CompositionPrincipes actifs
Vutrisiran (sous forme de vutrisiran sodique). Contient un petit acide ribonucléique interférent (pARNi) fabriqué par synthèse, modifié chimiquement.
Excipients
Dihydrogénophosphate de sodium dihydraté, phosphate disodique dihydraté, chlorure de sodium, eau pour préparations injectables, hydroxyde de sodium (pour l’ajustement du pH), acide phosphorique (pour l’ajustement du pH), contient 5,9 mg de sodium pour 1 mL.
Indications/Possibilités d’emploiAmvuttra est indiqué dans le traitement de l’amylose héréditaire à transthyrétine (amylose hATTR), chez les patients adultes atteints de polyneuropathie de stade 1 ou de stade 2.
Posologie/Mode d’emploiLe traitement doit être instauré sous la supervision d’un médecin expérimenté dans la prise en charge de l’amylose. Le traitement doit être démarré le plus tôt possible après l’apparition de la maladie afin d’empêcher le développement d’une invalidité.
Posologie usuelle
La posologie recommandée d’Amvuttra est de 25 mg administrés par injection sous-cutanée une fois tous les 3 mois.
Une supplémentation en vitamine A à une dose d’environ 2500 UI à 3000 UI par jour au maximum est recommandée pour les patients traités par Amvuttra (voir rubrique « Mises en garde et précautions »).
La décision de poursuivre le traitement chez les patients dont la maladie évolue en polyneuropathie de stade 3 doit être prise à la discrétion du médecin, après évaluation globale des bénéfices et des risques.
Patients présentant des troubles de la fonction hépatique
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance hépatique légère (bilirubine totale ≤ 1 x la limite supérieure de la normale (LSN) et aspartate aminotransférase (AST) > 1 x LSN, ou bilirubine totale > 1,0 à 1,5 x LSN et tout autre taux d’AST). Le vutrisiran n’a pas été étudié chez des patients présentant une insuffisance hépatique modérée ou sévère et ne doit être utilisé chez ces patients que si le bénéfice clinique attendu dépasse le risque potentiel encouru (voir rubrique « Pharmacocinétique »).
Patients présentant des troubles de la fonction rénale
Aucun ajustement posologique n’est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (débit de filtration glomérulaire estimé [DFGe] ≥ 30 à < 90 mL/min/1,73 m2). Le vutrisiran n’a pas été étudié chez les patients présentant une insuffisance rénale sévère ou une maladie rénale au stade terminal et ne doit être utilisé chez ces patients que si le bénéfice clinique attendu dépasse le risque potentiel encouru (voir rubrique « Pharmacocinétique »).
Patients âgés
Aucun ajustement posologique n’est nécessaire chez les patients âgés de 65 ans et plus (voir rubrique « Pharmacocinétique »).
Enfants et adolescents
La sécurité et l’efficacité d’Amvuttra chez les enfants ou les adolescents de moins de 18 ans n’ont pas encore été établies. Aucune donnée n’est disponible.
Prise retardée
En cas d’oubli d’une dose, Amvuttra doit être administré dès que possible. L’administration doit être reprise tous les 3 mois, à compter de la dernière dose administrée.
Mode d’administration
Amvuttra est destiné à une administration par voie sous-cutanée uniquement. Amvuttra peut être administré par un professionnel de santé, par le patient ou par un aidant.
Les patients ou les aidants peuvent administrer Amvuttra après avoir été formés par un professionnel de santé à la technique correcte d’injection sous-cutanée.
Ce médicament est prêt à l’emploi et à usage unique exclusivement.
Inspecter visuellement la solution pour vérifier l’absence de particules et de coloration anormale. Ne pas utiliser en présence de coloration anormale ou de particules.
Avant l’administration, si elle est conservée au froid, la seringue préremplie doit être réchauffée en laissant le carton à température ambiante pendant environ 30 minutes.
▪ L’injection sous-cutanée doit être administrée dans l’un des sites suivants : l’abdomen, les cuisses ou les bras. En cas d’injection dans le bras, celle-ci doit être effectuée par un professionnel de santé ou un aidant. Amvuttra ne doit pas être injecté dans du tissu cicatriciel ou dans des zones rougies, enflammées ou gonflées.
▪ En cas d’injection dans l’abdomen, la zone entourant le nombril doit être évitée.
Contre-indicationsHypersensibilité grave (p. ex. anaphylaxie) à la substance active ou à l’un des excipients mentionnés dans la rubrique « Composition ».
Mises en garde et précautionsCarence en vitamine A
En réduisant le taux de la protéine transthyrétine (TTR) sérique, le traitement par Amvuttra entraîne une diminution des taux sériques de vitamine A (rétinol) (voir rubrique « Propriétés/Effets »). Les taux sériques de vitamine A inférieurs à la limite inférieure de la normale doivent être corrigés et tout symptôme ou signe oculaire dû à une carence en vitamine A doit être évalué avant l’instauration du traitement par Amvuttra.
Les patients traités par Amvuttra doivent prendre une supplémentation orale quotidienne de vitamine A d’environ 2500 UI à 3000 UI maximum, afin de réduire le risque potentiel de symptômes oculaires dus à une carence en vitamine A. Une consultation ophtalmologique est recommandée chez les patients présentant des symptômes oculaires pouvant indiquer une carence en vitamine A, notamment une vision nocturne réduite ou une cécité nocturne, une sécheresse oculaire persistante, une inflammation oculaire, une inflammation ou une ulcération de la cornée, un épaississement ou une perforation de la cornée.
Au cours des 60 premiers jours de la grossesse, des taux de vitamine A trop élevés ou trop faibles peuvent être associés à un risque accru de malformation fœtale. Par conséquent, toute grossesse doit être exclue avant l’instauration d’un traitement par Amvuttra et les femmes en âge de procréer doivent utiliser une méthode de contraception efficace (voir rubrique « Grossesse, Allaitement »). Si une femme planifie une grossesse, Amvuttra et la supplémentation en vitamine A doivent être interrompus et les taux sériques de vitamine A doivent être surveillés et revenus à la normale avant la tentative de conception. Les taux sériques de vitamine A peuvent rester faibles pendant plus de 12 mois après la dernière dose d’Amvuttra.
En cas de grossesse non planifiée, le traitement par Amvuttra doit être interrompu (voir rubrique « Grossesse, Allaitement »). Aucune recommandation ne peut être donnée concernant la poursuite ou l’interruption de la supplémentation en vitamine A au cours du premier trimestre d’une grossesse non planifiée. Si la supplémentation en vitamine A est poursuivie, la dose quotidienne ne doit pas dépasser 3000 UI par jour, en raison du manque de données justifiant des doses plus élevées. Par la suite, une supplémentation en vitamine A de 2500 UI à 3000 UI par jour devra être reprise aux deuxième et troisième trimestres si les taux sériques de vitamine A ne sont pas encore revenus à la normale, en raison du risque accru de carence en vitamine A au troisième trimestre.
On ignore si la supplémentation en vitamine A pendant la grossesse sera suffisante pour prévenir le déficit en vitamine A si la femme enceinte continue de recevoir Amvuttra. Cependant, il n’est pas attendu que l’augmentation de la supplémentation en vitamine A à plus de 3000 UI par jour pendant la grossesse permette le rétablissement d’un taux plasmatique normal de rétinol en raison du mécanisme d’action d’Amvuttra. Une telle supplémentation peut être dangereuse pour la mère et le fœtus.
Autres composants
Ce médicament contient moins de 1 mmol (23 mg) de sodium par mL, c.-à-d. qu’il est essentiellement « sans sodium ».
InteractionsAucune étude clinique d’interaction n’a été réalisée. Le vutrisiran ne devrait pas causer d’interactions ni être influencé par des inhibiteurs ou des inducteurs des enzymes du cytochrome P450, ni moduler l’activité des transporteurs. Par conséquent, le vutrisiran ne devrait pas avoir d’interactions cliniquement significatives avec d’autres médicaments.
Grossesse, AllaitementFemmes en âge de procréer
Le traitement par Amvuttra diminue les taux sériques de vitamine A. Des taux de vitamine A trop élevés ou trop faibles peuvent être associés à une augmentation du risque de malformation fœtale. Par conséquent, toute grossesse doit être exclue avant l’instauration du traitement et les femmes en âge de procréer doivent utiliser une méthode de contraception efficace. Si une femme envisage une grossesse, Amvuttra et la supplémentation en vitamine A doivent être interrompus et les taux sériques de vitamine A doivent être contrôlés et revenus à la normale avant la tentative de conception (voir rubrique « Mises en garde et précautions »). Les taux sériques de vitamine A peuvent rester faibles pendant plus de 12 mois après la dernière dose du traitement.
Grossesse
Il n’existe pas de données sur l’utilisation d’Amvuttra chez la femme enceinte. Les études effectuées chez l’animal sont insuffisantes pour permettre de conclure sur la toxicité sur la reproduction (voir rubrique « Données précliniques »). En raison du risque de tératogénicité dû à des taux de vitamine A non équilibrés, Amvuttra ne doit pas être utilisé pendant la grossesse. Par mesure de précaution, les taux de vitamine A (voir rubrique « Mises en garde et précautions ») et d’hormone thyréostimulante ou thyréostimuline (TSH) doivent être mesurés au début de la grossesse. Une surveillance étroite du fœtus doit être effectuée en cas de grossesse non planifiée, en particulier durant le premier trimestre.
Allaitement
On ne sait pas si le vutrisiran est excrété dans le lait maternel. Il n’existe pas de données suffisantes sur l’excrétion du vutrisiran dans le lait animal (voir rubrique « Données précliniques »).
La décision d’interrompre l’allaitement ou d’interrompre le traitement par Amvuttra doit tenir compte du bénéfice de l’allaitement pour l’enfant par rapport au bénéfice du traitement pour la femme.
Fertilité
Il n’existe pas de données concernant l’effet d’Amvuttra sur la fertilité chez l’homme. Aucune incidence sur la fertilité mâle ou femelle n’a été détectée dans les études effectuées chez l’animal (voir rubrique « Données précliniques »).
Effet sur l’aptitude à la conduite et l’utilisation de machinesAmvuttra n’a aucune influence ou une influence négligeable sur l’aptitude à la conduite ou l’utilisation de machines.
Effets indésirablesRésumé du profil de sécurité
Au cours de la période de traitement de 18 mois de l’étude HELIOS-A, les effets indésirables les plus fréquemment signalés chez les patients traités par Amvuttra étaient des extrémités douloureuses (15 %) et des arthralgies (11 %).
Liste des effets indésirables
Les effets indésirables sont présentés par termes préférentiels et par classe de systèmes d’organes (SOC) selon la terminologie MedDRA. La fréquence des effets indésirables est exprimée selon les catégories suivantes :
«Très fréquent» (≥ 1/10)
«Fréquent» (≥ 1/100, < 1/10)
«Peu fréquent» (≥ 1/1000, < 1/100)
Tableau 1 : Effets indésirables signalés pour Amvuttra
|
Classe de systèmes d’organes
|
Effet indésirable
|
Fréquence
| |
Affections respiratoires, thoraciques et médiastinales
|
Dyspnéea
|
Fréquent
| |
Affections musculo-squelettiques et du tissu conjonctif
|
Arthralgie
|
Très fréquent
| |
Extrémités douloureuses
|
Très fréquent
| |
Troubles généraux et anomalies au site d’administration
|
Réaction au site d’injectionb
|
Fréquent
| |
Investigations
|
Phosphatase alcaline sanguine augmentée
|
Fréquent
| |
a
Inclut dyspnée, dyspnée d’effort et dyspnée paroxystique nocturne
b Les symptômes rapportés comprenaient des bleus, un érythème, une douleur, un prurit et une chaleur. Les réactions au site d’injection étaient légères, transitoires et n’ont pas conduit à l’interruption du traitement.
|
Description d’effets indésirables spécifiques et informations complémentaires
Immunogénicité
Au cours de la période de traitement de 18 mois de l’étude HELIOS-A, 4 (3,3 %) patients traités par Amvuttra ont développé des anticorps anti-médicament (ADA). Les titres d’ADA étaient faibles et transitoires et ne semblaient pas affecter l’efficacité clinique, le profil de sécurité, ou le profil pharmacocinétique ou pharmacodynamique du vutrisiran.
L’annonce d’effets secondaires présumés après l’autorisation est d’une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d’effet secondaire nouveau ou grave via le portail d’annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch
SurdosageEn cas de surdosage, il est recommandé de suivre le patient, si cela est indiqué sur le plan médical, afin de déceler tout signe ou symptôme de réactions indésirables et d’instaurer un traitement symptomatique adapté, si nécessaire.
Propriétés/EffetsCode ATC
N07XX18
Mécanisme d’action
Amvuttra contient du vutrisiran, un petit acide ribonucléique interférent (pARNi) double brin, stabilisé chimiquement, qui cible spécifiquement les ARN messagers (ARNm) mutés et sauvages de la transthyrétine (TTR) et qui est lié de manière covalente à un ligand contenant trois résidus de N-acétylgalactosamine (GalNAc) permettant de délivrer le pARNi aux hépatocytes.
Grâce à un processus naturel appelé interférence par ARN (ARNi), le vutrisiran provoque la dégradation catalytique de l’ARNm de la TTR dans le foie, ce qui entraîne une diminution des taux sériques de protéine TTR mutée et sauvage.
Pharmacodynamique
Le taux sérique moyen de TTR était réduit dès le jour 22, avec une réduction moyenne de la TTR de 73 %, proche de l’état d’équilibre, à la semaine 6. Avec des doses répétées de 25 mg tous les 3 mois, les réductions moyennes du taux sérique de TTR après 9 et 18 mois de traitement étaient de 83 % et 88 %, respectivement. Des réductions similaires de taux de TTR ont été observées indépendamment du génotype (V30M ou non V30M), de la prise antérieure d’un stabilisateur de TTR, du poids, du sexe, de l’âge ou de l’origine ethnique.
La TTR sérique est un transporteur de la protéine 4 de liaison au rétinol, qui est le principal transporteur de la vitamine A dans le sang. Amvuttra a réduit les taux de vitamine A avec des réductions moyennes maximales et minimales à l’état d’équilibre de 70 % et 63 %, respectivement (voir rubriques «Mises en garde et précautions» et «Interactions»).
Efficacité clinique
L’efficacité d’Amvuttra a été évaluée dans une étude clinique internationale, randomisée et en ouvert (HELIOS-A) chez des patients adultes atteints d’amylose hATTR avec polyneuropathie. Les patients ont été randomisés selon une proportion de 3:1 pour recevoir 25 mg d’Amvuttra (N = 122) par voie sous-cutanée une fois tous les 3 mois, ou 0,3 mg/kg de patisiran (N = 42) par voie intraveineuse une fois toutes les 3 semaines. La période de traitement de l’étude s’est déroulée sur 18 mois avec deux analyses, à 9 et 18 mois. Quatre-vingt-dix-sept pour cent (97 %) des patients traités par Amvuttra ont terminé au moins 18 mois des traitements attribués (vutrisiran ou patisiran). Les évaluations de l’efficacité étaient basées sur une comparaison entre le bras vutrisiran de l’étude et un groupe placebo externe (bras placebo de l’étude de phase 3 APOLLO) composé d’une population similaire de patients atteints d’amylose hATTR avec polyneuropathie. L’évaluation de la non-infériorité de la réduction du taux sérique de TTR était basée sur la comparaison entre le bras vutrisiran et le bras patisiran au sein de l’étude.
Parmi les patients ayant reçu Amvuttra, l’âge médian des patients à l’entrée dans l’étude était de 60 ans (intervalle de 34 à 80 ans), 38 % étaient âgés de 65 ans ou plus, et 65 % des patients étaient des hommes. Vingt-deux (22) variants différents de la TTR étaient représentés : V30M (44 %), T60A (13 %), E89Q (8 %), A97S (6 %), S50R (4 %), V122I (3 %), L58H (3 %) et Autres (18 %). Vingt pour cent (20 %) des patients présentaient le génotype V30M et des symptômes d’apparition précoce (< 50 ans). À l’entrée dans l’étude, 69 % des patients étaient atteints d’une maladie de stade 1 (capacité ambulatoire non altérée ; neuropathie sensitive et motrice légère autonome des membres inférieurs) et 31 % étaient atteints d’une maladie de stade 2 (aide à la marche nécessaire, déficience modérée des membres inférieurs, des membres supérieurs et du tronc). Il n’y avait pas de patients atteints d’une maladie de stade 3. Soixante-et-un pour cent (61 %) des patients avaient déjà été traités par des stabilisateurs de tétramère de TTR. Selon la classification de l’insuffisance cardiaque de la New York Heart Association (NYHA), 9 % des patients présentaient une insuffisance cardiaque de classe I et 35 % présentaient une insuffisance cardiaque de classe II. Trente-trois pour cent (33 %) des patients remplissaient les critères prédéfinis d’atteinte cardiaque (définie comme une épaisseur de la paroi du ventricule gauche (VG) ≥ 13 mm à l’entrée dans l’étude, sans antécédents d’hypertension ou de valvulopathie aortique).
Le critère d’évaluation principal de l’efficacité était le changement à 18 mois par rapport à sa valeur initiale du score modifié de déficience neuropathique mNIS+7 (modified Neuropathy Impairment Score). Ce critère d’évaluation est une mesure composite de la neuropathie motrice, sensitive et autonome comprenant des évaluations de la force motrice et des réflexes, des tests sensitifs quantitatifs, des études sur la conduction nerveuse, et de la tension artérielle orthostatique, avec un score allant de 0 à 304 points, où un score croissant indique une aggravation de la déficience.
Le changement, par rapport à sa valeur initiale, du score total au questionnaire de Norfolk sur la qualité de vie dans la neuropathie diabétique (QdV-ND) a été évalué comme un critère secondaire. Le questionnaire sur la QdV-ND de Norfolk (résultats déclarés par le patient) comprend des domaines relatifs aux petites fibres, aux grosses fibres, à la fonction nerveuse autonome, aux symptômes de polyneuropathie et aux activités de la vie quotidienne, le score total allant de - 4 à 136, où un score croissant indique une détérioration de la qualité de vie.
Les autres critères d’évaluation secondaires comprenaient la vitesse de marche (test de marche de 10 mètres), l’état nutritionnel (IMCm) et l’aptitude à accomplir les activités de la vie quotidienne et à participer à la vie sociale (selon l’échelle d’incapacité globale de Rasch [R-ODS]), selon les déclarations des patients.
Le traitement par Amvuttra dans l’étude HELIOS-A a montré des améliorations statistiquement significatives de tous les critères d’évaluation (Tableau 2 et Figure 1) mesurés entre l’entrée dans l’étude et les 9e et 18e mois, par rapport au groupe placebo externe de l’étude APOLLO (tous p < 0,0001).
La réduction moyenne en pourcentage du taux de TTR jusqu’au 18e mois était de 84,7 % pour le vutrisiran et de 80,6 % pour le patisiran. Le pourcentage de réduction des taux sériques de TTR dans le groupe vutrisiran était non inférieur (selon les critères prédéfinis) à celui du groupe patisiran au sein de l’étude jusqu’au 18e mois, avec une différence médiane de 5,3 % (IC à 95 % : 1,2 %, 9,3 %).
Tableau 2 : Résumé des résultats d’efficacité clinique de l’étude HELIOS-A
|
Critère d’évaluationa
|
Référence, moyenne
(E-T)
|
Évolution par rapport à la référence au 18e mois, moyenne des MC (ETM)
|
Amvuttra placebob
Différence entre les traitements, moyenne des MC (IC à 95 %)
|
Valeur p
| |
Amvuttra
N = 122
|
Placebob
N = 77
|
Amvuttra
|
Placebob
| |
9e mois
| |
mNIS+7c
|
60,6 (36,0)
|
74,6 (37,0)
|
-2,2 (1,4)
|
14,8 (2,0)
|
-17,0
(-21,8 ; -12,2)
|
p < 0,0001
| |
QdV-ND de Norfolkc
|
47,1 (26,3)
|
55,5 (24,3)
|
-3,3 (1,7)
|
12,9 (2,2)
|
-16,2
(-21,7 ; -10,8)
|
p < 0,0001
| |
Test de marche sur 10 mètres (m/s)d
|
1,01 (0,39)
|
0,79 (0,32)
|
0 (0,02)
|
-0,13 (0,03)
|
0,13
(0,07 ; 0,19)
|
p < 0,0001
| |
18e mois
| |
mNIS+7c
|
60,6 (36,0)
|
74,6 (37,0)
|
-0,5 (1,6)
|
28,1 (2,3)
|
-28,5
(-34,0 ; -23,1)
|
p < 0,0001
| |
QdV-ND de Norfolkc
|
47,1 (26,3)
|
55,5 (24,3)
|
-1,2 (1,8)
|
19,8 (2,6)
|
-21.0
(-27,1 ; -14,9)
|
p < 0,0001
| |
Test de marche sur 10 mètres (m/s)d
|
1,01 (0,39)
|
0,79 (0,32)
|
-0,02 (0,03)
|
-0,26 (0,04)
|
0,24
(0,15 ; 0,33)
|
p < 0,0001
| |
IMCme
|
1057,5 (233,8)
|
989,9 (214,2)
|
25,0 (9,5)
|
-115,7 (13,4)
|
140,7
(108,4 ; 172,9)
|
p < 0,0001
| |
R-ODSf
|
34,1 (11,0)
|
29,8 (10,8)
|
-1,5 (0,6)
|
-9,9 (0,8)
|
8,4
(6,5 ; 10,4)
|
p < 0,0001
|
Abréviations : IC = intervalle de confiance ; moyenne des MC = moyenne des moindres carrés ; IMCm = indice de masse corporelle modifié ; mNIS = modified Neuropathy Impairment Score ; (score de déficience neuropathique modifié) ; QoL-ND = Quality of Life - Diabetic Neuropathy (Qualité de vie – Neuropathie diabétique) ; E-T = écart type ; ETM = erreur type de la moyenne
a Tous les critères d’évaluation du 9e mois ont été analysés à l’aide de l’analyse de covariance (ANCOVA) avec la méthode d’imputation multiple (IM) et tous les critères d’évaluation du 18e mois ont été analysés à l’aide du modèle à effets mixtes à mesures répétées (mixed effect model repeated measures, MMRM)b Groupe placebo externe de l’étude contrôlée randomisée APOLLOc Un nombre moins élevé indique une déficience moindre/la présence de symptômes moins nombreux
d Un nombre plus élevé indique une incapacité moindre/déficience moindre
e IMCm : indice de masse corporelle (IMC ; kg/m2) multiplié par l’albumine sérique (g/L) ; un nombre plus élevé indique un meilleur état nutritionnel.
f Un nombre plus élevé indique une incapacité moindre/déficience moindre.
Figure 1 : Évolution du score mNIS+7 par rapport à l’entrée dans l’étude (9e et 18e mois)
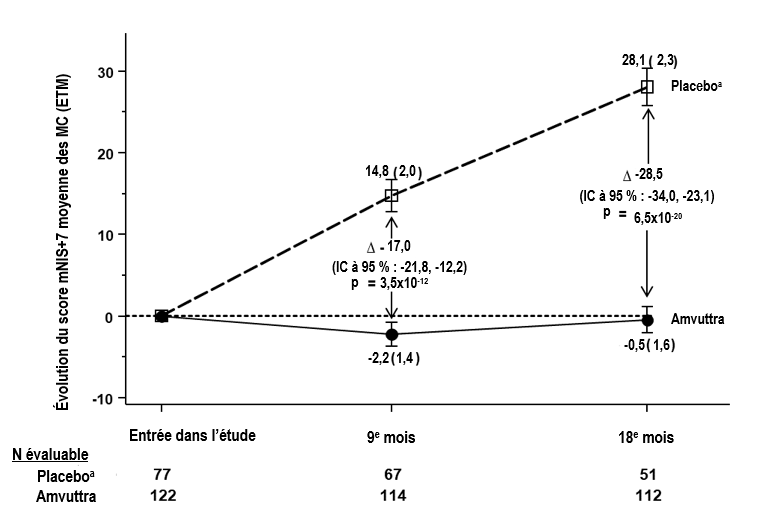
Une diminution du score mNIS+7 indique une amélioration
D indique la différence entre les groupes de traitement, représentée par la différence de moyenne des MC (IC à 95 %) pour AMVUTTRA - placebo externe.
Tous les critères d’évaluation du 9e mois ont été analysés à l’aide de l’analyse de la covariance (ANCOVA) avec la méthode d’imputation multiple (IM) et tous les critères d’évaluation du 18e mois ont été analysés à l’aide du modèle à effets mixtes à mesures répétées (MMRM)
a Groupe placebo externe de l’étude contrôlée randomisée APOLLO
Par rapport au placebo, les patients recevant Amvuttra ont connu des bénéfices similaires au niveau du score total mNIS+7 et du questionnaire de qualité de vie QdV-ND de Norfolk aux 9e et 18e mois dans tous les sous-groupes, y compris pour l’âge, le sexe, l’origine ethnique, l’origine géographique, le score sur l’échelle de déficience neurologique NIS, le génotype V30M, la prise antérieure d’un stabilisateur de TTR, le stade de la maladie et les patients présentant ou non des critères prédéfinis d’atteinte cardiaque.
La fraction N-terminale du peptide natriurétique de type B (NTproBNP) est un biomarqueur pronostique de dysfonction cardiaque. Les valeurs à l’entrée dans l’étude du NTproBNP (moyenne géométrique) étaient de 273 ng/L et 531 ng/L chez les patients traités par Amvuttra et par le placebo, respectivement. Au 18e mois, la moyenne géométrique des taux de NTproBNP a diminué de 6 % chez les patients sous Amvuttra, alors qu’une augmentation de 96 % a été observée chez les patients sous placebo.
Les échocardiogrammes évalués de façon centralisée ont montré des modifications de l’épaisseur de la paroi du VG (différence de moyenne des MC : -0,18 mm [IC à 95 % : -0,74 ; 0,38]) et de la déformation longitudinale (différence de moyenne des MC : -0,4 % [IC à 95 % : -1,2 ; 0,4]) avec le traitement par Amvuttra par rapport au placebo.
Malgré les valeurs observées pour le NTproBNP et l’épaisseur de la paroi du VG, un bénéfice clinique en ce qui concerne la cardiomyopathie reste à confirmer.
Population pédiatrique
Swissmedic a accordé une dérogation à l’obligation de soumettre les résultats d’études réalisées avec le vutrisiran dans tous les sous-groupes de la population pédiatrique en cas d’amylose hATTR (voir rubrique « Posologie/Mode d’emploi » pour les informations concernant l’usage pédiatrique).
PharmacocinétiqueLes propriétés pharmacocinétiques d’Amvuttra ont été caractérisées en mesurant les concentrations plasmatiques et urinaires de vutrisiran.
Absorption
Après administration sous-cutanée, le vutrisiran est rapidement absorbé avec un temps jusqu’à la concentration plasmatique maximale (tmax) de 3,0 heures (intervalle : 2,0 à 6,5). Au schéma posologique recommandé de 25 mg une fois tous les 3 mois par voie sous-cutanée, les concentrations maximales moyennes (% coefficient de variation [% CV]) à l’état d’équilibre (Cmax), et l’aire sous la courbe de la concentration en fonction du temps entre 0 et 24 heures (ASC0-24) étaient respectivement de 0,12 μg/mL (64,3 %), et 0,80 μg·h/mL (35,0 %). Il n’y a pas eu d’accumulation de vutrisiran dans le plasma après des administrations trimestrielles répétées.
Distribution
Le vutrisiran se lie à plus de 80 % aux protéines plasmatiques dans la plage de concentrations observées chez l’homme à la dose de 25 mg une fois tous les 3 mois par voie sous-cutanée. La liaison du vutrisiran aux protéines plasmatiques était dépendante de la concentration et diminuait avec l’augmentation des concentrations de vutrisiran (de 78 % à 0,5 µg/mL à 19 % à 50 µg/mL). L’estimation de la population pour le volume apparent de distribution du compartiment central (Vd/F) du vutrisiran chez l’homme était de 10,2 L (% d’erreur standard relative [RSE] = 5,71 %). Le vutrisiran est principalement distribué dans le foie après l’administration sous-cutanée.
Métabolisme
Le vutrisiran est métabolisé par des endonucléases et des exonucléases en courts fragments nucléotidiques de taille variable dans le foie. Il n’y a pas de métabolites majeurs circulant chez l’homme. Des études in vitro indiquent que le vutrisiran n’est pas métabolisé par les enzymes du CYP450.
Élimination
Après l’administration d’une dose unique de 25 mg par voie sous-cutanée, la clairance plasmatique apparente médiane était de 21,4 L/h (intervalle : 19,8 ; 30,0). La demi-vie d’élimination terminale (t1/2) médiane du vutrisiran était de 5,23 (intervalle : 2,24 ; 6,36) heures. Après l’administration d’une dose unique sous-cutanée de 5 à 300 mg, la fraction moyenne de substance active sous forme inchangée éliminée dans l’urine a varié de 15,4 à 25,4 % et la clairance rénale moyenne a varié de 4,45 à 5,74 L/h pour le vutrisiran.
Linéarité/non-linéarité
Après l’administration de doses sous-cutanées uniques comprises entre 5 et 300 mg, la Cmax du vutrisiran s’est avérée proportionnelle à la dose, tandis que l’aire sous la courbe de la concentration en fonction du temps à partir du moment de l’administration extrapolée à l’infini (ASCinf) et l’aire sous la courbe de la concentration en fonction du temps à partir du moment de l’administration jusqu’à la dernière concentration mesurable (ASCdern) étaient légèrement plus que proportionnelles à la dose.
Relations pharmacocinétique/pharmacodynamique
Des analyses pharmacocinétiques/pharmacodynamiques de population chez des sujets sains et des patients atteints d’amylose hATTR (n = 202) ont montré une relation dose-dépendante entre les concentrations hépatiques prédites de vutrisiran et les réductions du taux sérique de TTR. Les réductions médianes maximales, minimales et moyennes du taux de TTR prédites par le modèle à l’état d’équilibre étaient respectivement de 88 %, 86 % et 87 %, confirmant une variabilité minimale entre la Cmax et la Cmin pendant l’intervalle d’administration de 3 mois. L’analyse des covariables a indiqué une réduction similaire du taux de TTR chez les patients présentant une insuffisance rénale légère à modérée ou une insuffisance hépatique légère, ainsi que selon le sexe, l’origine ethnique, la prise antérieure de stabilisateurs de TTR, le génotype (V30M ou non V30M), l’âge et le poids.
Cinétique pour certains groupes de patients
Sexe et origine ethnique
Les études cliniques n’ont pas permis d’identifier de différences significatives à l’état d’équilibre au niveau des paramètres pharmacocinétiques ou de la réduction du taux de TTR, en fonction du sexe ou de l’origine ethnique.
Troubles de la fonction hépatique
Les analyses pharmacocinétiques et pharmacodynamiques de population n’ont indiqué aucune incidence d’une insuffisance hépatique légère (bilirubine totale ≤ 1 x LSN et AST > 1 x LSN, ou bilirubine totale > 1,0 à 1,5 x LSN et tout autre taux d’AST) sur l’exposition au vutrisiran ou la réduction du taux de TTR par rapport aux patients ayant une fonction hépatique normale. Le vutrisiran n’a pas été étudié chez des patients présentant une insuffisance hépatique modérée ou sévère.
Troubles de la fonction rénale
Les analyses pharmacocinétiques et pharmacodynamiques de population n’ont indiqué aucune incidence d’une insuffisance rénale légère ou modérée (TFGe ≥ 30 à < 90 mL/min/1,73 m2) sur l’exposition au vutrisiran ou la réduction du taux de TTR par rapport aux sujets ayant une fonction rénale normale. Le vutrisiran n’a pas été étudié chez les patients présentant une insuffisance rénale sévère ou une maladie rénale au stade terminal.
Patients âgés
Dans l’étude HELIOS-A, 46 patients (38 %) traités par vutrisiran étaient âgés de 65 ans ou plus et parmi ces patients, 7 (5,7 %) étaient âgés de 75 ans ou plus. Il n’y avait pas de différences significatives à l’état d’équilibre au niveau des paramètres pharmacocinétiques ou de la réduction du taux de TTR, entre les patients âgés de moins de 65 ans et ceux de 65 ans ou plus.
Données précliniquesL’administration sous-cutanée répétée une fois par mois de vutrisiran à ≥ 30 mg/kg chez le singe a produit les réductions durables attendues du taux de TTR circulant (jusqu’à 99 %) et de la vitamine A (jusqu’à 89 %) sans aucune toxicité apparente.
Après l’administration répétée d’une dose mensuelle pendant une période allant jusqu’à 6 mois chez le rat et 9 mois chez le singe, les modifications histologiques légères et cohérentes non défavorables dans le foie (hépatocytes, cellules de Kupffer), les reins (tubules rénaux), les ganglions lymphatiques et les sites d’injection (macrophages) ont reflété la distribution et l’accumulation principales du vutrisiran. Cependant, aucune toxicité n’a été identifiée pour une ASC plasmatique jusqu’à 1000 et 3000 fois plus élevée, normalisée à la dose trimestrielle et comparée à l’exposition anticipée à la dose maximale recommandée chez l’homme (DMRH).
Génotoxicité et carcinogénicité
Le vutrisiran n’a pas montré de potentiel génotoxique in vitro ou in vivo. Le vutrisiran n’a pas été cancérogène chez le rat et la souris mâle. Chez des souris femelles recevant le vutrisiran à la dose de 3, 9 ou 18 mg/kg une fois par mois, il a été observé une tendance dépendante de la dose statistiquement significative aux adénomes et carcinomes hépatocellulaires combinés, dont la pertinence chez l’homme n’est pas connue. Lorsque toutes les données toxicologiques sont prises en compte, le potentiel cancérogène du vutrisiran est considéré comme faible.
Toxicité sur la reproduction
Le vutrisiran n’est pas pharmacologiquement actif chez le rat et le lapin, ce qui limite la prédictivité de ces études. Néanmoins, une dose unique d’un orthologue du vutrisiran spécifique au rat n’a pas eu d’impact sur la fertilité et le développement embryonnaire précoce dans une étude combinée chez le rat.
Des administrations sous-cutanées hebdomadaires de vutrisiran n’ont pas eu d’effet sur la fertilité et le développement embryonnaire précoce à plus de 300 fois la DMRH normalisée. Dans une étude embryo-foetale avec administration sous-cutanée quotidienne de vutrisiran chez des rates gravides, des effets indésirables sur le poids corporel maternel, la consommation alimentaire, l’augmentation des naissances prématurées et la perte post-implantation ont été observés avec une dose sans effet nocif observable (DSENO) maternelle de 10 mg/kg/jour qui était plus de 300 fois la DMRH normalisée de 0,005 mg/kg/jour. Sur la base d’une réduction défavorable du poids corporel et de variations squelettiques accrues des fœtus à ≥ 10 mg/kg/jour, la DSENO fœtale du vutrisiran était de 3 mg/kg/jour, soit 97 fois la DMRH normalisée.
Dans une étude sur le développement embryo-fœtal chez des lapines gravides, aucun effet indésirable sur le développement embryo-fœtal n’a été observé à ≤ 30 mg/kg/jour de vutrisiran, soit plus de 1900 fois la DMRH normalisée.
Dans une étude de développement pré-/post-natal, l’administration de vutrisiran par voie sous-cutanée tous les 6 jours n’a eu aucun effet sur la croissance et le développement de la progéniture avec une DSENO de 20 mg/kg, soit plus de 90 fois la DMRH normalisée.
Remarques particulièresIncompatibilités
En l’absence d’études de compatibilité, ce médicament ne doit pas être mélangé avec d’autres médicaments.
Stabilité
Ce médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l’emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de 30°C. Ne pas congeler.
Tenir hors de portée des enfants.
Remarques concernant la manipulation
Veuillez tenir compte de l’annexe.
Numéro d’autorisation69074
PrésentationAMVUTTRA® 25 mg, solution injectable dans une seringue préremplie (en verre de type I) avec une aiguille en acier inoxydable de 29 Gauge dotée d’un protège-aiguille.
Chaque emballage contient 1 seringue préremplie à usage unique [B].
Titulaire de l’autorisationAlnylam Switzerland GmbH, Zoug
Mise à jour de l’informationJuin 2025
ANNEXE : Mode d’emploi
Amvuttra 25 mg solution injectable en seringue préremplie
Vutrisiran
Seringue préremplie unidose avec protège-aiguille
Veuillez lire ces instructions avant d’utiliser cette seringue préremplie.
Informations sur la seringue préremplie
La seringue préremplie (désignée par la « seringue ») est jetable et à usage unique.
Mode et voie d’administration
Chaque boîte contient une seringue d’Amvuttra à usage unique. Chaque seringue d’Amvuttra contient 25 mg de vutrisiran pour injection sous la peau (injection sous-cutanée) tous les trois mois.
Avant que vous ou votre aidant ne prépariez et injectiez une dose d’Amvuttra vous-mêmes, votre médecin, votre pharmacien ou votre infirmier/ère vous montrera ou montrera à votre aidant comment procéder. Pour des conseils supplémentaires et une assistance si nécessaire, contactez votre pharmacien, votre infirmier/ère ou votre médecin.
Conservez ce mode d’emploi jusqu’à ce que la seringue ait été utilisée.
Comment conserver Amvuttra
À conserver à une température ne dépassant pas 30 °C.
Ne pas congeler.
Tenir ce médicament hors de la vue et de la portée des enfants.
Mises en garde importantes
Ne pas utiliser le médicament si la boîte est endommagée ou semble avoir été ouverte.
Ne pas utiliser la seringue si elle est tombée sur une surface dure.
Ne pas toucher le piston avant d’être prêt(e) à injecter.
Ne retirer le capuchon de l’aiguille qu’immédiatement avant l’injection.
Ne replacer à aucun moment le capuchon sur la seringue.
|
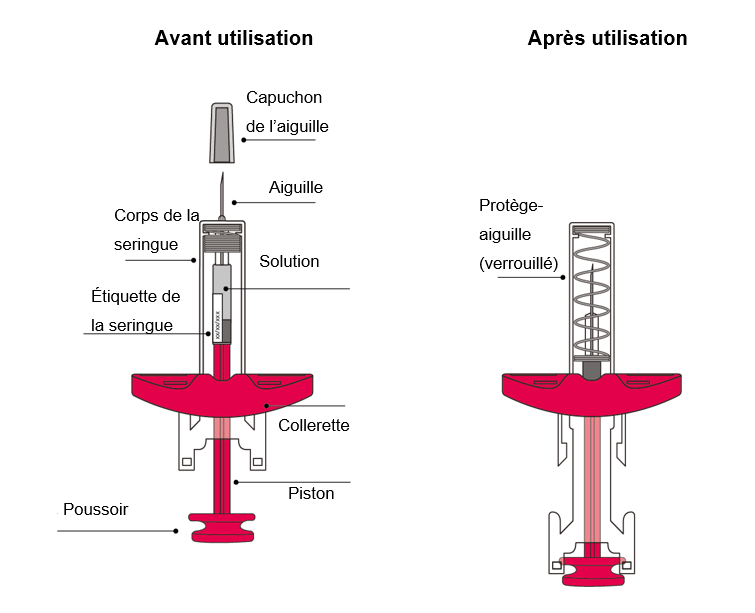
Comment se présente la seringue avant et après utilisation :
|
|
|
|
|
Étape 1 : rassembler le matériel
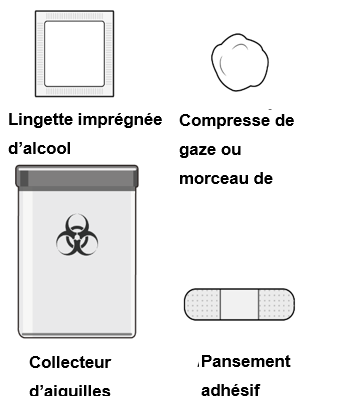
Rassembler le matériel ci-dessous (non fourni) et le placer sur une surface plane et propre :
·Lingette imprégnée d’alcool
·Compresse de gaze ou morceau de coton
·Pansement adhésif
·Collecteur d’aiguilles
|
|
| |
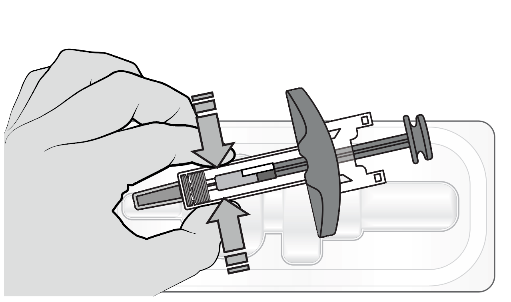
Étape 2 : préparer la seringue
Si le médicament est conservé au froid, laisser la seringue se réchauffer à température ambiante pendant au moins 30 minutes avant utilisation.
Ne pas réchauffer la seringue d’une autre façon, par exemple au micro-ondes, dans de l’eau chaude ou près d’une autre source de chaleur.
Retirer la seringue de l’emballage en saisissant le corps de la seringue.
Ne pas toucher le piston avant d’être prêt(e) à injecter.
Ne pas utiliser la seringue si elle est tombée sur une surface dure.
Ne retirer le capuchon de l’aiguille qu’immédiatement avant l’injection.
|
|
| |
Étape 3 : examiner la seringue
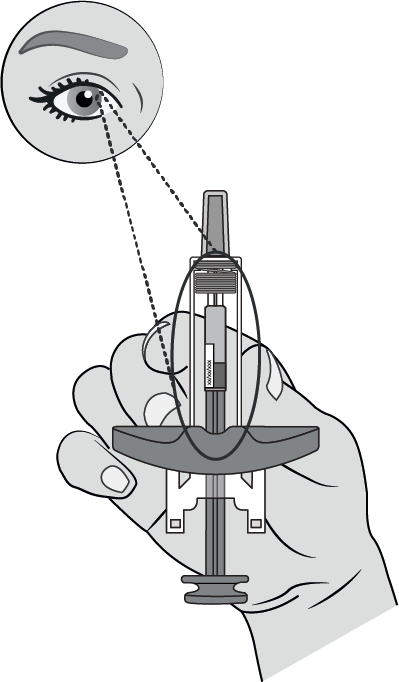
Vérifier ce qui suit :
ü La seringue n’est pas endommagée, fissurée ou ne présente pas de fuite.
ü Le capuchon de l’aiguille est intact et fixé à la seringue.
ü La solution dans la seringue est limpide et incolore à jaune.
ü La mention « Amvuttra 25 mg » figure sur l’étiquette de la seringue.
ü La date de péremption indiquée sur l’étiquette de la seringue.
Il est normal de voir des bulles d’air dans la seringue.
Ne pas utiliser la seringue si un quelconque problème est constaté lors du contrôle de la seringue et de la solution.
Ne pas utiliser si la date de péremption est dépassée.
Ne pas utiliser si la solution contient des particules, est trouble ou présente une couleur anormale.
Pour tout problème, contacter un professionnel de santé.
|
|
| |
Étape 4 : choisir le site d’injection
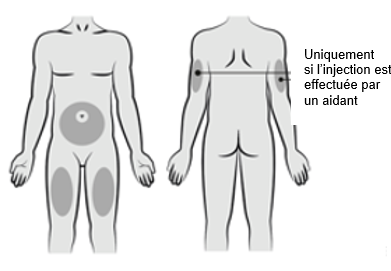
Choisir un site d’injection parmi les régions suivantes :
·Abdomen, à l’exception de la zone de 5 cm autour du nombril.
·Face avant de la cuisse.
·Si l’injection est effectuée par un tiers, la face arrière du haut du bras peut également être utilisée.
Ne pas injecter dans des zones présentant une sensibilité, une rougeur, un gonflement, une ecchymose ou une induration ni dans la zone de 5 cm autour du nombril.
|
|
| |
Étape 5 : préparer l’injection
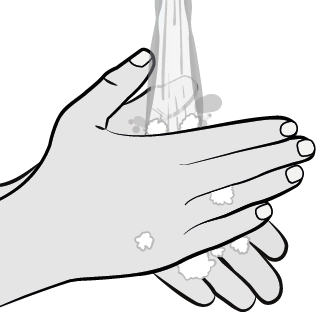
Se laver les mains à l’eau et au savon et les sécher soigneusement avec une serviette propre.
|
|
| |
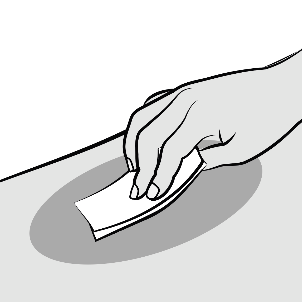
Nettoyer le site d’injection choisi à l’aide d’une lingette imprégnée d’alcool.
Laisser la peau sécher à l’air avant de procéder à l’injection. Éviter de toucher le site d’injection ou de souffler dessus après l’avoir nettoyé.
|
|
| |
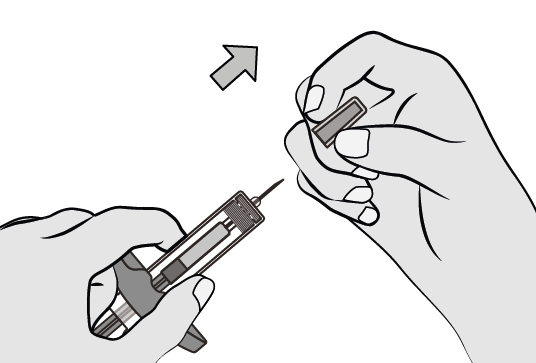
Étape 6 : retirer le capuchon de l’aiguille
Tenir le corps de la seringue d’une main.
De l’autre main, retirer le capuchon de l’aiguille en tirant droit et le jeter immédiatement.
Il est normal de voir une goutte de liquide à la pointe de l’aiguille.
Ne pas toucher l’aiguille ni la laisser toucher une surface quelconque.
Ne pas replacer le capuchon sur la seringue.
Ne pas tirer le piston.
Ne pas utiliser la seringue si elle est tombée sur une surface dure.
|
|
| |
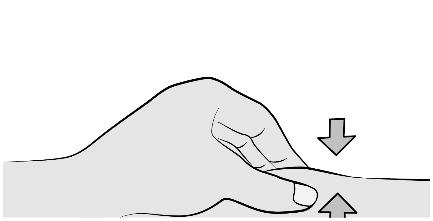
Étape 7 : insérer l’aiguille
De la main libre, pincer délicatement la peau nettoyée autour du site d’injection afin de créer une surélévation pour l’injection.
|
|
| |
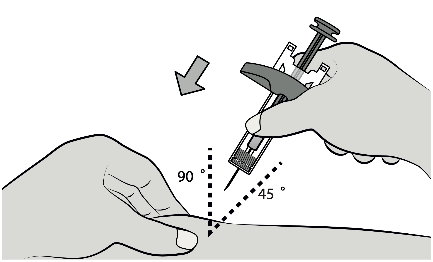
Insérer complètement l’aiguille dans la peau pincée à un angle de 45° à 90°.
|
|
| |
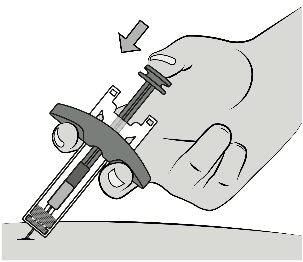
Étape 8 : injecter le médicament
À l’aide du poussoir, appuyer sur le piston avec deux doigts posés sur la collerette.
|
|
| |
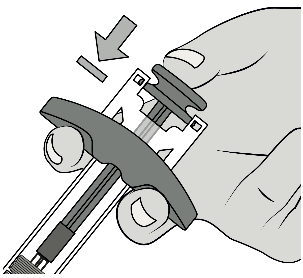
Appuyer à fond sur le piston, aussi loin que possible, afin d’injecter la totalité de la solution.
Le piston doit être enfoncé à fond pour que la dose soit administrée.
|
|
| |
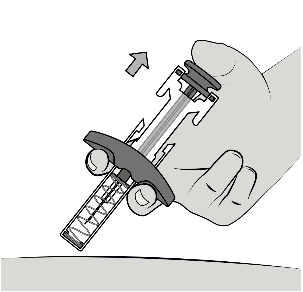
Étape 9 : relâcher le piston
Relâcher le piston afin que le protège-aiguille recouvre l’aiguille.
Retirer l’aiguille de la peau.
Ne pas bloquer le mouvement du piston.
Ne pas tirer le protège-aiguille vers le bas. Le protège-aiguille recouvre automatiquement l’aiguille.
|
|
| |
Étape 10 : contrôler le site d’injection
Il peut y avoir un peu de sang ou de liquide au niveau du site d’injection.
Dans ce cas, exercer une pression sur le site d’injection avec une compresse de gaze ou un morceau de coton jusqu’à ce que le saignement s’arrête.
Éviter de frotter le site d’injection.
|
| |
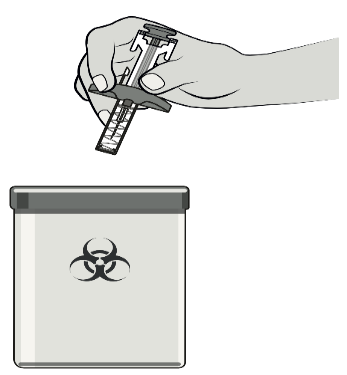
Étape 11 : jeter la seringue
Jeter immédiatement la seringue usagée dans un collecteur d’aiguilles.
Les seringues ne doivent être jetées que dans un collecteur d’aiguilles.
|
|
|
|