Propriétés/EffetsCode ATC
L04AG08
Mécanisme d'action
L'ocrélizumab est un anticorps monoclonal humanisé recombinant qui cible de manière sélective les cellules B exprimant le CD20.
Le CD20 est un antigène de surface trouvé sur les cellules pro-B, les cellules B matures et les cellules B à mémoire, mais qui n'est cependant pas exprimé par les cellules souches lymphoïdes et les plasmocytes.
Les mécanismes exacts par lesquels l'ocrélizumab exerce son effet thérapeutique clinique dans le cadre de la SEP ne sont pas entièrement connus, mais l'implication d'une immunomodulation suite à la réduction du nombre et de la fonction des cellules B exprimant le CD20 est supposée. Après sa liaison à la surface de la cellule, l'ocrélizumab diminue sélectivement les cellules B exprimant le CD20 par le biais d'une phagocytose cellulaire dépendante des anticorps (ADCP), d'une cytotoxicité cellulaire dépendante des anticorps (ADCC), d'une cytotoxicité dépendante du complément (CDC) et par apoptose. La capacité de restauration des cellules B et l'immunité humorale préexistante sont conservées. De plus, l'immunité innée et le nombre total de cellules T ne sont pas affectés.
Pharmacodynamique
Le traitement par Ocrevus entraîne une chute rapide des cellules B exprimant le CD19 dans le sang dans les 14 jours suivant le traitement (premier point d'évaluation), ce qui représente un effet pharmacologique attendu. Ce dernier persiste pendant la période de traitement par Ocrevus i.v. Le CD19 est utilisé pour la numération des cellules B car la présence d'Ocrevus perturbe la reconnaissance du CD20 lors du test.
Les études de phase III ont montré qu'un rétablissement du nombre de cellules B a lieu entre chaque dose d'Ocrevus i.v. chez jusqu'à 5 % des patients (> limite inférieure de la normale (LIN) ou valeur initiale), du moins à un certain moment. L'ampleur et la durée de la diminution des cellules B étaient stables dans les études portant sur la SEP-PP et la SEP-R.
La plus longue période de suivi après la dernière perfusion i.v. d'Ocrevus (phase II WA21493, n = 51) a montré que la durée médiane jusqu'à la restauration des cellules B (retour à la valeur initiale/LIN, en fonction de ce qui est survenu en premier) était de 72 semaines (plage de 27 – 175 semaines). 90 % des patients avaient rétabli leur population de cellules B au niveau de la LIN ou de la valeur initiale en l'espace d'environ 2,5 ans après la dernière perfusion i.v.
Efficacité clinique
Ocrevus i.v.
SEP d'évolution récurrente
L'efficacité et la sécurité d'Ocrevus ont été évaluées dans deux études cliniques randomisées, en double aveugle, à double placebo, contrôlées contre substance active (WA21092, WA21093) avec une conception identique chez des patients atteints de SEP d'évolution récurrente (selon les critères de McDonald 2010). La conception des études et les caractéristiques initiales de la population des études sont résumées dans le tableau 2.
Les caractéristiques démographiques et les données initiales étaient équilibrées dans les deux groupes de traitement. Les patients du groupe A ont reçu 600 mg d'Ocrevus tous les 6 mois (dose 1 sous forme de 2 perfusions i.v. de 300 mg chacune, à 2 semaines d'intervalle), les doses suivantes ont été administrées sous forme de perfusion i.v. unique de 600 mg. Les patients du groupe B ont reçu 44 μg d'interféron bêta-1a (Rebif®) par voie s.c. 3 fois par semaine.
Les résultats de ces études montrent qu'Ocrevus réduit significativement les poussées, l'activité subclinique de la maladie mesurée à l'IRM ainsi que la progression de la maladie, en comparaison avec 44 µg d'interféron bêta-1a par voie sous-cutanée.
Les résultats cliniques clés et les répercussions sur l'IRM sont indiqués dans le tableau 3 et la figure 1.
Tableau 2: Conception des études et caractéristiques démographiques
|
|
Étude 1
|
Étude 2
| |
Nom de l'étude
|
WA21092 (OPERA I) (n = 821)
|
WA21093 (OPERA II) (n = 835)
| |
Plan de l'étude
| |
Population de l'étude
|
Patients atteints de SEP d'évolution récurrente
| |
Anamnèse lors du screening
|
Au moins deux poussées au cours des deux années précédentes ou une poussée au cours de l'année précédente; EDSS entre 0 et 5,5 compris
| |
Durée de l'étude
|
2 ans (96 semaines)
| |
Groupes de traitement
|
Groupe A: Ocrevus 600 mg toutes les 24 semaines i.v.
Groupe B: interféron bêta-1a (Rebif®), 44 μg 3x / semaine s.c (IFN)
| |
Données initiales
|
Ocrevus 600 mg (n = 410)
|
IFN
44 µg
(n = 411)
|
Ocrevus 600 mg (n = 417)
|
IFN
44 µg
(n = 418)
| |
Âge moyen (années)
|
37,1
|
36,9
|
37,2
|
37,4
| |
Tranche d'âge (années) à l'inclusion dans l'étude
|
18 - 56
|
18 - 55
|
18 - 55
|
18 - 55
| |
Répartition des sexes (% d'hommes / % de femmes)
|
34,1 / 65,9
|
33,8 / 66,2
|
35,0 / 65,0
|
33,0 / 67,0
| |
Durée moyenne/médiane depuis le début des symptômes de SEP (années)
|
6,74 / 4,88
|
6,25 / 4,62
|
6,72 / 5,16
|
6,68 / 5,07
| |
Durée moyenne/médiane depuis le diagnostic (années)
|
3,82 / 1,53
|
3,71 / 1,57
|
4,15 / 2,10
|
4,13 / 1,84
| |
Nombre moyen de poussées au cours de la dernière année
|
1,31
|
1,33
|
1,32
|
1,34
| |
EDSS moyen (moyenne)*
|
2,82
|
2,71
|
2,73
|
2,79
| |
Patients naïfs de traitement modificateur de la SEP antérieur (%) **
|
73,4
|
71,0
|
72,7
|
74,9
| |
Nombre moyen de lésions rehaussées par le Gd en T1
|
1,69
|
1,87
|
1,82
|
1,95
| |
Nombre moyen de lésions hyperintenses en T2
|
51,04
|
51,06
|
49,26
|
51,01
|
* Expanded Disability Status Scale.
** Patients qui n'avaient pas de médication spécifique pour la SEP durant les 2 années précédant la randomisation.
Tableau 3: Résultats cliniques clés et critères d'évaluation à l'IRM des études WA21092 et WA21093
|
Critères d'évaluation
|
Étude 1: WA21092
(OPERA I)
|
Étude 2: WA21093
(OPERA II)
| |
Ocrevus 600 mg
(n = 410)
|
IFN 44 μg
(n = 411)
|
Ocrevus 600 mg
(n = 417)
|
IFN 44 μg
(n = 418)
| |
Critères d'évaluation cliniques
| |
Taux annualisé de poussées (critère d'évaluation primaire)
|
0,156
|
0,292
|
0,155
|
0,290
| |
Diminution relative
|
46 % (p < 0,0001)
|
47 % (p < 0,0001)
| |
Rapport des patients avec une progression du handicap confirmée à 12 semaines3
|
9,8 % Ocrevus vs 15,2 % IFN
| |
Réduction du risque (analyse groupée1)
|
40 %
(p = 0,0006)
| |
Réduction du risque (études individuelles2)
|
43 % (p = 0,0139)
|
37 % (p = 0,0169)
| |
Rapport des patients avec une progression du handicap confirmée à 24 semaines3
|
7,6 % Ocrevus vs 12,0 % IFN
| |
Réduction du risque (analyse groupée1)
|
40 %
(p = 0,0025)
| |
Réduction du risque (études individuelles2)
|
43 % (p = 0,0278)
|
37 % (p = 0,0370)
| |
Rapport des patients avec au moins une amélioration du handicap confirmée à 12 semaines4 (groupée)
|
20,7 % Ocrevus vs 15,6 % IFN
| |
Amélioration relative (analyse groupée1)
|
33 % (p = 0,0194)
| |
Amélioration relative (études individuelles2)
|
61 % (p = 0,0106)
|
14 % (p = 0,4019)
| |
Variation moyenne du Multiple Sclerosis Functional Composite (MSFC) par rapport à la valeur initiale
|
0,213
|
0,174
|
0,276
|
0,169
| |
Différence
|
0,039 (p = 0,3261)
|
0,107 (p = 0,0040)
| |
Rapport des patients sans activité de la maladie mesurable (NEDA)5
|
48 %
|
29 %
|
48 %
|
25 %
| |
Amélioration relative2
|
64 % (p < 0,0001)
|
89 % (p < 0,0001)
| |
Critères IRM
| |
Nombre moyen de lésions rehaussées par le Gd en T1 à l'IRM
|
0,016
|
0,286
|
0,021
|
0,416
| |
Diminution relative
|
94 % (p < 0,0001)
|
95 % (p < 0,0001)
| |
Nombre moyen de lésions hyperintenses en T2 nouvelles et/ou élargies à l'IRM
|
0,323
|
1,413
|
0,325
|
1,904
| |
Diminution relative
|
77 % (p < 0,0001)
|
83 % (p < 0,0001)
| |
Nombre moyen de nouvelles lésions hypointenses en T1 (chronic black holes) à l'IRM
|
0,420
|
0,982
|
0,449
|
1,255
| |
Diminution relative
|
57 % (p < 0,0001)
|
64 % (p < 0,0001)
| |
Pourcentage de variation du volume du cerveau entre les semaines 24 et 96
|
-0,572
|
-0,741
|
-0,638
|
-0,750
| |
Diminution relative de la perte de volume du cerveau
|
22,8 % (p = 0,0042)2
|
14,9 % (p = 0,0900)
| |
Qualité de vie
| |
Variation moyenne du SF-36 Physical Component Summary par rapport à la valeur initiale
|
0,036
|
-0,657
|
0,326
|
-0,833
| |
Différence
|
0,693 (p = 0,2193)
|
1,159 (p = 0,0404)6
|
1 Données prospectives groupées des études 1 & 2.
2 Analyse de la valeur de p non confirmatoire; ne faisait pas partie de la hiérarchie prédéterminée des tests.
3 Définie par une augmentation de ≥1,0 point par rapport à la valeur initiale du score de l'Expanded Disability Status Scale (EDSS) chez les patients présentant un score égal ou inférieur à 5,5, ou de ≥0,5 point lorsque le score initial était > 5,5, d'après les estimations de Kaplan-Meier à la semaine 96.
4 Définie par une diminution de ≥1,0 point par rapport à la valeur initiale du score de l'EDSS chez les patients présentant un score initial de l'EDSS ≥2 et ≤5,5, ou ≥0,5, lorsque le score initial était > 5,5. Les patients présentant un score initial < 2 n'ont pas été inclus dans l'analyse.
5 NEDA définie par l'absence de poussées, telles que définies par le protocole, de progression confirmée du handicap (CDP) et d'activité à l'IRM (que ce soit des lésions T1 rehaussées par le gadolinium ou des lésions T2 nouvelles ou élargies) au cours de la totalité des 96 semaines de traitement.
6 Résultats exploratoires sur la base de la population ITT totale.
Figure 1: Courbe de Kaplan-Meier du temps avant le début d'une progression du handicap confirmée persistant au moins 24 semaines, pour laquelle l'événement initial de la détérioration neurologique est survenu durant la phase de traitement en double aveugle (population en ITT groupée)*
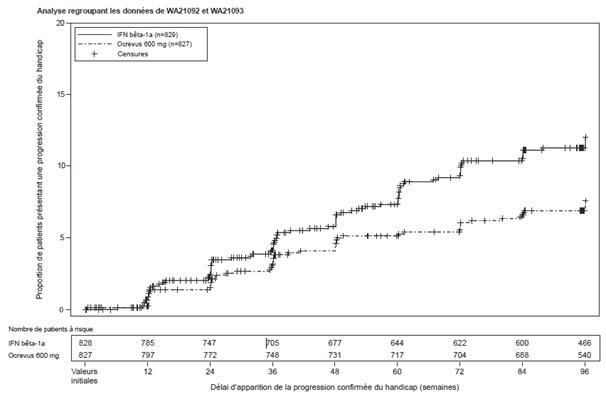
* Analyse groupée définie à l'avance d'OPERA I & II.
Les résultats des analyses groupées définies à l'avance du temps avant le début d'une progression du handicap confirmée persistant au moins 24 semaines ont montré qu'un traitement par ocrélizumab a entraîné une réduction du risque de 40 % en comparaison avec l'interféron bêta-1a (p = 0,0025).
Sous-étude sur le raccourcissement de la durée de perfusion
La sécurité de la perfusion plus rapide (2 heures) d'Ocrevus i.v. a été évaluée dans une sous-étude prospective, multicentrique, randomisée, en double aveugle, contrôlée, en bras parallèle de l'étude MA30143 (étude Ensemble) menée chez des patients atteints de sclérose en plaques récurrente-rémittente, naïfs d'autres traitements de fond de la maladie. La première dose d'Ocrevus i.v. a été administrée en deux perfusions de 300 mg (600 mg au total) séparées de 14 jours. Les patients ont été randomisés à partir de la deuxième dose (doses 2 à 6) selon un rapport 1:1, soit dans le groupe de perfusion classique avec Ocrevus i.v. (perfusé pendant 3,5 heures environ toutes les 24 semaines), soit dans le groupe de perfusion plus rapide avec Ocrevus i.v. (perfusé pendant 2 heures environ toutes les 24 semaines). La randomisation a été stratifiée par région et en fonction de la dose attribuée en premier aux patients par randomisation.
Le critère principal d'évaluation était la proportion de patients présentant des IRR survenant pendant ou dans les 24 heures suivant la première perfusion randomisée d'Ocrevus. L'analyse primaire a été réalisée lorsque 580 patients ont été randomisés. La proportion de patients présentant des IRR survenant pendant ou dans les 24 heures suivant la première perfusion randomisée était de 24,6% dans le groupe de perfusion plus rapide comparée à 23,1% dans le groupe de perfusion classique. La différence entre les groupes stratifiés était similaire. Globalement, pour toutes les doses randomisées, la majorité des IRR étaient légères ou modérées, et seules deux IRR étaient d'intensité sévère (une IRR sévère dans chaque groupe). Il n'y a pas eu de IRR mettant en jeu le pronostic vital, fatale ou grave.
SEP d'évolution primaire progressive
L'efficacité et la sécurité d'Ocrevus ont également été évaluées dans une étude clinique randomisée, en double aveugle, contrôlée contre placebo chez des patients atteints de SEP d'évolution primaire progressive (étude WA25046). La conception de l'étude et les caractéristiques initiales de la population de l'étude sont résumées dans le tableau 4.
Les caractéristiques démographiques et les données initiales étaient équilibrées dans les deux groupes de traitement.
Les patients du groupe Ocrevus (groupe A) ont reçu 600 mg tous les 6 mois (sous forme de 2 perfusions i.v. de 300 mg chacune à 2 semaines d'intervalle). Les patients du groupe B ont en revanche reçu un placebo. Les profils pharmacocinétiques et pharmacodynamiques des perfusions de 600 mg chez les patients atteints de SEP-R étaient cohérents avec ceux des perfusions de 2x 300 mg chez les patients atteints de SEP-PP. Les profils des réactions liées à la perfusion pour chaque perfusion étaient également similaires, que la dose de 600 mg ait été administrée sous forme de perfusion de 600 mg en une dose unique ou sous forme de deux perfusions séparées de 300 mg chacune à 2 semaines d'intervalle (voir «Effets indésirables, Réactions liées à la perfusion» et «Pharmacocinétique»). Étant donné qu'il y a dans l'ensemble eu plus de perfusions de 300 mg, le nombre total de réactions liées à la perfusion était plus élevé dans ce groupe. Après la dose 1 (dose initiale), il est par conséquent conseillé d'administrer Ocrevus sous forme de perfusion unique de 600 mg (voir tableau 1), afin de réduire le nombre total de perfusions et ainsi le nombre de réactions liées à la perfusion qui y sont associées (ainsi que l'exposition à la méthylprédnisolone prophylactique administrée de façon concomitante).
Tableau 4: Conception de l'étude et caractéristiques initiales de la population de l'étude WA25046
|
Nom de l'étude
|
Étude WA25046 ORATORIO (n = 732)
| |
Plan de l'étude
| |
Population de l'étude
|
Patients atteints de SEP d'évolution primaire progressive
| |
Durée de l'étude
|
Déterminée par les événements (au moins 120 semaines et 253 événements confirmés d'une progression du handicap)
Temps de suivi médian: Ocrevus 3,0 ans, placebo 2,8 ans
| |
Anamnèse lors du screening
|
Âge compris entre 18 et 55 ans,
EDSS de 3,0 – 6,5,
score ≥2,0 pour le système fonctionnel pyramidal sur la base des résultats des extrémités inférieures.
| |
Groupes de traitement
|
Groupe A: Ocrevus 600 mg
Groupe B: placebo, randomisé 2:1
| |
Données initiales
|
Ocrevus 600 mg (n = 488)
|
Placebo (n = 244)
| |
Âge moyen (années)
|
44,7
|
44,4
| |
Tranche d'âge (années) à l'inclusion dans l'étude
|
20 - 56
|
18 - 56
| |
Répartition des sexes (% d'hommes / % de femmes)
|
51,4 / 48,6
|
49,2 / 50,8
| |
Durée moyenne/médiane depuis le début des symptômes de SEP (en années)
|
6,7 / 6,0
|
6,1 / 5,5
| |
Durée moyenne/médiane de la maladie depuis le diagnostic de SEP-PP (en années)
|
2,9 / 1,6
|
2,8 / 1,3
| |
EDSS moyen (moyenne)
|
4,7
|
4,7
|
Les critères d'évaluation cliniques et les effets à l'IRM sont indiqués dans le tableau 5 et la figure 2.
Tableau 5: Critères d'évaluation cliniques et critères d'évaluation à l'IRM de l'étude WA25046 (SEP-PP)
|
|
Étude 3
| |
Critères d'évaluation
|
WA25046 (ORATORIO)
| |
Ocrevus 600 mg
(n = 488)
|
Placebo
(n = 244)
| |
Critères d'évaluation cliniques
| |
Critère d'efficacité primaire
Proportion de patients avec une progression du handicap confirmée à 12 semaines1 (critère d'évaluation primaire)
|
30,2 %
|
34,0 %
| |
Réduction du risque
|
24 %
(p = 0,0321)
| |
Proportion de patients avec une progression du handicap confirmée à 24 semaines1
|
28,3 %
|
32,7 %
| |
Réduction du risque
|
25 %
(p = 0,0365)
| |
Pourcentage de variation de la vitesse de marche sur 25 pieds du début de l'étude à la semaine 120
|
38,9
|
55,1
| |
Réduction relative du taux de progression de la vitesse de marche
|
29,4 %
(p = 0,0404)
| |
Critères d'évaluation à l'IRM
| |
Pourcentage de variation du volume des lésions hyperintenses en T2 du début de l'étude à la semaine 120
|
-3,4
|
7,4
| |
(p < 0,0001)
| |
Pourcentage de variation du volume du cerveau de la semaine 24 à la semaine 120
|
-0,902
|
-1,093
| |
Taux de la diminution relative de la perte de volume du cerveau
|
17,5 %
(p = 0,0206)
| |
Qualité de vie
| |
Variation moyenne du SF-36 Physical Component Summary par rapport à la valeur initiale
|
-0,731
|
-1,108
| |
Différence
|
0,377
(p = 0,6034)
|
1 Définie par une augmentation du score EDSS de ≥1,0 point par rapport à la valeur initiale obtenue chez les patients présentant un score initial égal ou inférieur à 5,5, ou ≥0,5 point si le score initial était > 5,5, d'après les estimations de Kaplan-Meier à la semaine 120.
Figure 2: Courbe de Kaplan-Meier du temps jusqu'au début d'une progression du handicap confirmée persistant au moins 12 semaines, pour laquelle l'événement initial de la détérioration neurologique est survenu durant la phase de traitement en double aveugle (population en ITT)*
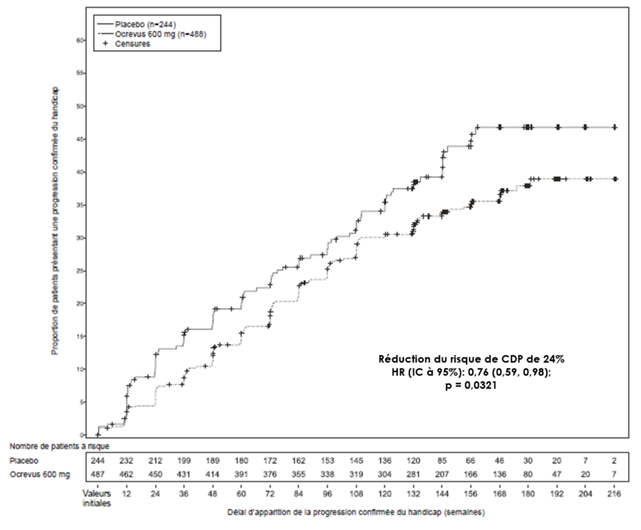
* Tous les patients de cette analyse ont fait l'objet d'un suivi d'au moins 120 semaines. L'analyse primaire est basée sur tous les événements survenus.
Des analyses post-hoc ont été réalisées durant la période contrôlée prolongée (Extended Controlled period [ECP]), qui a inclus un traitement en double aveugle et approximativement 9 mois supplémentaires de suivi contrôlé avant de poursuivre une période d'extention en ouvert (Open Label Extention [OLE]) ou jusqu'à l'arrêt du traitement à l'étude. La proportion de patients présentant une progression du handicap confirmée à 24 semaines avec un score EDSS ≥ 7,0 (24W-CDP de l'EDSS ≥ 7,0; délai jusqu'au recours nécessaire à une chaise roulante) était de 9,1 % dans le groupe sous placebo contre 4,8 % dans le groupe sous Ocrevus à la semaine 144, entraînant une réduction de 47 % du risque de devoir recourir à une chaise roulante (HR: 0,53, [0,31; 0,92]) pendant l'ECP. Comme ces résultats étaient de nature exploratoire et qu'ils incluaient des données après la levée de l'aveugle, ces résultats devraient être interprétés avec prudence.
Ocrevus s.c.
OCARINA II
L'étude CN42097 (OCARINA II) était une étude multicentrique, randomisée, en ouvert, en groupes parallèles, visant à évaluer la pharmacocinétique, la pharmacodynamique, la sécurité et l'immunogénicité, ainsi que les effets radiologiques et cliniques d'Ocrevus s.c., en comparaison à Ocrevus i.v., chez des patients atteints de SEP-R ou de SEP-PP. L'étude OCARINA II a été conçue pour démontrer la non-infériorité du traitement par Ocrevus s.c. par rapport à Ocrevus i.v. sur la base du critère d'évaluation PK primaire, à savoir l'aire sous la courbe de la concentration en fonction du temps (AUC) jusqu'à la semaine 12 suivant l'injection/la perfusion (AUCS1-12).
Au total, 236 patients atteints de SEP-R ou de SEP-PP (213 patients atteints de SEP-R, 23 patients atteints de SEP-PP) ont été randomisés selon un rapport de 1:1 dans le bras sous traitement s.c. ou le bras sous traitement i.v. Pendant la période contrôlée (du jour 0 à la semaine 24), les patients ont reçu soit une injection unique de 920 mg par voie s.c. au jour 1 de l'étude, soit deux perfusions i.v. de 300 mg aux jours 1 et 14 de l'étude. Après la période contrôlée, tous les patients ont eu la possibilité de recevoir des injections supplémentaires de 920 mg par voie s.c. aux semaines 24 et 48 (doses 2 et 3). Les patients ont été exclus s'ils avaient reçu un traitement antérieur par anticorps anti-CD20, y compris l'ocrélizumab, au cours des 24 derniers mois.
Les patients étaient âgés de 18 à 65 ans et présentaient un score à l'EDSS compris entre 0 et 6,5 lors de la sélection. Les caractéristiques démographiques étaient similaires et les caractéristiques à l'inclusion étaient bien équilibrées entre les deux groupes de traitement. L'âge moyen était de 39,9 ans dans le bras sous traitement s.c. et de 40,0 ans dans le bras sous traitement i.v. 34,7 % des patients étaient de sexe masculin dans le bras sous traitement s.c., contre 40,7 % dans le bras sous traitement i.v. La durée moyenne/médiane depuis le diagnostic de SEP était de 5,70/3,10 ans dans le bras sous traitement s.c. et de 4,78/2,35 ans dans le bras sous traitement i.v.
La non-infériorité de l'exposition à l'ocrélizumab après l'administration de 920 mg d'Ocrevus s.c., par rapport à l'administration de 600 mg d'Ocrevus i.v., a été démontrée sur la base du critère d'évaluation PK primaire, à savoir l'AUC jusqu'à la semaine 12 (AUCS1-12) après l'injection (voir «Pharmacocinétique»).
Immunogénicité
Les données d'immunogénicité dépendent fortement de la sensibilité et de la spécificité des tests utilisés pour la détection. De plus, la fréquence des résultats positifs observée avec une méthode de test donnée peut être influencée par différents facteurs, notamment la manipulation des échantillons, le moment du prélèvement des échantillons, une interférence médicamenteuse, les traitements concomitants et la maladie sous-jacente. La comparaison de l'incidence des anticorps dirigés contre Ocrevus avec l'incidence d'anticorps dirigés contre d'autres produits peut par conséquent induire en erreur.
Ocrevus i.v.
Les patients des études portant sur la SEP (WA21092, WA21093 et WA25046) ont été testés à plusieurs reprises (au début de l'étude, puis tous les 6 mois jusqu'à la fin du traitement et de l'étude) à la recherche d'éventuels anticorps anti-médicament (anti-drug antibodies, ADA). Chez les 1311 patients traités par l'ocrélizumab, 12 (~ 1 %) ont été testés positifs pour des ADA survenus au cours du traitement, dont 2 patients présentant des anticorps neutralisants. Les effets d'un développement d'ADA sous traitement sur la sécurité et l'efficacité ne peuvent pas être estimés en raison de la faible incidence de ces ADA associés à Ocrevus.
Ocrevus s.c.
Dans les études OCARINA II et OCARINA I, aucun patient n'a développé d'ADA liés au traitement et dirigés contre l'ocrélizumab.
L'incidence des anticorps anti-rHuPH20 (hyaluronidase) liés au traitement chez les patients traités par Ocrevus s.c. dans l'étude OCARINA I a été de 2,3 % (3/132). Aucun patient de l'étude OCARINA II n'a développé d'anticorps anti-rHuPH20 liés au traitement.
|