CompositionPrincipes actifs
Zilucoplan (sous forme de zilucoplan sodique)
Excipients
Dihydrogénophosphate sodique monohydraté
Phosphate disodique
Chlorure de sodium
Eau pour préparations injectables
Chaque seringue préremplie de 0,416 ml contient 1,72 – 2,05 mg de sodium.
Chaque seringue préremplie de 0,574 ml contient 2,39 – 2,85 mg de sodium.
Chaque seringue préremplie de 0,810 ml contient 3,37 – 4,01 mg de sodium.
Indications/Possibilités d’emploiZilbrysq est indiqué en association au traitement standard pour le traitement de la myasthénie auto-immune généralisée (MAg) chez les patients adultes présentant des anticorps anti-récepteurs à l'acétylcholine (R-ACh).
Posologie/Mode d’emploiZilbrysq est destiné à être utilisé sous la conduite et la surveillance de professionnels de santé, expérimentés dans la prise en charge des patients atteints de troubles neuromusculaires.
Avant de commencer le traitement par Zilbrysq, les patients doivent être vaccinés contre Neisseria meningitidis. Si le traitement doit commencer moins de 2 semaines après la vaccination antiméningococcique, le patient doit recevoir une antibioprophylaxie appropriée jusqu'à 2 semaines après la première dose de vaccination (voir rubriques «Contre-indications» et «Mises en garde et précautions»).
Posologie usuelle
La dose recommandée doit être administrée en une injection sous-cutanée quotidienne et approximativement à la même heure chaque jour.
Tableau 1: Dose quotidienne totale par catégorie de poids corporel
|
Poids corporel du patient
|
Dose*
|
Nombre de seringues préremplies par couleur
| |
Moins de 56 kg
|
16,6 mg
|
1 seringue préremplie (Rouge rubis)
| |
56 à moins de 77 kg
|
23 mg
|
1 seringue préremplie (Orange)
| |
77 kg et plus
|
32,4 mg
|
1 seringue préremplie (Bleu foncé)
|
* La dose recommandée correspond à approximativement 0,3 mg/kg
Le zilucoplan n'a pas été étudié chez les patients atteints d'une MAg de Classe V selon la Myasthenia Gravis Foundation of America (MGFA).
L'expérience est limitée chez les patients pesant moins de 43 kg et plus de 150 kg.
Oubli d'une dose
Si une dose de Zilbrysq est oubliée, elle doit être administrée le jour même, puis la dose normale prise le jour suivant. Ne pas administrer plus d'une dose par jour.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance hépatique légère à modérée. On ne dispose pas de données sur les patients présentant une insuffisance hépatique sévère (voir rubrique «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une insuffisance rénale. Il n'existe pas de données sur les patients nécessitant une dialyse (voir rubrique «Pharmacocinétique»).
Patients âgés
Aucun ajustement de la dose n'est nécessaire chez les patients âgés (voir rubrique «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité de Zilbrysq chez les enfants et les adolescents n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Zilbrysq est administré par injection sous-cutanée.
Les sites d'injection appropriés sont l'avant des cuisses, l'abdomen et l'arrière du bras (voir paragraphe «Pharmacocinétique» dans ce document et «Mode d'emploi» dans l'Information destinée aux patients).
Les sites d'injection doivent être alternés quotidiennement et les injections ne doivent pas être effectuées dans des zones où la peau est sensible, érythémateuse, meurtrie, indurée ou dans des zones où la peau présente des cicatrices ou des vergetures.
L'administration doit être effectuée par une personne ayant été formée à l'administration d'injections et conformément aux instructions détaillées fournies à la rubrique «Mode d'emploi» de l'Information destinée aux patients.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique «Composition».
Patients sans vaccination à jour contre Neisseria meningitidis (voir rubrique «Mises en garde et précautions»).
Patients présentant une infection non résolue à Neisseria meningitidis.
Mises en garde et précautionsInfections à Neisseria
Infection à méningocoque
En raison de son mécanisme d'action, l'utilisation de Zilbrysq peut augmenter la susceptibilité du patient aux infections à Neisseria meningitidis. Par mesure de précaution, tous les patients doivent être vaccinés contre les infections à méningocoque, au moins 2 semaines avant le début du traitement par Zilbrysq.
Si le traitement par Zilbrysq doit commencer moins de 2 semaines après la vaccination contre l'infection à méningocoque, le patient doit recevoir une antibioprophylaxie appropriée jusqu'à 2 semaines après la première dose de vaccination. Les vaccins anti-méningococciques réduisent mais n'éliminent pas complètement le risque d'infections à méningocoque.
Les vaccins contre les sérogroupes A, C, Y, W et B lorsque disponibles, sont recommandés pour prévenir les sérogroupes de méningocoques les plus fréquemment pathogènes. La vaccination et l'antibioprophylaxie doivent être effectuées conformément au plan de vaccination Suisse en vigueur.
Pendant le traitement par Zilbrysq, les patients doivent être surveillés pour détecter les signes et les symptômes d'une infection à méningocoque et être examinés immédiatement si une infection est suspectée. En cas de suspicion d'infection à méningocoque, des mesures appropriées, telles qu'un traitement par antibiotiques et l'arrêt du traitement par Zilbrysq, doivent être prises jusqu'à ce que l'infection à méningocoque puisse être exclue. Les patients doivent être informés qu'ils doivent consulter immédiatement un médecin en cas de signes ou de symptômes d'infection à méningocoque.
Les prescripteurs doivent être familiarisés avec les supports pédagogiques pour la prise en charge des infections à méningocoque et fournir une carte patient («Carte d'alerte patient») ainsi qu'un guide («Guide pour les patients/soignants») aux patients traités par zilucoplan.
Autres infections à Neisseria
Les patients traités par Zilbrysq peuvent également être sensibles aux infections causées par Neisseria sp. (autres que Neisseria meningitidis), comme les infections gonococciques. Les patients doivent être informés de l'importance de la prévention et du traitement de la gonorrhée.
Immunisation
Avant l'instauration d'un traitement par zilucoplan, il est recommandé d'initier les vaccinations chez les patients conformément aux recommandations actuelles.
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par seringue préremplie, c'est-à-dire qu'il est essentiellement «sans sodium».
InteractionsD'après les résultats des tests in vitro, aucune interaction cliniquement pertinente n'est attendue entre le zilucoplan et un inhibiteur ou un inducteur des principales enzymes CYP ou des transporteurs.
Le potentiel du zilucoplan à inhiber les enzymes CYP (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A et 4F2) et UGTs (1A1, 1A3, 1A4, 1A6, 1A9, 2B7 et 2B15) et les transporteurs (P-gp, BCRP, BSEP, MRP2, MRP3, MATE1, MATE2-k, NTCP, OCT1, OCT2, OAT1, OAT3, OATP1B1 et OATP1B3) a été étudié in vitro. De plus, le potentiel d'induction de CYP1A2, 2B6 et CYP3A4 par zilucoplan a été étudié.
Le zilucoplan inhibe MRP3 in vitro à des concentrations thérapeutiques; la pertinence clinique de cette inhibition est inconnue.
Compte tenu de l'effet inhibiteur potentiel du zilucoplan sur la cytotoxicité du rituximab dépendant du complément, le zilucoplan peut réduire les effets pharmacodynamiques attendus du rituximab.
Grossesse, allaitementGrossesse
Il n'existe pas de données sur l'utilisation de Zilbrysq chez la femme enceinte.
Les études effectuées chez l'animal ont montré une légère augmentation des pertes prénatales par rapport aux données de contrôle historiques, mais pas d'effets sur la parturition, le développement post-natal du nourrisson ni sur les pertes post-natales (voir section «Données précliniques»).
L'utilisation du zilucoplan pendant la grossesse et chez des femmes en âge de procréer et ne prenant pas de contraception est déconseillée et ne doit être envisagée que si le bénéfice clinique l'emporte sur les risques.
Allaitement
On ne sait pas si le zilucoplan est excrété dans le lait maternel ou absorbé par voie systémique après ingestion orale par les nouveau-nés/nourrissons. Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
La décision d'interrompre l'allaitement ou d'arrêter le traitement par le zilucoplan doit être prise en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme.
Fertilité
L'effet du zilucoplan sur la fertilité humaine n'a pas été évalué. Dans certaines études de fertilité et de toxicité à doses répétées sur des primates non humains, des résultats d'une pertinence clinique incertaine ont été observés sur les organes reproducteurs masculins et féminins (voir section «Données précliniques»).
Effet sur l’aptitude à la conduite et l’utilisation de machinesZilbrysq n'a aucune influence ou une influence négligeable sur l'aptitude à la conduite ou l'utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Un total de 115 patients ont été traités avec zilucoplan dans des études cliniques contrôlées par placebo dans MAg, représentant 26,4 patients-années d'exposition. Les effets indésirables les plus fréquemment rapportés étaient des réactions au site d'injection et des infections des voies respiratoires supérieures.
Liste des effets indésirables
Le Tableau 2 présente les effets indésirables du zilucoplan par fréquence selon la convention suivante: très fréquents (≥1/10), fréquents (≥1/100, < 1/10), peu fréquents (≥1/1 000, < 1/100), rares (≥1/10 000, < 1/1 000), très rares (< 1/10 000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
La fréquence des effets indésirables présentés dans le Tableau 2 a été établi sur la base des données groupées à partir d'études cliniques contrôlées par placebo (n=115) menées dans la MAg, à l'exception de la morphée, qui a été rapportée uniquement dans les études d'extension ouvertes à long terme (n=213) menées dans la MAg.
Tableau 2: Effets indésirables
|
Classe de systèmes d'organes
|
Fréquence
|
Effet indésirable
| |
Infections et infestations
|
Très fréquent
|
Infections des voies respiratoires supérieures (13,0 %)
| |
Affections gastro-intestinales
|
Fréquent
|
Diarrhée
| |
Affections de la peau et du tissu sous-cutané
|
Fréquent
|
Morphée a
| |
Troubles généraux et anomalies au site d'administration
|
Très fréquent
|
Réactions au site d'injection (25,2 %)
| |
Investigations
|
Fréquent
|
Augmentation de la lipase
| |
Fréquent
|
Augmentation de l'amylase
| |
Peu fréquent
|
Augmentation des éosinophiles sanguins
|
aLa morphée n'a été rapportée que dans les études cliniques à long terme en ouvert. La durée maximale de l'exposition au zilucoplan pendant les études cliniques à long terme était supérieure à 4 ans.
Description d'effets indésirables spécifiques et informations complémentaires
Réactions au site d'injection
Les réactions les plus fréquentes étaient: contusion, douleurs, nodules, prurit et hématome au site d'injection. Tous les cas étaient bénins, de gravité légère ou modérée, et moins de 3 % des réactions ont conduit à l'arrêt du traitement. Dans les études groupées contrôlées par placebo, des réactions au site d'injection ont été rapportées chez 25,2 % des patients traités par le zilucoplan et 15,5 % des patients traités par placebo.
Infections des voies respiratoires supérieures
Les infections les plus fréquentes étaient: rhinopharyngite, infection des voies respiratoires supérieures et sinusite. Plus de 95 % des cas étaient bénins, de gravité légère ou modérée, et n'ont pas conduit à l'arrêt du traitement. Dans les études groupées contrôlées par placebo, des infections des voies respiratoires supérieures ont été rapportées chez 13,0 % des patients traités par le zilucoplan et chez 7,8 % des patients traités par placebo.
Augmentation des enzymes pancréatiques
Des cas d'élévation de la lipase (5,2 %) et/ou de l'amylase (6,1 %) ont été observés. Ces élévations étaient transitoires et ont rarement conduit à l'arrêt du traitement. La majorité d'entre elles sont survenues dans les 2 mois suivant l'instauration du traitement par zilucoplan et sont revenues à la normale dans les 2 mois.
Augmentation des éosinophiles sanguins
Des élévations des éosinophiles sanguins ont été observées. Celles-ci étaient transitoires, n'ont pas conduit à l'arrêt du traitement et n'étaient pas associées à des dysfonctions d'organes cliniquement significatives.
Morphée
Des cas de morphée ont été observés après un traitement à long terme pendant l'étude d'extension en ouvert. La majorité des cas présentaient un délai d'apparition supérieur à un an après le début du traitement, étaient de sévérité légère à modérée et n'ont pas donné lieu à un arrêt du traitement
Immunogénicité
Comme tous les peptides thérapeutiques, le zilucoplan présente également un potentiel d'immunogénicité. La positivité aux anticorps anti-médicament dépend fortement de la sensibilité et de la spécificité du test utilisé. En outre, l'incidence observée de la présence d'anticorps anti-médicament peut être influencée par divers facteurs tels que la méthode de test, la manipulation des échantillons, le moment du prélèvement, les traitements médicamenteux concomitants et la maladie sous-jacente. Par conséquent, une comparaison de l'incidence des anticorps dirigés contre le zilucoplan avec l'incidence des anticorps dirigés contre d'autres principes actifs peut être trompeuse.
Dans l'étude MG0010, les patients étaient testés pour la positivité aux anticorps anti-médicament (AAM) et la positivité aux anticorps anti-polyéthylène glycol (PEG). Au total, 2 patients (2,3 %) dans chaque groupe, zilucoplan (0,3 mg/kg) et placebo, étaient positifs aux AAM. Au total, 8 patients (9,3 %) du groupe zilucoplan (0,3 mg/kg) et 6 patients (6,8 %) du groupe placebo ont été testés positifs aux anticorps anti-PEG. Les titres d'anticorps étaient faibles et n'indiquaient aucun lien entre un statut positif aux AAM ou un statut positif aux anticorps anti-PEG et l'incidence des effets indésirables.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans une étude sur des volontaires sains où 32 participants ont reçu des doses suprathérapeutiques de 0,6 mg/kg, administrées par voie sous-cutanée pendant 7 jours maximum, les données de sécurité étaient conformes au profil de sécurité à la dose recommandée.
En cas de surdosage, il est recommandé de surveiller étroitement le patient afin de détecter tout effet indésirable, et d'instaurer immédiatement des mesures de soutien appropriées.
Propriétés/EffetsCode ATC
L04AJ06
Mécanisme d'action
Le zilucoplan inhibe les effets de la protéine C5 du complément par un double mécanisme d'action. Il se lie spécifiquement à C5, inhibant ainsi son clivage par la C5 convertase en C5a et C5b, ce qui entraîne une régulation à la baisse de l'assemblage et de l'activité cytolytique du complexe d'attaque membranaire (CAM). De plus, en se liant à la fraction C5b de C5, le zilucoplan empêche stériquement la liaison de C5b à C6, ce qui empêche l'assemblage et l'activité ultérieurs du CAM, si du C5b est formé.
Pharmacodynamique
L'effet pharmacodynamique du zilucoplan a été analysé par l'aptitude d'inhibition ex vivo de la lyse des globules rouges de mouton (GRm) induite par le complément.
Les données des études de phase II et de phase III démontrent une inhibition rapide, complète (> 95 %) et durable du complément avec le zilucoplan lorsqu'il est dosé conformément au Tableau 1.
Efficacité clinique
L'innocuité et l'efficacité du zilucoplan ont été évaluées dans le cadre d'une étude multicentrique de 12 semaines, randomisée, en double aveugle, contrôlée par placebo MG0010 (RAISE) et de l'étude d'extension en ouvert MG0011 (RAISE-XT).
MG0010
Au total, 174 patients ont été inclus, âgés d'au moins 18 ans, présentant une myasthénie auto-immune généralisée (MAg) positive aux anticorps anti-récepteurs à l'acétylcholine, de classe II–IV selon la classification MGFA (légère à sévère), un score des activités de la vie quotidienne (MG-ADL, Myasthenia Gravis Activities of Daily Living) ≥6 et un score quantitatif de la myasthénie (QMG, Quantitative Myasthenia Gravis) ≥12 (voir Tableau 3).
Les patients ont été traités soit par le zilucoplan une fois par jour (dosé conformément au Tableau 1), soit par un placebo. Un traitement standard (SOC, Standard of Care ) stable était autorisé.
Le critère d'évaluation principal était l'évolution du score total MG-ADL (Myasthenia Gravis Activities of Daily Living Score) entre l'inclusion (change from baseline, CFB) et la semaine 12. Les principaux critères d'évaluation secondaires étaient l'évolution des scores totaux entre l'inclusion et la semaine 12: score QMG (Quantitative Myasthenia Gravis), score MGC (Myasthenia Gravis Composite) et MG-QoL15r (Myasthenia Gravis Quality of Life).
Tableau 3: Caractéristiques démographiques et cliniques des patients à l'inclusion dans l'étude MG0010
|
|
Zilucoplan (n= 86)
|
Placebo (n = 88)
| |
Âge, années, moyenne (ET)
|
52,6 (14,6)
|
53,3 (15,7)
| |
Âge au moment de l'apparition, années, moyenne (ET)
|
43,5 (17,4)
|
44,0 (18,7)
| |
Sexe, homme, n (%)
|
34 (39,5)
|
41 (46,6)
| |
Score MG-ADL à l'inclusion, moyenne (ET)
|
10,3 (2,5)
|
10,9 (3,4)
| |
Score QMG à l'inclusion, moyenne (ET)
|
18,7 (3,6)
|
19,4 (4,5)
| |
Score MGC à l'inclusion, moyenne (ET)
|
20,1 (6,0)
|
21,6 (7,2)
| |
Score MG-QoL 15r à l'inclusion, moyenne (ET)
|
18,6 (6,6)
|
18,9 (6,8)
| |
Réfractaire au traitement, oui, n (%)
|
44 (51,2)
|
44 (50,0)
| |
Durée de la maladie, années, moyenne (ET)
|
9,3 (9,5)
|
9,0 (10,4)
|
La majorité des participants recevaient déjà au début de l'étude un traitement par parasympathomimétiques (84,5 %) corticoïdes systémiques (63,2 %) et immunosuppresseurs (51,1 %).
L'effet du traitement dans le groupe zilucoplan pour les 4 critères d'évaluation a débuté rapidement à la semaine 1, a augmenté jusqu'à la semaine 4 et s'est maintenu jusqu'à la semaine 12.
À la semaine 12, une amélioration cliniquement pertinente et hautement statistiquement significative du score total MG-ADL (Figure 1) et du score total QMG a été observée pour le zilucoplan par rapport au placebo.
Figure 1: Evolution du score total MG-ADL depuis l'inclusion (CFB)
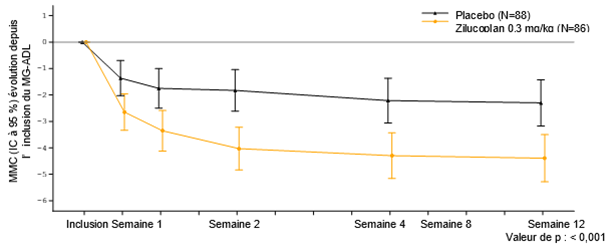
Analyse basée sur un modèle MMRM ANCOVA
Variation cliniquement pertinente= variation de 2 points du score MG-ADL
Le Tableau 4 présente l'évolution entre l'inclusion et la semaine 12 des scores totaux pour MG-ADL, QMG, MGC et MG-QoL15r.
Tableau 4: Evolution des scores totaux MG-ADL, QMG, MGC et MG-QoL15r entre l'inclusion et la semaine 12
|
Critères d'évaluation: Evolution des scores totaux entre l'inclusion et la semaine 12: Moyenne des MC (IC à 95 %)
|
Zilucoplan (n = 86)
|
Placebo (n = 88)
|
Variation du score sous zilucoplan versus placebo selon la méthode des moindres carrés (IC à 95 %)
|
p-value*
| |
MG-ADL
|
-4,39 (-5,28, -3,50)
|
-2,30 (-3,17, -1,43)
|
-2,09 (-3,24, -0,95)
|
< 0,001
| |
QMG
|
-6,19 (-7,29, -5,08)
|
-3,25 (-4,32, -2,17)
|
-2,94 (-4,39, -1,49)
|
< 0,001
| |
MGC
|
-8,62 (-10,22, -7,01)
|
-5,42 (-6,98, -3,86)
|
-3,20 (-5,24, -1,16)
|
0,0023
| |
MG-QoL15r
|
-5,65 (-7,17, -4,12)
|
-3,16 (-4,65, -1,67)
|
-2,49 (-4,45, -0,54)
|
0,0128
|
* Analyse basée sur un modèle ANCOVA MMRM
À la semaine 12, la proportion cumulative de patients ayant eu besoin d'un traitement de secours (immunoglobuline G par voie intraveineuse ou échange de plasma) était plus faible dans le groupe zilucoplan (5 %) que dans le groupe placebo (12 %).
MG0011
Deux cents patients ayant participé à une étude de phase II contrôlée par placebo (MG0009) ou l'étude de phase III (MG0010), ont continué dans l'étude de suivi en ouvert MG0011, dans le cadre de laquelle tous les patients recevaient du zilucoplan (dosé conformément au Tableau 1). L'objectif principal était d'obtenir des données de sécurité à long terme et, secondairement, d'observer l'évolution des critères d'efficacité par rapport à l'inclusion des scores MG-ADL, QMG, MGC et MG-QoL15r à la semaine 12 de la phase d'extension en ouvert (correspondant à la semaine 24 de la participation totale à l'étude).
Figure 2: Evolution moyenne du score total MG-ADL entre l'inclusion dans l'étude en double aveugle et la semaine 60
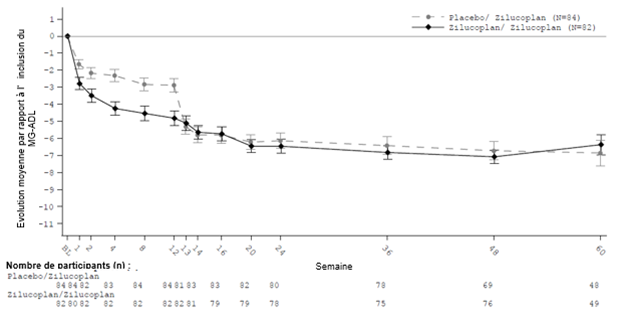
Tableau 5: Evolution moyenne des scores totaux pour MG-ADL, QMG, MGC et MG-QoL15r à la semaine 24 (semaine 12 de MG0011) et à la semaine 60 (semaine 48 de MG0011) par rapport à l'inclusion dans l'étude en double aveugle (MG0010)
|
Critères d'évaluation: Evolution du score total à la semaine 24 et à la semaine 60 par rapport à l'inclusion: Moyenne des MC (IC à 95 %)
|
Zilucoplan (n=82)
|
Placebo/zilucoplan (n=84)
| |
MG-ADL
|
| |
Semaine 24
|
-5,46 (0,59)
|
-5,20 (0,52)
| |
Semaine 60
|
-5,16 (0,61)
|
-4,37 (0,54)
| |
QMG
|
| |
Semaine 24
|
-7,10 (0,80)
|
-7,19 (0,69)
| |
Semaine 60
|
-6,44 (0,83)
|
-6,15 (0,71)
| |
MGC
|
| |
Semaine 24
|
-10,37 (1,15)
|
-11,12 (1,00)
| |
Semaine 60
|
-8,89 (1,20)
|
-9,01 (1,04)
| |
MG-QoL15r
|
| |
Semaine 24
|
-8,09 (0,96)
|
-7,96 (0,89)
| |
Semaine 60
|
-7,22 (0,99)
|
-6,09 (0,91)
|
Analyse basée sur un modèle de régression ANCOVA MMRM où le recours au traitement de secours et l'arrêt sont imputés comme échecs de traitement. Le pire score possible est imputé aux décès (par ex., score de 24 pour MG-ADL).
ET = erreur type
PharmacocinétiqueLes propriétés pharmacocinétiques du zilucoplan et de ses principaux métabolites circulants (RA102758 et RA103488) ont été étudiées chez des sujets adultes sains et des patients atteints de MAg.
Absorption
Après l'administration sous-cutanée quotidienne unique et multiple de la dose de zilucoplan recommandée (Tableau 1) chez des sujets sains, le zilucoplan a atteint sa concentration plasmatique maximale généralement entre 3 et 6 heures après administration de la dose.
Dans l'étude MG0010 menée chez des patients atteints de MAg, après une administration sous-cutanée quotidienne répétée de la dose de zilucoplan recommandée, les concentrations plasmatiques de zilucoplan étaient constantes, les concentrations minimales à l'état d'équilibre étant atteintes à la semaine 4 et maintenues jusqu'à la semaine 12.
Les expositions après l'administration sous-cutanée de doses uniques de zilucoplan dans l'abdomen, la cuisse ou la partie supérieure du bras étaient comparables.
Distribution
Le zilucoplan et ses deux métabolites principaux sont fortement liés aux protéines plasmatiques (> 99 %). Le volume moyen de distribution pour le zilucoplan (Vc/F) à partir d'une analyse pharmacocinétique de population est de 3,51 L.
Le zilucoplan n'est pas un substrat des transporteurs (P-gp, BCRP, OATP1B1 et OATP1B3).
Métabolisme
Le zilucoplan n'est pas un substrat des principales enzymes CYP (1A2, 2B6, 2C8, 2C9, 2C19, 2D6 et 3A). Dans le plasma, deux métabolites principaux, RA103488 et RA102758, ont été détectés. La formation de RA103488 est principalement due au cytochrome CYP450 4F2. RA103488 a une activité pharmacologique similaire à celle du zilucoplan, mais est présent à une concentration beaucoup plus faible par rapport au zilucoplan. La contribution de RA103488 à l'activité pharmacologique est faible. RA102758 est pharmacologiquement inactif.
Élimination
En tant que peptide, le zilucoplan devrait être dégradé en petits peptides et en acides aminés par les voies cataboliques. La demi-vie moyenne d'élimination terminale du plasma était d'environ 172 heures (7 à 8 jours). La demi-vie était de 220 heures et 96 heures pour le métabolite actif (RA103488) et le métabolite inactif majeur (RA102758), respectivement. L'excrétion du zilucoplan et de ses métabolites mesurée dans l'urine et les fèces était négligeable.
Linéarité/non-linéarité
Dans l'analyse pharmacocinétique de population (doses correspondant à 0,05 à 0,6 mg/kg), la pharmacocinétique du zilucoplan se caractérise par une concentration disponible du médicament au niveau de la cible, avec une augmentation de l'exposition inférieure à la proportionnalité des doses en cas de doses croissantes, même après comparaison de l'administration de doses multiples avec des doses uniques.
Anticorps
Les incidences d'anticorps anti-médicament AAM et d'anticorps anti-PEG dans l'étude de phase III chez les patients atteints de MAg étaient comparables entre le groupe de traitement par zilucoplan et le groupe de traitement par le placebo (voir rubrique «Effets indésirables»).
Le statut des anticorps AAM et des anticorps anti-PEG des patients traités par le zilucoplan n'a pas eu d'incidence sur les concentrations de zilucoplan.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Les effets de l'insuffisance hépatique modérée sur la pharmacocinétique du zilucoplan et de ses métabolites ont été étudiés dans une étude de phase I en ouvert, où une dose unique de zilucoplan à 0,3 mg/kg a été administrée à des sujets sains et à des sujets atteints d'insuffisance hépatique modérée.
L'exposition systémique au zilucoplan était inférieure de 24 % chez les sujets présentant une insuffisance hépatique modérée par rapport aux sujets sains, ce qui concorde avec une exposition systémique et une exposition maximale plus élevées pour les deux métabolites chez les sujets présentant une insuffisance hépatique par rapport aux sujets sains. L'exposition maximale au zilucoplan ainsi que la demi-vie terminale étaient comparables dans les deux groupes. Une analyse pharmacodynamique n'a pas identifié de différences significatives dans les taux de complément ou l'inhibition de l'activité du complément entre les deux groupes. Sur la base de ces résultats, aucun ajustement de dose n'est requis chez les patients présentant une insuffisance hépatique légère ou modérée.
Troubles de la fonction rénale
L'effet de l'insuffisance rénale sur la pharmacocinétique du zilucoplan et de ses métabolites a été étudié dans une étude de phase I en ouvert, où une dose unique de zilucoplan à 0,3 mg/kg a été administrée à des sujets sains et à des sujets atteints d'insuffisance rénale grave.
Sur la base des résultats pharmacocinétiques, aucun ajustement posologique n'est nécessaire chez les patients atteints d'insuffisance rénale.
Patients âgés
D'après une analyse pharmacocinétique de population, l'âge n'a pas influencé la pharmacocinétique du zilucoplan. Aucun ajustement de la dose n'est nécessaire.
Groupe ethnique
Dans une étude clinique de phase 1 menée chez des sujets sains caucasiens et japonais, le profil pharmacocinétique du zilucoplan et de ses deux métabolites principaux a été comparé après une dose unique de 0,3 mg/ et après des doses multiples de 0,3 mg/kg pendant 14 jours. Les résultats étaient généralement similaires entre les deux groupes.
L'analyse pharmacocinétique de population a démontré qu'il n'y a pas de différences entre les différentes catégories ethniques (Noirs/Afro-américains, Asiatiques/Japonais et Caucasiens). Aucun ajustement de la dose n'est nécessaire.
Sexe
Dans l'analyse pharmacocinétique de population, aucune différence de pharmacocinétique entre les sexes n'a été observée. Aucun ajustement de la dose n'est nécessaire.
Poids
L'analyse pharmacocinétique de population a montré que le poids corporel exerce une influence significative sur la pharmacocinétique du zilucoplan. La posologie du zilucoplan est basée sur les catégories de poids corporel (voir rubrique «Posologie/Mode d'emploi»). Aucun ajustement posologique supplémentaire n'est nécessaire.
Données précliniquesToxicité en administration répétée
Dans les études de toxicité à doses répétées menées sur des primates non humains, une dégénérescence vésiculaire/hyperplasie des cellules épithéliales et des infiltrats de cellules mononucléaires ont été observés dans divers tissus à une exposition cliniquement significative. Dans le pancréas, cela s'est parfois manifesté sous forme d'une dégénérescence des cellules acineuses pancréatiques, et parfois sous forme d'une fibrose et d'une dégénérescence /régénérescence canalaire, et s'est accompagné d'une élévation des concentrations plasmatiques d'amylase et de lipase. Les résultats chez les primates non humains sont d'une pertinence clinique incertaine et certains sont potentiellement liés à des infections secondaires à l'effet pharmacologique du zilucoplan, mais d'autres mécanismes ne peuvent être exclus.
Génotoxicité
Le zilucoplan a montré des résultats négatifs au test de mutagénicité in vitro (test d'Ames), au test d'aberration chromosomique in vitro, et au test du micronoyau in vivo sur des cellules de moelle osseuse de rat.
Carcinogénicité
Aucune étude de carcinogénicité n'a été réalisée avec le zilucoplan.
Toxicité sur la reproduction
Dans une étude de fertilité chez le singe mâle, une dégénérescence/déplétion minime à légère de la lignée germinale a été observée à des expositions cliniquement pertinentes, mais la gravité n'a pas augmenté avec la dose. Aucun impact sur la spermatogenèse n'a été observé. Dans les organes reproducteurs femelles (vagin, col de l'utérus, utérus), des infiltrats de cellules mononucléaires avec dégénérescence épithéliale et métaplasie épidermoïde du col de l'utérus ont été observés.
L'administration sous-cutanée de zilucoplan (0, 1, 2 ou 4 mg/kg/jour) à des singes gravides pendant toute la période de gestation a conduit avec tous les dosages à une légère augmentation des pertes prénatales, sans toxicité pour la mère. La dose testée la plus faible a été associée à une exposition maternelle (ASC) correspondant à la dose maximale recommandée chez l'homme de 32,4 mg/jour. Chez les primates non humains, aucun effet n'a été constaté sur la parturition, le développement post-natal du nourrisson, ni sur les pertes post-natales.
Remarques particulièresIncompatibilités
Non pertinent.
Stabilité
36 mois
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Conserver au réfrigérateur (2-8 °C).
Ne pas congeler.
Conserver la seringue préremplie dans son emballage extérieur pour protéger son contenu de la lumière.
Conserver hors de portée des enfants.
Les patients peuvent conserver la seringue préremplie de Zilbrysq à température ambiante dans son emballage d'origine jusqu'à 30 °C pendant une période unique de 3 mois maximum, à l'abri de la lumière. Une fois que Zilbrysq a été conservé à température ambiante, il ne doit pas être remis au réfrigérateur et doit être jeté s'il n'est pas utilisé dans les 3 mois ou avant la date de péremption, selon la première éventualité.
Numéro d’autorisation69066 (Swissmedic)
PrésentationZILBRYSQ 16,6 mg solution injectable en seringue préremplie (A)
0,416 ml de solution injectable en seringue préremplie avec piston rouge rubis
7 seringues préremplies
ZILBRYSQ 23 mg solution injectable en seringue préremplie (A)
0,574 ml de solution injectable en seringue préremplie avec piston orange
7 seringues préremplies
ZILBRYSQ 32,4 mg solution injectable en seringue préremplie (A)
0,810 ml de solution injectable en seringue préremplie avec piston bleu foncé
7 seringues préremplies
Titulaire de l’autorisationUCB-Pharma AG
Mise à jour de l’informationJuin 2024
|