CompositionPrincipes actifs
Atogépant
Excipients
Copovidone K 28, tocofersolan, mannitol (E 421), cellulose microcristalline, chlorure de sodium, croscarmellose sodique, dioxyde de silicium hautement dispersé, fumarate de stéaryle de sodium.
1 comprimé de 10 mg ou 60 mg contient respectivement 5,26 mg et 31,48 mg de sodium.
Indications/Possibilités d’emploiTraitement prophylactique de la migraine chez l'adulte, pour autant que celui-ci soit indiqué.
Posologie/Mode d’emploiL'indication de la thérapie doit être posée par un médecin ayant de l'expérience dans le domaine du traitement de la migraine et accompagnée par celui-ci dans la suite du traitement.
Posologie usuelle
La dose orale recommandée d'AQUIPTA est de 60 mg une fois par jour.
Ajustement de la posologie en raison d'interactions
Les ajustuements de la posologie en cas d'administration concomitante de certains médicaments sont présentés dans le tableau 1 (voir «Interactions»).
Tableau 1: Ajustements de la posologie en raison d'interactions pour certains groupes de patients
|
Ajustements de la dose
|
Dose recommandée (une fois par jour)
| |
Inhibiteurs puissants du CYP3A4
|
10 mg
| |
Inhibiteurs des OATP
|
10 mg
| |
Insuffisance rénale sévère et insuffisance rénale terminale (CLcr <30 ml/min)
|
10 mg
|
Instructions posologiques particulières
Patients âgés
La modélisation pharmacocinétique de population suggère qu'il n'y a pas de différences pharmacocinétiques cliniquement significatives entre les sujets âgés (≥65 ans) et les sujets plus jeunes participant à l'étude. Aucun ajustement de la posologie d'AQUIPTA n'est nécessaire chez les patients âgés.
Patients présentant des troubles de la fonction hépatique
L'utilisation d'AQUIPTA est à éviter chez les patients atteints d'insuffisance hépatique sévère. Aucun ajustement de la posologie n'est recommandé chez les patients atteints d'insuffisance hépatique légère ou modérée (voir «Pharmacocinétique»).
Patients présentant des troubles de la fonction rénale
Chez les patients atteints d'insuffisance rénale sévère (CLcr 15-29 ml/min) et chez les patients atteints d'insuffisance rénale terminale (end-stage renal disease, ESRD) (CLcr < 15 ml/min), la dose recommandée d'AQUIPTA est de 10 mg une fois par jour. Chez les patients atteints d'ESRD et recevant une dialyse intermittente, AQUIPTA doit être pris de préférence après la dialyse. Aucun ajustement de la posologie n'est recommandé chez les patients atteints d'insuffisance rénale légère ou modérée (voir «Pharmacocinétique»).
Enfants et adolescents
La sécurité et l'efficacité d'AQUIPTA chez les enfants et les adolescents de moins de 18 ans n'ont pas encore été prouvées. Aucune donnée n'est disponible. AQUIPTA n'est pas autorisé pour une utilisation dans la population pédiatrique.
Retard dans l'administration de la dose
Lorsqu'une dose a été oubliée, elle doit être prise le plus rapidement possible. Lorsqu'une dose été oubliée pendant une journée entière, il ne faut pas prendre la dose oubliée mais prendre la dose suivante comme prévu.
Mode d'administration
AQUIPTA doit être pris par voie orale une fois par jour avec ou sans repas.
Contre-indicationsHypersensibilité au principe actif ou à l'un des excipients (voir «Composition») (voir «Mises en garde et précautions»).
Mises en garde et précautionsRéactions d'hypersensibilité
Des réactions d'hypersensibilité, notamment une anaphylaxie, une dyspnée, une éruption cutanée, un prurit, de l'urticaire et un œdème facial, ont été signalées lors de l'emploi d'AQUIPTA (voir «Effets indésirables»). Certaines réactions d'hypersensibilité peuvent survenir plusieurs jours après l'administration. En cas de réaction d'hypersensibilité, AQUIPTA doit être arrêté et un traitement approprié doit être instauré.
Troubles de la fonction hépatique
AQUIPTA n'est pas recommandé chez les patients dont la fonction hépatique est fortement réduite (voir «Posologie/Mode d'emploi»).
Patients non évalués dans les études cliniques de phase 3
Les patients présentant des maladies cardiovasculaires ou cérébrovasculaires cliniquement pertinentes, telles qu'une cardiopathie ischémique, des troubles du rythme ou de la conduction cardiaque, un infarctus du myocarde, un accident ischémique transitoire, une insuffisance cardiaque ou une hypertension artérielle non contrôlée, ont été exclus des études pivots. Aucune donnée de sécurité n'est disponible pour ces patients.
Céphalées liées à une surconsommation de médicaments (MOH, medication overuse headache)
Une utilisation excessive de médicaments pour le traitement aigu des céphalées peut les aggraver. Bien qu'il n'existe aucune preuve que la prise d'atogépant une fois par jour en traitement préventif puisse entraîner des MOH, un diagnostic de MOH doit être suspecté chez les patients qui ont régulièrement ou quotidiennement des céphalées malgré (ou à cause de) la prise régulière de médicaments pour le traitement aigu. Si c'est le cas ou si l'on soupçonne une telle éventualité, il convient d'arrêter le médicament pour le traitement aigu.
Sodium
AQUIPTA 10 mg comprimés contient moins de 1 mmol de sodium (23 mg) par comprimé, c'est-à-dire qu'il est essentiellement «sans sodium».
AQUIPTA 60 mg comprimés contient 31,48 mg de sodium par comprimé, ce qui équivaut à 1,6 % de l'apport alimentaire quotidien maximal en sodium de 2 g recommandé par l'OMS pour un adulte.
InteractionsEffet d'autres médicaments sur AQUIPTA
Atogepant est principalement éliminé par voie métabolique, principalement par le CYP3A4.
In vitro, l'atogépant est un substrat de la Pgp, de la BCRP, d'OATP1B1, d'OATP1B3 et d'OAT1. L'atogépant n'est pas un substrat de la d'OAT3, d'OCT2 ni de MATE1.
Des études d'interactions ont été menées avec les médicaments administrés de manière concomitante et listés dans le tableau ci-dessous.
Tableau 2. Effets cliniques d'autres médicaments sur l'atogépant
|
Médication concomitante (enzyme ou transporteur)
|
Dosage de la médication concomitante
|
Dosage d'atogépant
|
GMRa (90% CIb)
|
Dose recommandée pour l'atogépant
| |
Cmax
|
AUC
| |
Itraconazole
(Inhibiteur puissant du CYP3A4)
|
200 mg 1x par jour pendant 7 jours
|
60 mg en dose unique
|
2,15 (1,95, 2,37
|
5,51 (5,09, 5,96)
|
10 mg une fois par jour
| |
Rifampicine
(Inhibiteur d'OATP)
|
600 mg en dose unique
|
60 mg en dose unique
|
2,23 (1,99, 2,50)
|
2,85 (2,60, 3,12)
|
10 mg une fois par jour
| |
Rifampicine
(Inducteur puissant du CYP3A4)
|
600 mg 1x par jour pendant 7 jours
|
60 mg en dose unique
|
0,70 (0,60, 0,81)
|
0,39 (0,35, 0,44)
|
Aucun ajustement de la dose d'atogépant n'est recommandé
| |
Topiramate
(inducteur faible du CYP3A4)
|
100 mg 2x par jour pendant 5 jours
|
60 mg 1x par jour pendant 17 jours
|
0,76 (0,68, 0,85)
|
0,75 (0,69, 0,81)
| |
Quinidine
(inhibiteur de la Pgp)
|
648 mg 2x par jour pendant 4 jours
|
60 mg en dose unique
|
1,04 (0,89, 1,22)
|
1,26 (1,11, 1,43)
| |
Esoméprazole
(inhibiteur de la pompe à protons)
|
40 mg 1x par jour pendant 7 jours
|
60 mg en dose unique
|
0,77 (0,68, 0,86)
|
0,92 (0,84, 1,01)
| |
Famotidine
(Antagoniste du récepteur H2)
|
2x 20 mg
|
60 mg en dose unique
|
0,51
(0,41, 0,63)
|
0,79
(0,67, 0,93)
| |
Sumatriptan
(Agoniste du récepteur 5-HT1B/1D)
|
100 mg en dose unique
|
60 mg en dose unique
|
0,78 (0,69, 0,89)
|
0,95 (0,86, 1,05)
| |
Ubrogépant
(Antagoniste du récepteur CGRP)
|
100 mg le jour 1 et tous les 3 jours du jour 7-28
|
60 mg 1x par jour du jour 2-28
|
1,04 (0,94, 1,15)
|
1,04 (0,98, 1,12)
| |
Naproxène
(AINS)
|
500 mg en dose unique
|
60 mg en dose unique
|
1,00 (0,91, 1,11)
|
0,99 (0,92, 1,06)
| |
Paracétamol
(analgésique, anti-pyrétique)
|
1000 mg en dose unique
|
60 mg en dose unique
|
1,00 (0,90, 1,11)
|
1,13 (1,04, 1,22)
|
aGMR – Rapport entre les moyennes géométriques défini comme l'exposition (concentration maximale ou aire sous la courbe AUC) à l'atogépant quand il est utilisé avec une médication concomitante, divisé par l'exposition à l'atogépant sans médication concomitante.
bCI = Intervalle de confiance
Effet d'AQUIPTA sur d'autres médicaments
In vitro, l'atogépant n'est pas un inhibiteur des CYPs 3A4, 1A2, 2B6, 2C8, 2C9, 2C19 ou 2D6 à des concentrations cliniquement pertinentes. L'atogépant n'inhibe pas la MAO-A ni l'UGT1A1 à des concentrations cliniquement pertinentes. Il est peu probable que l'atogépant provoque des interactions médicamenteuses cliniquement significatives en inhibant le CYP450, la MAO-A ou l'UGT1A1. L'atogépant n'est pas un inducteur des CYP1A2, CYP2B6 ou CYP3A4 à des concentrations cliniquement pertinentes.
In vitro, l'atogépant n'est pas un inhibiteur de la Pgp, de la BCRP, d'OAT1, d'OAT3, de la NTCP, de la BSEP, de la MRP3 ni de la MRP4 à des concentrations cliniquement pertinentes. L'atogépant est un inhibiteur faible d'OATP1B1, d'OATP1B3, d'OCT1 et de MATE1. Il est peu probable que l'atogépant provoque des interactions médicamenteuses avec ces transporteurs.
Des études d'interactions ont été menées avec les médicaments administrés de manière concomitante et listés dans le tableau ci-dessous.
Tableau 3. Effets cliniques d'atogépant sur d'autres médicaments
|
Médication concomitante (enzyme ou transporteur)
|
Dosage de la médication concomitante
|
Dosage d'atogépant
|
GMRa (90% CIb)
|
Dose recommandée pour le médicament concomitant
| |
Cmax
|
AUC
| |
Topiramate
(faible inducteur du CYP3A4)
|
100 mg 2x par jour pendant 11 jours
|
60 mg 1x par jour pendant 7 jours
|
0,94 (0,87, 1,01)
|
0,94 (0,88, 1,01)
|
Aucun ajustement de la dose du médicament concomitant n'est recommandé
| |
Sumatriptan
(Agoniste du récepteur 5-HT1B/1D)
|
100 mg en dose unique
|
60 mg en dose unique
|
0,95 (0,85, 1,07)
|
1,02 (0,97, 1,08)
| |
Ubrogépant
(Antagoniste du récepteur CGRP)
|
100 mg le jour 1 et tous les 3 jours du jour 7-28
|
60 mg 1x par jour du jour 2-28
|
1,26 (1,06, 1,49)
|
1,19 (1,09, 1,30)
| |
Ethinylestradiol (œstrogène)
|
0,03 mg en dose unique
|
60 mg 1x par jour pendant 17 jours
|
0,90 (0,84, 0,96)
|
1,00 (0,96, 1,05)
| |
Lévonorgestrel (progestatif)
|
0,15 mg en dose unique
|
60 mg 1x par jour pendant 17 jours
|
1,09 (1,03, 1,17)
|
1,19 (1,13, 1,26)
| |
Naproxène
(AINS)
|
500 mg en dose unique
|
60 mg en dose unique
|
0,94 (0,90, 0,97)
|
0,98 (0,96, 1,00)
| |
Paracétamol
(analgésique, anti-pyrétique)
|
1000 mg en dose unique
|
60 mg en dose unique
|
0,89 (0,81, 0,97)
|
0,94 (0,89, 0,99)
|
aGMR – Rapport entre les moyennes géométriques défini comme l'exposition (concentration maximale ou aire sous la courbe AUC) à la médication concomitante quand elle est utilisée avec l'atogépant, divisé par l'exposition à la médication concomitante sans l'atogépant.
bCI = Intervalle de confiance
Grossesse, allaitementGrossesse
Il existe des données limitées sur l'utilisation de l'atogépant chez la femme enceinte. Les expérimentations animales ont révélé une toxicité sur la reproduction (voir «Données précliniques»). L'utilisation d'AQUIPTA pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception n'est pas recommandée.
Allaitement
On ne sait pas si l'atogépant passe dans le lait maternel. Les données toxicologiques disponibles issues des études effectuées chez l'animal ont mis en évidence que l'atogépant passe dans le lait maternel. Un risque pour les nouveaux-nés/enfants ne peut être exclu. Une décision doit être prise pour savoir si l'allaitement doit être interrompu ou si le traitement par AQUIPTA doit être interrompu ou abandonné. Cette décision doit tenir compte à la fois des bénéfices de l'allaitement pour l'enfant et des bénéfices du traitement pour la femme.
Fertilité
Il n'existe pas de données concernant l'effet de l'atogépant sur la fertilité humaine. Les études effectuées chez l'animal n'ont pas montré d'effet du traitement par l'atogépant sur la fertilité féminine ou masculine.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude correspondante n'a été effectuée. AQUIPTA peut toutefois provoquer une fatigue/somnolence chez certains patients. Les patients doivent faire preuve de prudence avant de conduire ou d'utiliser des machines tant qu'ils ne sont pas suffisamment certains qu'AQUIPTA n'altère pas leurs capacités.
Effets indésirablesRésumé du profil de sécurité
La sécurité d'AQUIPTA a été évaluée chez 2'657 patients migraineux ayant reçu au moins une dose d'AQUIPTA. Parmi eux, 1'225 patients ont été traités par AQUIPTA pendant au moins 6 mois et 826 patients l'ont été pendant 12 mois.
Dans les études cliniques contrôlées contre placebo d'une durée de 12 semaines, 678 patients ont reçu au moins une dose d'AQUIPTA 60 mg une fois par jour et 663 patients ont reçu le placebo.
Les effets indésirables les plus fréquemment rapportés étaient: nausées (9 %), constipation (8 %) et fatigue/somnolence (5 %). La majorité des effets indésirables étaient d'intensité légère ou modérée. Les nausées (0,4 %) étaient l'effet indésirable ayant entraîné le plus fréquemment l'arrêt du traitement.
Liste des effets indésirables
Les effets indésirables sont présentés ci-après par classes d'organes et par fréquence.
Les catégories de fréquence sont définies comme suit: très fréquents (≥1/10); fréquents (≥1/100, < 1/10); occasionnels (≥1/1000, < 1/100); rares (≥1/10'000, < 1/1000); très rares (< 1/10'000), fréquence inconnue (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes d'organes
|
Fréquence
|
Effets indésirables
| |
Affections du système immunitaire
|
Fréquents
|
Réactions d'hypersensibilité*** (p.ex. dyspnée, éruption cutanée, prurit, urticaire, œdème facial)
| |
Rares
|
Anaphylaxie***
| |
Troubles du métabolisme et de la nutrition
|
Fréquents
|
Perte d'appétit
Perte de poids*
| |
Affections gastro-intestinales
|
Fréquents
|
Nausées, constipation
| |
Affections hépatobiliaires
|
Occasionnels
|
Augmentation de l'ALAT/ASAT**
| |
Troubles généraux et anomalies au site d'administration
|
Fréquents
|
Fatigue/somnolence
|
* Définie dans les études cliniques comme une perte de poids d'au moins 7 % à tout moment.
** Des cas d'augmentations de l'ALAT et de l'ASAT (définies comme des valeurs ≥3 × la limite supérieure de la normale [upper limit of normal, ULN]) présentant une association temporelle avec l'administration d'atogépant ont été observés dans les études cliniques, y compris des cas avec dechallenge positif éventuel qui ont régréssé dans les 8 semaines suivant l'arrêt du traitement. Cependant, la fréquence globale était comparable dans les groupes de traitement par atogépant et par placebo.
*** Voir «Réactions d'hypersensibilité» sous «Mises en garde et précautions».
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDans les études cliniques, l'atogépant a été administré en doses uniques allant jusqu'à 300 mg et en doses répétées allant jusqu'à 170 mg une fois par jour. Les effets indésirables étaient comparables à ceux observés aux doses plus faibles et aucune toxicité spécifique n'a été identifiée. Il n'existe pas d'antidote connu à AQUIPTA. Le traitement en cas de surdosage d'AQUIPTA doit consister en une prise en charge générale incluant la surveillance des paramètres vitaux et l'observation de l'état clinique du patient.
Propriétés/EffetsCode ATC
N02CD07
Mécanisme d'action/pharmacodynamie
L'atogépant est un antagoniste du récepteur du peptide lié au gène de la calcitonine (CGRP) qui bloque la liaison du CGRP au récepteur et s'oppose à la fonction du récepteur du CGRP. Le CGRP est un neuropeptide qui a été associé à la pathophysiologie de la migraine. Dans le système trigéminovasculaire, le CGRP module la signalisation nociceptive et l'inflammation et agit en outre comme un vasodilatateur.
Électrophysiologie cardiaque
À une dose 5 fois supérieure à la dose quotidienne maximale recommandée, AQUIPTA ne prolonge pas l'intervalle QT.
Efficacité clinique
AQUIPTA a été évalué dans deux études d'homologation pour la prophylaxie de la migraine sur l'ensemble du spectre de la migraine chronique et épisodique. Dans l'étude sur la migraine épisodique (ADVANCE) les patients qui répondaient aux critères International Classification of Headache Disorders (ICHD) pour le diagnostic de migraine avec ou sans aura ont été inclus. L'étude sur la migraine chronique (PROGRESS) a inclus des patients qui répondaient également aux critères ICHD de migraine chronique. Les patients ayant des antécédents d'infarctus du myocarde, d'accident vasculaire cérébral ou d'accident ischémique transitoire survenus au cours des six mois précédant la sélection ont été exclus des deux études.
Migraine épisodique
AQUIPTA a été évalué pour la prophylaxie de la migraine épisodique (4 à 14 jours de migraine par mois) dans une étude multicentrique randomisée en double aveugle, contrôlée contre placebo (ADVANCE). Les patients ont été randomisés pour recevoir AQUIPTA 60 mg (N = 235) ou le placebo (N = 223) une fois par jour pendant 12 semaines. Le traitement aigu des céphalées (c'est-à-dire les triptans, les dérivés de l'ergotamine, les AINS, le paracétamol et les opioïdes) était autorisé pour les patients.
Au total, 88 % des patients ont terminé la période d'étude en double aveugle de 12 semaines. L'âge moyen des patients était de 42 ans (entre 18 et 73 ans); 89 % étaient de sexe féminin et 83 % étaient caucasiens. La fréquence moyenne des jours de migraine lors de l'inclusion était d'environ 8 jours de migraine par mois et était comparable dans les deux groupes de traitement.
Le critère d'évaluation principal de l'efficacité était la variation par rapport à l'inclusion du nombre moyen de jours de migraine par mois (JMM) pendant la période de traitement de 12 semaines.
Le traitement par AQUIPTA a démontré des améliorations statistiquement significatives des résultats d'efficacité primaires et secondaires contrôlés par multiplicité dans l'étude ADVANCE par rapport au placebo, comme résumé dans le tableau 4.
Tableau 4. Critères d'efficacité dans l'étude ADVANCE
|
|
AQUIPTA 60 mg
n = 226
|
Placebo
n = 216
| |
Nombre de jours de migraine par mois (JMM) pendant 12 semaines
| |
Inclusion
|
7,8
|
7,5
| |
Variation moyenne par rapport à l'inclusion
|
-4,1
|
-2,5
| |
Différence par rapport au placebo
|
-1,7
|
| |
Valeur p
|
< 0,001
|
| |
Nombre de jours de céphalées par mois pendant 12 semaines
| |
Inclusion
|
9,0
|
8,5
| |
Variation moyenne par rapport à l'inclusion
|
-4,2
|
-2,5
| |
Différence par rapport au placebo
|
-1,7
|
| |
Valeur p
|
< 0,001
|
| |
Nombre de jours d'utilisation d'un traitement de crise par mois pendant 12 semaines
| |
Inclusion
|
6,9
|
6,5
| |
Variation moyenne par rapport à l'inclusion
|
-3,8
|
-2,3
| |
Différence par rapport au placebo
|
-1,4
|
| |
Valeur p
|
< 0,001
|
| |
Répondeurs ayant obtenu une réduction ≥50 % du nombre de JMM pendant 12 semaines
| |
Répondeurs (%)
|
59
|
29
| |
Odds Ratio (IC à 95 %)
|
3,55 (2,39; 5,28)
|
| |
Valeur p
|
< 0,001
|
| |
MSQ v.2.1 RFRa à la semaine 12
| |
Inclusion
|
46,6
|
46,6
| |
Variation moyenne par rapport à l'inclusion
|
31,0
|
20,0
| |
Différence par rapport au placebo
|
11,0
|
| |
Valeur p
|
< 0,001
|
|
aScore sur le Questionnaire Migraine Specific Quality of Life (Qualité de vie spécifique à la migraine) version 2.1 dans la section Role Function Restrictive
La figure 1 présente la variation moyenne du nombre de jours de migraine par mois (JMM) par rapport à l'inclusion dans l'étude ADVANCE. Comparativement aux patients qui recevaient le placebo, il a été observé chez les patients traités par AQUIPTA 60 mg une fois par jour une diminution moyenne plus importante du nombre de jours de migraine par mois par rapport à l'inclusion pendant la période de traitement de 12 semaines. Comparativement au placebo, AQUIPTA 60 mg, pris une fois par jour, a induit une diminution moyenne plus importante du nombre de jours de migraine par mois par rapport à l'inclusion pendant le premier mois du traitement.
Figure 1: Variation du nombre de jours de migraine par mois (JMM) par rapport à l'inclusion dans l'étude ADVANCE
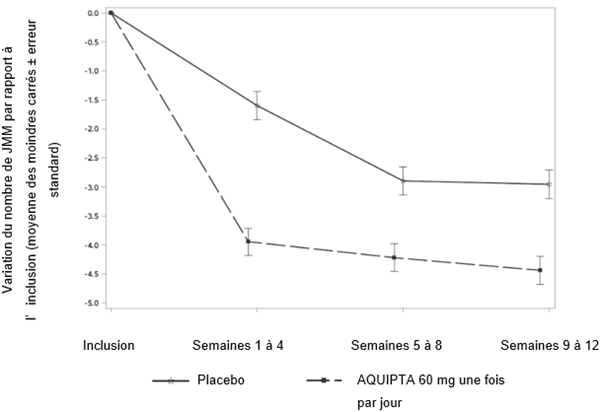
Efficacité à long terme
Dans une étude en ouvert dans laquelle des patients présentant une migraine épisodique ont reçu AQUIPTA 60 mg une fois par jour, l'efficacité a été maintenue jusqu'à un an. 68,4 % des patients ont terminé la période de traitement. La réduction du nombre moyen de jours de migraine par mois (méthode des moindres carrés) pendant le premier mois (semaines 1 à 4) était de 3,8 jours, avec une amélioration jusqu'à une réduction moyenne (méthode des moindres carrés) de 5,2 jours pendant le dernier mois (semaines 49 à 52). Environ 84 %, 70 % et 48% des patients ont mentionné une réduction ≥50%, ≥75 % et de 100 % respectivement du nombre de jours de migraine par mois aux semaines 49 à 52.
Patients chez lesquels des traitements prophylactiques oraux par des médicaments de deux à quatre classes différentes ont précédemment échoué
Dans l'étude ELEVATE, 315 patients adultes souffrant de migraine épisodique qui n'avaient pas répondu précédemment à des traitements prophylactiques oraux par des médicaments de deux à quatre classes différentes (p.ex. topiramate, antidépresseurs tricycliques, bêtabloquants) en termes d'efficacité et/ou de tolérance ont été randomisés (1:1) pour recevoir soit de l'atogépant à raison de 60 mg (n = 157) soit un placebo (n = 158) pendant douze semaines. Les résultats de cette étude étaient cohérents avec les principaux résultats d'études précédentes sur l'efficacité en cas de migraine épisodique et étaient statistiquement significatifs s'agissant des critères principaux et secondaires d'évaluation de l'efficacité, y compris plusieurs questionnaires destinés aux patients pour évaluer les capacités fonctionnelles. Le nombre de jours de migraine par mois (JMM) a baissé de 4,2 jours sous atogépant alors qu'il a diminué de 1,9 jours sous placebo (p < 0,001). Au total, 50,6 % (78/154) des patients du groupe sous atogépant ont obtenu une réduction du nombre de JMM d'au moins 50 % par rapport à la valeur initiale, contre 18,1 % (28/155) des patients sous placebo (odds ratio [IC à 95 %]: 4,82 [2,85; 8,14]; p < 0,001).
Migraine chronique
AQUIPTA a été évalué pour la prophylaxie de la migraine chronique (15 jours ou plus de céphalées par mois avec au moins 8 jours de migraine) dans une étude multicentrique, randomisée en double aveugle, contrôlée contre placebo (PROGRESS). Les patients ont été randomisés pour recevoir AQUIPTA 60 mg (N = 262) ou le placebo (N = 259) une fois par jour pendant 12 semaines L'utilisation concomitante d'un médicament pour la prophylaxie de la migraine (par exemple, amitriptyline, propranolol, topiramate) était autorisée dans un sous-groupe de patients (11 %). Le traitement aigu des céphalées (c'est-à-dire les triptans, les dérivés de l'ergotamine, les AINS, le paracétamol et les opioïdes) était autorisé pour les patients. Ont également été inclus des patients présentant une utilisation excessive de médicaments de la médication aiguë et des céphalées dues à l'utilisation excessive de médicaments..
Au total, 463 patients (89 %) ont terminé la période d'étude en double aveugle de 12 semaines. L'âge moyen des patients était de 42 ans (entre 18 et 74 ans); 87 % étaient de sexe féminin et 59 % étaient caucasiens. La fréquence moyenne des jours de migraine lors de l'inclusion était d'environ 19 jours de migraine par mois et était comparable dans les deux groupes de traitement.
Le critère d'évaluation principal de l'efficacité était la variation par rapport à l'inclusion du nombre moyen de jours de migraine par mois (JMM) par rapport à l'inclusion pendant la période de traitement de 12 semaines.
Le traitement par AQUIPTA a démontré une amélioration statistiquement significative des résultats d'efficacité primaires et secondaires contrôlés par multiplicité dans l'étude PROGRESS par rapport au placebo, comme résumé dans le tableau 5.
Tableau 5. Critères d'efficacité dans l'étude PROGRESS
|
|
AQUIPTA 60 mg
n = 257
|
Placebo
n = 249
| |
Nombre de jours de migraine par mois (JMM) pendant 12 semaines
| |
Inclusion
|
19,2
|
19,0
| |
Variation moyenne par rapport à l'inclusion
|
-6,8
|
-5,1
| |
Différence par rapport au placebo
|
-1,7
|
| |
Valeur p
|
0,002
|
| |
Nombre de jours de céphalées par mois pendant 12 semaines
| |
Inclusion
|
21,5
|
21,4
| |
Variation moyenne par rapport à l'inclusion
|
-6,9
|
-5,2
| |
Différence par rapport au placebo
|
-1,7
|
| |
Valeur p
|
0,002
|
| |
Nombre de jours d'utilisation d'un traitement de crise par mois pendant 12 semaines
| |
Inclusion
|
15,5
|
15,3
| |
Variation moyenne par rapport à l'inclusion
|
-6,2
|
-4,1
| |
Différence par rapport au placebo
|
-2,1
|
| |
Valeur p
|
0,002
|
| |
Répondeurs ayant obtenu une réduction ≥50 % du nombre de JMM pendant 12 semaines
| |
Répondeurs (%)
|
40
|
27
| |
Différence par rapport au placebo (%)
|
14
|
| |
Valeur p
|
0,002
|
| |
MSQ v.2.1 RFRa à la semaine 12
| |
Inclusion
|
43,3
|
44,1
| |
Variation moyenne par rapport à l'inclusion
|
23,1
|
17,3
| |
Différence par rapport au placebo
|
5,8
|
| |
Valeur p
|
0,002
|
|
aScore sur le Questionnaire Migraine Specific Quality of Life (Qualité de vie spécifique à la migraine) version 2.1 dans la section Role Function Restrictive
La figure 2 présente la variation moyenne du nombre de jours de migraine par mois (JMM) par rapport à l'inclusion dans l'étude PROGRESS. Les patients traités par AQUIPTA 60 mg une fois par jour ont présenté une réduction moyenne plus importante du nombre de jours de migraine par mois par rapport à l'inclusion pendant la période de traitement de 12 semaines, par rapport aux patients ayant reçu un placebo.
Figure 2: Variation du nombre de jours de migraine par mois (JMM) par rapport à l'inclusion dans l'étude PROGRESS
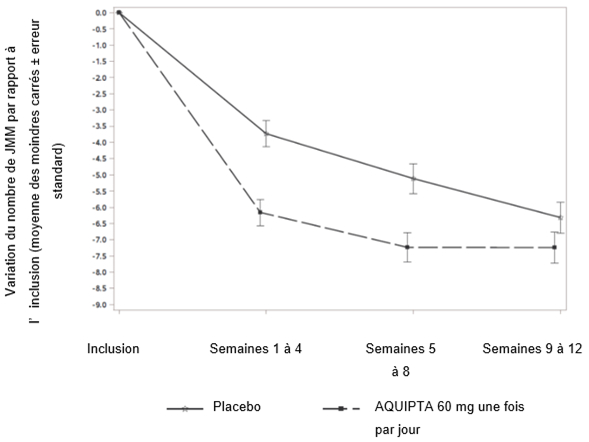
Pédiatrie
Il n'existe pas de données sur l'utilisation chez les enfants et les adolescents de moins de 18 ans.
PharmacocinétiqueAbsorption
Après administration orale d'AQUIPTA, l'atogépant est rapidement absorbé et les valeurs Tmax se situent dans la médiane entre 1 et 2 heures. Après une dose unique quotidienne, l'atogépant présente un profil pharmacocinétique proportionnel à la dose jusqu'à la dose de 170 mg (environ 3 fois la dose maximale recommandée), sans accumulation.
Influence des aliments
Lorsque AQUIPTA était administré avec un repas riche en graisses, l'influence des aliments n'était pas significative (l'ASC et la Cmax étaient diminuées d'environ 18 % et 22 % respectivement, sans influence sur da durée médiane jusqu'à la concentration plasmatique maximale de l'atogépant). Dans les études d'efficacité clinique, AQUIPTA était pris au cours ou en dehors des repas.
Distribution
La liaison de l'atogépant aux protéines plasmatiques ne dépendait pas de la concentration dans la plage de 0,1 à 10 µM; la fraction non liée d'atogépant dans le plasma humain était d'environ 4,7 %. Après administration par voie orale, le volume apparent de distribution (Vz/F) moyen de l'atogépant est d'environ 292 litres.
Métabolisme
L'atogépant est principalement éliminé par voie métabolique, principalement par le CYP3A4.
La molécule mère (atogépant) et un métabolite glucurono-conjugué (M23) étaient les composants circulant le plus fréquemment dans le plasma humain.
Élimination
La demi-vie d'élimination de l'atogépant est de 11 heures environ. La clairance orale apparente moyenne (CL/F) de l'atogépant est d'environ 19 litres/heure. Après administration d'une dose orale unique de 50 mg 14Catogépant chez des hommes volontaires sains, 42 % et 5 % de la dose ont été éliminés sous forme d'atogépant inchangé dans les fèces et les urines respectivement.
Cinétique pour certains groupes de patients
Sur la base d'une analyse pharmacocinétique de population, l'âge (18 à 78 ans), le sexe, l'ethnie (caucasienne vs. japonaise ou chinoise) et le poids corporel (40,7 à 196 kg) n'ont pas eu d'influence significative sur la pharmacocinétique (Cmax et ASC) de l'atogépant. Par conséquent, aucun ajustement de la dose n'est nécessaire en raison de ces facteurs.
Troubles de la fonction rénale
L'élimination par voie rénale ne joue qu'un rôle mineur dans la clairance de l'atogépant. Sur la base d'une analyse pharmacocinétique de population, les paramètres pharmacocinétiques de l'atogépant ne sont pas significativement différents chez les patients présentant une insuffisance rénale légère ou modérée (CLcr de 30 à 89 mL/min) par rapport à ceux ayant une fonction rénale normale (CLcr ≥90 mL/min). Du fait de l'absence d'étude chez les patients présentant une insuffisance rénale sévère ou terminale (end-stage renal disease, ESRD; CLcr < 30 ml/min), l'utilisation de l'atogépant 10 mg est recommandée chez ces patients.
Troubles de la fonction hépatique
L'exposition totale à l'atogépant était augmentée de respectivement 24 %, 15 % et 38 % chez les patients présentant une insuffisance hépatique préexistante légère (classe A de Child Pugh), modérée (classe B de Child Pugh) ou sévère (classe C de Child Pugh). Cependant, l'exposition à l'atogépant non lié était environ 3 fois plus élevée chez les patients atteints d'insuffisance hépatique sévère. L'utilisation d'AQUIPTA est contre-indiquée chez les patients atteints d'insuffisance hépatique sévère.
Données précliniquesSur la base des études conventionnelles de pharmacologie de sécurité, de toxicité à doses répétées, de génotoxicité, de cancérogénicité et de fertilité, les données précliniques ne révèlent aucun risque particulier pour l'homme.
Toxicité sur la reproduction
L'administration par voie orale d'atogépant (0, 5, 15, 125 ou 750 mg/kg/jour) à des rates gestantes pendant l'organogenèse a entraîné une diminution du poids corporel des fœtus et une augmentation de l'incidence des modifications squelettiques chez les fœtus aux deux plus fortes doses étudiées (125 et 750 mg/kg), ce qui n'était pas lié à une toxicité maternelle. À la dose sans effets indésirables sur le développement embryofœtal (15 mg/kg/jour), l'exposition plasmatique (ASC) était environ 4 fois plus élevée que chez les personnes recevant une dose de 60 mg/jour.
L'administration par voie orale d'atogépant (0, 30, 90 ou 130 mg/kg/jour) à des lapines gestantes pendant l'organogenèse a entraîné une augmentation de l'incidence des modifications viscérales et squelettiques à la dose la plus élevée (130 mg/kg/jour). À la dose sans effets indésirables sur le développement des lapins (90 mg/kg/jour), l'exposition plasmatique (ASC) était environ 3 fois plus élevée que chez les personnes recevant une dose de 60 mg/jour.
L'administration par voie orale d'atogépant (0, 15, 45 ou 125 mg/kg/jour) à des rates pendant la gestation et l'allaitement a entraîné une diminution du poids corporel de la progéniture à la dose la plus élevée (125 mg/kg/jour). L'exposition plasmatique (ASC) à la plus forte dose étudiée sans effet sur le développement de la progéniture (45 mg/kg/jour) était environ 5 fois supérieure à celle observée chez l'homme à la dose de 60 mg/jour.
Examens de toxicité chez les animaux juvéniles
L'administration d'atogépant sous forme d'une dose orale de 10, 30 ou 200 mg/kg/jour une fois par jour chez de jeunes rats du jour 28 au jour 70 après la naissance n'a pas été associée à des effets indésirables sur leur développement.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur l'emballage.
Remarques particulières concernant le stockage
Ne pas conserver au-dessus de + 30 °C.
Conserver hors de portée des enfants.
Numéro d’autorisation69128 (Swissmedic)
PrésentationAQUIPTA 10 mg: blisters de 28 comprimés (B)
AQUIPTA 60 mg: blisters de 28 comprimés (B)
Titulaire de l’autorisationAbbVie AG, 6330 Cham
Mise à jour de l’informationNovembre 2024
|