anticorps anti-HB) ne doivent pas être traités par Ocrevus (voir «Contre-indications»). Les patients dont la sérologie est positive (c'est-à-dire dont les résultats sont négatifs pour l'antigène HBsAg, positifs pour les anticorps HB Core [HBcAb+] et porteurs du HBV [positifs pour l'antigène de surface, HBsAg+]) doivent consulter un spécialiste des affections du foie avant de commencer le traitement. Par ailleurs, ils doivent être surveillés et soignés selon les standards médicaux locaux, afin de prévenir une réactivation de l'hépatite B.
Neutropénie tardive
Des cas de neutropénies tardives ont été rapportés. Bien que certains cas étaient de grade 3 ou 4, la majorité des cas étaient de grade 1 ou 2. Les cas de neutropénies tardives ont été rapportés au moins 4 semaines après la dernière perfusion d'Ocrevus. Il est recommandé de mesurer les neutrophiles sanguins chez les patients ayant des signes et symptômes d'infection (voir «Effets indésirables»).
Traitement immunosuppresseur avant, pendant ou après le traitement par Ocrevus
Dans le cas d'autres maladies auto-immunes, l'utilisation concomitante d'Ocrevus et de médicaments immunosuppresseurs (p.ex. corticostéroïdes chroniques, médicaments antirhumatismaux modificateurs de la maladie [DMARD] biologiques ou non, mycophénolate mofétil, cyclophosphamide, azathioprine) a entraîné une augmentation des infections sévères, y compris des infections opportunistes. Les infections comprenaient entre autres la pneumonie atypique et la pneumonie à Pneumocystis jiroveci (pneumocystose), la pneumonie varicelleuse, la tuberculose et l'histoplasmose. Certaines de ces infections ont dans de rares cas conduit au décès. Une analyse exploratoire a identifié les facteurs associés à un risque d'infection sévère suivants: doses d'Ocrevus plus élevées que celles recommandées pour la SEP, autres comorbidités, utilisation chronique d'immunosuppresseurs ou de corticostéroïdes ainsi qu'une origine ethnique asiatique. L'utilisation concomitante d'autres immunosuppresseurs et d'Ocrevus, à l'exception des corticostéroïdes pour le traitement symptomatique des poussées, n'est pas recommandée.
Lors de l'instauration d'un traitement par Ocrevus à la suite d'une thérapie immunosuppressive ou lors de l'instauration d'un traitement immunosuppresseur après un traitement par Ocrevus, il faut penser à la possibilité d'effets pharmacodynamiques de superposition (interactions) (voir «Mécanisme d'action/pharmacodynamique»). La prudence est de mise lors de la prescription d'Ocrevus, en tenant compte de la pharmacodynamique d'autres médicaments contre la SEP modifiant le cours de la maladie. Ocrevus n'a pas été étudié en association avec d'autres médicaments contre la SEP modifiant le cours de la maladie.
Vaccinations
Le médecin doit contrôler le statut des vaccinations de ses patients, tenir compte des recommandations régionales en vigueur en matière de vaccinations de protection et pratiquer les vaccinations de rappel importantes avant d'instaurer un traitement par Ocrevus. Les vaccinations doivent être achevées au moins 6 semaines avant la première perfusion d'Ocrevus.
La sécurité d'une immunisation par des vaccins vivants ou des vaccins vivants atténués à la suite d'un traitement par Ocrevus n'a pas fait l'objet d'études. Ce type de vaccination n'est pas recommandé pendant le traitement et jusqu'à la reconstitution (réplétion) des cellules B (le temps moyen écoulé jusqu'à la réplétion des cellules B était de 72 semaines, voir «Mécanisme d'action/pharmacodynamique»).
Après un traitement par Ocrevus i.v. de deux ans, la proportion des patients ayant des titres d'anticorps positifs contre S. pneumoniae et contre les virus des oreillons, de la rubéole et de la varicelle était semblable à celle d'avant le début du traitement.
Dans une étude randomisée, ouverte (groupes parallèles: Ocrevus i.v. versus aucun traitement immunomodulateur ou un autre traitement immunomodulateur), les patients atteints de SEP-R et traités par Ocrevus i.v. ont développé une réponse immunitaire humorale en partie nettement diminuée contre la toxine tétanique (réponse IgG positive chez 23,9 % contre 54,5 % des patients), le vaccin polysaccharide anti-pneumococcique 23-valent (réponse immunitaire positive réduite de 2/3 au maximum, une nouvelle vaccination de rappel n'a pas entraîné d'augmentation significative), le néoantigène hémocyanine de patelle et les vaccins contre la grippe saisonnière (les titres séroprotecteurs pour les vaccins contre la grippe saisonnière ont varié entre 55,6 et 80 % contre 75 et 97 %).
Il est recommandé pour toutes les vaccinations, sauf celles avec des vaccins vivants ou des vaccins vivants atténués, de respecter les recommandations locales en matière de vaccinations (y compris avec le vaccin inactivé contre la grippe saisonnière). Il faut envisager de doser le titre d'anticorps induit par le vaccin pour vérifier si les personnes vaccinées peuvent développer une réponse immunitaire protectrice, car l'efficacité de la vaccination est diminuée dans certaines circonstances.
Exposition in utero à l'ocrélizumab et vaccination des nouveau-nés et des nourrissons avec des vaccins vivants ou des vaccins vivants atténués
En raison d'une déplétion potentielle en cellules B chez les nouveau-nés et les nourrissons dont les mères ont reçu Ocrevus pendant la grossesse, une surveillance de la déplétion en cellules B doit être mise en place chez ceux-ci. Il convient de déterminer le nombre de cellules B CD19 positives chez les nouveau-nés et les nourrissons avant de les vacciner. Une vaccination avec des vaccins vivants ou des vaccins vivants atténués ne devra être pratiquée qu'après normalisation complète du nombre de cellules B. La sécurité et le moment de l'immunisation doivent être discutés avec le pédiatre de l'enfant.
Tumeurs malignes
Dans le cadre d'études cliniques, des cas d'affections malignes (dont 6 cas de carcinome mammaire sous Ocrevus, aucun cas dans les bras témoins (Rebif® ou placebo) des études contrôlées) ont été rapportés. L'incidence était cohérente avec les taux attendus dans la population générale des patients atteints de SEP.
À l'exception des patients avec un carcinome basocellulaire, les patients avec une affection maligne active (y compris les patients activement surveillés en ce qui concerne la récidive d'une affection maligne) ne doivent pas être traités par Ocrevus (voir «Contre-indications»). Chez les patients avec des facteurs de risque connus de malignité, le rapport bénéfice-risque d'Ocrevus doit être soigneusement examiné et une surveillance tumorale doit être effectuée avant et pendant le traitement.
Réactions cutanées
Des cas de pyoderma gangrenosum ont été décrits au cours du traitement par Ocrevus. D'autres réactions cutanées sévères telles qu'une nécrolyse épidermique toxique (syndrome de Lyell) et un syndrome de Stevens-Johnson ont également été observées avec d'autres anticorps anti-CD20. Une biopsie cutanée est utile pour différencier les différentes réactions cutanées et définir le traitement ultérieur. Un arrêt du traitement doit être envisagé au cas où un tel événement surviendrait.
Autres remarques
Ocrevus, solution à diluer pour perfusion contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
Ocrevus, solution injectable contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement «sans sodium».
Dans le cadre des mesures destinées à minimiser les risques, Roche met à disposition des supports de formation et des brochures d'information à l'intention des professionnels de la santé et des patients.
InteractionsAucune étude formelle sur les interactions médicamenteuses n'a été réalisée. Le risque d'interaction avec des médicaments utilisés de façon concomitante ne peut être exclu.
Grossesse, allaitementFemmes en âge de procréer
Le traitement ne doit pas être instauré pendant la grossesse (voir «Contre-indications»). Les femmes en âge de procréer doivent utiliser une méthode de contraception fiable pendant le traitement par Ocrevus et jusqu'à 6 mois après l'administration de la dernière dose d'Ocrevus (voir «Pharmacocinétique, Élimination»).
Grossesse
Ocrevus est un anticorps monoclonal humanisé d'une immunoglobuline de sous-type G1; il est bien connu que les immunoglobulines passent la barrière placentaire. Les expérimentations animales n'ont révélé aucun effet tératogène, une toxicité de reproduction a toutefois été observée (voir «Données précliniques»).
On ne dispose pas de suffisamment de données bien contrôlées provenant d'études menées chez des femmes enceintes; on sait toutefois que les nourrissons dont la mère a reçu d'autres anticorps anti-CD20 pendant la grossesse présentent une diminution temporaire des cellules B périphériques et une lymphopénie. Une réduction des cellules B in utero a également été observée dans les études d'expérimentation animale.
Chez les nouveau-nés et nourrissons exposés in utero à Ocrevus, il est recommandé de reporter les vaccinations avec des vaccins vivants ou des vaccins vivants atténués à une date ultérieure. Le nombre de cellules B chez les nouveau-nés et les nourrissons exposés in utero à Ocrevus n'a pas été examiné dans le cadre d'études cliniques. La durée potentielle de la déplétion en cellules B chez les nouveau-nés et les nourrissons n'est pas connue (voir «Mises en garde et précautions, Vaccinations»).
Chez les nouveau-nés exposés in utero dont le nombre de cellules B ne se situe pas dans la norme, le report de vaccinations avec des vaccins vivants ou des vaccins vivants atténués doit être envisagé.
Ocrevus ne doit pas être utilisé pendant la grossesse, à moins que le bénéfice potentiel pour la mère ne prédomine sur le risque potentiel encouru par le fœtus.
Travail et accouchement
La sécurité d'emploi d'Ocervus n'a pas été évaluée pendant le travail et l'accouchement.
Allaitement
On ignore toujours si Ocrevus est excrété dans le lait maternel humain, ou s'il a des conséquences sur la production de lait et pour le nourrisson. Au cours d'études menées chez l'animal, il a été démontré que l'ocrélizumab était excrété dans le lait maternel («Données précliniques»). Les femmes doivent être enjointes de cesser l'allaitement pendant le traitement par ocrélizumab parce que l'IgG humaine est excrétée dans le lait maternel et que l'on ignore l'importance de la diminution des cellules B lors de l'ingestion d'Ocrevus.
Fertilité
Les données précliniques issues des études de fertilité chez le singe Cynomolgus mâle et femelle n'ont pas révélé de risque particulier pour l'être humain.
Effet sur l’aptitude à la conduite et l’utilisation de machinesOcrevus n'a lui-même aucune influence ou une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
L'influence de la prémédication avec des antihistaminiques doit cependant être prise en compte. Après des réactions à la perfusion, il faut attendre que l'état du patient soit stabilisé avant que des véhicules ou machines ne soient utilisés.
Effets indésirablesÉtudes cliniques
Le profil de sécurité de l'ocrélizumab repose sur les données de patients atteints de SEP-R et de SEP-PP ayant reçu de l'ocrélizumab par voie intraveineuse ou sous-cutanée.
La sécurité d'Ocrevus a été évaluée sur la base d'études cliniques pivots portant sur la SEP menées chez 1311 patients traités par Ocrevus i.v., dont 825 patients atteints de sclérose en plaques récurrente (SEP-R) dans deux études cliniques identiques contrôlées contre substance active, et 486 patients atteints de sclérose en plaques primaire progressive (SEP-PP) dans une étude contrôlée contre placebo (voir «Propriétés/Effets, Efficacité clinique»). Les effets indésirables (EI) les plus fréquemment signalés étaient des réactions liées à la perfusion et des infections des voies respiratoires.
Les catégories de fréquence sont définies comme suit: très fréquents (≥1/10), fréquents (< 1/10, ≥1/100), occasionnels (< 1/100, ≥1/1000), rares (< 1/1000, ≥1/10000), très rares (< 1/10000) et fréquence inconnue (ne peut pas être estimée sur la base des données disponibles). Les effets indésirables sont indiqués par fréquence en ordre décroissant.
Résumé des EI survenus sous Ocrevus i.v. lors de SEP-R ou de SEP-PP
Infections et infestations
Très fréquents: infections des voies respiratoires supérieures, (SEP-R: 15,2 %; SEP-PP: 12,1 %), rhinopharyngite (SEP-PP: 24,1 %; SEP-R: 14,9 %), grippe (SEP-PP: 11,7 %; SEP-R: 4,6%).
Fréquents: bronchite, sinusite, gastro-entérite, infection virale, herpès buccal, infection des voies respiratoires, cellulite, zona, conjonctivite.
Fréquence inconnue (ne peut pas être estimée sur la base des données disponibles): LEMP* et colite à médiation immunitaire* (voir «Mises en garde et précautions»).
* Observée après commercialisation.
Affections hématologiques et du système lymphatique
Fréquents: neutropénie.
Fréquence inconnue (ne peut pas être estimée sur la base des données disponibles): neutropénie tardive*.
* Observée après commercialisation.
Affections du système immunitaire
Fréquence inconnue (ne peut pas être estimée sur la base des données disponibles): hypersensibilité* (voir «Mises en garde et précautions»).
* Observée après commercialisation.
Affections respiratoires, thoraciques et médiastinales
Fréquents: toux, catarrhe.
Affections de la peau et du tissu sous-cutané
Fréquence inconnue (ne peut être estimée sur la base des données disponibles): pyoderma gangrenosum* (voir «Mises en garde et précautions»).
* Observé après commercialisation.
Investigations
Très fréquents: taux d'IgM sériques réduits (SEP-R: 16,5 %, SEP-PP: 15,5 %).
Fréquents: taux d'IgG sériques réduits.
Lésions, intoxications et complications d'interventions
Très fréquents: réactions liées à la perfusion (SEP-PP: 40,1 %; SEP-R: 34,3 %) (les symptômes signalés jusqu'à 24 h après une perfusion en tant que réactions liées à la perfusion sont ci-dessous décrits comme «réactions liées à la perfusion»).
Ocrevus s.c.
La sécurité d'Ocrevus s.c. a été évaluée sur la base d'études cliniques portant sur la SEP menées chez 312 patients traités par Ocrevus s.c., parmi lesquels des patients de l'étude pivot OCARINA II et des patients de l'étude OCARINA I. Sur ces 312 patients, 181 patients de l'étude OCARINA II et 118 patients de l'étude OCARINA I ont reçu au moins une dose de 920 mg d'Ocrevus s.c.
Les données de sécurité portant sur Ocrevus s.c. étaient cohérentes avec le profil de sécurité d'Ocrevus i.v. connu décrit ci-dessus, à l'exception d'IR très fréquentes en tant qu'EI lors de l'administration sous-cutanée.
Description de certains effets indésirables provenant des études cliniques
Réactions liées à la perfusion (IRR) lors de l'administration d' Ocrevus i.v.
Les symptômes survenus dans le cadre de réactions liées à la perfusion dans les études portant sur la SEP-R et la SEP-PP comprenaient entre autres: démangeaisons, éruption cutanée, urticaire, érythème, bouffées de chaleur, hypotension, fièvre, fatigue, céphalées, vertiges, irritations de la gorge, douleurs oro-pharyngées, dyspnée, œdème de la gorge ou du larynx, nausées, tachycardie. Dans les études cliniques contrôlées, il n'y a pas eu de réaction fatale liée à la perfusion.
Dans les études cliniques contrôlées contre substance active (SEP-R), les réactions liées à la perfusion représentaient l'événement indésirable le plus fréquent chez les patients traités par Ocrevus i.v. à la dose de 600 mg, avec une incidence totale de 34,3 % contre 9,9 % dans le groupe traité par interféron bêta-1a (perfusion d'un placebo). L'incidence des réactions liées à la perfusion était la plus élevée pour la perfusion 1 lors de la dose initiale/dose 1 (27,5 %) et a diminué au cours du temps à < 10 % à la dose 4. La majorité des réactions liées à la perfusion ont été légères (21,7 %) à modérées (10,1 %) dans les deux groupes de traitement; 2,4 % ont eu des réactions sévères liées à la perfusion et 0,1 % des réactions liées à la perfusion mettant en jeu le pronostic vital (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»).
Dans l'étude clinique contrôlée contre placebo (SEP-PP), l'EI le plus fréquent était les réactions liées à la perfusion avec une incidence de 40,1 % en comparaison avec 25,5 % dans le groupe placebo. L'incidence des réactions liées à la perfusion était la plus élevée pour la perfusion 1 lors de la dose initiale/dose 1 (27,4 %) et a diminué lors doses suivantes à < 10 % à la dose 4. Le pourcentage de patients ayant présenté des réactions liées à la perfusion était dans chaque groupe plus élevé lors de la première perfusion de chaque dose que lors de la deuxième perfusion de la même dose. La majorité des réactions liées à la perfusion sous Ocrevus ont été légères (26,7 %) à modérées (11,9 %); 1,4 % ont eu des réactions sévères liées à la perfusion et aucun patient n'a eu de réaction liée à la perfusion mettant en jeu le pronostic vital (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»).
Perfusion plus rapide alternative du traitement d'entretien
Dans une étude (MA30143, sous-étude sur le raccourcissement de la durée de perfusion) conçue pour décrire le profil de sécurité de perfusions plus rapides (2 heures) d'Ocrevus chez les patients atteints de sclérose en plaques récurrente-rémittente, l'incidence, l'intensité, et le type de symptômes des IRR étaient comparables à ceux observés lors de perfusions administrées pendant environ 3,5 heures (voir «Efficacité clinique»). Le nombre global d'interventions nécessaires était faible dans les deux groupes de perfusion; cependant, plus d'interventions (ralentissements ou interruptions temporaires de la perfusion) ont été nécessaires pour prendre en charge les symptômes des IRR dans le groupe de perfusions plus rapides (2 heures) en comparaison au groupe de perfusions de 3,5 heures (8,7% vs 4,8%).
Réactions liées à l'injection (IR) lors de l'administration d'Ocrevus s.c.
Sur la base des symptômes observés, les IR sont classées en IR systémiques et IR locales.
Dans l'étude OCARINA II (patients non précédemment traités par l'ocrélizumab), les symptômes les plus fréquents rapportés dans le cadre d'IR systémiques et locales étaient les suivants: céphalées (2,5 %), nausées (1,7 %), érythème au site d'injection (29,7 %), douleurs au site d'injection (14,4 %), gonflement au site d'injection (8,5 %) et prurit au site d'injection (6,8 %). 118 patients ont reçu la première injection. Des IR sont survenues chez 48,3 % de ces patients après la première injection. Sur ces 118 patients, respectivement 11,0 % et 45,8 % ont présenté au moins un événement d'IR systémique et d'IR locale. Parmi les patients présentant des IR, celles-ci sont apparues dans les 24 heures suivant la fin de l'injection, et non pendant l'injection, chez la majorité des patients (82,5 %). Toutes les IR étaient non graves et de sévérité légère (71,9 %) ou modérée (28,1 %). La durée médiane a été de 3 jours pour les IR systémiques et de 4 jours pour les IR locales. Les IR ont totalement disparu chez tous les patients, 26,3 % ayant nécessité un traitement symptomatique.
Dans l'étude OCARINA I, 125 patients ont reçu une ou plusieurs injections de 1200 mg d'Ocrevus s.c. Sur les 125 patients ayant reçu la première injection, 16,0 % ont présenté au moins un événement d'IR systémique et 64,0 % ont présenté au moins un événement d'IR locale. Sur les 104 patients ayant reçu la deuxième injection, l'incidence des IR systémiques et des IR locales a diminué respectivement à 7,7 % et 37,5 %. Toutes les IR étaient non graves et toutes sauf une étaient de sévérité légère à modérée lors de la première injection. Toutes les IR étaient non graves et de sévérité légère à modérée lors de la deuxième injection. Parmi les patients ayant présenté une IR, 21,2 % ont eu besoin d'un traitement symptomatique après la première injection et 17,9 % après la deuxième injection.
Infections
Le traitement par Ocrevus n'a pas été associé à une augmentation des infections sévères (chez les patients atteints de SEP-R, le taux d'infections sévères était plus bas (Ocrevus 1,3 %) qu'avec l'interféron bêta-1a (2,9 %), et chez les patients atteints de SEP-PP le taux était comparable à celui du placebo (6,2 % versus 6,7 %)).
Dans les études cliniques contrôlées contre substance active (SEP-R) avec Ocrevus i.v. et l'étude clinique contrôlée contre placebo (SEP-PP), les infections des voies respiratoires et les infections à herpès (toutes deux majoritairement légères à modérées) ont été plus fréquemment observées dans le bras de traitement par Ocrevus.
La proportion globale des patients atteints par une infection sévère était comparable dans les groupes Ocrevus et contrôles (placebo et IFN) avec les fréquences correspondantes semblables. Les infections potentiellement fatales (grade 4) étaient très peu nombreuses sous Ocrevus, mais elles étaient plus fréquentes que dans les groupes contrôles (0,2 % sous OCR versus 0 % sous IFN lors de SEP-R et 1,6 % sous OCR versus 0,4 % sous placebo lors de SEP-PP). Ces infections n'ont entraîné aucune restriction au niveau du traitement. Il existe un risque accru de pneumonie par aspiration chez les patients atteints de SEP-PP avec troubles de la déglutition. Un traitement par Ocrevus peut encore augmenter le risque d'une pneumonie sévère chez ces patients. Les médecins doivent immédiatement prendre les mesures appropriées chez les patients atteints de pneumonie.
Infections des voies respiratoires
Le pourcentage d'infections des voies respiratoires était plus élevé chez les patients traités par Ocrevus que chez ceux traités par l'interféron bêta-1a ou le placebo. Les infections étaient en majeure partie légères à modérées et étaient principalement des infections des voies respiratoires supérieures (y compris des rhinopharyngites) et des bronchites. Il y a eu des cas de pneumonie avec issue fatale sous Ocrevus. La fréquence des pneumonies avec issue fatale rapportées sous traitement par Ocrevus est de l'ordre de la fréquence des pneumonies avec issue fatale observées chez les patients recevant un placebo dans d'autres études portant sur la SEP.
Herpès
Dans les études cliniques contrôlées contre substance active (SEP-R) avec Ocrevus i.v., les infections à herpès ont été constatées plus fréquemment chez les patients traités par Ocrevus que chez ceux traités par l'interféron bêta-1a; ces infections comprennent le zona (2,1 % vs 1,0 %), l'Herpes simplex (0,7 % vs 0,1 %), l'herpès buccal (3,0 % vs 2,2 %), l'herpès génital (0,1 % vs 0 %) et les infections généralisées dues au virus de l'herpès (0,1 % vs 0 %). Ces infections étaient principalement légères à modérées et les patients se sont remis après un traitement standard. Il n'y a eu aucun rapport d'herpès disséminé.
Dans l'étude clinique contrôlée contre placebo (SEP-PP) avec Ocrevus i.v., un pourcentage plus élevé de patients avec une infection à Herpes simplex a été observé dans le bras de traitement par Ocrevus (2,7 % vs 0,8 %).
Infections sévères dans les études portant sur des maladies auto-immunes autres que la SEP
L'utilisation concomitante d'Ocrevus avec d'autres médicaments immunosuppresseurs (p.ex. stéroïdes chroniques, médicaments antirhumatismaux modificateurs de la maladie [DMARD] biologiques ou non, mycophénolate mofétil, cyclophosphamide, azathioprine) a été évaluée dans le cadre d'autres maladies auto-immunes.
La majorité des données disponibles sont issues d'études réalisées chez des patients atteints de polyarthrite rhumatoïde (PR), où un déséquilibre d'infections sévères, y compris, mais sans se limiter à, la pneumonie atypique et la pneumonie à Pneumocystis jiroveci (pneumocystose), la pneumonie varicelleuse, la tuberculose et l'histoplasmose, a été observé dans le groupe Ocrevus-immunosuppresseur. Dans de rares cas, certaines de ces infections ont eu une issue fatale. Des infections sévères ont été plus fréquemment rapportées dans le groupe avec la dose de 1000 mg que dans le groupe avec la dose de 400 mg ou le groupe placebo avec un immunosuppresseur.
Dans ces études, les facteurs de risque d'infections sévères comprenaient: autres comorbidités, utilisation chronique d'immunosuppresseurs/stéroïdes et une origine ethnique asiatique.
Anomalies des paramètres biologiques
Immunoglobulines
Durant la période d'évaluation contrôlée des études menées avec Ocrevus i.v., le traitement par Ocrevus a entraîné une diminution des immunoglobulines totales, principalement due à une réduction des IgM.
Dans les études contrôlées contre substance active (SEP-R), les pourcentages initiaux de patients avec des valeurs d'IgG, IgA et IgM plus basses que la limite inférieure de la normale (< LIN) dans le bras sous Ocrevus i.v. étaient de respectivement 0,5 %, 1,5 % et 0,1 %. Après un traitement de 96 semaines, les pourcentages de patients traités par Ocrevus i.v. avec des valeurs d'IgG, IgA et IgM < LIN étaient respectivement de 1,5 %, 2,4 % et 16,5 %.
Dans l'étude contrôlée contre placebo (SEP-PP) les pourcentages initiaux de patients avec des valeurs d'IgG, IgA et IgM < LIN dans le bras sous Ocrevus i.v. étaient respectivement de 0,0 %, 0,2 % et 0,2 %. Après un traitement de 120 semaines, les pourcentages de patients traités par Ocrevus i.v. avec des valeurs d'IgG, IgA et IgM < LIN étaient respectivement de 1,1 %, 0,5 % et 15,5 %.
Les données cumulées des études cliniques pivots portant sur Ocrevus i.v. (SEP-R et SEP-PP) et de leurs extensions en ouvert (durée d'exposition allant jusqu'à sept ans environ) semblent montrer une relation entre la réduction des taux d'immunoglobulines et les infections sévères (IS) et ce, le plus clairement pour les IgG (chez 0,5 % des patients, une IS est survenue durant la période où les taux d'IgG étaient inférieurs à la LIN). Le type, la sévérité, le temps de latence, la durée et les conséquences de l'IS, observés pendant les épisodes où les taux d'immunoglobulines étaient inférieurs à la LIN, concordent avec les données généralement observées pour les IS chez les patients traités par Ocrevus.
Neutrophiles
Dans l'ensemble, une diminution du nombre de neutrophiles était dans la plupart des cas temporaire (uniquement observée une fois chez un patient traité par Ocrevus i.v.) et d'un degré de sévérité de 1 ou 2. Une neutropénie peut survenir plusieurs mois après l'administration d'Ocrevus i.v. (voir «Mises en garde et précautions»).
Durant la période de traitement contrôlée contre substance active (SEP-R), le nombre de neutrophiles a diminué chez 14,7 % des patients sous Ocrevus i.v. et chez 40,9 % des patients traités par l'interféron bêta-1a. Dans l'étude clinique contrôlée contre placebo (SEP-PP), le pourcentage de patients Ocrevus avec un nombre de neutrophiles réduit était légèrement plus élevé (12,9 %) que celui des patients placebo (10,0 %); parmi ces patients, environ 1 % des patients du groupe Ocrevus avaient une neutropénie de grade 4, en comparaison avec 0 % dans le groupe placebo.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSignes et symptômes
Il n'existe qu'une expérience limitée issue des études cliniques en ce qui concerne des doses plus élevées d'Ocrevus que celle autorisée. La dose la plus élevée testée jusqu'à présent chez les patients atteints de SEP était de 2000 mg, sous forme de 2 perfusions de 1000 mg chacune administrées par voie i.v. à 2 semaines d'intervalle (étude de détermination de la dose de phase II dans la SEP-RR), et de 1200 mg sous forme d'injection s.c. (étude de détermination de la dose de phase Ib). Les effets indésirables correspondaient au profil de sécurité d'Ocrevus des études cliniques pivots.
Traitement
Il n'existe aucun antidote spécifique dans le cas d'un surdosage. La perfusion ou l'injection doit être immédiatement interrompue et le patient doit être surveillé pour détecter d'éventuelles réactions liées à la perfusion ou des réactions liées à l'injection (voir «Mises en garde et précautions, Réactions liées à la perfusion et réactions liées à l'injection»).
Propriétés/EffetsCode ATC
L04AG08
Mécanisme d'action
L'ocrélizumab est un anticorps monoclonal humanisé recombinant qui cible de manière sélective les cellules B exprimant le CD20.
Le CD20 est un antigène de surface trouvé sur les cellules pro-B, les cellules B matures et les cellules B à mémoire, mais qui n'est cependant pas exprimé par les cellules souches lymphoïdes et les plasmocytes.
Les mécanismes exacts par lesquels l'ocrélizumab exerce son effet thérapeutique clinique dans le cadre de la SEP ne sont pas entièrement connus, mais l'implication d'une immunomodulation suite à la réduction du nombre et de la fonction des cellules B exprimant le CD20 est supposée. Après sa liaison à la surface de la cellule, l'ocrélizumab diminue sélectivement les cellules B exprimant le CD20 par le biais d'une phagocytose cellulaire dépendante des anticorps (ADCP), d'une cytotoxicité cellulaire dépendante des anticorps (ADCC), d'une cytotoxicité dépendante du complément (CDC) et par apoptose. La capacité de restauration des cellules B et l'immunité humorale préexistante sont conservées. De plus, l'immunité innée et le nombre total de cellules T ne sont pas affectés.
Pharmacodynamique
Le traitement par Ocrevus entraîne une chute rapide des cellules B exprimant le CD19 dans le sang dans les 14 jours suivant le traitement (premier point d'évaluation), ce qui représente un effet pharmacologique attendu. Ce dernier persiste pendant la période de traitement par Ocrevus i.v. Le CD19 est utilisé pour la numération des cellules B car la présence d'Ocrevus perturbe la reconnaissance du CD20 lors du test.
Les études de phase III ont montré qu'un rétablissement du nombre de cellules B a lieu entre chaque dose d'Ocrevus i.v. chez jusqu'à 5 % des patients (> limite inférieure de la normale (LIN) ou valeur initiale), du moins à un certain moment. L'ampleur et la durée de la diminution des cellules B étaient stables dans les études portant sur la SEP-PP et la SEP-R.
La plus longue période de suivi après la dernière perfusion i.v. d'Ocrevus (phase II WA21493, n = 51) a montré que la durée médiane jusqu'à la restauration des cellules B (retour à la valeur initiale/LIN, en fonction de ce qui est survenu en premier) était de 72 semaines (plage de 27 – 175 semaines). 90 % des patients avaient rétabli leur population de cellules B au niveau de la LIN ou de la valeur initiale en l'espace d'environ 2,5 ans après la dernière perfusion i.v.
Efficacité clinique
Ocrevus i.v.
SEP d'évolution récurrente
L'efficacité et la sécurité d'Ocrevus ont été évaluées dans deux études cliniques randomisées, en double aveugle, à double placebo, contrôlées contre substance active (WA21092, WA21093) avec une conception identique chez des patients atteints de SEP d'évolution récurrente (selon les critères de McDonald 2010). La conception des études et les caractéristiques initiales de la population des études sont résumées dans le tableau 2.
Les caractéristiques démographiques et les données initiales étaient équilibrées dans les deux groupes de traitement. Les patients du groupe A ont reçu 600 mg d'Ocrevus tous les 6 mois (dose 1 sous forme de 2 perfusions i.v. de 300 mg chacune, à 2 semaines d'intervalle), les doses suivantes ont été administrées sous forme de perfusion i.v. unique de 600 mg. Les patients du groupe B ont reçu 44 μg d'interféron bêta-1a (Rebif®) par voie s.c. 3 fois par semaine.
Les résultats de ces études montrent qu'Ocrevus réduit significativement les poussées, l'activité subclinique de la maladie mesurée à l'IRM ainsi que la progression de la maladie, en comparaison avec 44 µg d'interféron bêta-1a par voie sous-cutanée.
Les résultats cliniques clés et les répercussions sur l'IRM sont indiqués dans le tableau 3 et la figure 1.
Tableau 2: Conception des études et caractéristiques démographiques
|
|
Étude 1
|
Étude 2
| |
Nom de l'étude
|
WA21092 (OPERA I) (n = 821)
|
WA21093 (OPERA II) (n = 835)
| |
Plan de l'étude
| |
Population de l'étude
|
Patients atteints de SEP d'évolution récurrente
| |
Anamnèse lors du screening
|
Au moins deux poussées au cours des deux années précédentes ou une poussée au cours de l'année précédente; EDSS entre 0 et 5,5 compris
| |
Durée de l'étude
|
2 ans (96 semaines)
| |
Groupes de traitement
|
Groupe A: Ocrevus 600 mg toutes les 24 semaines i.v.
Groupe B: interféron bêta-1a (Rebif®), 44 μg 3x / semaine s.c (IFN)
| |
Données initiales
|
Ocrevus 600 mg (n = 410)
|
IFN
44 µg
(n = 411)
|
Ocrevus 600 mg (n = 417)
|
IFN
44 µg
(n = 418)
| |
Âge moyen (années)
|
37,1
|
36,9
|
37,2
|
37,4
| |
Tranche d'âge (années) à l'inclusion dans l'étude
|
18 - 56
|
18 - 55
|
18 - 55
|
18 - 55
| |
Répartition des sexes (% d'hommes / % de femmes)
|
34,1 / 65,9
|
33,8 / 66,2
|
35,0 / 65,0
|
33,0 / 67,0
| |
Durée moyenne/médiane depuis le début des symptômes de SEP (années)
|
6,74 / 4,88
|
6,25 / 4,62
|
6,72 / 5,16
|
6,68 / 5,07
| |
Durée moyenne/médiane depuis le diagnostic (années)
|
3,82 / 1,53
|
3,71 / 1,57
|
4,15 / 2,10
|
4,13 / 1,84
| |
Nombre moyen de poussées au cours de la dernière année
|
1,31
|
1,33
|
1,32
|
1,34
| |
EDSS moyen (moyenne)*
|
2,82
|
2,71
|
2,73
|
2,79
| |
Patients naïfs de traitement modificateur de la SEP antérieur (%) **
|
73,4
|
71,0
|
72,7
|
74,9
| |
Nombre moyen de lésions rehaussées par le Gd en T1
|
1,69
|
1,87
|
1,82
|
1,95
| |
Nombre moyen de lésions hyperintenses en T2
|
51,04
|
51,06
|
49,26
|
51,01
|
* Expanded Disability Status Scale.
** Patients qui n'avaient pas de médication spécifique pour la SEP durant les 2 années précédant la randomisation.
Tableau 3: Résultats cliniques clés et critères d'évaluation à l'IRM des études WA21092 et WA21093
|
Critères d'évaluation
|
Étude 1: WA21092
(OPERA I)
|
Étude 2: WA21093
(OPERA II)
| |
Ocrevus 600 mg
(n = 410)
|
IFN 44 μg
(n = 411)
|
Ocrevus 600 mg
(n = 417)
|
IFN 44 μg
(n = 418)
| |
Critères d'évaluation cliniques
| |
Taux annualisé de poussées (critère d'évaluation primaire)
|
0,156
|
0,292
|
0,155
|
0,290
| |
Diminution relative
|
46 % (p < 0,0001)
|
47 % (p < 0,0001)
| |
Rapport des patients avec une progression du handicap confirmée à 12 semaines3
|
9,8 % Ocrevus vs 15,2 % IFN
| |
Réduction du risque (analyse groupée1)
|
40 %
(p = 0,0006)
| |
Réduction du risque (études individuelles2)
|
43 % (p = 0,0139)
|
37 % (p = 0,0169)
| |
Rapport des patients avec une progression du handicap confirmée à 24 semaines3
|
7,6 % Ocrevus vs 12,0 % IFN
| |
Réduction du risque (analyse groupée1)
|
40 %
(p = 0,0025)
| |
Réduction du risque (études individuelles2)
|
43 % (p = 0,0278)
|
37 % (p = 0,0370)
| |
Rapport des patients avec au moins une amélioration du handicap confirmée à 12 semaines4 (groupée)
|
20,7 % Ocrevus vs 15,6 % IFN
| |
Amélioration relative (analyse groupée1)
|
33 % (p = 0,0194)
| |
Amélioration relative (études individuelles2)
|
61 % (p = 0,0106)
|
14 % (p = 0,4019)
| |
Variation moyenne du Multiple Sclerosis Functional Composite (MSFC) par rapport à la valeur initiale
|
0,213
|
0,174
|
0,276
|
0,169
| |
Différence
|
0,039 (p = 0,3261)
|
0,107 (p = 0,0040)
| |
Rapport des patients sans activité de la maladie mesurable (NEDA)5
|
48 %
|
29 %
|
48 %
|
25 %
| |
Amélioration relative2
|
64 % (p < 0,0001)
|
89 % (p < 0,0001)
| |
Critères IRM
| |
Nombre moyen de lésions rehaussées par le Gd en T1 à l'IRM
|
0,016
|
0,286
|
0,021
|
0,416
| |
Diminution relative
|
94 % (p < 0,0001)
|
95 % (p < 0,0001)
| |
Nombre moyen de lésions hyperintenses en T2 nouvelles et/ou élargies à l'IRM
|
0,323
|
1,413
|
0,325
|
1,904
| |
Diminution relative
|
77 % (p < 0,0001)
|
83 % (p < 0,0001)
| |
Nombre moyen de nouvelles lésions hypointenses en T1 (chronic black holes) à l'IRM
|
0,420
|
0,982
|
0,449
|
1,255
| |
Diminution relative
|
57 % (p < 0,0001)
|
64 % (p < 0,0001)
| |
Pourcentage de variation du volume du cerveau entre les semaines 24 et 96
|
-0,572
|
-0,741
|
-0,638
|
-0,750
| |
Diminution relative de la perte de volume du cerveau
|
22,8 % (p = 0,0042)2
|
14,9 % (p = 0,0900)
| |
Qualité de vie
| |
Variation moyenne du SF-36 Physical Component Summary par rapport à la valeur initiale
|
0,036
|
-0,657
|
0,326
|
-0,833
| |
Différence
|
0,693 (p = 0,2193)
|
1,159 (p = 0,0404)6
|
1 Données prospectives groupées des études 1 & 2.
2 Analyse de la valeur de p non confirmatoire; ne faisait pas partie de la hiérarchie prédéterminée des tests.
3 Définie par une augmentation de ≥1,0 point par rapport à la valeur initiale du score de l'Expanded Disability Status Scale (EDSS) chez les patients présentant un score égal ou inférieur à 5,5, ou de ≥0,5 point lorsque le score initial était > 5,5, d'après les estimations de Kaplan-Meier à la semaine 96.
4 Définie par une diminution de ≥1,0 point par rapport à la valeur initiale du score de l'EDSS chez les patients présentant un score initial de l'EDSS ≥2 et ≤5,5, ou ≥0,5, lorsque le score initial était > 5,5. Les patients présentant un score initial < 2 n'ont pas été inclus dans l'analyse.
5 NEDA définie par l'absence de poussées, telles que définies par le protocole, de progression confirmée du handicap (CDP) et d'activité à l'IRM (que ce soit des lésions T1 rehaussées par le gadolinium ou des lésions T2 nouvelles ou élargies) au cours de la totalité des 96 semaines de traitement.
6 Résultats exploratoires sur la base de la population ITT totale.
Figure 1: Courbe de Kaplan-Meier du temps avant le début d'une progression du handicap confirmée persistant au moins 24 semaines, pour laquelle l'événement initial de la détérioration neurologique est survenu durant la phase de traitement en double aveugle (population en ITT groupée)*
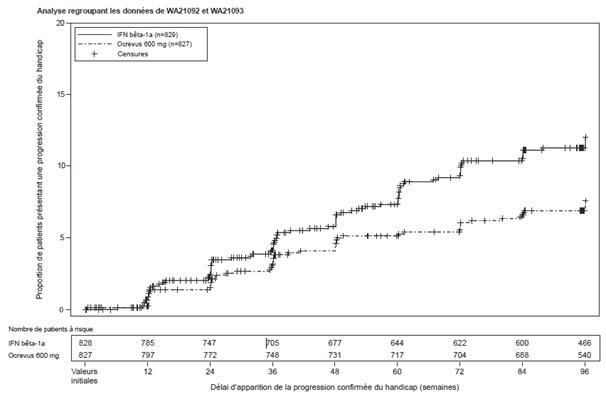
* Analyse groupée définie à l'avance d'OPERA I & II.
Les résultats des analyses groupées définies à l'avance du temps avant le début d'une progression du handicap confirmée persistant au moins 24 semaines ont montré qu'un traitement par ocrélizumab a entraîné une réduction du risque de 40 % en comparaison avec l'interféron bêta-1a (p = 0,0025).
Sous-étude sur le raccourcissement de la durée de perfusion
La sécurité de la perfusion plus rapide (2 heures) d'Ocrevus i.v. a été évaluée dans une sous-étude prospective, multicentrique, randomisée, en double aveugle, contrôlée, en bras parallèle de l'étude MA30143 (étude Ensemble) menée chez des patients atteints de sclérose en plaques récurrente-rémittente, naïfs d'autres traitements de fond de la maladie. La première dose d'Ocrevus i.v. a été administrée en deux perfusions de 300 mg (600 mg au total) séparées de 14 jours. Les patients ont été randomisés à partir de la deuxième dose (doses 2 à 6) selon un rapport 1:1, soit dans le groupe de perfusion classique avec Ocrevus i.v. (perfusé pendant 3,5 heures environ toutes les 24 semaines), soit dans le groupe de perfusion plus rapide avec Ocrevus i.v. (perfusé pendant 2 heures environ toutes les 24 semaines). La randomisation a été stratifiée par région et en fonction de la dose attribuée en premier aux patients par randomisation.
Le critère principal d'évaluation était la proportion de patients présentant des IRR survenant pendant ou dans les 24 heures suivant la première perfusion randomisée d'Ocrevus. L'analyse primaire a été réalisée lorsque 580 patients ont été randomisés. La proportion de patients présentant des IRR survenant pendant ou dans les 24 heures suivant la première perfusion randomisée était de 24,6% dans le groupe de perfusion plus rapide comparée à 23,1% dans le groupe de perfusion classique. La différence entre les groupes stratifiés était similaire. Globalement, pour toutes les doses randomisées, la majorité des IRR étaient légères ou modérées, et seules deux IRR étaient d'intensité sévère (une IRR sévère dans chaque groupe). Il n'y a pas eu de IRR mettant en jeu le pronostic vital, fatale ou grave.
SEP d'évolution primaire progressive
L'efficacité et la sécurité d'Ocrevus ont également été évaluées dans une étude clinique randomisée, en double aveugle, contrôlée contre placebo chez des patients atteints de SEP d'évolution primaire progressive (étude WA25046). La conception de l'étude et les caractéristiques initiales de la population de l'étude sont résumées dans le tableau 4.
Les caractéristiques démographiques et les données initiales étaient équilibrées dans les deux groupes de traitement.
Les patients du groupe Ocrevus (groupe A) ont reçu 600 mg tous les 6 mois (sous forme de 2 perfusions i.v. de 300 mg chacune à 2 semaines d'intervalle). Les patients du groupe B ont en revanche reçu un placebo. Les profils pharmacocinétiques et pharmacodynamiques des perfusions de 600 mg chez les patients atteints de SEP-R étaient cohérents avec ceux des perfusions de 2x 300 mg chez les patients atteints de SEP-PP. Les profils des réactions liées à la perfusion pour chaque perfusion étaient également similaires, que la dose de 600 mg ait été administrée sous forme de perfusion de 600 mg en une dose unique ou sous forme de deux perfusions séparées de 300 mg chacune à 2 semaines d'intervalle (voir «Effets indésirables, Réactions liées à la perfusion» et «Pharmacocinétique»). Étant donné qu'il y a dans l'ensemble eu plus de perfusions de 300 mg, le nombre total de réactions liées à la perfusion était plus élevé dans ce groupe. Après la dose 1 (dose initiale), il est par conséquent conseillé d'administrer Ocrevus sous forme de perfusion unique de 600 mg (voir tableau 1), afin de réduire le nombre total de perfusions et ainsi le nombre de réactions liées à la perfusion qui y sont associées (ainsi que l'exposition à la méthylprédnisolone prophylactique administrée de façon concomitante).
Tableau 4: Conception de l'étude et caractéristiques initiales de la population de l'étude WA25046
|
Nom de l'étude
|
Étude WA25046 ORATORIO (n = 732)
| |
Plan de l'étude
| |
Population de l'étude
|
Patients atteints de SEP d'évolution primaire progressive
| |
Durée de l'étude
|
Déterminée par les événements (au moins 120 semaines et 253 événements confirmés d'une progression du handicap)
Temps de suivi médian: Ocrevus 3,0 ans, placebo 2,8 ans
| |
Anamnèse lors du screening
|
Âge compris entre 18 et 55 ans,
EDSS de 3,0 – 6,5,
score ≥2,0 pour le système fonctionnel pyramidal sur la base des résultats des extrémités inférieures.
| |
Groupes de traitement
|
Groupe A: Ocrevus 600 mg
Groupe B: placebo, randomisé 2:1
| |
Données initiales
|
Ocrevus 600 mg (n = 488)
|
Placebo (n = 244)
| |
Âge moyen (années)
|
44,7
|
44,4
| |
Tranche d'âge (années) à l'inclusion dans l'étude
|
20 - 56
|
18 - 56
| |
Répartition des sexes (% d'hommes / % de femmes)
|
51,4 / 48,6
|
49,2 / 50,8
| |
Durée moyenne/médiane depuis le début des symptômes de SEP (en années)
|
6,7 / 6,0
|
6,1 / 5,5
| |
Durée moyenne/médiane de la maladie depuis le diagnostic de SEP-PP (en années)
|
2,9 / 1,6
|
2,8 / 1,3
| |
EDSS moyen (moyenne)
|
4,7
|
4,7
|
Les critères d'évaluation cliniques et les effets à l'IRM sont indiqués dans le tableau 5 et la figure 2.
Tableau 5: Critères d'évaluation cliniques et critères d'évaluation à l'IRM de l'étude WA25046 (SEP-PP)
|
|
Étude 3
| |
Critères d'évaluation
|
WA25046 (ORATORIO)
| |
Ocrevus 600 mg
(n = 488)
|
Placebo
(n = 244)
| |
Critères d'évaluation cliniques
| |
Critère d'efficacité primaire
Proportion de patients avec une progression du handicap confirmée à 12 semaines1 (critère d'évaluation primaire)
|
30,2 %
|
34,0 %
| |
Réduction du risque
|
24 %
(p = 0,0321)
| |
Proportion de patients avec une progression du handicap confirmée à 24 semaines1
|
28,3 %
|
32,7 %
| |
Réduction du risque
|
25 %
(p = 0,0365)
| |
Pourcentage de variation de la vitesse de marche sur 25 pieds du début de l'étude à la semaine 120
|
38,9
|
55,1
| |
Réduction relative du taux de progression de la vitesse de marche
|
29,4 %
(p = 0,0404)
| |
Critères d'évaluation à l'IRM
| |
Pourcentage de variation du volume des lésions hyperintenses en T2 du début de l'étude à la semaine 120
|
-3,4
|
7,4
| |
(p < 0,0001)
| |
Pourcentage de variation du volume du cerveau de la semaine 24 à la semaine 120
|
-0,902
|
-1,093
| |
Taux de la diminution relative de la perte de volume du cerveau
|
17,5 %
(p = 0,0206)
| |
Qualité de vie
| |
Variation moyenne du SF-36 Physical Component Summary par rapport à la valeur initiale
|
-0,731
|
-1,108
| |
Différence
|
0,377
(p = 0,6034)
|
1 Définie par une augmentation du score EDSS de ≥1,0 point par rapport à la valeur initiale obtenue chez les patients présentant un score initial égal ou inférieur à 5,5, ou ≥0,5 point si le score initial était > 5,5, d'après les estimations de Kaplan-Meier à la semaine 120.
Figure 2: Courbe de Kaplan-Meier du temps jusqu'au début d'une progression du handicap confirmée persistant au moins 12 semaines, pour laquelle l'événement initial de la détérioration neurologique est survenu durant la phase de traitement en double aveugle (population en ITT)*
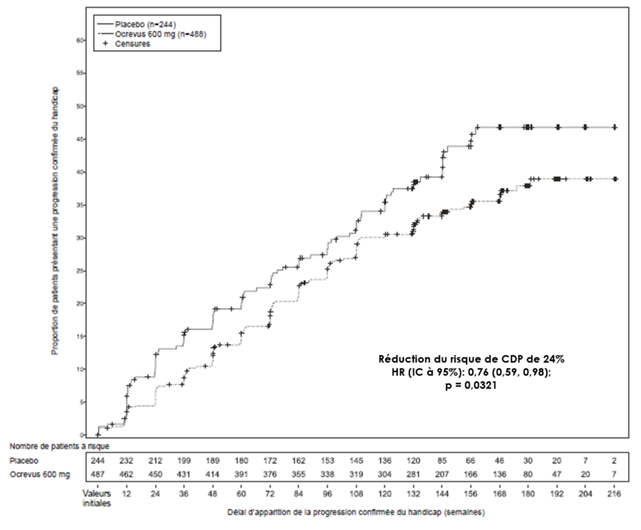
* Tous les patients de cette analyse ont fait l'objet d'un suivi d'au moins 120 semaines. L'analyse primaire est basée sur tous les événements survenus.
Des analyses post-hoc ont été réalisées durant la période contrôlée prolongée (Extended Controlled period [ECP]), qui a inclus un traitement en double aveugle et approximativement 9 mois supplémentaires de suivi contrôlé avant de poursuivre une période d'extention en ouvert (Open Label Extention [OLE]) ou jusqu'à l'arrêt du traitement à l'étude. La proportion de patients présentant une progression du handicap confirmée à 24 semaines avec un score EDSS ≥ 7,0 (24W-CDP de l'EDSS ≥ 7,0; délai jusqu'au recours nécessaire à une chaise roulante) était de 9,1 % dans le groupe sous placebo contre 4,8 % dans le groupe sous Ocrevus à la semaine 144, entraînant une réduction de 47 % du risque de devoir recourir à une chaise roulante (HR: 0,53, [0,31; 0,92]) pendant l'ECP. Comme ces résultats étaient de nature exploratoire et qu'ils incluaient des données après la levée de l'aveugle, ces résultats devraient être interprétés avec prudence.
Ocrevus s.c.
OCARINA II
L'étude CN42097 (OCARINA II) était une étude multicentrique, randomisée, en ouvert, en groupes parallèles, visant à évaluer la pharmacocinétique, la pharmacodynamique, la sécurité et l'immunogénicité, ainsi que les effets radiologiques et cliniques d'Ocrevus s.c., en comparaison à Ocrevus i.v., chez des patients atteints de SEP-R ou de SEP-PP. L'étude OCARINA II a été conçue pour démontrer la non-infériorité du traitement par Ocrevus s.c. par rapport à Ocrevus i.v. sur la base du critère d'évaluation PK primaire, à savoir l'aire sous la courbe de la concentration en fonction du temps (AUC) jusqu'à la semaine 12 suivant l'injection/la perfusion (AUCS1-12).
Au total, 236 patients atteints de SEP-R ou de SEP-PP (213 patients atteints de SEP-R, 23 patients atteints de SEP-PP) ont été randomisés selon un rapport de 1:1 dans le bras sous traitement s.c. ou le bras sous traitement i.v. Pendant la période contrôlée (du jour 0 à la semaine 24), les patients ont reçu soit une injection unique de 920 mg par voie s.c. au jour 1 de l'étude, soit deux perfusions i.v. de 300 mg aux jours 1 et 14 de l'étude. Après la période contrôlée, tous les patients ont eu la possibilité de recevoir des injections supplémentaires de 920 mg par voie s.c. aux semaines 24 et 48 (doses 2 et 3). Les patients ont été exclus s'ils avaient reçu un traitement antérieur par anticorps anti-CD20, y compris l'ocrélizumab, au cours des 24 derniers mois.
Les patients étaient âgés de 18 à 65 ans et présentaient un score à l'EDSS compris entre 0 et 6,5 lors de la sélection. Les caractéristiques démographiques étaient similaires et les caractéristiques à l'inclusion étaient bien équilibrées entre les deux groupes de traitement. L'âge moyen était de 39,9 ans dans le bras sous traitement s.c. et de 40,0 ans dans le bras sous traitement i.v. 34,7 % des patients étaient de sexe masculin dans le bras sous traitement s.c., contre 40,7 % dans le bras sous traitement i.v. La durée moyenne/médiane depuis le diagnostic de SEP était de 5,70/3,10 ans dans le bras sous traitement s.c. et de 4,78/2,35 ans dans le bras sous traitement i.v.
La non-infériorité de l'exposition à l'ocrélizumab après l'administration de 920 mg d'Ocrevus s.c., par rapport à l'administration de 600 mg d'Ocrevus i.v., a été démontrée sur la base du critère d'évaluation PK primaire, à savoir l'AUC jusqu'à la semaine 12 (AUCS1-12) après l'injection (voir «Pharmacocinétique»).
Immunogénicité
Les données d'immunogénicité dépendent fortement de la sensibilité et de la spécificité des tests utilisés pour la détection. De plus, la fréquence des résultats positifs observée avec une méthode de test donnée peut être influencée par différents facteurs, notamment la manipulation des échantillons, le moment du prélèvement des échantillons, une interférence médicamenteuse, les traitements concomitants et la maladie sous-jacente. La comparaison de l'incidence des anticorps dirigés contre Ocrevus avec l'incidence d'anticorps dirigés contre d'autres produits peut par conséquent induire en erreur.
Ocrevus i.v.
Les patients des études portant sur la SEP (WA21092, WA21093 et WA25046) ont été testés à plusieurs reprises (au début de l'étude, puis tous les 6 mois jusqu'à la fin du traitement et de l'étude) à la recherche d'éventuels anticorps anti-médicament (anti-drug antibodies, ADA). Chez les 1311 patients traités par l'ocrélizumab, 12 (~ 1 %) ont été testés positifs pour des ADA survenus au cours du traitement, dont 2 patients présentant des anticorps neutralisants. Les effets d'un développement d'ADA sous traitement sur la sécurité et l'efficacité ne peuvent pas être estimés en raison de la faible incidence de ces ADA associés à Ocrevus.
Ocrevus s.c.
Dans les études OCARINA II et OCARINA I, aucun patient n'a développé d'ADA liés au traitement et dirigés contre l'ocrélizumab.
L'incidence des anticorps anti-rHuPH20 (hyaluronidase) liés au traitement chez les patients traités par Ocrevus s.c. dans l'étude OCARINA I a été de 2,3 % (3/132). Aucun patient de l'étude OCARINA II n'a développé d'anticorps anti-rHuPH20 liés au traitement.
PharmacocinétiqueDans les études portant sur la SEP, la pharmacocinétique d'Ocrevus a été examinée dans une analyse pharmacocinétique de population. À des doses faibles, la clairance d'Ocrevus était dépendante de la concentration. Dans la gamme des doses thérapeutiques, la pharmacocinétique d'Ocrevus est linéaire.
Ocrevus i.v.
L'exposition totale (l'aire sous la courbe (AUC) durant l'intervalle de 24 semaines entre les doses) était comparable entre l'étude portant sur la SEP-PP avec 2x 300 mg et l'étude portant sur la SEP-R avec 1x 600 mg, ce qui était attendu, des doses identiques de 600 mg étant administrées par voie i.v. L'AUC après la quatrième dose de 600 mg d'Ocrevus était de 3510 µg/ml•jour et la concentration maximale moyenne (Cmax) de 212 µg/ml pour la SEP-R (perfusion de 600 mg) et 141 µg/ml pour la SEP-PP (300 mg / perfusion).
Ocrevus s.c.
Après l'administration de 920 mg d'Ocrevus s.c., l'exposition moyenne prévue (AUC sur l'intervalle posologique de 24 semaines) était de 3730 μg/ml•jour.
Le critère d'évaluation PK primaire dans l'étude OCARINA II, l'AUCS1-12, après l'administration de 920 mg d'Ocrevus s.c. s'est avéré non inférieur à l'administration d'Ocrevus i.v. Le rapport des moyennes géométrique (GMR, Geometric Mean Ratio) de l'AUCS1-12 était de 1,29 (IC à 90 %: 1,23-1,35).
Absorption
Ocrevus est administré par perfusion i.v. ou par injection. La biodisponibilité estimée après administration s.c. de 920 mg d'Ocrevus s.c. était de 81 %. La Cmax moyenne a été de 132 µg/ml et le tmax a été atteint après environ 4 jours (intervalle: de 2 à 13 jours).
Distribution
L'estimation du volume de distribution central issue de la pharmacocinétique de population était de 2,78 l. Le volume périphérique a été estimé à 2,68 l et la clairance intercompartimentale à 0,294 l/jour.
Métabolisme
Le métabolisme d'Ocrevus n'a pas fait l'objet d'un examen particulier, les anticorps étant principalement éliminés par catabolisme.
Élimination
La clairance était constante et a été estimée à 0,17 l/jour. La demi-vie terminale était de 26 jours.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Aucune étude pharmacocinétique formelle n'a été réalisée chez ces patients. Les patients atteints d'insuffisance hépatique légère ont été inclus dans les études cliniques et aucune modification de la pharmacocinétique d'Ocrevus n'a été observée chez ces patients.
Troubles de la fonction rénale
Aucune étude pharmacocinétique formelle n'a été réalisée. Les patients atteints d'insuffisance rénale légère ont été inclus dans les études cliniques et aucune modification de la pharmacocinétique d'Ocrevus n'a été observée chez ces patients.
Patients âgés
Aucune étude n'a été réalisée pour évaluer la pharmacocinétique d'Ocrevus chez les patients âgés (≥58 ans).
Enfants et adolescents
Aucune étude n'a été réalisée pour évaluer la pharmacocinétique d'Ocrevus chez les enfants et les adolescents (< 18 ans).
Données précliniquesPharmacologie de sécurité
Les données précliniques n'ont révélé aucun danger particulier pour l'homme sur la base des études conventionnelles sur la sécurité pharmacologique et la toxicité des doses aiguës et répétées.
Génotoxicité
Aucune étude n'a été réalisée pour déterminer la génotoxicité.
Cancérogénicité
Aucune étude n'a été réalisée pour déterminer la cancérogénicité.
Toxicité sur la reproduction
Les données précliniques, basées sur les études portant sur la fertilité masculine et féminine chez le singe, n'ont détecté aucun danger particulier pour l'être humain.
Dans une étude sur le développement embryo-fœtal chez le singe, aucun signe de toxicité maternelle, tératogénicité ou embryotoxicité n'ont été trouvés après l'administration de 75/100 mg/kg (dose de charge/dose à l'étude) d'Ocrevus. Comme les molécules IgG passent la barrière placentaire, Ocrevus entraîne une diminution du nombre de cellules B dans les fœtus des singes traités.
Dans une étude sur le développement pré- et postnatal chez le singe, l'administration d'Ocrevus (dose de charge/dose à l'étude de 15/20 et 75/100 mg/kg, correspondant à une dose équivalente à environ 3000 mg chez l'homme (env. 5x la dose clinique) et 15 000 mg (env. 25x la dose clinique)) a été associée à une glomérulopathie (7 animaux sur 24), la formation de follicules lymphoïdes dans la moelle osseuse (9 animaux sur 24) et une inflammation lymphoplasmocytaire dans les reins (2 animaux sur 24). Le poids des testicules des nouveau-nés à la naissance était significativement réduit dans le groupe 75/100 mg/kg en comparaison avec le groupe contrôle. Il y a eu deux cas de décès (2 animaux sur 24) dans l'étude. L'un des cas était la conséquence d'une faiblesse due à une naissance prématurée combinée à une infection opportuniste, l'autre à une méningo-encéphalite infectieuse du nouveau-né avec implication du cervelet due à une infection active (mastite) de la mère. L'évolution des infections de ces deux nouveau-nés a probablement été influencé par la déplétion des cellules B. Les nouveau-nés de mères ayant reçu Ocrevus ont présenté des populations de cellules B épuisées durant la phase postnatale.
Des quantités mesurables (env. 0,2 % du taux sérique à l'état d'équilibre) d'Ocrevus ont été détectées dans le lait maternel durant la période d'allaitement.
Données particulières
L'administration sous-cutanée d'ocrélizumab avec la hyaluronidase a été bien tolérée chez le rat et le cochon nain dans les études évaluant la tolérance locale.
Aucune étude de carcinogénicité, de génotoxicité ou de fertilité n'a été réalisée avec la hyaluronidase humaine recombinante. Des études de toxicologie sur la reproduction effectuées avec la rHuPH20 chez la souris, au cours desquelles la dose sans effet (NEL, No Effect Dosis) était > 1100 fois plus élevée que la dose clinique recommandée, ont révélé des pertes embryo-fœtales, sans signe de tératogénicité.
Remarques particulièresOcrevus i.v.
Incompatibilités
Aucune incompatibilité entre Ocrevus i.v. et les poches en polychlorure de vinyle (PVC) ou en polyoléfine (PO) ou les sets de perfusion i.v n'a été observée.
Aucun autre agent de dilution que celui mentionné sous «Remarques concernant la manipulation» ne doit être utilisé pour Ocrevus i.v. car une telle utilisation n'a pas été évaluée.
Stabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité de la solution pour perfusion diluée
Cette préparation ne contient pas d'agent conservateur et est destinée exclusivement à un usage unique.
La solution pour perfusion reconstituée doit être utilisée immédiatement. Si elle n'est pas utilisée immédiatement, elle peut être conservée pendant 24 heures au maximum à une température comprise entre 2 et 8 °C, et pendant 8 heures à température ambiante.
Si une solution pour perfusion i.v. n'est pas complètement utilisée dans la même journée, la solution restante doit être éliminée.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver au réfrigérateur (2-8 °C).
Ne pas agiter. Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Remarques concernant la manipulation
Les perfusions d'Ocrevus i.v. doivent être préparées par un professionnel de la santé, dans des conditions d'asepsie. Une aiguille et une seringue stériles doivent être utilisées pour préparer la solution diluée pour perfusion.
Ocrevus i.v. peut contenir de fines particules translucides et/ou réfléchissantes avec une opalescence accrue. La solution ne doit pas être utilisée lorsqu'elle présente une coloration ou des particules étrangères se déplaçant librement.
Ocrevus i.v. doit être dilué avant l'administration. Les solutions d'Ocrevus i.v. destinées à l'administration i.v. sont constituées par dilution du médicament dans une poche de perfusion contenant du chlorure de sodium à 0,9 % (300 mg / 250 ml ou 600 mg / 500 ml), pour obtenir une concentration finale du médicament d'environ 1,2 mg/ml.
La solution pour perfusion diluée doit être administrée avec un set de perfusion équipé d'un filtre en ligne de 0,2 ou 0,22 micromètres.
Le contenu de la poche de perfusion doit être amené à température ambiante avant de réaliser la perfusion i.v. afin d'éviter une réaction indésirable à la perfusion due à une température trop basse de la solution.
Ocrevus s.c.
Incompatibilités
Aucune incompatibilité entre Ocrevus s.c. et le polypropylène (PP), le polycarbonate (PC), le polyéthylène (PE), le chlorure de polyvinyle (PVC), le polyuréthane (PUR) et l'acier inoxydable n'a été observée.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Conserver hors de portée des enfants.
Conserver au réfrigérateur (2-8 °C).
Ne pas agiter. Ne pas congeler.
Conserver le récipient dans son carton pour le protéger de la lumière.
Si nécessaire, le flacon non ouvert peut être conservé à une température ≤25 °C jusqu'à 12 heures.
Les flacons non ouverts peuvent être sortis et remis au réfrigérateur plusieurs fois, c.-à-d. qu'ils peuvent être conservés sans réfrigération à une température ≤25 °C pendant une durée cumulée maximale de 12 heures.
Stockage de la seringue
·D'un point de vue microbiologique, le produit doit être utilisé immédiatement après le transfert du flacon dans la seringue, car il ne contient pas de conservateur antimicrobien. S'il n'est pas utilisé immédiatement, les délais d'utilisation et les conditions de stockage avant l'emploi relèvent de la responsabilité de l'utilisateur, mais, de manière générale, l'entreposage ne devrait pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C.
·Si la préparation s'est déroulée dans des conditions aseptiques contrôlées et validées, la seringue fermée peut être conservée au réfrigérateur jusqu'à 30 jours à une température comprise entre 2 °C et 8 °C et ensuite jusqu'à 8 heures à la lumière du jour diffuse à une température ≤30 °C.
Remarques concernant la manipulation
Ocrevus s.c. doit être préparé par un professionnel de la santé, dans des conditions d'asepsie.
Ocrevus s.c. est une solution prête à l'emploi à usage unique, destinée exclusivement à l'injection sous-cutanée; ce médicament ne doit pas être dilué ou mélangé à d'autres médicaments.
La solution d'Ocrevus s.c. doit être inspectée visuellement pour vérifier qu'il n'y a pas de particules ou de changement de coloration. Ne pas utiliser la solution en cas de changement de coloration ou en présence de particules étrangères.
Préparation de la seringue
Avant l'utilisation, sortir le flacon du réfrigérateur pour laisser la solution revenir à température ambiante.
Prélever l'intégralité du contenu de la solution d'Ocrevus s.c. du flacon avec une seringue et une aiguille de transfert (21 G recommandé).
Retirer l'aiguille de transfert et fixer un set de perfusion s.c. (p.ex. avec une aiguille à ailettes/papillon) muni d'une aiguille de 24-26 G pour l'injection. Utiliser un set de perfusion s.c. dont le volume résiduel NE dépasse PAS 0,8 ml.
Remplir la tubulure de perfusion s.c. avec la solution médicamenteuse pour éliminer l'air dans la tubulure et arrêter avant que le liquide n'atteigne l'aiguille.
Veiller à ce que la seringue contienne exactement 23 ml de solution médicamenteuse après le remplissage de la tubulure et l'expulsion du volume excédentaire.
Injecter immédiatement le médicament pour éviter toute obstruction de l'aiguille.
Il est recommandé d'administrer immédiatement le médicament, car Ocrevus s.c. ne contient pas de conservateur antimicrobien. Dans le cas où la dose n'est pas administrée immédiatement, voir «Stockage de la seringue» ci-dessous. NE PAS conserver la seringue si elle a déjà été préparée et fixée au set de perfusion s.c. déjà rempli.
Stockage de la seringue
·Si la dose n'est pas administrée immédiatement, utiliser une technique aseptique pour prélever l'intégralité du contenu d'Ocrevus s.c. du flacon dans la seringue afin de prendre en compte le volume de la dose (23 ml) et le volume de remplissage du set de perfusion s.c. Remplacer l'aiguille de transfert par le capuchon de la seringue. NE PAS fixer le set de perfusion s.c. pour le stockage.
·Si la seringue a été conservée au réfrigérateur, elle doit revenir à température ambiante avant l'administration du médicament.
Ocrevus i.v. et s.c.
Élimination du médicament non utilisé/périmé
Le rejet de médicaments dans l'environnement doit être réduit au minimum. Ne pas jeter le médicament avec les eaux usées ou avec les déchets ménagers. Utilisez des «systèmes collecteurs» établis s'ils sont disponibles sur votre site.
Les points suivants doivent être strictement respectés en ce qui concerne l'utilisation et l'élimination de seringues et d'autres objets tranchants à usage médical:
·Les aiguilles et les seringues ne doivent en aucun cas être réutilisées.
·Placez toutes les aiguilles et les seringues dans un récipient pour objets pointus (récipient jetable permettant d'éviter les blessures par piqûre).
Le médicament non utilisé et/ou les déchets doivent être éliminés conformément aux exigences nationales. Les aiguilles et les seringues ne doivent en aucun cas être réutilisées.
Numéro d’autorisation66185, 68988 (Swissmedic).
PrésentationOcrevus, flacon de 10 ml de solution à diluer pour perfusion (300 mg/10 ml) (i.v.): 1 [A]
Ocrevus sous-cutané, flacon de 23 ml de solution injectable (920 mg/23 ml) (s.c.): 1 [A]
Titulaire de l’autorisationRoche Pharma (Suisse) SA, Bâle.
Mise à jour de l’informationAoût 2024.
|