CompositionPrincipes actifs
Sitagliptine sous forme de chlorhydrate monohydraté de sitagliptine, chlorhydrate de metformine.
Excipients
Hypromellose, stéarate de magnésium, hydrogénophosphate de calcium, cellulose microcristalline, stéarylfumarate de sodium, croscarmellose sodique (E468).
Enrobage pour les comprimés à 50/500 mg et 100/1000 mg:
Poly(alcool vinylique), dioxyde de titane (E171), macrogol 4000, talc, oxyde de fer jaune (E172), oxyde de fer rouge (E172).
Enrobage pour les comprimés à 50/1000 mg:
Poly(alcool vinylique), dioxyde de titane (E171), macrogol 4000, talc.
Chaque comprimé à libération modifiée à 50/500 mg et 50/1000 mg contient 2.15 mg de sodium.
Chaque comprimé à libération modifiée à 100/1000 mg contient 1.43 mg de sodium.
Indications/Possibilités d’emploiSitagliptin-Metformin-Mepha retard est indiqué en tant que traitement complémentaire, en plus d'un régime alimentaire et d'une activité physique régulière, pour équilibrer la glycémie chez les patients diabétiques de type 2 lorsqu'un contrôle suffisant de la glycémie n'est atteint ni avec la metformine seule, ni avec la sitagliptine seule, ainsi que chez les patients déjà traités par une association de sitagliptine et de metformine.
Sitagliptin-Metformin-Mepha retard est également indiqué en association avec une sulfonylurée (triple association thérapeutique), en plus d'un régime alimentaire et d'une activité physique régulière, chez les patients atteints de diabète de type 2 chez lesquels un contrôle suffisant de la glycémie n'est pas atteint par un traitement associant deux des trois principes actifs suivants: metformine, sitagliptine ou sulfonylurée.
Lorsqu'un régime alimentaire, une augmentation de l'activité physique et l'insuline ne permettent pas à eux seuls de diminuer suffisamment la glycémie: en association avec une insulinothérapie.
Posologie/Mode d’emploiGénéralités
La dose du traitement anti-hyperglycémiant par Sitagliptin-Metformin-Mepha retard doit être définie de façon individualisée en fonction du traitement actuel du patient, de l'efficacité et de la tolérance, en veillant à ne pas dépasser la dose maximale recommandée de 100 mg de sitagliptine par jour.
Sitagliptin-Metformin-Mepha retard doit être pris une fois par jour avec un repas, de préférence le soir.
La dose doit être augmentée progressivement afin de réduire le risque de troubles gastro-intestinaux dus à la metformine.
Pour que les propriétés de libération retardée restent préservées, les comprimés doivent être avalés entiers sans être divisés, cassés, broyés ou mâchés.
Les concentrations plasmatiques de metformine augmentent lors d'une prise de Sitagliptin-Metformin-Mepha retard avec des aliments. Des cas de comprimés de chlorhydrate de sitagliptine/metformine à libération retardée non entièrement dissouts, excrétés dans les selles, ont été rapportés. On ne sait pas si la matière retrouvée dans les selles contient de la substance active. L'équilibre glycémique doit donc être contrôlé de manière particulièrement attentive chez les patients observant de manière répétée des restes de comprimés dans leurs selles.
Des modifications de traitements chez le diabète de type 2 exigent toujours de la précaution et une surveillance adéquate en raison des modifications possibles du contrôle glycémique. Ceci s'applique aussi au passage de comprimés de metformine à libération non retardée à des comprimés de metformine à libération retardée tels que p.ex. Sitagliptin-Metformin-Mepha retard.
Recommandations posologiques
La dose initiale de Sitagliptin-Metformin-Mepha retard doit se baser sur le traitement déjà suivi.
Sitagliptin-Metformin-Mepha retard est pris une fois par jour avec un repas, de préférence le soir. Les comprimés Sitagliptin-Metformin-Mepha retard à libération modifiée sont disponibles dans les dosages suivants:
50 mg de sitagliptine/500 mg de chlorhydrate de metformine à libération retardée
50 mg de sitagliptine/1000 mg de chlorhydrate de metformine à libération retardée
100 mg de sitagliptine/1000 mg de chlorhydrate de metformine à libération retardée
Les patients utilisant les comprimés de 50 mg de sitagliptine/500 mg de metformine à libération retardée ou les comprimés de 50 mg de sitagliptine/1000 mg de metformine à libération retardée doivent prendre leur dose quotidienne en une seule prise de deux comprimés à la fois. Les comprimés de 100 mg de sitagliptine/1000 mg de metformine à libération retardée doivent être pris en un seul comprimé une fois par jour.
Patients chez lesquels un traitement à la metformine seule ne permet pas d'atteindre un équilibre glycémique suffisant
Chez les patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule, la dose quotidienne initiale recommandée de Sitagliptin-Metformin-Mepha retard est de 100 mg de sitagliptine au total plus la dose de metformine prescrite dans le cadre de l'ancien traitement.
Patients chez lesquels un traitement à la sitagliptine seule ne permet pas d'atteindre un équilibre glycémique suffisant
Chez les patients n'ayant pas atteint un équilibre glycémique suffisant sous sitagliptine seule, la dose quotidienne initiale recommandée de Sitagliptin-Metformin-Mepha retard est de 100 mg de sitagliptine au total et de 1000 mg de chlorhydrate de metformine. La dose de metformine peut être ajustée selon le besoin conformément au contrôle glycémique atteint. Une augmentation progressive de la dose devrait être envisagée pour réduire les effets indésirables gastro-intestinaux éventuellement causés par la metformine. Les patients sous sitagliptine seule dont la dose a été ajustée en raison d'un trouble de la fonction rénale ne doivent pas passer à Sitagliptin-Metformin-Mepha retard (voir la section «Contre-indications»).
Patients passant à Sitagliptin-Metformin-Mepha retard après un traitement associé de sitagliptine et metformine
Les patients passant à Sitagliptin-Metformin-Mepha retard après avoir été traités avec une association de sitagliptine et de metformine peuvent continuer à prendre la même dose initiale de sitagliptine et de metformine qu'auparavant sous la forme de l'association fixe Sitagliptin-Metformin-Mepha retard.
Patients chez lesquels un traitement associant deux des trois antidiabétiques metformine, sitagliptine ou sulfonylurée ne permet pas d'atteindre un équilibre glycémique suffisant
La dose initiale usuelle de Sitagliptin-Metformin-Mepha retard doit contenir au total 100 mg de sitagliptine par jour. Pour la définition de la dose initiale de la composante de metformine, il faut tenir compte de la glycémie actuelle et de la dose de metformine utilisée jusque-là le cas échéant. La dose de metformine doit être augmentée de façon progressive pour éviter des symptômes gastro-intestinaux dus à la metformine. Chez les patients qui prennent déjà ou doivent commencer à prendre une sulfonylurée, il est recommandé d'utiliser une dose réduite de la sulfonylurée afin de réduire le risque d'hypoglycémie induite par la sulfonylurée (voir «Mises en garde et précautions»).
Patients chez lesquels un traitement associant deux des trois antidiabétiques sitagliptine, metformine ou insuline ne permet pas d'atteindre un équilibre glycémique suffisant
La dose initiale usuelle de Sitagliptin-Metformin-Mepha retard doit contenir au total 100 mg de sitagliptine par jour. Pour la définition de la dose initiale de la composante de metformine, il faut tenir compte de la glycémie actuelle et de la dose de metformine utilisée jusque-là le cas échéant. La dose de metformine doit être augmentée de façon progressive pour éviter d'éventuels symptômes gastro-intestinaux dus à la metformine. Chez les patients qui utilisent déjà ou doivent commencer à utiliser de l'insuline, il est recommandé d'utiliser une dose réduite d'insuline afin de diminuer le risque d'hypoglycémie (voir «Mises en garde et précautions»).
Aucune étude spécifique n'a été effectuée sur la sécurité et l'efficacité de chlorhydrate de sitagliptine/metformine comprimés à libération retardée chez les patients traités antérieurement par d'autres antidiabétiques oraux, qui sont ensuite passés à chlorhydrate de sitagliptine/metformine comprimés à libération retardée. Tout changement du traitement du diabète de type 2 doit être réalisé avec prudence et être soigneusement contrôlé, étant donné que des fluctuations de l'équilibre glycémique peuvent se produire.
Utilisation chez des patients souffrant d'un trouble de la fonction rénale
On évaluera la fonction rénale avant le début du traitement avec Sitagliptin-Metformin-Mepha retard et ensuite à intervalles réguliers.
Sitagliptin-Metformin-Mepha retard est contre-indiqué chez les patients dont le débit de filtration glomérulaire estimé (DFGe) est <30 ml/min/1,73 m2. Sitagliptin-Metformin-Mepha retard doit être arrêté si le DFGe du patient s'abaisse plus tard en-dessous de 30 ml/min/1,73 m2 (voir «Contre-indications» et «Mises en garde et précautions»).
Chez des patients avec un DFGe ≥30 ml/min/1,73 m2 et <45 ml/min/1,73 m2 le commencement d'un traitement par Sitagliptin-Metformin-Mepha retard n'est pas recommandé. Chez des patients qui prennent Sitagliptin-Metformin-Mepha retard et chez lesquels le DFGe s'abaisse plus tard en-dessous de 45 ml/min/1,73 m2, le bénéfice et le risque de la continuation du traitement sont à évaluer et la dose de la sitagliptine est à limiter à 50 mg une fois par jour.
Utilisation chez les enfants et les adolescents
Sitagliptin-Metformin-Mepha retard ne doit pas être utilisé chez les enfants et les adolescents âgés de 10 à 17 ans en raison d'un manque d'efficacité. Chlorhydrate de sitagliptine/metformine comprimés à libération retardée n'ont pas été étudiés chez les patients pédiatriques de moins de 10 ans (voir «Efficacité clinique»).
Utilisation chez les patients âgés
La sitagliptine et la metformine étant essentiellement éliminées par voie rénale, Sitagliptin-Metformin-Mepha retard doit être utilisé avec d'autant plus de prudence que le patient est âgé. Un contrôle de la fonction rénale est en particulier nécessaire chez les patients âgés afin de prévenir une acidose lactique due à la metformine (voir la section «Mises en garde et précautions», Chlorhydrate de metformine, Acidose lactique).
Contre-indicationsSitagliptin-Metformin-Mepha retard est contre-indiqué chez les patients présentant les particularités suivantes:
1.Trouble sévère de la fonction rénale (DFGe <30 ml/min/1,73 m2) (voir «Mises en garde et précautions», Chlorhydrate de metformine, Trouble de la fonction rénale).
2.Maladies aiguës susceptibles d'entraîner une détérioration de la fonction rénale, par exemple problèmes cardio-vasculaires (y compris infarctus du myocarde), septicémie, déshydratation (diarrhée, vomissements répétés), infections graves, par ex. des voies urinaires, forte fièvre.
3.Hypersensibilité connue à la sitagliptine, au chlorhydrate de metformine ou à un quelconque composant de Sitagliptin-Metformin-Mepha retard. (Voir les sections «Mises en garde et précautions», Réactions d'hypersensibilité, et «Effets indésirables», Expériences post-commercialisation).
4.Acidose métabolique aiguë ou chronique, y compris acidocétose diabétique avec ou sans coma.
5.L'application intravasculaire de produits de contraste contenant de l'iode pour des examens radiologiques peut entraîner une défaillance rénale et, de ce fait, causer une accumulation de metformine et une acidose lactique. Le traitement par la metformine doit être suspendu 48 h avant un tel examen si la clairance de la créatinine est <60 ml/min ou le DFGe <60 ml/min/1,73 m2. Le traitement avec la metformine ne pourra être poursuivi que si un contrôle de la fonction rénale réalisé 48 h après l'examen avec le produit de contraste ne montre pas de détérioration supplémentaire (voir «Mises en garde et précautions»).
Mises en garde et précautionsSitagliptin-Metformin-Mepha retard
Sitagliptin-Metformin-Mepha retard ne devrait pas être utilisé chez les patients atteints de diabète de type 1 ou pour le traitement de l'acidocétose diabétique.
Pancréatite: Des cas de pancréatite aiguë, y compris des cas de pancréatite hémorragique ou nécrosante avec issue fatale et non fatale (voir «Effets indésirables»), ont été observés chez des patients sous sitagliptine. Les patients doivent être informés sur les symptômes caractéristiques d'une pancréatite aiguë: douleurs abdominales sévères, persistantes. Une amélioration de la pancréatite a été observée après l'arrêt du traitement par la sitagliptine. En cas de suspicion de pancréatite, on arrêtera le traitement par Sitagliptin-Metformin-Mepha retard et par d'autres médicaments potentiellement suspects.
Contrôle de la fonction rénale: La sitagliptine et la metformine sont essentiellement éliminées par voie rénale. Avant le début d'un traitement avec Sitagliptin-Metformin-Mepha retard, la fonction rénale doit être examinée car le risque d'accumulation de la metformine et d'acidose lactique augmente avec la limitation de la fonction rénale. Sitagliptin-Metformin-Mepha retard est contre-indiqué chez les patients qui présentent un trouble grave de la fonction rénale (DFGe <30 ml/min/1,73 m2).
La fonction rénale doit être déterminée à intervalles réguliers à nouveau sous traitement par Sitagliptin-Metformin-Mepha retard, soit au minimum:
·une fois par an chez les patients dont la fonction rénale est normale;
·au moins tous les 3 à 6 mois chez les patients dont le DFGe est ≥45 - 59 ml/min/1,73 m2, ainsi que chez les patients âgés;
·au moins tous les 3 mois chez les patients dont le DFGe est ≥30 - 44 ml/min/1,73 m2.
La prise de Sitagliptin-Metformin-Mepha retard soit être arrêtée immédiatement si le DFGe diminue en-dessous de 30 ml/min/1,73 m2.
En association avec des agents susceptibles de provoquer une hypoglycémie: On sait que des antidiabétiques de la classe des sulfonylurées ou de l'insuline peuvent provoquer des hypoglycémies. Pour cette raison, il peut être nécessaire de réduire la posologie de l'insuline ou de la sulfonylurée en cas d'association avec Sitagliptin-Metformin-Mepha retard.
Sitagliptine
Myopathie/rhabdomyolyse
En rapport avec l'utilisation de chlorhydrate de sitagliptine/metformine comprimés à libération retardée des myopathies ont été signalées qui se manifestent par des myalgies, une faiblesse musculaire ou une sensibilité musculaire, accompagnées d'un taux de créatine kinase fortement augmenté (CK, jusqu'à dix fois la limite supérieure de la norme). La myopathie peut parfois se manifester sous la forme d'une rhabdomyolyse, accompagnée ou non d'une défaillance rénale aiguë suite à une myoglobinurie et des décès ont eu lieu dans de rares cas.
La prescription de Sitagliptin-Metformin-Mepha retard à des patients présentant des facteurs prédisposant à une rhabdomyolyse requiert une prudence particulière. Une détermination du taux de créatine kinase devrait être réalisée avant le début du traitement dans les situations suivantes:
·Insuffisance rénale.
·Hypothyroïdie non contrôlée.
·Antécédents personnels ou familiaux de maladies musculaires héréditaires.
·Antécédents de toxicité musculaire avec une statine ou un fibrate.
·Dépendance à l'alcool.
·Chez les personnes âgées (≥65 ans), la nécessité d'une telle mesure devrait être envisagée en présence d'autres facteurs prédisposant à une rhabdomyolyse.
·Chez les personnes de sexe féminin, la nécessité d'une telle mesure devrait être envisagée en présence d'autres facteurs prédisposant à une rhabdomyolyse.
Dans ces situations, le risque d'un traitement devrait être considéré dans l'optique du bénéfice escompté.
En association avec des agents susceptibles de provoquer une hypoglycémie: Dans les études cliniques sur l'administration de sitagliptine seule ou en association avec des principes actifs non associés à une hypoglycémie (par exemple metformine ou agonistes du PPARγ (thiazolidinediones)), l'incidence des hypoglycémies en rapport avec la prise de sitagliptine a été similaire à celle observée sous placebo.
Réactions d'hypersensibilité
Il existe des rapports post-commercialisation de réactions sérieuses d'hypersensibilité chez des patients traités à la sitagliptine (l'un des principes actifs de Sitagliptin-Metformin-Mepha retard). Ces réactions ont englobé: anaphylaxie, angio-œdème et anomalies cutanées exfoliatives, y compris syndrome de Stevens-Johnson. Ces réactions se sont produites dans les 3 premiers mois du traitement à la sitagliptine, parfois même après la première dose. Étant donné que ces rapports proviennent d'une population de taille inconnue, il est impossible de fournir des indications fiables sur la fréquence de ces événements. Lors d'une suspicion de réaction d'hypersensibilité, le traitement par Sitagliptin-Metformin-Mepha retard doit être arrêté (Voir la section «Contre-indications» et «Effets indésirables», Expériences post-commercialisation).
Pemphigoïde bulleuse
Après commercialisation, des cas de pemphigoïde bulleuse ayant nécessité une hospitalisation ont été rapportés avec l'utilisation d'inhibiteurs de la dipeptidylpeptidase-4 (DPP-4). Dans les cas rapportés, les patients ont récupéré généralement sous traitement immunosuppresseur topique ou systémique et après arrêt de la prise de l'inhibiteur de la DPP-4. Il faut signaler aux patients de contacter leur médecin en cas d'apparition de vésicules ou d'érosions cutanées sous traitement par Sitagliptin-Metformin-Mepha retard. En cas de suspicion de pemphigoïde bulleuse, le traitement par Sitagliptin-Metformin-Mepha retard devrait être arrêté. Il faudrait considérer d'adresser le patient à un dermatologue pour confirmer le diagnostic et instaurer un traitement adéquat.
Chlorhydrate de metformine
Acidose lactique: L'acidose lactique est une complication métabolique rare mais grave pouvant être provoquée par une accumulation de metformine pendant le traitement par Sitagliptin-Metformin-Mepha retard (chlorhydrate de sitagliptine/metformine à libération retardée). L'acidose lactique est mortelle dans environ 50% des cas. L'acidose lactique peut aussi se produire sous différentes conditions physiopathologiques, y compris le diabète sucré, et chaque fois qu'une hypoperfusion et une hypoxie tissulaire se développent dans une mesure significative. L'acidose lactique est caractérisée par un taux sanguin accru de lactate (>5 mmol/l), un pH sanguin abaissé, des troubles électrolytiques avec trou anionique augmenté et une valeur accrue du rapport lactate/pyruvate. Lorsque la metformine est à l'origine d'une acidose lactique, on trouve généralement des taux plasmatiques de metformine supérieurs à 5 µg/ml.
L'incidence de l'acidose lactique chez les patients traités à la metformine est très faible (environ 0,03 cas sur 1000 patient-années, avec environ 0,015 cas de décès sur 1000 patient-années). Dans des études cliniques sur plus de 20'000 patient-années d'exposition à la metformine, aucune acidose lactique n'a été rapportée. Les cas signalés se sont produits essentiellement chez des patients diabétiques avec une grave limitation de la fonction rénale ou une défaillance rénale chronique. Les patients insuffisants cardiaques qui nécessitent un traitement médicamenteux – en particulier les patients présentant une insuffisance cardiaque instable ou aiguë avec hypoperfusion et hypoxie – ont un risque accru de développer une acidose lactique. Le risque d'acidose lactique augmente avec le degré de l'insuffisance rénale et avec l'âge du patient. Ce risque peut donc significativement être abaissé par des contrôles réguliers de la fonction rénale chez les patients sous metformine, ainsi qu'en utilisant la plus faible dose efficace de metformine. La fonction rénale doit être surveillée soigneusement en particulier pendant le traitement des patients âgés (voir Utilisation chez des personnes âgées, Chlorhydrate de metformine). La metformine ne doit pas non plus être utilisée en présence de toute condition médicale associée à une hypoxie, une déshydratation ou une septicémie. Une fonction hépatique insuffisante pouvant significativement ralentir la dégradation du lactate, un traitement à la metformine doit en général être évité chez tous les patients chez lesquels le tableau clinique ou les résultats de laboratoire font suspecter une maladie hépatique.
Avant de prendre de la metformine, les patients doivent être mis en garde contre une consommation excessive aiguë ou chronique d'alcool, étant donné que l'alcool renforce les effets du chlorhydrate de metformine sur le métabolisme du lactate. En outre, le traitement à la metformine doit être suspendu transitoirement avant l'administration intravasculaire de substances de contraste et avant une intervention chirurgicale.
Le début de l'acidose lactique est souvent insidieux, avec des symptômes d'abord peu spécifiques tels que malaise, myalgies, détresse respiratoire, somnolence accrue et douleurs abdominales non spécifiques. Lors d'une acidose prononcée, on observera éventuellement une hypothermie, une hypotension et une brady-arythmie résistante. Le patient et le médecin traitant doivent être conscients de la signification éventuelle de tels symptômes et le patient doit être instruit de contacter le médecin immédiatement si de tels symptômes se manifestent. Dans un tel cas, le traitement à la metformine doit être arrêté et le patient doit être hospitalisé. Les électrolytes et cétones sériques, la glycémie et le pH sanguin peuvent être utiles pour le diagnostic, de même que les taux de lactate et de metformine.
Chez les patients sous metformine, des taux plasmatiques à jeun de lactate situés au-dessus de la normale mais au-dessous de 5 mmol/l n'évoquent pas nécessairement une acidose lactique menaçante et peuvent être expliqués par d'autres mécanismes tels que, par exemple, un diabète mal contrôlé ou un excès de poids, ou encore par un effort physique intensif ou des problèmes techniques lors de la manipulation des échantillons.
La possibilité d'une acidose lactique doit être considérée chez tout patient diabétique présentant une acidose métabolique en l'absence d'indices d'une acidocétose (cétonurie et cétonémie).
L'acidose lactique est une urgence médicale qui doit être traitée à l'hôpital. Chez un patient sous metformine qui présente une acidose lactique, le médicament doit être arrêté immédiatement et les mesures de soutien usuelles doivent être prises. Étant donné que le chlorhydrate de metformine et le lactate sont dialysables (avec une clairance allant jusqu'à 170 ml/min dans de bonnes conditions hémodynamiques), une hémodialyse immédiate est recommandée. Un tel traitement permet souvent une régression rapide des symptômes et un net rétablissement du patient (voir la section «Contre-indications»).
Une fois qu'un patient est stabilisé sous metformine, les symptômes gastro-intestinaux dus à la metformine – fréquents au début du traitement – sont très rares. Si des symptômes gastro-intestinaux apparaissent encore ultérieurement, ils peuvent être dus à une acidose lactique ou à une autre maladie sérieuse.
Hypoglycémie: Chez les patients traités à la metformine seule, aucune hypoglycémie n'est observée dans le cadre d'une utilisation normale. Dans des cas exceptionnels, une hypoglycémie peut se développer en association avec la metformine, par ex. en cas d'apport calorique insuffisant ou de dépense énergétique fortement accrue (grand effort physique), ainsi qu'en cas de consommation concomitante d'alcool ou d'autres substances hypoglycémiantes (par exemple sulfonylurées et insuline).
Les patients âgés, faibles ou présentant une malnutrition ou souffrant d'une insuffisance surrénalienne ou hypophysaire ou d'une intoxication alcoolique sont particulièrement prédisposés à subir des effets hypoglycémiques. L'hypoglycémie est parfois difficile à reconnaître chez les personnes âgées et chez les personnes prenant des antagonistes des récepteurs bêta-adrénergiques.
Prise concomitante d'autres médicaments pouvant influencer la fonction rénale ou la disponibilité de la metformine: Les médications concomitantes susceptibles d'influencer la fonction rénale ou de modifier significativement l'hémodynamique ou la disponibilité de la metformine – par exemple médicaments cationiques éliminés par sécrétion tubulaire dans le rein (voir la section «Interactions», Chlorhydrate de metformine) – doivent être utilisées avec prudence.
Utilisation de produits de contraste iodés: Les produits de contraste iodés administrés par voie intravasculaire pour des examens radiologiques peuvent entraîner une défaillance rénale. Comme cela peut mener à une accumulation de metformine et à une acidose lactique, le traitement par la metformine chez des patients dont le DFGe est <60 ml/min/1.73 m2 doit donc être suspendu à temps (environ 2 jours) et ne doit être repris que si la fonction rénale ne s'est pas détériorée 2 jours après l'examen avec le produit de contraste.
Hypoxie: Des défaillances cardio-vasculaires d'origines variées, insuffisances cardiaques aiguës, infarctus myocardiques aigus et autres états associés à une hypoxie ont été mis en rapport avec le développement d'une acidose lactique. Ces états peuvent aussi provoquer une azotémie prérénale. Si de tels événements se produisent pendant le traitement par Sitagliptin-Metformin-Mepha retard, la prise du médicament doit immédiatement être arrêtée.
Opérations: La prise de Sitagliptin-Metformin-Mepha retard doit transitoirement être suspendue pour les interventions chirurgicales (à l'exception d'interventions mineures permettant un apport de nourriture et de liquides sans restrictions) et n'être reprise qu'une fois qu'une absorption normale de nourriture est à nouveau possible et que la fonction rénale a été jugée suffisante (voir «Posologie/Mode d'emploi»).
Consommation d'alcool: On sait que l'alcool renforce les effets de la metformine sur le métabolisme du lactate. Les patients qui prennent Sitagliptin-Metformin-Mepha retard doivent donc être mis en garde contre une consommation excessive – chronique ou aiguë – d'alcool.
Insuffisance hépatique: L'insuffisance hépatique ayant été mise en rapport avec certains cas d'acidose lactique, Sitagliptin-Metformin-Mepha retard ne doit pas être utilisé chez les patients chez lesquels le tableau clinique ou les résultats de laboratoire font soupçonner une maladie hépatique.
Taux sanguin de vitamine B12: Au cours d'un traitement à long terme par la metformine, le taux sanguin de vitamine B12 peut diminuer (voir «Effets indésirables»). Le risque d'une carence en vitamine B12 augmente avec la dose de metformine et la durée du traitement. Il est recommandé de contrôler le taux sérique de vitamine B12 à intervalles réguliers (par ex. chaque année), en particulier chez les patients souffrant d'anémie ou de neuropathie périphérique.
Modifications de l'état clinique des patients dont le diabète de type 2 était antérieurement sous contrôle: Un patient dont le diabète de type 2 était antérieurement bien contrôlé sous Sitagliptin-Metformin-Mepha retard mais qui présente soudainement des valeurs de laboratoire anormales ou devient cliniquement malade (souvent de façon vague et mal définie) doit immédiatement être examiné pour identifier ou exclure une éventuelle acidocétose ou acidose lactique.
L'examen doit englober les électrolytes sériques, les cétones sériques et la glycémie ainsi que, si indiqué, le pH sanguin et les taux de lactate, de pyruvate et de metformine. En cas de suspicion d'acidose métabolique – quelle qu'en soit la genèse – la prise de Sitagliptin-Metformin-Mepha retard doit être immédiatement arrêtée et le patient doit être hospitalisé.
Perte de contrôle de la glycémie: Chez les patients qui maintiennent un bon équilibre glycémique sous un quelconque antidiabétique, il est possible d'observer un dérèglement transitoire de l'équilibre glycémique dans des situations de stress telles que fièvre, traumatisme, infection ou opération.
Dans de tels cas, il sera éventuellement nécessaire de suspendre la prise de Sitagliptin-Metformin-Mepha retard et d'administrer transitoirement de l'insuline à la place. Le traitement par Sitagliptin-Metformin-Mepha retard pourra ensuite être poursuivi après la fin de l'épisode aigu.
Utilisation chez les personnes âgées
Sitagliptin-Metformin-Mepha retard
La sitagliptine et la metformine sont essentiellement éliminées par voie rénale. Sitagliptin-Metformin-Mepha retard doit par conséquent être utilisé avec d'autant plus de prudence que le patient est âgé. Un contrôle de la fonction rénale est en particulier nécessaire chez les patients âgés afin de prévenir une acidose lactique due à la metformine (voir la section «Mises en garde et précautions», Chlorhydrate de metformine, Acidose lactique).
Sitagliptine
Dans les études cliniques, la sécurité et l'efficacité de la sitagliptine ont été similaires chez les patients âgés (≥65 ans) et les patients plus jeunes (<65 ans).
Chlorhydrate de metformine
Les études cliniques contrôlées sur la metformine n'ont pas inclus suffisamment de patients âgés pour permettre de trancher définitivement la question de savoir si ces patients réagissent autrement que les patients plus jeunes. Toutefois, d'autres expériences cliniques n'ont révélé aucune différence entre patients âgés et patients plus jeunes.
Sitagliptin-Metformin-Mepha retard doit en principe être pris avec le repas du soir. Une prise avec le repas permet de réduire en partie les effets indésirables gastro-intestinaux de la metformine. Il faut aussi compter que la biodisponibilité de la metformine à libération retardée est réduite dans le cas d'une prise à jeun, ce qui pourrait rendre le contrôle du diabète plus difficile. Il faut savoir que la biodisponibilité de la metformine à libération non retardée, à contratio de celle de la metformine à libération retardée, est réduite lors d'une prise avec de la nourriture.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par comprimé à libération modifiée, c.àd. qu'il est essentiellement «sans sodium».
InteractionsSitagliptine et metformine
Chez les patients diabétiques de type 2, un traitement associé avec des doses multiples de sitagliptine (50 mg 2x par jour) et de metformine (1000 mg 2x par jour) n'a influencé significativement ni la pharmacocinétique de la sitagliptine ni celle de la metformine.
Aucune étude d'interaction pharmacocinétique n'a été effectuée sur chlorhydrate de sitagliptine/metformine comprimés à libération retardée. Toutefois, de telles études ont été réalisées sur les principes actifs individuels de Sitagliptin-Metformin-Mepha retard, c'est-à-dire sur la sitagliptine et sur la metformine.
Sitagliptine
Des études d'interaction pharmacologique avec la sitagliptine n'ont montré aucune influence cliniquement significative sur la pharmacocinétique des médicaments suivants: metformine, rosiglitazone, glibenclamide, simvastatine, warfarine et contraceptifs oraux. Ces données laissent supposer que la sitagliptine n'inhibe pas les isoenzymes CYP3A4, 2C8 et 2C9. Des données obtenues in vitro laissent également supposer que la sitagliptine n'inhibe pas les CYP2D6, 1A2, 2C19 ou 2B6 et n'induit pas le CYP3A4.
Des analyses pharmacocinétiques de population auprès de patients diabétiques de type 2 n'ont constaté aucune influence significative de la médication concomitante sur la pharmacocinétique de la sitagliptine. Ces analyses se sont penchées sur les médicaments usuels utilisés chez les patients diabétiques de type 2.
L'administration simultanée de sitagliptine et de digoxine a provoqué une légère augmentation de l'aire sous la courbe (AUC, 11%) ainsi que de la concentration plasmatique maximale moyenne (Cmax, 18%) de la digoxine. Ces modifications ne sont pas cliniquement significatives. Les patients prenant de la digoxine devraient être surveillés en conséquence.
L'administration simultanée d'une dose orale unique de 100 mg de sitagliptine et d'une dose unique orale de 600 mg de ciclosporine, un inhibiteur puissant de la glycoprotéine-p, a provoqué une augmentation de l'AUC de la sitagliptine de près de 29% ainsi qu'une hausse de la Cmax de la sitagliptine de 68%. Les modifications observées au niveau de la pharmacocinétique de la sitagliptine ne sont pas considérées comme cliniquement significatives.
Chlorhydrate de metformine
Glibenclamide: Dans une étude d'interaction avec administration unique à des patients diabétiques de type 2, la prise simultanée de metformine et de glibenclamide n'a modifié ni la pharmacocinétique ni la pharmacodynamie de la metformine.
Des valeurs réduites de l'AUC et de la Cmax du glibenclamide ont été observées mais de façon fortement variable. Vu l'administration unique des médicaments dans cette étude et vu l'absence d'une corrélation entre le taux sanguin de glibenclamide et les effets pharmacodynamiques, il est impossible de se prononcer clairement sur la signification clinique de cette interaction.
Furosémide: Une étude d'interaction avec administration unique de metformine et de furosémide chez des volontaires sains a montré que les paramètres pharmacocinétiques des deux principes actifs ont été influencés. Le furosémide a fait augmenter la Cmax de la metformine de 22% dans le plasma et le sang et l'AUC de la metformine de 15% dans le sang, sans exercer une influence significative sur la clairance rénale de la metformine. Lors d'une administration concomitante de metformine, la Cmax et l'AUC du furosémide ont été réduites respectivement de 31% et de 12% par rapport à la monothérapie. La demi-vie terminale a été raccourcie de 32% sans influence significative sur la clairance rénale du furosémide. On ne dispose pas de données sur l'interaction entre metformine et furosémide dans le cadre d'un traitement prolongé.
Nifédipine: Une étude d'interaction avec administration unique de metformine et de nifédipine chez des volontaires sains a montré que l'administration concomitante de nifédipine faisait augmenter la Cmax et l'AUC de la metformine respectivement de 20% et de 9% dans le plasma et que la metformine était davantage éliminée dans l'urine.
Les valeurs de Tmax et de demi-vie n'ont pas été influencées. La nifédipine semble accélérer l'absorption de la metformine. La metformine n'exerce guère d'influence sur la nifédipine.
L'administration concomitante de médicaments qui interfèrent avec les systèmes de transport tubulaires rénaux impliqués dans l'élimination rénale de la metformine (par ex. le transporteur de cations organiques 2 [OCT2] / les inhibiteurs des transporteurs d'extrusion de multiples médicaments et toxines [MATE] tels que la ranolazine, le vandétanib, le dolutégravir et la cimétidine), pourrait augmenter l'exposition systémique de la metformine et, par conséquent, le risque d'une acidose lactique. Dans des études d'interaction avec administration unique et répétée de metformine en association avec la cimétidine à des sujets sains, les concentrations maximales de metformine dans le plasma et le sang complet ont augmenté de 60% et l'AUC plasmatique et l'AUC pour le sang complet de la metformine ont augmenté de 40%. Le bénéfice et les risques de l'utilisation concomitante de ces substances avec la metformine doivent donc être soigneusement évalués.
Autres: Certains médicaments peuvent déclencher une hyperglycémie et perturber ainsi l'équilibre glycémique. Ces médicaments englobent par exemple les diurétiques thiazidiques et autres, les corticostéroïdes, les phénothiazines, les médicaments pour la glande thyroïde, les œstrogènes, les contraceptifs oraux, la phénytoïne, la niacine, les sympathicomimétiques, les bloqueurs des canaux calciques et l'isoniazide. Si l'un de ces médicaments est administré à un patient traité par Sitagliptin-Metformin-Mepha retard, ce patient doit être surveillé de près pour assurer un équilibre glycémique adéquat.
Dans des études d'interaction avec administration unique chez des volontaires sains, la pharmacocinétique de la metformine et du propranolol ainsi que la pharmacocinétique de la metformine et de l'ibuprofène n'ont pas été influencées lors d'une administration des associations correspondantes.
La metformine ne se lie presque pas aux protéines plasmatiques et n'interagit donc guère – contrairement aux sulfonylurées, qui se lient fortement aux protéines sériques – avec les médicaments fortement liés aux protéines, par exemple les salicylates, les sulfamides, le chloramphénicol et le probénécide.
Grossesse, allaitementEmploi pendant la grossesse
Aucune étude adéquate et sévèrement contrôlée n'a été réalisée chez les femmes enceintes sur chlorhydrate de sitagliptine/metformine comprimés à libération retardée ou ses composants individuels. C'est pourquoi la sécurité de Sitagliptin-Metformin-Mepha retard chez les femmes enceintes n'est pas connue.
La prise de Sitagliptin-Metformin-Mepha retard, ainsi que celle d'autres antidiabétiques oraux, n'est pas recommandée durant la grossesse.
Emploi en période d'allaitement
Aucune étude n'a été effectuée sur des animaux en lactation avec les principes actifs associés de Sitagliptin-Metformin-Mepha retard. Dans les études sur les principes actifs individuels, aussi bien la sitagliptine que la metformine ont été excrétées dans le lait des rates en lactation. On ignore si la sitagliptine passe également dans le lait maternel humain. C'est pourquoi Sitagliptin-Metformin-Mepha retard ne devrait pas être utilisé en période d'allaitement.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude concernant les effets de chlorhydrate de sitagliptine/metformine comprimés à libération retardée sur l'aptitude à conduire et à utiliser des machines n'a été menée. On ne s'attend toutefois pas à ce que Sitagliptin-Metformin-Mepha retard influence les aptitudes à la conduite et l'utilisation de machines.
Effets indésirablesLa bioéquivalence entre chlorhydrate de sitagliptine/metformine comprimés à libération retardée et une co-administration de sitagliptine et de metformine à libération retardée a été démontrée (voir «Pharmacocinétique»).
Dans les études cliniques contrôlées par placebo, le traitement associé à la sitagliptine et à la metformine à libération non retardée a généralement été bien toléré par les patients souffrant d'un diabète de type 2. L'incidence globale des effets indésirables chez les patients sous traitement associé à la sitagliptine et à la metformine a été similaire à celle observée sous placebo et metformine.
Les incidences des effets indésirables sont définies comme suit: fréquents ≥1/100, <1/10; occasionnels ≥1/1000, <1/100; rares ≥1/10000, <1/1000; très rares <1/10000.
Traitement d'association à la sitagliptine et à la metformine
Une étude de 24 semaines avec contrôle par placebo, effectuée auprès de patients chez lesquels un régime alimentaire et un exercice physique régulier n'avaient pas permis d'obtenir un équilibre glycémique suffisant, a examiné le traitement par la sitagliptine 50 mg 2x par jour en association avec la metformine 500 ou 1000 mg 2x par jour. Les effets indésirables en rapport avec le traitement associé qui se sont produits chez ≥1% des patients (et ont été plus fréquents que dans le groupe recevant le placebo) ont été les suivants:
Troubles du métabolisme et de la nutrition:
Fréquents: hypoglycémie.
Troubles du système nerveux
Fréquents: céphalées.
Troubles gastro-intestinaux
Fréquents: diarrhées, nausées, dyspepsie, ballonnements, vomissements.
Dans une étude de 24 semaines avec contrôle par placebo, examinant la sitagliptine administrée en plus d'un traitement déjà commencé à la metformine, 464 patients traités à la metformine ont reçu 100 mg de sitagliptine une fois par jour, tandis que 237 autres patients traités à la metformine ont reçu un placebo.
Les effets indésirables en rapport avec le traitement associé qui se sont produits chez ≥1% des patients (et ont été plus fréquents que dans le groupe recevant le placebo) ont été les suivants:
Troubles gastro-intestinaux
Fréquents: nausées.
Hypoglycémie et troubles gastro-intestinaux
Dans les études contrôlées par placebo sur la sitagliptine et la metformine en tant que traitement associé, l'incidence des hypoglycémies (indépendamment de l'évaluation de la causalité par le médecin investigateur) dans le cadre d'une prise de sitagliptine en association avec de la metformine a été comparable à celle sous metformine plus placebo.
L'incidence des troubles gastro-intestinaux chez les patients traités par l'association sitagliptine-metformine a été comparable à celle chez les patients sous metformine seule (voir le Tableau 1).
Tableau 1:
Hypoglycémie et troubles gastro-intestinaux (indépendamment de l'évaluation de la causalité par le médecin investigateur) chez les patients sous traitement associé
|
|
Nombre de patients (%)
| |
Étude sur la sitagliptine et la metformine chez des patients dont l'équilibre glycémique était insuffisant avec un régime alimentaire et un exercice physique régulier comme seuls traitements
|
Étude sur la sitagliptine en tant que traitement complémentaire en plus de la metformine
| |
Placebo
|
Sitagliptine
100 mg 1x par jour
|
Metformine
500 ou
1000 mg 2x par jour ††
|
Sitagliptine
50 mg 2x par jour +
metformine
500 ou
1000 mg 2x par jour ††
|
Placebo et metformine
≥1500 mg par jour
|
Sitagliptine 100 mg 1x par jour et metformine
≥1500 mg par jour
| |
N= 176
|
N= 179
|
N= 364
|
N= 372
|
N= 237
|
N= 464
| |
Hypoglycémie
|
1 (0.6)
|
1 (0.6)
|
3 (0.8)
|
6 (1.6)
|
5 (2.1)
|
6 (1.3)
| |
Diarrhées
|
7 (4.0)
|
5 (2.8)
|
28 (7.7)
|
28 (7.5)
|
6 (2.5)
|
11 (2.4)
| |
Nausées
|
2 (1.1)
|
2 (1.1)
|
20 (5.5)
|
18 (4.8)
|
2 (0.8)
|
6 (1.3)
| |
Vomissements
|
1 (0.6)
|
0 (0.0)
|
2 (0.5)
|
8 (2.1)
|
2 (0.8)
|
5 (1.1)
| |
Douleurs abdominales†
|
4 (2.3)
|
6 (3.4)
|
14 (3.8)
|
11(3.0)
|
9 (3.8)
|
10 (2.2)
|
† Dans l'étude auprès de patients insuffisamment contrôlés par un régime alimentaire et un exercice physique régulier, les symptômes abdominaux ont également été comptés en tant que douleurs abdominales.
†† Données groupées pour les patients ayant reçu de plus faibles / de plus fortes doses de metformine.
Dans toutes les études, tous les effets indésirables considérés comme des épisodes d'hypoglycémie – avec ou sans confirmation par un contrôle de la glycémie – ont été pris en compte.
Sitagliptine en association avec la metformine et une sulfonylurée
Dans une étude de 24 semaines, effectuée avec contrôle par placebo, les participants ont reçu une dose quotidienne de 100 mg de sitagliptine en plus d'un traitement déjà en cours avec des doses quotidiennes de ≥4 mg de glimépiride et de ≥1500 mg de metformine. Les effets indésirables médicamenteux suivants ont été observés chez ≥1% des patients sous sitagliptine (n= 116) et plus fréquemment que chez les patients sous placebo (n= 113):
Troubles du métabolisme et de la nutrition
Fréquents: hypoglycémie.
Troubles gastro-intestinaux
Fréquents: constipation.
Aucune altération cliniquement significative des fonctions vitales ou de l'ECG (y compris intervalle QT) n'a été observée pendant le traitement à la sitagliptine associée à la metformine.
Sitagliptine en association avec la metformine et l'insuline
Dans une étude de 24 semaines avec contrôle par placebo, examinant la sitagliptine 100 mg ajoutée à un traitement en cours associant la metformine ≥1500 mg par jour et une dose stable d'insuline, l'hypoglycémie a été la seule réaction indésirable observée sous sitagliptine chez ≥1% des patients (n= 229) et plus souvent que sous placebo (n= 233) (sitagliptine 15,3%, placebo 8,2%). Ces incidences correspondent à tous les effets indésirables indépendamment de la présence ou non d'un rapport de causalité selon l'appréciation du médecin investigateur. En particulier pour les taux d'hypoglycémie, il faut considérer que tous les patients étaient traités non seulement avec la sitagliptine ou avec le placebo, mais aussi avec l'insuline et dans certains cas également avec la metformine. Dans une autre étude de 24 semaines chez des patients qui, pendant une intensification de l'insuline (avec ou sans metformine) ont reçu de la sitagliptine en traitement complémentaire, les effets indésirables qui ont été observés chez ≥1% des patients traités par sitagliptine et metformine (N= 285) et qui étaient plus fréquents que chez les patients sous placebo et metformine (N= 283), étaient: constipation (sitagliptine et metformine, 1,4%; placebo et metformine, 1,1%), diarrhée (4,9%; 2,5%), vomissements (3,2%; 1,1%), œdème périphérique (2,1%; 1,4%), fièvre (1,1%; 0,4%), bronchite (1,4%; 1,1%), cellulite (1,4%; 1,1%), pharyngite (1,8%; 1,1%), infection des voies respiratoires supérieures (4,2%; 1,4%), diminution de la clairance de la créatinine (1,1%; 0,0%), douleurs musculo-squelettiques (1,4%; 1,1%), myalgie (1,1%; 0,7%), néphrolithiase (1,1%; 0,4%) et toux (2,8%; 1,8%). Ces incidences correspondent à tous les effets indésirables indépendamment de la présence ou non d'un rapport de causalité selon l'appréciation des médecins investigateurs. De plus, l'incidence d'hypoglycémie était de 24,9% chez les patients traités par sitagliptine, metformine et insuline et de 37,8% chez les patients sous placebo, metformine et insuline.
Effets indésirables connus sous sitagliptine
Chez les patients prenant de la sitagliptine, aucun effet indésirable médicamenteux n'a été observé à une incidence ≥1%.
Effets indésirables connus sous metformine
Troubles de la circulation sanguine et lymphatique
Fréquents: carence en vitamine B12. Chez les patients souffrant d'anémie mégaloblastique, il est recommandé de prendre une telle étiologie en considération (voir «Mises en garde et précautions»).
Cas isolés de leucopénie, de thrombopénie et d'anémie hémolytique.
Troubles du métabolisme et de la nutrition
Très rares: acidose lactique (incidence: 3 cas/100'000 patient-années, cf. la section «Mises en garde et précautions»).
Troubles du système nerveux
Fréquents: goût métallique dans la bouche (3%).
Troubles gastro-intestinaux
Fréquents: troubles gastro-intestinaux tels que nausées, vomissements, diarrhées, douleurs abdominales, perte d'appétit.
Ces symptômes se manifestent généralement au début du traitement et régressent en général spontanément par la suite.
Troubles hépato-biliaires
Dans des cas isolés: valeurs anormales des tests hépatiques fonctionnels, par exemple taux accrus de transaminases ou hépatite (réversible après l'arrêt d'administration de la metformine).
Troubles cutanés et des tissus sous-cutanés
Très rares: réactions cutanées telles qu'érythème, prurit, urticaire.
Étude de sécurité cardiovasculaire TECOS: L'étude TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) incluait une population en intention de traiter dont 7332 patients traités par 100 mg de sitagliptine par jour (ou 50 mg par jour si le débit de filtration glomérulaire estimé [DFGe] se situait entre ≥30 et <50 ml/min/1,73 m2 au début de l'étude) et 7339 patients traités par placebo. Les deux traitements étaient ajoutés au traitement conventionnel. La population étudiée comprenait au total 2004 patients âgés de ≥75 ans (970 traités par la sitagliptine et 1034 par placebo). La fréquence des événements indésirables sévères était généralement comparable chez les patients sous sitagliptine et ceux sous placebo. Une évaluation des complications liées au diabète prédéfinies a révélé des incidences semblables entre les groupes, y compris au niveau des infections (18,4% des patients traités par la sitagliptine et 17,7% des patients traités par placebo) et de l'insuffisance rénale (1,4% des patients traités par la sitagliptine et 1,5% des patients traités par placebo). Le profil des événements indésirables chez les patients âgés de ≥75 ans était généralement similaire à celui de la population totale.
Chez les patients recevant de l'insuline et/ou une sulfonylurée au début de l'étude, l'incidence d'une hypoglycémie sévère était de 2,7% chez les patients traités par la sitagliptine et de 2,5% chez les patients traités par placebo. Chez les patients n' utilisant pas d'insuline et/ou pas de sulfonylurée au début de l'étude, l'incidence d'une hypoglycémie sévère était de 1,0% chez les patients traités par la sitagliptine et de 0,7% chez les patients traités par placebo. L'incidence des pancréatites confirmées par un comité de vérification était de 0,3% chez les patients traités par la sitagliptine et de 0,2% chez les patients traités par placebo. L'incidence des tumeurs malignes confirmées par un comité de vérification était de 3,7% chez les patients traités par la sitagliptine et de 4,0% chez ceux traités par placebo.
Pancréatite: Dans une analyse cumulée de 19 études en double aveugle totalisant 10'246 patients randomisés sous sitagliptine 100 mg par jour (n = 5429) ou sous un traitement de contrôle correspondant (agent actif ou placebo) (n = 4817), l'incidence de pancréatite aiguë a été de 0,1 sur 100 patients-années dans tous les groupes (4 patients avec 1 événement sur 4708 patients-années sous sitagliptine, 4 patients avec 1 événement sur 3942 patients-années sous traitement de contrôle) (voir aussi Étude de sécurité cardiovasculaire TECOS et «Mises en garde et précautions», Pancréatite).
Expériences post-commercialisation
Des réactions indésirables additionnelles ont été signalées après la commercialisation de chlorhydrate de sitagliptine/metformine comprimés à libération retardée ou de la sitagliptine, l'un des composants de Sitagliptin-Metformin-Mepha retard. Ces effets indésirables ont été rapportés lors de l'utilisation de chlorhydrate de sitagliptine/metformine comprimés à libération retardée ou de sitagliptine, seuls et/ou en association avec d'autres substances anti-hyperglycémiantes. Vu que ces données reposent sur des rapports spontanés issus d'une population de taille inconnue, il est impossible d'en déterminer l'incidence ou la causalité: réactions d'hypersensibilité, y compris anaphylaxie, angio-œdème, éruption cutanée, urticaire, vasculite cutanée et anomalies cutanées exfoliatives, y compris syndrome de Stevens-Johnson (voir les sections «Contre-indications» et «Mises en garde et précautions», Réactions d'hypersensibilité); pancréatite aiguë, y compris pancréatite hémorragique avec issue fatale et non fatale et pancréatite nécrosante (voir aussi «Mises en garde et précautions»); détérioration de la fonction rénale, y compris insuffisance rénale aiguë (dans certains cas, une dialyse s'avère nécessaire); rhabdomyolyse; pemphigoïde bulleuse (voir «Mises en garde et précautions»); infections des voies respiratoires supérieures, nasopharyngite; constipation, vomissements; céphalées; arthralgie, myalgie, douleur des extrémités, douleur dorsale; prurit; lithiase biliaire et cholécystite.
Investigations
Sitagliptine
L'incidence des anomalies de paramètres de laboratoire a été similaire entre le groupe sous sitagliptine et metformine et le groupe témoin sous placebo et metformine.
Plusieurs études cliniques ont constaté une légère augmentation du taux de leucocytes (environ 200 cellules par µl par rapport au placebo; valeur initiale moyenne de leucocytes: environ 6600 cellules par µl), en raison d'une légère augmentation du nombre de neutrophiles. Cette observation a été faite dans la plupart des études, mais non dans toutes. Ces anomalies des paramètres de laboratoire ont été jugées sans importance clinique.
Chlorhydrate de metformine
Au cours d'un traitement à long terme par la metformine, le taux sanguin de vitamine B12 peut diminuer (voir «Effets indésirables»). Le risque d'une carence en vitamine B12 augmente avec la dose de metformine et la durée du traitement. Il est recommandé de contrôler le taux sérique de vitamine B12 à intervalles réguliers (par ex. chaque année), en particulier chez les patients souffrant d'anémie ou de neuropathie périphérique.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageSitagliptine
Dans les études cliniques contrôlées auprès de volontaires sains, des doses uniques de sitagliptine atteignant jusqu'à 800 mg ont généralement été bien tolérées.
Pour une dose de 800 mg de sitagliptine, une étude a observé des sus-décalages minimaux de l'intervalle QT, cependant jugés comme sans signification clinique.
Dans les études cliniques il n'existe pas de données sur les doses supérieures à 800 mg.
Dans des études de phase I avec administrations multiples de sitagliptine à des doses allant jusqu'à 600 mg par jour pendant ≤10 jours et à des doses allant jusqu'à 400 mg par jour pendant des périodes couvrant jusqu'à 28 jours, aucun effet indésirable dépendant de la dose n'a été observé cliniquement.
Dans le cas d'un surdosage, il convient de prendre les mesures de soutien habituelles, par exemple l'élimination des substances actives encore non absorbées dans le tractus gastro-intestinal, la surveillance clinique (y compris par ECG) et, si nécessaire, l'instauration d'un traitement de soutien.
La sitagliptine n'est que modérément dialysable. Au cours d'études cliniques, près de 13,5% de la dose a été éliminée durant une hémodialyse de trois à quatre heures. Une hémodialyse plus longue peut être envisagée si elle est cliniquement utile. On ignore si la sitagliptine peut être dialysée par dialyse péritonéale.
Chlorhydrate de metformine
Des surdosages de metformine se sont déjà produits, même à des doses de plus de 50 grammes. Une hypoglycémie a été observée dans environ 10% des cas, cependant sans qu'un rapport causal avec la metformine ait pu être démontré. Dans environ 32% des cas de surdosage à la metformine, une acidose lactique a été constatée (voir la section «Mises en garde et précautions», Chlorhydrate de metformine). La metformine étant dialysable (avec une clairance allant jusqu'à 170 ml/min dans de bonnes conditions hémodynamiques), une hémodialyse est recommandée pour éliminer la metformine accumulée.
Propriétés/EffetsCode ATC
A10BD07
Mécanisme d'action
La sitagliptine appartient à une classe d'antidiabétiques oraux, les inhibiteurs de la dipeptidylpeptidase-4 (DPP-4), qui améliorent le contrôle de la glycémie chez les patients atteints de diabète de type 2 en augmentant les concentrations des incrétines actives. Les incrétines, y compris le GLP-1 (Glucagon-like Peptide-1) et le GIP (Glucose-dependent insulinotropic Peptide), sont libérées toute la journée à partir des intestins. Leur concentration augmente lors de la prise de nourriture. Les incrétines font partie d'un système endogène qui intervient dans la régulation physiologique de l'homéostasie du glucose. Lorsque la glycémie est normale ou élevée, le GLP-1 et le GIP favorisent la synthèse d'insuline et la sécrétion d'insuline à partir des cellules bêta du pancréas par des signaux intracellulaires impliquant l'AMP cyclique. Le traitement du diabète de type 2 au GLP-1 ou aux inhibiteurs de la DPP-4 dans des modèles animaux a montré que la sensibilité des cellules bêta au glucose est améliorée et que la biosynthèse ainsi que la sécrétion d'insuline sont stimulées. En présence de concentrations plus élevées d'insuline, l'absorption tissulaire de glucose augmente. Parallèlement, le GLP-1 diminue la sécrétion de glucagon à partir des cellules alpha du pancréas. De faibles concentrations de glucagon accompagnées de concentrations d'insuline élevées diminuent la production de glucose dans le foie, ce qui entraîne à son tour une baisse de la glycémie. Les effets du GLP-1 et du GIP sont dépendants du glucose. Lors d'une glycémie basse, le GLP-1 ne stimule ni la sécrétion d'insuline ni la suppression de la sécrétion de glucagon. Grâce au GLP-1 et au GIP, la sécrétion d'insuline est augmentée lorsque la glycémie dépasse les valeurs normales. Le GLP-1 n'affecte pas la réaction normale du glucagon lors d'hypoglycémie. L'activité du GLP-1 et du GIP est régulée par l'enzyme DPP-4, qui hydrolyse rapidement les incrétines et les transforme en substances inactives. La sitagliptine empêche l'hydrolyse des incrétines par la DPP-4, ce qui augmente les concentrations plasmatiques des formes actives du GLP-1 et du GIP. Suite à l'augmentation de la concentration d'incrétines actives, la sitagliptine provoque une hausse de la sécrétion d'insuline et une baisse de la concentration de glucagon, en fonction de la concentration de glucose. Chez les patients atteints de diabète de type 2 et présentant une hyperglycémie, ces modifications de la concentration d'insuline et de glucagon entraînent une baisse de la concentration de l'hémoglobine A1c (HbA1c) ainsi qu'une diminution des glycémies postprandiales et à jeun. Ce mécanisme dépendant du glucose se distingue de celui des sulfonylurées. Celles-ci provoquent une sécrétion d'insuline même en présence d'une glycémie basse, ce qui peut provoquer une hypoglycémie chez les patients diabétiques de type 2 et les sujets sains. Alors que la sitagliptine est un inhibiteur puissant et très sélectif de l'enzyme DPP-4, ses concentrations thérapeutiques n'inhibent pas les enzymes apparentées DPP-8 et DPP-9, très similaires.
Chlorhydrate de metformine
La metformine est un antidiabétique qui améliore la tolérance au glucose chez les patients souffrant de diabète de type 2 et réduit aussi bien la glycémie basale que la glycémie postprandiale. Les mécanismes d'action pharmacologiques de la metformine se distinguent de ceux des autres classes d'antidiabétiques oraux. La metformine réduit aussi bien la synthèse hépatique de glucose que l'absorption intestinale de glucose; elle améliore la sensibilité à l'insuline en augmentant l'assimilation périphérique et l'utilisation du glucose. Contrairement aux sulfonylurées, la metformine ne provoque pas d'hypoglycémie chez les patients diabétiques de type 2 ou chez les sujets sains, sauf dans des cas très particuliers (voir la section «Mises en garde et précautions», Chlorhydrate de metformine). Elle ne provoque pas non plus d'hyperinsulinémie. Pendant un traitement à la metformine, la sécrétion d'insuline reste inchangée tandis que les taux d'insuline à jeun et la réponse insulinique dans le plasma au cours de la journée peuvent même baisser.
Pharmacodynamique
Non pertinent.
Efficacité clinique
Études cliniques
Aucune étude clinique n'a été effectuée sur l'efficacité et la sécurité de chlorhydrate de sitagliptine/metformine comprimés à libération retardée. Des études cliniques avec administration concomitante de sitagliptine et de metformine à libération non retardée ont constaté une amélioration significative du contrôle de l'équilibre glycémique chez les patients adultes souffrant d'un diabète de type 2. La bioéquivalence entre chlorhydrate de sitagliptine/metformine comprimés à libération retardée et un traitement consistant en une administration concomitante de sitagliptine et de comprimés de metformine à libération retardée a été démontrée pour toutes les puissances de dosage.
Dans une étude comparative, l'administration une fois par jour de metformine à libération retardée a permis pour tous les paramètres glycémiques évalués une efficacité comparable à celle de l'administration usuellement prescrite de metformine à libération non retardée deux fois par jour.
Sitagliptine et metformine à libération non retardée chez les patients diabétiques de type 2
Une étude en double aveugle de 24 semaines, randomisée, avec contrôle par placebo, a examiné la sécurité et l'efficacité de l'association de sitagliptine et de metformine chez 1091 patients diabétiques de type 2 qui présentaient un équilibre glycémique insuffisant malgré un régime alimentaire et un exercice physique régulier.
Chez les patients ayant un contrôle glycémique insuffisant sous régime alimentaire plus exercice physique, l'association sitagliptine-metformine a permis des améliorations significatives en ce qui concerne le taux d'HbA1c, le taux plasmatique de glucose à jeun (FPG) et la valeur postprandiale à 2 h du taux plasmatique de glucose (PPG) par rapport au placebo, à la metformine seule et à la sitagliptine seule (p <0,001; Tableau 2, Figure 1). Une amélioration avec réduction presque maximale du FPG a été atteinte au premier bilan à 3 semaines (correspondant au premier bilan après le début du traitement) et a pu être maintenue pendant toute la durée de cette étude de 24 semaines. En comparaison avec le groupe sous placebo, les patients qui avaient eu des valeurs initiales plus élevées d'HbA1c ont bénéficié d'une réduction moyenne plus importante du taux d'HbA1c.
L'amélioration du taux d'HbA1c n'a pas été influencée par le sexe, l'âge, la race ou la valeur initiale du BMI. Les examens de la fonction des cellules bêta, HOMA-β, de même que le rapport pro-insuline/insuline, ont également montré une amélioration sous l'association sitagliptine-metformine en comparaison avec chacune des substances seules. Les paramètres lipidiques n'ont pas été influencés. Les réductions moyennes du taux d'HbA1c observées dans le sous-groupe des patients sans traitement antidiabétique avant l'étude ont été: -1,06% sous sitagliptine 100 mg une fois par jour (n= 88); -1,09% sous metformine 500 mg deux fois par jour (n= 90); -1,24% sous metformine 1000 mg deux fois par jour (n= 87); -1,59% sous sitagliptine 50 mg deux fois par jour et metformine 500 mg deux fois par jour (n= 100); -1,94% sous sitagliptine 50 mg deux fois par jour et metformine 1000 mg deux fois par jour (n= 86); -0,17% sous placebo (n= 83).
Fig. 1: Variation moyenne du taux d'HbA1c sur 24 semaines par rapport aux valeurs initiales, sous sitagliptine seule, metformine seule et sous association des deux substances chez des patients diabétiques de type 2†
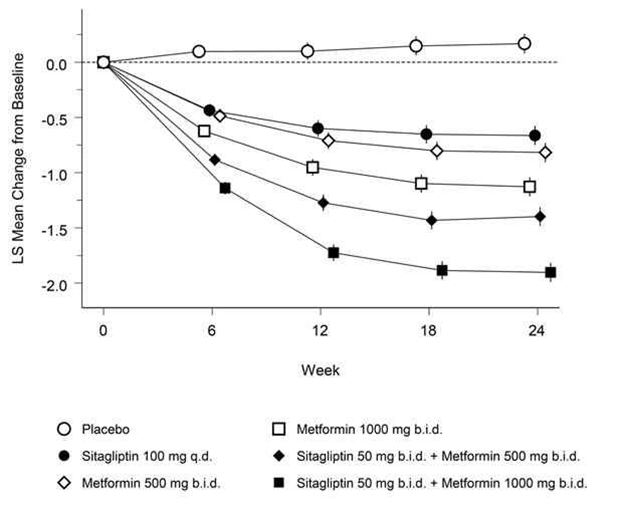
† Population de tous les patients traités (analyse en intention de traitement, ITT), moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
Tableau 2:
Paramètres glycémiques et poids corporel lors de l'examen final à 24 semaines†
|
|
Placebo
|
Sitagliptine
100 mg
1x par jour
|
Metformine
500 mg 2x par jour
|
Sitagliptine
50 mg 2x par jour +
metformine
500 mg 2x par jour
|
Metformine
1000 mg 2x par jour
|
Sitagliptine
50 mg 2x par jour +
metformine
1000 mg 2x par jour
| |
HbA1c (%)
|
N= 165
|
N= 175
|
N= 178
|
N= 183
|
N= 177
|
N= 178
| |
Valeur initiale (moyenne)
|
8,68
|
8,87
|
8,90
|
8,79
|
8,68
|
8,76
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
0,17
|
-0,66
|
-0,82
|
-1,40
|
-1,13
|
-1,90
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
-
|
-0,83§
|
-0,99§
|
-1,57§
|
-1,30§
|
-2,07§
| |
Patients (%) avec taux d'HbA1c <7%
|
15 (9,1)
|
35 (20,0)
|
41 (23,0)
|
79 (43,2)
|
68 (38,4)
|
118 (66,3)
| |
FPG (mmol/l)
|
N= 169
|
N= 178
|
N= 179
|
N= 183
|
N= 179
|
N= 180
| |
Valeur initiale (moyenne)
|
10,91
|
11,19
|
11,40
|
11,33
|
10,94
|
10,93
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
0,32
|
-0,97
|
-1,52
|
-2,62
|
-1,63
|
-3,55
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
-
|
-1,29§
|
-1,84§
|
-2,94§
|
-1,95§
|
-3,87§
| |
PPG à 2 h (mmol/l)
|
N= 129
|
N= 136
|
N= 141
|
N= 147
|
N= 138
|
N= 152
| |
Valeur initiale (moyenne)
|
15,38
|
15,86
|
16,26
|
16,21
|
15,74
|
15,94
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
0,02
|
-2,88
|
-2,97
|
-5,14
|
-4,33
|
-6,48
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
|
-2,9§
|
-2,98§
|
-5,16§
|
-4,35§
|
-6,49§
| |
Poids corporel (kg)**
|
N= 167
|
N= 175
|
N= 179
|
N= 184
|
N= 175
|
N= 178
| |
Valeur initiale (moyenne)
|
90,1
|
85,9
|
88,1
|
90,0
|
89,4
|
88,2
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,9
|
0,0
|
-0,9
|
-0,6
|
-1,1
|
-1,3
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
|
0,9¶
|
0,1#
|
0,4#
|
-0,1#
|
-0,3#
|
† All Patients Treated Population (une analyse en intention de traitement, ITT).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
§ p <0,001 par rapport au placebo.
** Population de tous les patients tels que traités (all Patients as Treated, APaT) à l'exclusion des données après un traitement glycémique d'urgence.
¶ p = 0,005 par rapport au placebo.
# Statistiquement non significatif (p ≥0,05) par rapport au placebo.
En outre, cette étude a inclus des patients (n = 117) présentant une forte hyperglycémie (HbA1c >11% ou glycémie >15,56 mmol/l), traités deux fois par jour à la sitagliptine 50 mg et à la metformine 1000 mg. Aucun contrôle avec placebo n'a été fait pour ce groupe de patients. Dans ce groupe de patients, le taux initial moyen d'HbA1c était de 11,15%, le FPG de 17,47 mmol et la valeur de PPG à deux heures de 24,5 mmol/l. Au bout de 24 semaines, les réductions suivantes ont été constatées par rapport aux valeurs initiales: -2,9% pour le taux d'HbA1c, -7,04 mmol/l pour le FPG et –11,55 mmol/l pour le PPG à 2 heures. Dans cette cohorte, une augmentation de 1,3 kg du poids corporel a été constatée à 24 semaines.
Traitement complémentaire à la sitagliptine chez les patients n'atteignant pas un bon équilibre glycémique sous metformine seule
Deux études cliniques effectuées en double aveugle avec contrôle par placebo auprès de patients diabétiques de type 2 ont évalué la sécurité et l'efficacité d'une association sitagliptine-metformine à libération non retardée. Dans les deux études, des patients présentant une glycémie mal contrôlée ont poursuivi leurs prises de metformine ≥1500 mg tout en prenant de façon randomisée soit 100 mg de sitagliptine, soit un placebo en plus du traitement en cours à la metformine.
Dans une étude, 701 patients ont été traités 24 semaines soit avec 100 mg de sitagliptine, soit avec un placebo. L'ajout de la sitagliptine au traitement en cours à la metformine à libération non retardée a permis une amélioration significative du taux d'HbA1c (-0,65%), du FPG (-1,41 mmol/l) et de la glycémie postprandiale à 2 h (-2,81 mmol/l) en comparaison avec l'ajout du placebo au traitement en cours à la metformine (voir la Fig. 2 et Tableau 3). L'amélioration du taux d'HbA1C par rapport au placebo n'a pas été influencée par le taux initial d'HbA1C, les traitements antidiabétiques antérieurs, le sexe, l'âge, le BMI initial, le temps écoulé depuis le diagnostic du diabète, la présence d'un syndrome métabolique ou les indices standard de l'insulinorésistance (HOMA-IR) ou de la sécrétion d'insuline (HOMA-β).
Fig. 2: Modification moyenne du taux d'HbA1c à 24 semaines par rapport à la valeur initiale chez des patients diabétiques de type 2 traités avec une dose quotidienne de 100 mg de sitagliptine en association avec de la metformine ou avec un placebo en plus de la metformine† ‡
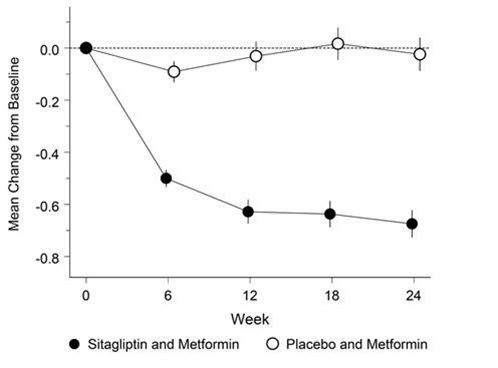
† Patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule.
‡ All Patients Treated Population. Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
Tableau 3:
Paramètres glycémiques et poids corporel lors de l'examen final (étude de 24 semaines)
Traitement complémentaire à la sitagliptine chez des patients n'ayant pas atteint un bon équilibre glycémique sous metformine seule†
|
|
Sitagliptine 100 mg par jour + metformine
|
Placebo +
metformine
| |
HbA1c (%)
|
N= 453
|
N= 224
| |
Valeur initiale (moyenne)
|
7,96
|
8,03
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,67
|
-0,02
| |
Différence par rapport au placebo + metformine (moyenne ajustée ‡)
|
-0,65§
|
| |
Patients (%) avec HbA1c <7%
|
213 (47,0)
|
41 (18,3)
| |
FPG (mmol/l)
|
N= 454
|
N= 226
| |
Valeur initiale (moyenne)
|
9,44
|
9,64
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,94
|
0,47
| |
Différence par rapport au placebo + metformine (moyenne ajustée‡)
|
-1,41§
|
| |
PPG à 2 h (mmol/l)
|
N= 387
|
N= 182
| |
Valeur initiale (moyenne)
|
15,25
|
15,13
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-3,44
|
-0,63
| |
Différence par rapport au placebo + metformine (moyenne ajustée‡)
|
-2,81§
|
| |
Poids corporel (kg)**
|
N= 399
|
N= 169
| |
Valeur initiale (moyenne)
|
86,9
|
87,6
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,7
|
-0,6
| |
Différence par rapport au placebo + metformine (moyenne ajustée‡)
|
-0,1¶
|
|
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antidiabétique antérieur et de la valeur initiale.
§ p <0,001 par rapport au placebo + metformine.
** All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
¶ Statistiquement non significatif (p≥0,05) par rapport au placebo + metformine.
Fig. 3: Profils glycémiques de 24 h chez des patients diabétiques de type 2 au bout de 4 semaines de traitement à la sitagliptine 50 mg 2x par jour associée à la metformine ou au placebo associé à la metformine†
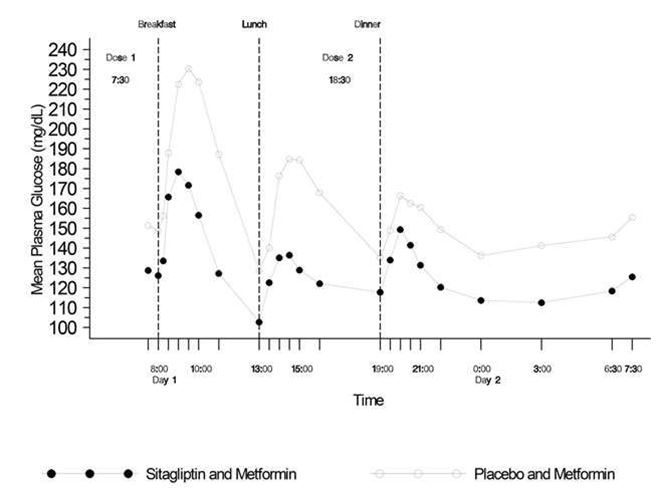
† Patients n'ayant pas atteint un équilibre glycémique suffisant sous metformine seule.
Ajout de sitagliptine au traitement de patients n'ayant pas atteint un équilibre glycémique suffisant avec l'association de metformine à libération non retardée et de glimépiride
Une étude randomisée de 24 semaines, effectuée en double aveugle avec contrôle par placebo auprès de 441 patients diabétiques de type 2, a évalué l'efficacité de la sitagliptine à la dose de 100 mg une fois par jour en association avec le glimépiride (en monothérapie ou en association avec la metformine). Dans cette étude, 220 patients suivaient déjà un traitement associant le glimépiride (≥4 mg par jour) et la metformine (≥1500 mg par jour).
L'association de sitagliptine, de glimépiride et de metformine a permis en comparaison avec le placebo une réduction significative du taux d'HbA1c par rapport aux valeurs initiales (-0,89%) et de la glycémie à jeun (FPG -1,15 mmol/l) (voir le Tableau 4). Les patients ayant présenté initialement un taux plus élevé d'HbA1c ont atteint une plus forte réduction moyenne du taux d'HbA1c par rapport au placebo.
Tableau 4:
Paramètres glycémiques* et poids corporel à l'examen final (étude de 24 semaines) chez les patients traités à la sitagliptine en plus du traitement associant le glimépiride et la metformine†
|
|
Sitagliptine 100 mg
+ glimépiride
+ metformine
|
Placebo
+ glimépiride
+ metformine
| |
HbA1c (%)
|
N= 115
|
N= 105
| |
Valeur initiale (moyenne)
|
8,27
|
8,28
| |
Variation vs valeur initiale (moyenne ajustée‡)
|
-0,59
|
0,30
| |
Variation vs placebo (moyenne ajustée‡)
|
-0,89§
|
| |
Patients (%) avec un taux d'HbA1c <7%
|
26 (22,6)
|
1 (1,0)
| |
FPG (mmol/l)
|
N= 115
|
N= 109
| |
Valeur initiale (moyenne)
|
9,96
|
9,94
| |
Variation vs valeur initiale (moyenne ajustée‡)
|
-0,43
|
0,72
| |
Variation vs placebo (moyenne ajustée‡)
|
-1,15§
|
| |
Poids corporel (kg)**
|
N= 102
|
N= 74
| |
Valeur initiale (moyenne)
|
86,5
|
84,6
| |
Variation vs valeur initiale (moyenne ajustée‡)
|
0,4
|
-0,7
| |
Variation vs placebo (moyenne ajustée‡)
|
1,1††
|
|
* Paramètres glycémiques primaires et secondaires: HbA1c et glycémie à jeun
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement antérieur et de la valeur initiale.
§ p <0,001 versus placebo.
** All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
†† p = 0,007 versus placebo
Sitagliptine en tant que traitement complémentaire chez les patients insuffisamment traités avec une association de metformine à libération non retardée et d'insuline
641 patients diabétiques de type 2 ont participé à une étude randomisée de 24 semaines, réalisée en double aveugle avec contrôle par placebo, dans laquelle on a évalué l'efficacité de la sitagliptine 100 mg une fois par jour en association avec une dose stable d'insuline. Environ 75% des patients prenaient également de la metformine. Les patients sous insulinothérapie (insuline prête à l'emploi avec une durée d'action intermédiaire ou longue) en association ou non avec de la metformine ont été randomisés pour recevoir 100 mg de sitagliptine ou un placebo en tant que traitement complémentaire. Les critères glycémiques évalués ont inclus le taux de HbA1c, la glycémie à jeun et la glycémie postprandiale (PPG) au bout de 2 heures.
L'association de sitagliptine, de metformine et d'insuline a permis en comparaison avec le placebo une amélioration significative du taux de HbA1c, de la glycémie à jeun et de la PPG au bout de 2 heures (Tableau 5). L'amélioration du taux de HbA1c par rapport au placebo a été concordante dans tous les sous-groupes. Ces sous-groupes étaient définis en fonction de l'âge, du sexe, de la race, du BMI initial, de la durée du diabète depuis le diagnostic, de la présence ou non d'un syndrome métabolique et des indices standard d'insulinorésistance (HOMA-IR) et d'insulinosécrétion (HOMA-β). Une modification significative du poids corporel par rapport à la valeur initiale n'a été observée dans aucun des groupes.
Tableau 5:
Paramètres glycémiques et poids corporel lors de l'examen final (étude de 24 semaines) après la prise de sitagliptine en tant que traitement complémentaire en plus du traitement associant la metformine et une dose stable d'insuline†
|
|
Sitagliptine 100 mg
+ insuline
+ metformine
|
Placebo
+ insuline
+ metformine
| |
HbA1c (%)
|
N= 223
|
N= 229
| |
Valeur initiale (moyenne)
|
8,73
|
8,60
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,66
|
-0,13
| |
Différence par rapport au placebo (moyenne ajustée‡,§)
|
-0,53**
|
| |
Patients (%) avec HbA1c <7%
|
32 (14,3)
|
12 (5,2)
| |
FPG (mmol/l)
|
N= 225
|
N= 229
| |
Valeur initiale (moyenne)
|
9,5
|
9,7
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-1,2
|
-0,2
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
-1,0**
|
| |
PPG à 2 h (mmol/l)
|
N= 182
|
N= 189
| |
Valeur initiale (moyenne)
|
15,4
|
15,4
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-2,1
|
0,1
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
-2,2**
|
| |
Poids corporel (kg)¶
|
N= 201
|
N= 200
| |
Valeur initiale (moyenne)
|
87,9
|
88,0
| |
Modification par rapport à la valeur initiale (moyenne ajustée‡)
|
-0,1
|
0,0
| |
Différence par rapport au placebo (moyenne ajustée‡)
|
0,1#
|
|
† All Patients treated Population (une analyse Intention-to-treat).
‡ Moyennes des moindres carrés, ajustées en fonction du traitement à l'insuline (prête à l'emploi ou non [de durée d'action intermédiaire ou longue]) lors de la visite 1 et en fonction de la valeur initiale.
§ Stratification en fonction de l'insuline; l'interaction n'était pas significative (p >0,10).
** p <0,001 versus placebo.
¶ All Patients as Treated (APaT) Population, à l'exclusion des données après un traitement glycémique d'urgence.
# Statistiquement non significatif (p ≥0,05) par rapport au placebo.
Dans une autre étude randomisée de 24 semaines, effectuée en double aveugle avec contrôle contre placebo, destinée à analyser l'effet de la sitagliptine en traitement concomitant complémentaire sur les économies d'insuline, 660 patients ayant un contrôle glycémique insuffisant sous insuline glargine avec ou sans metformine (≥1500 mg par jour) ont été randomisés en deux groupes: 100 mg sitagliptine (N= 330) ou placebo (N= 330). L'administration avait lieu une fois par jour parallèlement à une intensification du traitement par insuline. Chez les patients traités par metformine, le taux initial d'HbA1c était de 8,70% et la dose initiale d'insuline de 37 UI/jour. Les patients devaient titrer leur dose d'insuline glargine selon la glycémie à jeun obtenue après piqûre du doigt. Les critères d'évaluation secondaires de l'étude comprenaient l'HbA1c et la glycémie à jeun (FPG).
Chez les patients sous metformine, la hausse de la dose quotidienne d'insuline à semaine 24 était plus faible chez les patients traités par sitagliptine (19 UI/jour, N= 285) que chez les patients sous placebo (24 UI/jour, N= 283). Cette différence était statistiquement significative (p= 0,007).
Concernant les critères d'évaluation secondaires de l'étude, une réduction du taux d'HbA1c de -1,35% a été constatée chez les patients traités par sitagliptine, metformine et insuline contre -0,90% chez les patients sous placebo, metformine et insuline, soit une différence de -0,45% [IC à 95%: -0,62; -0,29]. La réduction du FPG chez les patients traités par sitagliptine, metformine et insuline était de -3,0 mmol/l contre -2,4 mmol/l chez les patients sous placebo, metformine et insuline, soit une différence de -0,7 mmol/l [IC à 95%: -1,0; -0,3].
Étude contrôlée avec substance active (glipizide) en association avec la metformine à libération non retardée
Les effets à long terme ont été examinés dans une étude randomisée de 52 semaines, effectuée en double aveugle avec contrôle avec glipizide auprès de patients diabétiques de type 2. Des patients n'ayant pas atteint un bon équilibre glycémique sous metformine (≥1500 mg par jour) ont reçu soit 100 mg de sitagliptine (n= 588), soit du glipizide (n= 584) pendant 52 semaines. Les patients sous glipizide ont reçu une dose initiale de 5 mg par jour, puis la dose a été augmentée progressivement de façon sélective au cours des 18 semaines suivantes sans provoquer d'hypoglycémie significative, jusqu'à l'obtention d'un glucose plasmatique à jeun de <110 mg/dl. Une dose maximale de 20 mg par jour était autorisée pour un équilibre glycémique optimal. Par la suite, la dose de glipizide a été maintenue constante. La dose moyenne de glipizide après l'adaptation initiale a été de 10,3 mg.
Les deux traitements ont conduit à une amélioration statistiquement significative de l'équilibre glycémique en comparaison avec les valeurs initiales. À 52 semaines, la réduction du taux d'HbA1c par rapport à la valeur initiale était de 0,67% chez les patients traités par 100 mg de sitagliptine par jour et de 0,67% chez les patients traités au glipizide. Ces résultats ont confirmé l'équivalence des deux principes actifs. La réduction de la glycémie à jeun a été de 0,56 mmol/l sous sitagliptine et de 0,42 mmol/l sous glipizide. Dans une analyse post hoc, les patients des deux groupes qui avaient présenté un taux initial plus élevé d'HbA1c (≥9%) ont bénéficié d'une réduction plus importante du taux d'HbA1c par rapport aux valeurs initiales (sitagliptine -1,68%; glipizide -1,76%). Le traitement à la sitagliptine a permis une réduction du rapport pro-insuline/insuline, un marqueur de la synthèse et de la sécrétion d'insuline. Sous glipizide, le rapport pro-insuline/insuline s'est aggravé. La fréquence des hypoglycémies a été significativement plus faible dans le groupe sous sitagliptine (4,9%) que dans le groupe sous glipizide (32,0%). Le poids corporel a baissé de façon significative chez les patients traités à la sitagliptine mais a augmenté significativement chez les patients traités au glipizide (-1,5 kg vs +1,1 kg).
Chlorhydrate de metformine
L'étude prospective randomisée UKPDS a confirmé les avantages à long terme d'un contrôle intensif de la glycémie dans le diabète de type 2. L'analyse des résultats – obtenus chez des patients présentant une surcharge pondérale et traités à la metformine après qu'un régime alimentaire se fut avéré insuffisant à lui seul – a révélé:
·Dans le groupe sous metformine, une réduction significative du risque absolu de subir une complication en rapport avec le diabète (29,8 événements sur 1000 patients-années) par rapport au régime alimentaire seul (43,3 événements sur 1000 patient-années), p = 0,0023, ainsi que par rapport aux groupes combinés sous monothérapie aux sulfonylurées ou à l'insuline (40,1 événements sur 1000 patient-année), p= 0,0034.
·Une réduction significative du risque absolu de mortalité en rapport avec le diabète: metformine 7,5 décès sur 1000 patient-années, régime alimentaire seul 12,7 décès sur 1000 patient-années, p= 0,017.
·Une réduction significative du risque absolu de mortalité totale: metformine 13,5 décès sur 1000 patient-années par rapport au régime alimentaire seul 20,6 sur 1000 patient-années (p= 0,011) et par rapport aux groupes combinés de monothérapie aux sulfonylurées ou à l'insuline 18,9 sur 1000 patient-années (p= 0,021).
·Une réduction significative du risque absolu de subir un infarctus du myocarde: metformine 11 événements sur 1000 patient-années, régime alimentaire seul 18 décès sur 1000 patient-années (p= 0,01).
Étude de sécurité cardiovasculaire TECOS
L'étude TECOS (Trial Evaluating Cardiovascular Outcomes with Sitagliptin) était randomisée et incluait 14'671 patients de la population en intention de traiter présentant des valeurs HbA1c de ≥6,5 à 8,0% et une maladie cardiovasculaire documentée. Les patients ont reçu soit 100 mg par jour de sitagliptine (7332) (ou 50 mg par jour si le DFGe initial était ≥30 et <50 ml/min/1,73 m2) soit un placebo (7339), donné en complément du traitement conventionnel avec des valeurs cibles correspondant aux standards régionaux en termes d'HbA1c et de facteurs de risque cardiovasculaire. Les patients présentant un DFGe <30 ml/min/1,73 m2 n'ont pas été inclus dans l'étude. L'étude incluait 2004 patients âgés de ≥75 ans et 3324 patients insuffisants rénaux (DFGe <60 ml/min/1,73 m2).
Les patients du groupe sitagliptine ont reçu moins d'antihyperglycémiants que ceux du groupe placebo (Hazard Ratio 0,72; IC à 95%, 0,68-0,77; p ≤0,001). Par ailleurs, les patients ne recevant pas d'insuline au début de l'étude étaient moins susceptibles de démarrer une insulinothérapie chronique (Hazard Ratio 0,70; IC à 95%, 0,63-0,79; p <0,001).
Le critère d'évaluation cardiovasculaire primaire était composé de la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal, d'un accident vasculaire cérébral (AVC) non fatal ou d'une hospitalisation en raison d'une angine de poitrine instable. Les critères d'évaluation cardiovasculaires secondaires étaient la première survenue d'un décès d'origine cardiovasculaire, d'un infarctus du myocarde non fatal ou d'un AVC non fatal; la première survenue d'un des composants du critère d'évaluation primaire; la mortalité totale; et les hospitalisations en raison d'une insuffisance cardiaque.
Après une période d'observation médiane de trois ans, le risque d'événements cardiovasculaires indésirables sévères ou d'hospitalisation en raison d'une insuffisance cardiaque n'a pas augmenté lorsque la sitagliptine était administrée chez les patients diabétiques de type 2 en complément du traitement conventionnel (tableau 6).
Tableau 6:
Taux d'événements cardiovasculaires et événements secondaires importants
|
|
Sitagliptine 100 mg
|
Placebo
|
Hazard Ratio
(IC à 95%)
|
Valeur p†
| |
N (%)
|
Taux d'incidence pour 100 patients-années*
|
N (%)
|
Taux d'incidence pour 100 patients-années
| |
Analyse de la population en intention de traiter
| |
Nombre de patients
|
7332
|
7339
|
0,98 (0,89–1,08)
|
<0,001
| |
Critère d'évaluation primaire composite
(décès cardiovasculaire, infarctus du myocarde non fatal, AVC non fatal ou hospitalisation en raison d'une angine de poitrine instable)
|
839 (11,4)
|
4,1
|
851 (11,6)
|
4,2
| |
Critère d'évaluation secondaire composite
(décès cardiovasculaire, infarctus du myocarde non fatal ou AVC non fatal)
|
745 (10,2)
|
3,6
|
746 (10,2)
|
3,6
|
0,99 (0,89–1,10)
|
<0,001
| |
Événements secondaires
| |
Décès cardiovasculaires
|
380 (5,2)
|
1,7
|
366 (5,0)
|
1,7
|
1,03 (0,89-1,19)
|
0,711
| |
Tous les infarctus du myocarde (fatals et non fatals)
|
300 (4,1)
|
1,4
|
316 (4,3)
|
1,5
|
0,95 (0,81–1,11)
|
0,487
| |
Tous les AVC (fatals et non fatals)
|
178 (2,4)
|
0,8
|
183 (2,5)
|
0,9
|
0,97 (0,79–1,19)
|
0,760
| |
Hospitalisations en raison d'une angine de poitrine instable
|
116 (1,6)
|
0,5
|
129 (1,8)
|
0,6
|
0,90 (0,70–1,16)
|
0,419
| |
Décès toutes causes
|
547 (7,5)
|
2,5
|
537 (7,3)
|
2,5
|
1,01 (0,90–1,14)
|
0,875
| |
Hospitalisations en raison d'une insuffisance cardiaque‡
|
228 (3,1)
|
1,1
|
229 (3,1)
|
1,1
|
1,00 (0,83–1,20)
|
0,983
|
* Le taux d'incidence pour 100 patients-années a été calculé selon la formule 100× (nombre total de patients présentant ≥1 événement pendant la période d'exposition décisive sur le nombre total de patients-années de l'observation).
† D'après le modèle de Cox stratifié par région. Pour les critères d'évaluation composites, les valeurs p correspondent à un test de non-infériorité dont l'objectif est de montrer que le Hazard Ratio est inférieur à 1,3. Pour tous les autres critères d'évaluation, les valeurs p correspondent à un test de différence des Hazard Rates.
‡ L'analyse des hospitalisations en raison d'une insuffisance cardiaque a été ajustée pour l'antécédent d'insuffisance cardiaque à l'inclusion dans l'étude.
Ajout de sitagliptine chez des patients pédiatriques présentant un contrôle glycémique insuffisant avec de la metformine avec ou sans insuline
Au total, 220 patients pédiatriques âgés de 10 à 17 ans atteints de diabète de type 2 et présentant un contrôle glycémique insuffisant avec de la metformine avec ou sans insuline ont participé à deux études parallèles randomisées, en double aveugle, contrôlées par placebo sur 54 semaines. L'ajout de 100 mg de sitagliptine (administrée sous forme de sitagliptine + metformine ou sitagliptine + metformine à libération retardée (XR)) a été comparé à l'ajout d'un placebo à la metformine ou à la metformine XR.
Les résultats des deux études pédiatriques pour le critère d'évaluation primaire (modification de l'HbA1c jusqu'à la Semaine 20) étaient discordants. Une analyse cumulée a même montré une supériorité du traitement par sitagliptine + metformine/sitagliptine + metformine XR par rapport à la metformine à la Semaine 20, cependant, cet effet n'était plus présent à la Semaine 54. Sitagliptin-Metformin-Mepha retard ne doit pas être utilisé chez les patients pédiatriques âgés de 10 à 17 ans en raison d'une efficacité insuffisamment documentée de sitagliptine/metformine comprimés à libération retardée chez les patients pédiatriques âgés de 10 à 17 ans.
Dans des études cliniques menée avec la sitagliptine chez les patients pédiatriques atteints de diabète de type 2 et âgés de 10 à 17 ans, le profil d'effets indésirables était généralement comparable à celui observé chez les adultes. Chez les patients pédiatriques traités avec ou sans insuline basale, la sitagliptine était liée à un risque accru d'hypoglycémie.
PharmacocinétiqueLes résultats d'une étude auprès de volontaires sains ont montré que les comprimés associés de chlorhydrate de sitagliptine/metformine comprimés à libération retardée 50 mg/500 mg et 100 mg/1000 mg sont bioéquivalents à une prise simultanée des doses correspondantes de sitagliptine (comprimés contenant de la sitagliptine) et chlorhydrate de metformine à libération retardée sous forme de comprimés séparés.
De même, la bioéquivalence entre deux comprimés de chlorhydrate de sitagliptine/metformine comprimés à libération retardée 50 mg/500 mg et un comprimé de chlorhydrate de sitagliptine/metformine comprimés à libération retardée 100 mg/1000 mg a été démontrée.
Dans une étude croisée auprès de volontaires sains, l'AUC et la Cmax de la sitagliptine et l'AUC de la metformine étaient similaires après l'administration d'un seul comprimé à libération retardée de chlorhydrate de sitagliptine/metformine 50 mg/500 mg (formule testée) et après l'administration d'un seul comprimé de chlorhydrate de sitagliptine/metformine (phosphate de sitagliptine/chlorhydrate de metformine à libération non retardée) de 50 mg/500 mg. Après administration d'un seul comprimé à libération retardée de chlorhydrate de sitagliptine/metformine 50 mg/500 mg (formule testée), la valeur moyenne de Cmax de la metformine était inférieure de 30% et la valeur médiane de Tmax était retardée de 4 heures par rapport aux valeurs correspondantes observées après l'administration d'un seul comprimé sans libération retardée de chlorhydrate de sitagliptine/metformine 50 mg/500 mg. Ceci est en accord avec les caractéristiques attendues de la libération retardée de la metformine de chlorhydrate de sitagliptine/metformine comprimés à libération retardée.
Après administration de deux comprimés à libération retardée de chlorhydrate de sitagliptine/metformine 50 mg/1000 mg une fois par jour avec le repas du soir pendant 7 jours chez des volontaires sains adultes, la sitagliptine et la metformine ont atteint l'état d'équilibre le jour 4 et le jour 5, respectivement. Les valeurs médianes de Tmax pour la sitagliptine et la metformine à l'état d'équilibre étaient d'environ 3 et 8 heures, respectivement, après la prise. Les valeurs médianes de Tmax pour la sitagliptine et la metformine après administration d'un seul comprimé sans libération retardée de chlorhydrate de sitagliptine/metformine étaient de 3 et de 3,5 heures, respectivement, après la prise.
Les données pharmacocinétiques caractérisant la distribution, le métabolisme et l'élimination de la metformine et de la sitagliptine proviennent d'études avec administration des monosubstances de metformine à libération non retardée et de sitagliptine; elles sont applicables à chlorhydrate de sitagliptine/metformine comprimés à libération retardée.
Absorption
Sitagliptin-Metformin-Mepha retard
Après administration de comprimés à libération retardée de chlorhydrate de sitagliptine/metformine avec un petit-déjeuner riche en graisses, l'AUC de la sitagliptine était inchangée. La Cmax moyenne était diminuée de 17%, tandis que la valeur médiane de Tmax était inchangée par rapport à l'état à jeun. Après administration de chlorhydrate de sitagliptine/metformine comprimés à libération retardée avec un petit-déjeuner riche en graisses, l'AUC de la metformine était augmentée de 62%, la Cmax de la metformine était diminuée de 9% et la valeur médiane de Tmax de la metformine était retardée de 2 heures par rapport à l'état à jeun.
Sitagliptine
La biodisponibilité absolue de la sitagliptine se monte à environ 87%. La prise simultanée d'un repas riche en graisses n'a aucune influence sur la pharmacocinétique de la sitagliptine.
Distribution
Sitagliptine
Le volume de distribution moyen à l'état stationnaire après une dose intraveineuse unique de 100 mg de sitagliptine chez des volontaires sains se monte à environ 198 litres. La fraction de sitagliptine liée de manière réversible aux protéines plasmatiques est faible (38%).
Chlorhydrate de metformine
Aucune étude n'a été effectuée sur la distribution de la metformine à libération retardée. Le volume de distribution apparent (V/F) de la metformine après la prise de doses orales uniques de 850 mg de chlorhydrate de metformine à libération non retardée a été en moyenne de 654 ± 358 l. Contrairement aux sulfonylurées, qui se lient à plus de 90% aux protéines plasmatiques, la metformine ne présente guère de liaison aux protéines plasmatiques. La metformine se répartit dans les érythrocytes, probablement en fonction du temps. Aux schémas posologiques et dosages cliniques normaux des comprimés de chlorhydrate de metformine, les concentrations plasmatiques de metformine à l'état stationnaire – normalement inférieures à 1 µg/ml – sont atteintes en l'espace de 24 à 48 heures. Dans des études cliniques contrôlées sur la metformine, les taux maximaux de metformine n'ont jamais dépassé 5 µg/ml même aux doses maximales.
Métabolisme
Sitagliptine
La sitagliptine est éliminée en première ligne sous forme inchangée dans l'urine et seule une faible fraction est métabolisée. Environ 79% de la sitagliptine sont éliminés dans l'urine sous forme inchangée.
Après une dose orale de [14C]sitagliptine, près de 16% de la radioactivité a été éliminée sous forme de métabolites de la sitagliptine. Six métabolites ont été détectés sous forme de traces. On ne pense pas que ces métabolites ont contribué à l'activité inhibitrice plasmatique de la DPP-4 par la sitagliptine. Des études in vitro ont pu montrer que le CYP3A4, avec la participation du CYP2C8, est la principale enzyme pour le métabolisme limité de la sitagliptine.
Chlorhydrate de metformine
Des études avec des doses intraveineuses uniques chez des volontaires sains montrent que la metformine est éliminée dans l'urine sous forme inchangée, sans être métabolisée dans le foie (aucun métabolite n'a été trouvé chez l'homme) ni éliminée dans la bile.
Élimination
Sitagliptine
Après l'administration d'une dose orale de [14C]sitagliptine à des volontaires sains, près de 100% de la radioactivité administrée a été retrouvée dans les selles (13%) ou l'urine (87%) en l'espace d'une semaine. La demi-vie terminale apparente t½ après une dose orale de 100 mg de sitagliptine était de près de 12,4 heures, et la clairance rénale se montait à près de 350 ml/min.
L'élimination de la sitagliptine se fait principalement par les reins et implique une sécrétion tubulaire active. La sitagliptine est un substrat pour le transporteur d'anions organiques 3 de l'homme (hOAT-3), qui est probablement impliqué dans l'élimination rénale de la sitagliptine. L'importance clinique du hOAT-3 pour le transport de la sitagliptine n'a pas encore pu être démontrée. La sitagliptine est également un substrat de la p-glycoprotéine, qui pourrait aussi être impliquée dans le mécanisme de l'élimination rénale de la sitagliptine. La ciclosporine, un inhibiteur de la glycoprotéine p, n'a toutefois pas permis de diminuer la clairance rénale de la sitagliptine.
Chlorhydrate de metformine
La clairance rénale est environ 3,5 fois supérieure à la clairance de la créatinine, ce qui permet de supposer que la sécrétion tubulaire est la voie d'élimination la plus importante de la metformine. Après une administration orale de metformine à libération non retardée, environ 90% de la dose assimilée sont éliminés dans les premières 24 h par voie rénale, avec une demi-vie d'élimination plasmatique de 6,2 heures environ. Dans le sang, la demi-vie d'élimination est d'environ 17,6 heures, ce qui permet de supposer que la masse érythrocytaire pourrait être un compartiment de distribution.
Trouble de la fonction rénale
Sitagliptine
Chez les patients présentant un trouble modéré de la fonction rénale, avec un DFGe de 30 à <45 ml/min/1.73 m2, une multiplication par deux environ de l'AUC plasmatique de la sitagliptine a été observée, et chez les patients présentant un trouble sévère de la fonction rénale (DFGe <30 ml/min/1.73 m2), y compris les patients atteints d'insuffisance rénale terminale (end-stage renal disease, ESRD) sous hémodialyse, une augmentation d'environ quatre fois a été observée par rapport aux volontaires dont la fonction rénale était normale.
Chlorhydrate de metformine
Chez les patients présentant une diminution de la fonction rénale, la demi-vie plasmatique et sanguine de la metformine à libération non retardée est prolongée et la clairance rénale est diminuée (voir «Contre-indications» et «Mises en garde et précautions»).
Trouble de la fonction hépatique
Sitagliptine
Chez les patients présentant un trouble modéré de la fonction hépatique (Child-Pugh Score 7 à 9), les valeurs moyennes de l'AUC et de la Cmax de la sitagliptine avaient augmenté de 21% et de 13% par rapport au groupe de contrôle après la prise d'une dose unique de 100 mg de sitagliptine. Ces effets ne sont pas considérés comme cliniquement significatifs.
Il n'existe aucune donnée clinique relative aux patients présentant un trouble sévère de la fonction hépatique (Child-Pugh Score >9). Toutefois, étant donné que la sitagliptine est surtout éliminée par les reins, un trouble sévère de la fonction hépatique n'influence probablement pas la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Il n'existe aucune étude pharmacocinétique sur la metformine chez les patients présentant un trouble de la fonction hépatique.
Utilisation chez les personnes âgées
Sitagliptine
Selon les données de phase I et II d'une analyse pharmacocinétique de population, l'âge n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine. Les personnes âgées (de 65 ans à 80 ans) ont présenté, par rapport à des personnes plus jeunes, des concentrations plasmatiques de sitagliptine supérieures de près de 19%.
Chlorhydrate de metformine
Il n'existe que peu de données d'études pharmacocinétiques contrôlées sur la metformine à libération non retardée chez les sujets âgés sains. Elles suggèrent que chez les personnes âgées saines, la clairance plasmatique totale de la metformine est réduite, la demi-vie prolongée et la Cmax accrue en comparaison avec les sujets sains encore jeunes.
Ces données font supposer que l'influence de l'âge des patients sur la pharmacocinétique de la metformine provient essentiellement d'altérations de la fonction rénale (voir l'information professionnelle de GLUCOPHAGE1).
1 GLUCOPHAGE® est une marque déposée de Merck Sante S.A.S, société partenaire de Merck KgaA à Darmstadt, Allemagne.
Emploi chez les enfants et les adolescents
La pharmacocinétique de la sitagliptine (à dose unique de 50 mg, 100 mg ou 200 mg) a été étudiée chez des patients pédiatriques (âgés de 10 à 17 ans) atteints de diabète de type 2. Dans cette population, l'AUC de la dose ajustée de sitagliptine dans le plasma était environ de 18% inférieure à celle des patients adultes atteints de diabète de type 2 pour une dose de 100 mg.
Aucune étude n'a été effectuée avec la sitagliptine chez des patients pédiatriques âgés de moins de 10 ans.
Sexe
Sitagliptine
Une analyse pharmacocinétique de données de phase I et une analyse de pharmacocinétique de population basée sur des données de phase I et II ont montré que le sexe n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Les paramètres pharmacocinétiques de la metformine n'ont pas été notablement différents entre les sujets sains et les patients diabétiques de type 2, lorsqu'ils ont été analysés en fonction du sexe des patients. De même, des études cliniques contrôlées auprès de patients diabétiques de type 2 ont permis de montrer que les effets anti-hyperglycémiants de la metformine sont similaires chez l'homme et la femme.
Race
Sitagliptine
Une analyse pharmacocinétique basée sur des données de phase I et une analyse pharmacocinétique de population basée sur des données de phase I et II, ayant inclus des Caucasiens, des Latino-américains, des Afro-américains, des Asiatiques et des membres d'autres groupes ethniques ont montré que l'appartenance ethnique n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Chlorhydrate de metformine
Il n'existe aucune étude sur les paramètres pharmacocinétiques de la metformine en fonction de la race. Dans des études cliniques contrôlées sur la metformine chez des patients diabétiques de type 2, les effets anti-hyperglycémiants ont été comparables entre les Caucasiens (n= 249), les Afro-américains (n= 51) et les Latino-américains (n= 24).
Indice de masse corporelle (Body-Mass-Index, BMI)
Sitagliptine
Une analyse pharmacocinétique de données de phase I et une analyse de pharmacocinétique de population sur la base de données de phase I et II ont montré que le BMI n'a pas d'effet cliniquement significatif sur la pharmacocinétique de la sitagliptine.
Données précliniquesSitagliptine et metformine
Des études précliniques toxicocinétiques et de toxicité orale ont été effectuées chez le chien avec la sitagliptine et la metformine sous forme d'association fixe à libération non retardée.
Dans une étude de toxicité orale de 16 semaines, des chiennes ont été traitées à la metformine 20 mg/kg/jour seule ou associée à 2, 10 ou 50 mg/kg/jour de sitagliptine. Une ataxie transitoire et/ou un tremblement ont été observés dans le groupe sous forte dose. Ces signes ont été jugés imputables à la sitagliptine, étant donné qu'ils avaient été observés dans d'autres études sur des chiens sous sitagliptine seule dosée à 50 mg/kg/jour. Dans cette étude, le niveau d'effet nul (no-effect level) pour les effets imputables au traitement a été de 10 mg/kg/jour de sitagliptine et de 20 mg/kg/jour de metformine, ce qui correspond chez l'homme à 6 fois l'exposition systémique à une dose journalière de 100 mg de sitagliptine et à 2,5 fois l'exposition systémique à une dose journalière de 2000 mg de metformine.
Aucune étude préclinique n'a été effectuée avec le médicament associé chlorhydrate de sitagliptine/metformine comprimés à libération retardée sur le potentiel cancérigène, la mutagénicité, la perturbation de la fertilité ou les effets sur la reproduction. Les données suivantes proviennent d'études séparées sur la sitagliptine seule et la metformine seule.
Sitagliptine
Lors d'études non cliniques chez des rats, des effets toxiques de la sitagliptine n'ont été observés qu'à des expositions largement supérieures à la dose humaine, de sorte qu'ils ne doivent pas être pris en compte pour l'utilisation normale chez l'être humain. Au cours d'études précliniques sur la sécurité, la sitagliptine ne s'est avérée ni génotoxique, ni mutagène.
Dans une série d'études sur la toxicité avec des administrations répétées du médicament à des chiens, des doses de 2, 10 et 50 mg/kg/jour ont été examinées pendant une période pouvant aller jusqu'à 53 semaines. Après 53 semaines de traitement avec une dose de 10 mg/kg/jour, ce qui correspond à six fois la dose recommandée pour l'adulte, de 100 mg/jour, aucun effet n'a été constaté. Chez des chiens ayant reçu la dose de 50 mg/kg/jour de sitagliptine (environ 26 fois l'exposition humaine), des symptômes physiques associés au traitement sont apparus passagèrement, par exemple une respiration avec la bouche ouverte, une salivation, des vomissements blanchâtres et mousseux, une ataxie, des tremblements, une diminution des activités et/ou une position du corps courbée. Lors des études sur la toxicité, de 14 et 27 semaines, avec une dose de 50 mg/kg/jour, une dégénérescence très légère à légère de la musculature squelettique a pu en outre être observée au niveau histologique. Au cours des études sur la toxicité de 53 semaines toutefois, aucune dégénérescence de la musculature squelettique n'a pu être constatée, ce qui laisse supposer qu'il s'agit d'un effet non reproductible ou que la modification n'apparaît plus avec un traitement prolongé. L'exposition systémique dans le domaine NOEL était lors de l'étude à 53 semaines (10 mg/kg/jour) 5 fois plus élevée que dans l'étude à 27 semaines (2 mg/kg/jour).
D'autres études de toxicité orale sur 3 mois ont été effectuées chez des singes rhésus et cynomolgus. L'étude de 3 mois chez le singe rhésus a examiné le potentiel de lésions cutanées ou de toxicité rénale de la sitagliptine, l'évaluation s'est limitée à la peau et aux reins. Les animaux ont reçu jusqu'à 100 mg de sitagliptine/kg/jour (28 fois l'exposition systémique d'une dose quotidienne de 100 mg). Il n'y a pas eu d'observations ante mortem ou post mortem dues au traitement.
Dans l'étude de toxicité de 3 mois menée chez le singe cynomolgus, les évaluations de routine complètes ont été effectuées. Les animaux ont reçu jusqu'à 100 mg de sitagliptine/kg/jour (27 fois l'exposition systémique d'une dose quotidienne de 100 mg). Il n'y a pas eu d'observations ante mortem ou post mortem dues au traitement.
La sitagliptine s'est avérée non cancérigène chez des souris ayant reçu pendant deux ans la dose maximale tolérée, de 500 mg/kg/jour, par voie orale. Une étude sur la cancérogénicité, de deux ans, a été réalisée avec des rats du sexe masculin et féminin, qui ont reçu des doses orales de 50, 150 et 500 mg/kg/jour de sitagliptine. Une augmentation des cas d'adénomes hépatiques et de cancers a été observée chez les rats du sexe masculin et une hausse des cas de cancers du foie a été constatée chez des rats du sexe féminin ayant reçu des doses élevées. Sur la base de la dose journalière recommandée chez l'être humain, de 100 mg/jour, cette dose correspondait à 58 fois l'exposition humaine chez des rats. Cette dose était associée, lors d'essais chez les rats, à une hépatotoxicité. Le No-observed-Effect-Level pour l'induction d'une néoplasie hépatique était de 150 mg/kg/jour, ce qui correspond à 19 fois l'exposition humaine à la dose recommandée de 100 mg. Étant donné qu'il a pu être montré que, chez le rat, l'induction d'une néoplasie hépatique était en corrélation avec l'hépatotoxicité, on peut considérer que la fréquence plus élevée des tumeurs hépatiques chez les rats était une conséquence de la toxicité hépatique chronique sous cette dose élevée. La signification clinique de ce résultat pour l'homme n'est pas connue.
Aucun effet négatif sur la fertilité de rats du sexe masculin et féminin ayant reçu de la sitagliptine par voie orale avant ou durant l'accouplement, à des doses pouvant aller jusqu'à 1000 mg/kg/jour (correspondant à près de cent fois l'exposition chez l'être humain, sur la base de la dose journalière recommandée, de 100 mg/jour pour l'adulte) n'a été observé.
Des études de toxicologie de la reproduction réalisées chez des rats ayant reçu des doses orales de 1000 mg/kg/jour ont montré une légère augmentation associée au traitement de la fréquence de malformations fœtales des côtes (côtes manquantes, hypoplasie et côtes déformées) dans la descendance. Le No-observed-Effect-Level pour les troubles du développement était de 250 mg/kg/jour (ce qui correspond à 32 fois l'exposition chez l'être humain, sur la base de la dose journalière recommandée, de 100 mg/jour chez l'adulte). La sitagliptine est excrétée dans le lait de rates allaitantes. Aucune étude n'a été effectuée avec la sitagliptine, la metformine et une sulfonylurée.
Chlorhydrate de metformine
Les données précliniques d'études conventionnelles de pharmacologie de sécurité, d'études de toxicité avec administrations répétées du médicament et d'études sur la génotoxicité, le potentiel carcinogène et la toxicité sur la reproduction montrent qu'aucun danger spécifique ne découle d'une prise de la metformine chez l'homme.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver dans l'emballage d'origine. Ne pas conserver au-dessus de 30°C.
Conserver hors de la portée des enfants.
Numéro d’autorisation69033 (Swissmedic).
PrésentationSitagliptin-Metformin-Mepha retard 50 mg/500 mg: plaquettes thermoformées de 60 et 160 comprimés à libération modifiée (B)
Sitagliptin-Metformin-Mepha retard 50 mg/1000 mg: plaquettes thermoformées de 60 et 160 comprimés à libération modifiée (B)
Sitagliptin-Metformin-Mepha retard 100 mg/1000 mg: plaquettes thermoformées de 30 et 90 comprimés à libération modifiée (B)
Titulaire de l’autorisationMepha Pharma AG, Basel.
Mise à jour de l’informationOctobre 2023
Numéro de version interne: 3.1
|