CompositionPrincipes actifs
Tramétinib sous forme de diméthylsulfoxyde de tramétinib
Excipients
Noyau des comprimés:
Mannitol, cellulose microcristalline, hypromellose, croscarmellose sodique, stéarate de magnésium, laurylsulfate de sodium, silice colloïdale,
Enrobage:
Dioxyde de titane (E 171), polyéthylèneglycol, oxyde de fer jaune (E 172, pour les comprimés de 0,5 mg), polysorbate 80 et oxyde de fer rouge (E 172, pour les comprimés de 2 mg).
La teneur totale en sodium est de 0,199 mg pour les comprimés de 0,5 mg et de 0,232 mg pour les comprimés de 2 mg.
Poudre pour solution buvable:
Sulfobutylbétadex sodique (9400 mg), sucralose (E 955), acide citrique monohydraté (E 330), monohydrogénophosphate de sodium, sorbate de potassium (E 202), parahydroxybenzoate de méthyle (E 218, 75,20 mg), arôme de fraise.
1 flacon contient au maximum 198 mg de sodium et 58 mg de potassium.
Chaque ml de solution reconstituée contient 100 mg de sulfobutylbétadex sodique, 0,58 mg de potassium, 1,98 mg de sodium et 0,8 mg de parahydroxybenzoate de sodium (E 218).
Indications/Possibilités d’emploiIndication autorisée de manière temporaire
Gliome de bas grade (Low Grade Gliom, LGG)
Mekinist en association avec le dabrafénib est indiqué pour le traitement de patients pédiatriques à partir de 1 an, atteints d'un gliome de bas grade (Low Grade Gliom, LGG) portant la mutation BRAF V600E, qui ont besoin d'un traitement systémique.
En raison de données insuffisantes au moment de l'examen de la demande, cette/ces indication(s) est/sont autorisées de manière temporaire (Art. 9a de la loi sur les produits thérapeutiques). L'autorisation temporaire est impérativement liée au respect de conditions dans le temps imparti. Une fois les conditions remplies, l'autorisation temporaire peut être convertie en autorisation régulière.
Indications autorisées de manière régulière
Mélanome non résécable ou métastatique
Mekinist en association avec le dabrafénib est indiqué pour le traitement des patients adultes atteints d'un mélanome non résécable ou métastatique exprimant une mutation V600 du gène BRAF (V600E/K).
Traitement adjuvant du mélanome
Mekinist en association avec le dabrafénib est indiqué pour le traitement adjuvant des patients atteints d'un mélanome de stade III exprimant la mutation BRAF V600 après résection complète.
Cancer bronchique non à petites cellules, avancé ou métastatique
Mekinist en association avec le dabrafénib peut être utilisé pour le traitement de patients adultes atteints d'un cancer bronchique non à petites cellules (NSCLC; non-small cell lung cancer) métastatique exprimant la mutation BRAF V600E.
Posologie/Mode d’emploiLe traitement par Mekinist doit être instauré et surveillé par un médecin ayant l'expérience de l'utilisation des médicaments oncologiques.
Mekinist est disponible sous deux formes galéniques: en comprimés et en poudre pour solution buvable.
Avant la prise de Mekinist en association avec le dabrafénib, la présence d'une mutation BRAF V600 doit être confirmée conformément à l'indication autorisée à l'aide d'un test validé. Mekinist en association avec le dabrafénib ne doit pas être utilisé chez les patients atteints de tumeurs à gène BRAF de type sauvage (voir «Efficacité clinique»).
Posologie usuelle
Comprimés pelliculés
Chez les patients adultes qui utilisent Mekinist en association avec le dabrafénib, la dose recommandée de Mekinist comprimés pelliculés est de 2 mg par voie orale une fois par jour et la dose recommandée de dabrafénib est de 150 mg par voie orale deux fois par jour, indépendamment du poids corporel (voir «Efficacité clinique»).
Ajustement de la posologie du fait d'effets indésirables/d'interactions
En cas de réactions indésirables, une interruption du traitement, une réduction de la dose ou un arrêt du traitement peut s'avérer nécessaire (voir Tableaux 1 et 2).
Tableau 1: Recommandations relatives à la réduction des doses de Mekinist en association avec le dabrafénib chez les patients adultes
|
Réductions de dose
|
Dose de Mekinist
|
Dose de dabrafénib
| |
Dose initiale
|
2 mg par voie orale une fois par jour
|
150 mg par voie orale deux fois par jour
| |
Première réduction de la dose
|
1,5 mg par voie orale une fois par jour
|
100 mg par voie orale deux fois par jour
| |
Deuxième réduction de la dose
|
1 mg par voie orale une fois par jour
|
75 mg par voie orale deux fois par jour
| |
Troisième réduction de la dose
|
pas de réduction supplémentaire
|
50 mg par voie orale deux fois par jour
|
Les recommandations relatives aux ajustements posologiques du dabrafénib sont à consulter dans l'information professionnelle du dabrafénib (voir «Posologie/Mode d'emploi»). Mekinist 1 mg en comprimés pelliculés par voie orale une fois par jour doit être définitivement arrêté s'il n'est pas toléré.
Poudre pour solution buvable
Les doses et réductions de dose recommandées pour Mekinist poudre pour solution buvable sont basées sur le poids corporel (Tableau 2).
Tableau 2: Doses en fonction du poids et réductions de dose recommandées pour Mekinist poudre pour solution buvable
|
Poids corporel (kilogrammes)
|
Dose recommandée
Quantité totale de solution buvable (teneur en tramétinib)
|
Réductions de dose
| |
Première réduction de dose (une fois par jour)
|
Deuxième réduction de dose (une fois par jour)
| |
8 kg
|
6 ml (0,3 mg)
|
5 ml
|
3 ml
| |
9 kg
|
7 ml (0,35 mg)
|
5 ml
|
4 ml
| |
10 kg
|
7 ml (0,35 mg)
|
5 ml
|
4 ml
| |
11 kg
|
8 ml (0,4 mg)
|
6 ml
|
4 ml
| |
12 à 13 kg
|
9 ml (0,45 mg)
|
7 ml
|
5 ml
| |
14 à 17 kg
|
11 ml (0,55 mg)
|
8 ml
|
6 ml
| |
18 à 21 kg
|
14 ml (0,7 mg)
|
11 ml
|
7 ml
| |
22 à 25 kg
|
17 ml (0,85 mg)
|
13 ml
|
9 ml
| |
26 à 29 kg
|
18 ml (0,9 mg)
|
14 ml
|
9 ml
| |
30 à 33 kg
|
20 ml (1 mg)
|
15 ml
|
10 ml
| |
34 à 37 kg
|
23 ml (1,15 mg)
|
17 ml
|
12 ml
| |
38 à 41 kg
|
25 ml (1,25 mg)
|
19 ml
|
13 ml
| |
42 à 45 kg
|
28 ml (1,4 mg)
|
21 ml
|
14 ml
| |
46 à 50 kg
|
32 ml (1,6 mg)
|
24 ml
|
16 ml
| |
≥51 kg
|
40 ml (2 mg)
|
30 ml
|
20 ml
| |
Arrêter définitivement lorsque deux réductions de dose ne sont pas tolérées.
|
Mekinist solution buvable doit être définitivement arrêté s'il n'est pas toléré après la deuxième réduction de dose.
Tableau 3: Modifications de dose recommandées pour Mekinist en cas d'effets secondaires
|
Sévérité de l'effet secondaire [voir Mises en garde et précautions]a
|
Modification de la dose de Mekinistb
| |
Hémorragie
| |
·Degré de sévérité 3
|
Interrompre Mekinist
·Après amélioration, reprendre Mekinist à une dose inférieure
·Si aucune amélioration, arrêter Mekinist définitivement
| |
·Degré de sévérité 4
|
Arrêter Mekinist définitivement
| |
Thrombo-embolie veineuse
| |
·Thrombose veineuse profonde (TVP) ou embolie pulmonaire (EP) non compliquée
|
Interrompre Mekinist pendant 3 semaines au plus
·Après amélioration au grade 0–1, reprendre Mekinist à une dose inférieure
·Si aucune amélioration, arrêter Mekinist définitivement
| |
·EP menaçant le pronostic vital
|
Arrêter Mekinist définitivement
| |
Cardiomyopathie
| |
·Réduction absolue, asymptomatique de la fraction d'éjection du ventricule gauche (FEVG) de 10% ou plus par rapport à la valeur initiale et en dessous de la limite inférieure de la normale (LIN) de l'institution
|
Interrompre Mekinist pendant 4 semaines au plus
·Si la FEVG revient à une valeur normale, reprendre le traitement par Mekinist à une dose inférieure
·Si la FEVG ne s'est pas améliorée à une valeur normale, Mekinist doit être définitivement arrêté
| |
·Cardiomyopathie symptomatique
·Baisse absolue de la FEVG de plus de 20% par rapport à la valeur initiale, qui se situe sous la LIN
|
Arrêter Mekinist définitivement
| |
Toxicités oculaires
| |
·Décollement de l'épithélium pigmentaire rétinien (EPR)
|
Interrompre Mekinist pendant 3 semaines au plus
·Après amélioration, reprendre Mekinist à une dose égale ou inférieure
·Si aucune amélioration, arrêter Mekinist définitivement ou reprendre Mekinist a une dose inférieure
| |
·Occlusion veineuse rétinienne (OVC)
|
Arrêter Mekinist définitivement
| |
Poumons
| |
·Pneumopathie interstitielle (PI)/pneumopathie inflammatoire
|
Arrêter Mekinist définitivement
| |
Réactions fébriles
| |
·Fièvre de 38 à 40 °C (ou premier symptôme en cas de récidive)
|
Interrompre Mekinist jusqu'à la baisse de la fièvre et reprendre Mekinist à une dose égale ou inférieure
| |
·Fièvre supérieure à 40 °C
·Fièvre, compliquée par des frissons, une hypotension, une déshydratation ou une défaillance rénale
|
·Interrompre Mekinist jusqu'à la baisse des réactions fébriles pendant au moins 24 heures, et reprendre Mekinist a une dose inférieure.
Ou
·Arrêter Mekinist définitivement
| |
Toxicités cutanées
| |
·Degré de sévérité 2 si pas tolérable
·Degré de sévérité 3 ou 4
|
Interrompre Mekinist pendant 3 semaines au plus
·En cas d'amélioration, reprendre Mekinist à une dose inférieure
·Si aucune amélioration, arrêter Mekinist définitivement
| |
·Effets secondaires cutanés sévères (SCAR)
|
Arrêter Mekinist définitivement
| |
Autres effets secondaires
| |
·Degré de sévérité 2 si pas tolérable
·Degré de sévérité 3
|
Interrompre Mekinist
·En cas d'amélioration à un grade 0–1, poursuivre à une dose inférieure
·Si aucune amélioration, arrêter définitivement
| |
·Première manifestation d'un degré de sévérité 4
|
·Interrompre Mekinist jusqu'à amélioration à un degré de sévérité 0-1, puis reprendre le traitement à une dose inférieure
Ou
·Arrêter Mekinist définitivement
| |
·Récidive d'un degré de sévérité 4
|
Arrêter Mekinist définitivement
|
a Degré de sévérité des événements cliniques indésirables selon les critères communs de terminologie pour les événements indésirables (Common Terminology Criteria for Adverse Events [CTCAE]), Version 4.0.
b Voir les tableaux 1 et 2 pour les réductions de dose recommandées pour Mekinist.
c Pour les effets secondaires suivants, aucun ajustement de la dose de Mekinist n'est recommandé lorsqu'il est administré en association avec le dabrafénib: affections malignes non cutanées et uvéite. Un ajustement de la dose de Mekinist n'est pas nécessaire en cas de nouvelle tumeur maligne primaire de la peau.
Suivez l'information professionnelle du dabrafénib pour les adaptations de dose en cas d'effets secondaires liés au dabrafénib.
Dès que les effets indésirables chez un patient sont efficacement contrôlés, une nouvelle augmentation progressive de la dose peut être envisagée en respectant les paliers utilisés au moment de la réduction de la dose. La posologie de Mekinist ne doit pas dépasser 2 mg une fois par jour.
Schéma d'administration
Mekinist et le dabrafénib doivent être pris sans nourriture, à savoir au moins une heure avant ou deux heures après un repas (voir «Pharmacocinétique»).
Lorsque Mekinist et le dabrafénib sont pris en association, la seule dose quotidienne de Mekinist doit être prise à la même heure chaque jour, avec la dose de dabrafénib du matin ou du soir. La pharmacocinétique de la prise en soirée de Mekinist n'a pas été étudiée, de sorte qu'une prise matinale doit être préférée.
Poudre pour solution buvable
Mekinist poudre pour solution buvable consiste en un flacon contenant une poudre, que le patient peut utiliser après préparation ou en tant que solution prête à l'emploi. Une fois la solution préparée, elle doit être utilisée dans les 35 jours. Toute solution non utilisée 35 jours après la reconstitution doit être éliminée.
Lors de l'utilisation de Mekinist sous forme de solution buvable, le médecin doit parcourir l'information destinée aux patients et les indications de préparation et de prise de Mekinist et en parler avec le patient ou le/les soignant(s). Le médecin doit s'assurer que le patient ou le/les soignant(s) ont compris comment mélanger Mekinist poudre pour solution buvable avec de l'eau et administrer la dose quotidienne correcte.
Un mode d'emploi complet et illustré pour la préparation de la solution buvable est fourni à la rubrique «Instructions d'utilisation et de manipulation».
Durée du traitement
Chez les patients présentant un mélanome non opérable ou métastasé ou un cancer du poumon non à petites cellules métastasé, il est recommandé de poursuivre le traitement jusqu'à progression de la maladie ou survenue d'effets secondaires inacceptables.
Dans le traitement adjuvant d'un mélanome, la durée du traitement est limitée à 1 an au maximum.
Chez les patients pédiatriques atteints de LGG, il est recommandé de poursuivre le traitement jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable. Il convient de noter que dans les études cliniques portant sur des patients pédiatriques avec LGG, des différences notables ont été observées dans l'évaluation de l'efficacité entre les examens indépendants et les centres d'examen, notamment pour la détermination de la progression de la maladie (voir également «Efficacité clinique»). De plus, on ne dispose que de données limitées sur l'utilisation à long terme et la durée optimale du traitement par Mekinist en association avec le dabrafénib dans la population pédiatrique (voir «Mises en garde et précautions»). La durée médiane de traitement dans les études cliniques pertinentes se situait à 24 mois environ (voir «Efficacité clinique»). D'autre part, on ne dispose que de données limitées concernant des patients avec LGG, âgés de plus de 18 ans, qui ont besoin d'un premier traitement systémique. La poursuite du traitement jusqu'à l'âge adulte ne doit se faire que sur la base de l'évaluation du rapport bénéfices-risques par le médecin.
Traitement de la pyrexie: les traitements doivent être interrompus (Mekinist s'il est utilisé en monothérapie, ou tant Mekinist que le dabrafénib si l'on utilise l'association) lorsque la température corporelle du patient est ≥38 °C (≥100,4 °F). En cas de récidive, le traitement peut également être interrompu aux premiers symptômes de pyrexie. Un traitement par un antipyrétique tel que l'ibuprofène ou le paracétamol doit être initié. Il convient de surveiller les patients quant aux signes et symptômes d'une infection (voir rubrique «Mises en garde et précautions»). Le traitement par Mekinist ou tant par Mekinist que par le dabrafénib, lorsqu'ils sont utilisés en association, peut être repris en cas d'absence de symptôme pendant au moins 24 heures: (1) à la même posologie ou (2) à une posologie réduite si la pyrexie était récidivante et/ou accompagnée d'autres symptômes sévères tels que déshydratation, hypotension ou insuffisance rénale. L'utilisation de corticoïdes par voie orale doit être envisagée dans le cas où les antipyrétiques ne sont pas suffisamment efficaces.
Lorsque des réactions de toxicité dépendantes du traitement se présentent sous traitement simultané avec Mekinist et le dabrafénib, les doses des deux médicaments doivent être réduites simultanément ou les deux traitements interrompus ou totalement arrêtés, les exceptions ci-dessous étant applicables.
Exceptions pour lesquelles seule une modification posologique du Mekinist est nécessaire:
·Fraction d'éjection du ventricule gauche (FEVG) réduite
·Occlusion veineuse rétinienne (OVR), décollement de l'épithélium pigmentaire de la rétine/décollement de la rétine (retinal pigment epithelial detachment, RPED) et choriorétinopathie
·Pneumonie et pneumopathie interstitielle diffuse (PID)
Traitement de la réduction de la FEVG/du dysfonctionnement ventriculaire gauche
La fraction d'éjection du ventricule gauche (FEVG) doit être évaluée avant le commencement du traitement avec Mekinist en association avec le dabrafénib, ainsi qu'un mois après le commencement du traitement et ensuite, tous les 3 mois pendant le traitement, par échographie du cœur ou par MUGA (multi-gated acquisition [angiographie isotopique]).
Chez les patients qui sont traités avec Mekinist en association avec le dabrafénib, le traitement avec Mekinist doit être interrompu chez les patients avec une diminution absolue et asymptomatique de la FEVG > 10% par rapport au début du traitement et une fraction d'éjection inférieure à la limite inférieure de la normale (LIN) pour l'établissement concerné (voir «Mises en garde et précautions»). Aucune adaptation posologique du dabrafénib n'est requise. Lorsque la FEVG est rétablie, le traitement par Mekinist peut être repris, mais à une dose réduite d'un palier et sous une surveillance étroite. Lors d'un dysfonctionnement cardiaque ventriculaire gauche de degré 3 ou 4 ou lorsque la FEVG ne peut pas être rétablie, Mekinist doit être définitivement arrêté.
Traitement de l'occlusion veineuse rétinienne (OVR), du décollement de l'épithélium pigmentaire de la rétine/décollement de la rétine (RPED [retinal pigment epithelial detachment]) et de la choriorétinopathie
Si des patients sous traitement avec Mekinist signalent l'apparition de nouveaux troubles de la vision, tels que des troubles de la vision centrale, une vision floue ou une perte d'acuité visuelle, une évaluation ophtalmologique urgente est nécessaire. Chez les patients pour lesquels une OVR est diagnostiquée, le traitement avec Mekinist doit être définitivement arrêté. Le traitement par dabrafénib peut être maintenu au même dosage. Si un RPED ou une choriorétinopathie est diagnostiqué, il convient de suivre le plan d'ajustement posologique pour Mekinist du tableau 3 - le traitement par dabrafénib est poursuivi avec la même dose (voir «Mises en garde et précautions»).
Traitement de la pneumonie et de la pneumopathie interstitielle diffuse (PID)
Lors de signes de pneumonie et de PID, il convient de suivre seulement le plan d'ajustement posologique pour Mekinist du tableau 3; aucune modification de la dose de dabrafénib n'est nécessaire.
Traitement associé
Les données sur la modification posologique du dabrafénib se trouvent dans l'information sur le médicament détaillée du dabrafénib.
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucune adaptation posologique n'est nécessaire chez les patients atteints d'insuffisance hépatique légère. Dans une analyse pharmacocinétique de population, la clairance orale du tramétinib, et ainsi également l'exposition, n'étaient pas significativement différentes entre les patients avec une insuffisance hépatique légère et les patients avec un foie sain. Les données disponibles concernant les personnes présentant une insuffisance hépatique modérée à sévère indiquent une influence limitée sur l'exposition au tramétinib (voir «Pharmacocinétique»). Mekinist doit être utilisé avec prudence chez les personnes présentant une insuffisance hépatique modérée à sévère.
Patients présentant des troubles de la fonction rénale
Aucune adaptation posologique n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. Une insuffisance rénale légère à modérée n'a eu aucun effet sur la pharmacocinétique de population du tramétinib (voir «Pharmacocinétique»).
L'absence de données concernant Mekinist chez les patients ayant une insuffisance rénale sévère ne permet pas d'établir de recommandations d'adaptation de la posologie.
Patients âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés de plus de 65 ans (voir «Pharmacocinétique»).
Enfants et adolescents
L'utilisation de Mekinist chez les patients pédiatriques âgés de moins de 1 an n'est pas autorisée. En raison de la toxicité rénale du dabrafénib dans les études de toxicité juvénile chez le rat, ces patients ont été exclus des études cliniques portant sur Mekinist en association avec le dabrafénib. Pour les enfants âgés de 1 à 2 ans, on ne dispose que de données limitées concernant la sécurité et l'efficacité (voir «Mises en garde et précautions»).
Prise retardée
Si une dose de Mekinist a été oubliée, elle ne doit pas être rattrapée s'il reste moins de 12 heures avant la prise de la dose suivante prévue. Si une dose de dabrafénib a été oubliée, elle ne doit pas être rattrapée s'il reste moins de 6 heures avant la prise de la dose suivante prévue.
Contre-indicationsHypersensibilité au principe actif ou à l'un des autres composants.
Mises en garde et précautionsLe taux de réponse au traitement combiné chez les patients chez lesquels une progression de la maladie a été observée lors d'un traitement préalable avec un inhibiteur de BRAF a été nettement plus faible que chez les patients n'ayant pas reçu un tel traitement préalable. Par conséquent, chez les patients chez lesquels une progression de la maladie a été observée lors d'un traitement préalable avec un inhibiteur de BRAF, d'autres options thérapeutiques doivent être envisagées avant d'initier le traitement par l'association.
Des informations supplémentaires concernant les mises en garde et précautions dans le contexte d'un traitement avec le dabrafénib se trouvent dans l'information professionnelle du dabrafénib.
Nouvelles tumeurs malignes
Lors de l'association de Mekinist et du dabrafénib, de nouvelles tumeurs malignes, cutanées ou non cutanées, peuvent survenir.
Hyperglycémie
Dans le cadre d'études cliniques dans lesquelles Mekinist avait été administré en association avec le dabrafénib, 15% des patients avec des antécédents de diabète ayant reçu une association de Mekinist et de dabrafénib ont eu besoin d'un traitement hypoglycémiant plus intensif. Des hyperglycémies de grades 3 et 4 sont survenues chez 2% des patients. Veuillez surveiller le taux de glucose sérique au début du traitement et si cela est approprié d'un point de vue clinique lorsque Mekinist est administré en association avec le dabrafénib à des patients présentant déjà un diabète existant ou une hyperglycémie.
Pneumopathie interstitielle diffuse (PID)
Dans différentes études cliniques portant sur Mekinist, une PID ou une pneumopathie inflammatoire sont survenues chez 2% des patients. Chez les patients avec apparition ou progression de symptômes pulmonaires, dont une toux, une dyspnée, une hypoxie, un épanchement pleural ou des infiltrats, Mekinist doit être arrêté jusqu'à l'évaluation clinique. Mekinist doit être définitivement arrêté chez les patients ayant reçu un diagnostic de PID ou de pneumopathie inflammatoire liée au traitement.
Carcinome épidermoïde cutané (cutaneous Squamous Cell Carcinoma, cuSCC)
Des cas de cuSCC (dont également ceux qui sont classifiés dans les sous-types kératoacanthome ou kératoacanthome mixte) ont été rapportés chez des patients qui sont traités avec Mekinist en association avec le dabrafénib.
Dans une étude de phase 3, un cuSCC a été rapporté chez 2% des patients qui avaient reçu Mekinist en association avec le dabrafénib, et chez 9% des patients qui avaient reçu le dabrafénib en monothérapie. La durée moyenne jusqu'au diagnostic de la première apparition de cuSCC dans le bras de traitement combiné a été de 222 jours (avec une plage allant de 56 à 328 jours) et de 57 jours (avec une plage de 9 à 169 jours) dans le bras dabrafénib en monothérapie.
Un examen de la peau doit être effectué avant le début du traitement avec le dabrafénib, pendant le traitement avec le dabrafénib, tous les 2 mois pendant tout le traitement. La surveillance doit être poursuivie tous les 2 à 3 mois pendant 6 mois après la fin du traitement avec le dabrafénib ou jusqu'à ce qu'un autre traitement antinéoplasique soit commencé.
Les cas de cuSCC doivent être pris en charge par une exérèse chirurgicale et ne nécessitent pas de modification du traitement. Des instructions doivent être données aux patients d'informer immédiatement leur médecin si de nouvelles lésions se forment.
Hémorragies
Des événements hémorragiques, dont des hémorragies graves, dans des zones corporelles et organes critiques sont survenus chez des patients qui ont utilisé Mekinist en association avec le dabrafénib (voir «Effets indésirables»). Lors d'études cliniques avec cette association, des hémorragies ont été rapportées chez 17% des patients; 3% des patients ont souffert d'hémorragies gastro-intestinales. Six des 559 patients (1%) atteints d'un mélanome non résécable ou métastatique qui recevaient Mekinist en association avec le dabrafénib ont souffert d'hémorragies intracrâniennes d'issue fatale. Trois cas ont été observés dans l'étude MEK115306 (COMBId) et trois dans l'étude MEK116513 (COMBIv). Lors du traitement adjuvant du mélanome, aucun évènement hémorragique d'issue fatale n'est survenu en phase III de l'étude.
Deux des 93 participants (2%) ayant reçu Mekinist en association avec le dabrafénib dans une étude de phase II sur le cancer bronchique non à petites cellules (NSCLC, non-small cell lung cancer) ont souffert d'hémorragies d'issue fatale (un cas d'hémorragie sous-arachnoïdienne et un cas d'hémorragie rétropéritonéale).
Lorsque des patients développent les symptômes d'une hémorragie intracrânienne, une intervention médicale est nécessaire dans les plus brefs délais.
Le risque d'événements hémorragiques peut être majoré par l'utilisation concomitante d'un traitement antiplaquettaire ou anticoagulant. En cas d'hémorragie, les patients doivent être traités selon l'indication clinique.
En cas d'hémorragies de grade 4, Mekinist doit être définitivement arrêté; il en est de même pour les hémorragies de grade 3 qui ne s'améliorent pas.
Nouveau mélanome primitif
Dans une étude de phase III, un cas de nouveau mélanome primitif chez un patient qui recevait Mekinist en association avec le dabrafénib a été mentionné, lequel mélanome est apparu 110 jours après le début de l'étude (< 1%). Les cas de nouveau mélanome primitif peuvent être pris en charge par une exérèse chirurgicale et ne nécessitent pas de modification du traitement. La surveillance des lésions cutanées doit être effectuée de la même manière que pour le cuSCC.
Tumeur maligne secondaire/récidivante non cutanée
Des expériences in vitro ont montré une activation paradoxale de la voie de signalisation MAPkinase dans les cellules BRAF de type sauvage avec mutations RAS dans le cas d'une exposition à des inhibiteurs de BRAF, ce qui peut conduire à un risque accru de tumeurs malignes non cutanées chez les patients traités avec le dabrafénib. Sous inhibiteurs de BRAF, des cas de tumeurs malignes dépendantes de RAS ont été constatés. Les patients doivent être surveillés selon la nécessité clinique. Avant la poursuite du traitement avec le dabrafénib chez les patients atteints d'une tumeur maligne non cutanée avec mutation RAS, les avantages et risques doivent être évalués. Sous le traitement combiné de Mekinist avec le dabrafénib, aucun ajustement de la dose de Mekinist n'est nécessaire.
Après l'arrêt du dabrafénib, la surveillance des tumeurs malignes secondaires/récidivantes non cutanées doit être poursuivie pendant au moins 6 mois ou jusqu'à ce qu'un autre traitement antinéoplasique soit commencé.
Pyrexie
Dans les études cliniques sur Mekinist en association avec le dabrafénib, la survenue de pyrexie a été rapportée. La majorité des évènements de pyrexie est survenue au cours du premier mois du traitement. Environ un tiers des patients sous le traitement combiné, chez lesquels une pyrexie s'est développée, a présenté trois événements ou plus. La pyrexie peut être accompagnée de frissons importants, de déshydratation et d'hypotension, ce qui peut conduire dans certains cas à une insuffisance rénale aiguë. Pendant et après des évènements sévères de pyrexie, la créatinine sérique et la fonction rénale doivent être surveillées. Des évènements fébriles non infectieux graves ont été observés. Dans les études cliniques, ces évènements ont bien répondu aux interruptions et/ou aux réductions posologiques, ainsi qu'aux soins de support (notamment l'administration d'antipyrétiques non stéroïdiens et stéroïdiens). Une comparaison portant sur 1810 patients de plusieurs études, qui étaient traités par l'association, a montré une réduction de l'incidence de la pyrexie sévère et d'autres événements indésirables liés à la pyrexie lorsque le traitement tant par Mekinist que par le dabrafénib a été interrompu, comparé à l'interruption du dabrafénib seul. Une interruption de Mekinist, mais également du dabrafénib est donc recommandée lorsque la température corporelle du patient est ≥38 °C (≥100,4 °F). En cas de récidive, le traitement peut également être interrompu aux premiers symptômes de pyrexie (voir les rubriques «Posologie/Mode d'emploi» et «Efficacité clinique»).
Voir l'information professionnelle du dabrafénib pour de plus amples informations concernant le traitement de la pyrexie (voir «Posologie/mode d'emploi»).
Colite et perforation de la paroi de l'estomac ou de l'intestin (perforation gastrique/intestinale)
Des cas de colite et de perforation de la paroi de l'estomac ou de l'intestin (perforation gastrique/intestinale), y compris des cas ayant une issue fatale, sont survenus chez des patients qui prenaient Mekinist en monothérapie ou en association avec le dabrafénib (voir «Effets indésirables»). Le traitement par Mekinist en monothérapie ou en association avec le dabrafénib doit être utilisé avec précaution chez les patients présentant des facteurs de risque de perforation gastrique/intestinale, y compris des antécédents de diverticulite ou de métastases du tractus gastro-intestinal. L'utilisation concomitante de médicaments connus pour être associés à un risque de perforation gastro-intestinale (p.ex. bévacizumab, aflibercept et d'autres inhibiteurs de l'angiogenèse) doit par ailleurs être évitée.
Une prise en charge médicale immédiate est nécessaire lorsqu'un patient présente des symptômes de colite ou de perforation gastrique ou intestinale.
Réduction de la FEVG/Dysfonction ventriculaire gauche
Sous Mekinist, en association avec le dabrafénib, une diminution de la FEVG a été mentionnée (voir «Effets indésirables»). Dans les études cliniques, la durée médiane jusqu'au début de la première apparition d'un dysfonctionnement ventriculaire gauche, d'une insuffisance cardiaque et d'une chute de la FEVG, chez les patients traités par Mekinist en association avec le dabrafénib, se situait entre deux et cinq mois. La prudence s'impose lors de l'utilisation de Mekinist chez des patients souffrant de maladies susceptibles de limiter la fonction ventriculaire gauche. La FEVG doit être évaluée chez tous les patients avant le traitement par Mekinist, un mois après le commencement du traitement et ensuite, environ tous les 3 mois pendant le traitement (voir «Posologie/Mode d'emploi»).
ECG
Mekinist en monothérapie n'influence pas ou seulement faiblement l'intervalle QT. Par contre, sous Mekinist, une diminution de la fréquence cardiaque, ainsi qu'un allongement de l'intervalle PR sont observés.
L'association de Mekinist avec le dabrafénib n'a pas fait l'objet d'une étude ciblée concernant l'intervalle QTc.
Troubles de la vision
Sous Mekinist, des maladies en relation avec des troubles de la vision, en particulier le décollement de l'épithélium pigmentaire de la rétine/décollement de la rétine (RPED), la choriorétinopathie et l'occlusion veineuse rétinienne (OVR), ont été observées. Dans les études cliniques avec Mekinist, des symptômes tels qu'une vision floue, une acuité visuelle diminuée et d'autres troubles visuels ont été rapportés. Mekinist n'est pas recommandé chez les patients ayant des antécédents d'OVR.
Au début et pendant le traitement par Mekinist, un examen ophtalmologique complet doit être effectué en cas de nécessité clinique. Si des patients sous traitement par Mekinist signalent l'apparition de troubles de la vision, une évaluation ophtalmologique doit être rapidement réalisée. Si une anomalie rétinienne est diagnostiquée, le traitement par Mekinist doit être immédiatement interrompu et un renvoi chez un spécialiste de la rétine doit être envisagé. En cas de diagnostic de RPED ou de choriorétinopathie, procéder selon le schéma de modification posologique des tableaux 1 et 2 de la rubrique «Posologie/Mode d'emploi». Chez les patients chez lesquels une OVR est diagnostiquée, le traitement par Mekinist doit être définitivement arrêté.
Éruption cutanée
Dans les études cliniques sur Mekinist en association avec le dabrafénib, une éruption cutanée s'est développée chez environ 20–30% des patients (voir «Effets indésirables»). Ces cas étaient majoritairement de degré de sévérité 1 ou 2 et n'ont exigé ni une interruption du traitement, ni une réduction de la posologie.
Réactions cutanées indésirables sévères
Des cas de réactions cutanées indésirables sévères (SCARS, severe adverse skin reactions) ont été rapportés sous traitement par Mekinist en association avec le dabrafénib. Il s'agit notamment du syndrome de Stevens-Johnson et de la réaction médicamenteuse avec symptômes systémiques et éosinophilie (DRESS, drug reaction with systemic symptoms and eosinophilia), qui peuvent engager le pronostic vital ou être d'issue fatale. Les patients doivent donc être informés des différents signes et symptômes avant le début du traitement. Pendant le traitement, les patients doivent faire l'objet d'une étroite surveillance pour détecter les réactions cutanées. Si des signes et des symptômes d'une réaction cutanée indésirable sévère surviennent, le traitement par Mekinist et le dabrafénib doit être arrêté.
Thrombose veineuse profonde (TVP)/embolie pulmonaire (EP)
Des TVP et EP peuvent survenir lors de l'utilisation de Mekinist en association avec le dabrafénib. Lorsque les patients développent les symptômes d'une embolie pulmonaire ou d'une thrombose veineuse profonde, ils doivent consulter immédiatement un médecin.
Lymphohistiocytose hémophagocytaire (LHH)
Une LHH a été observée lors du suivi post-marketing de Mekinist en association avec Tafinlar (dabrafénib). La LHH est un syndrome potentiellement mortel s'accompagnant d'une activation pathologique des défenses immunitaires. En l'absence de diagnostic et de traitement précoces, la LHH a fréquemment une évolution létale. Cette maladie se caractérise par des signes et symptômes cliniques d'une inflammation systémique sévère, tels que fièvre, éruption cutanée, hépatosplénomégalie, cytopénies, lymphadénopathie, symptômes neurologiques, taux élevé de ferritine sérique, hypertriglycéridémie ainsi que troubles de la fonction hépatique et de la coagulation. Les symptômes surviennent généralement dans les deux mois suivant le début du traitement, mais peuvent également se manifester plus tard. En cas de suspicion de LHH, le traitement doit être interrompu. Après confirmation du diagnostic de LHH, le traitement doit être arrêté et un traitement adapté de la LHH instauré.
Fertilité
Aucune donnée n'est disponible concernant les effets du tramétinib sur la fertilité humaine. Des études de fertilité sur des animaux n'ont certes pas été réalisées, des effets indésirables ont cependant pu être observés sur les organes reproducteurs femelles (voir «Données précliniques»). Il est possible que le tramétinib influence la fertilité humaine.
Population pédiatrique
Les patients pédiatriques en développement peuvent devoir suivre un traitement par Mekinist en association avec le dabrafénib à long terme. Les deux médicaments agissent simultanément sur une chaine de transduction de signal, qui joue un rôle important dans la régulation des développements cellulaire et tissulaire (voir «Mécanisme d'action»). Pour cette raison, les données concernant la population pédiatrique disponibles présentent des limitations (voir également «Posologie/Mode d'emploi»).
On ne dispose pas de données suffisantes concernant la sécurité et l'efficacité de l'utilisation à long terme et de la durée optimale du traitement par Mekinist en association avec le dabrafénib dans la population pédiatrique. Dans les études cliniques pertinentes, les durées médianes du traitement et du suivi se situaient respectivement, à 24 et 26 mois (voir «Efficacité clinique»). Les conséquences à long terme de la prise de poids indésirable très fréquemment observée sous l'association sont peu claires à ce jour. Bien que jusqu'ici, aucune augmentation des fréquences de néoplasies malignes secondaires n'ait été rapportée dans la population pédiatrique, une évaluation finale n'est aujourd'hui pas possible (voir «Effets indésirables»).
Pour les enfants âgés de 1 à 2 ans, on ne dispose que de données limitées concernant la sécurité et l'efficacité.
La contribution de Mekinist et du dabrafénib à l'efficacité de l'association chez les patients pédiatriques atteints de LGG n'est pas claire, parce qu'aucune comparaison directe entre l'association et les monothérapies n'existe pour une population de patients de taille suffisante.
Patients exclus des études cliniques
Les critères d'exclusion communs à toutes les études sur Mekinist étaient: BRAF de type sauvage ou mutation non V600; antécédents ou signes de risque cardiovasculaire; antécédents de pneumopathie inflammatoire ou de pneumopathie interstitielle; antécédents ou signe d'un risque de rétinopathie. Les critères d'exclusion spécifiques d'étude sont donnés dans la rubrique «Efficacité clinique».
Excipients
Sulfobutylbétadex sodique
Ce médicament contient 100 mg de cyclodextrine par ml. N'utilisez pas ce médicament chez les enfants de moins de 2 ans sauf si votre médecin vous l'a recommandé.
Parahydroxybenzoate de méthyle (E 218)
Peut engendrer des réactions allergiques, parfois retardées.
Potassium
Ce médicament contient du potassium, mais moins de 1 mmol (39 mg) de potassium par 2 mg de tramétinib (dose maximale), c.-à-d. qu'il est essentiellement «sans potassium».
Sodium
Comprimés pelliculés
Ce médicament contient moins de 1 mmol de sodium (23 mg) par comprimé pelliculé, c.-à-d. qu'il est essentiellement «sans sodium».
Poudre pour solution buvable
Ce médicament contient 79,2 mg de sodium (constituant principal du sel de cuisine/sel de table) par 2 mg de tramétinib (dose maximale). Cela équivaut à 4% de l'apport alimentaire quotidien maximal recommandé de sodium pour un adulte.
InteractionsVeuillez vous reporter à l'information professionnelle du dabrafénib pour de plus amples informations sur les interactions en relation avec le dabrafénib.
Interactions pharmacocinétiques
Le tramétinib étant essentiellement métabolisé par une désacétylation induite par des enzymes hydrolytiques (par exemple, carboxylestérases), il est peu vraisemblable que sa pharmacocinétique soit influencée par des interactions métaboliques avec d'autres principes actifs. L'exposition à des doses répétées de tramétinib n'a pas été influencée par l'administration simultanée d'un inducteur de CYP3A4 (voir «Pharmacocinétique»).
Interactions pharmacodynamiques
Selon les données in vitro et in vivo, il est peu vraisemblable que le tramétinib influence la pharmacocinétique d'autres médicaments par interaction avec les enzymes ou transporteurs du CYP (voir «Pharmacocinétique»).
L'administration répétée d'une dose de 2 mg de tramétinib une fois par jour n'a eu aucune influence sur la Cmax et l'ASC d'une dose unique de dabrafénib, un substrat du CYP2C8/CYP3A4.
Autres interactions
Association avec le dabrafénib
L'administration simultanée de doses répétées de tramétinib 2 mg une fois par jour et de dabrafénib 150 mg deux fois par jour n'a pas conduit à des modifications cliniquement significatives des Cmax et ASC du tramétinib et du dabrafénib, avec augmentation de la Cmax et de l'ASC du dabrafénib de respectivement 23 et 16%. Grâce à une analyse pharmacocinétique de population, un léger recul de la biodisponibilité du tramétinib, correspondant à une diminution de 12% de l'ASC, a été estimé pour l'administration simultanée de tramétinib et de dabrafénib. Pour les recommandations concernant les interactions du dabrafénib avec d'autres médicaments, voir l'information professionnelle du dabrafénib.
Effet de MEKINIST sur d'autres médicaments
Les données découlant de recherches in vitro et in vivo suggèrent que le tramétinib n'influence vraisemblablement pas la pharmacocinétique d'autres médicaments. D'après les résultats d'études in vitro, le tramétinib n'est pas un inhibiteur du CYP1A2, CYP2A6, CYP2B6, CYP2D6 et CYP3A4. In vitro, le tramétinib s'est révélé être un inhibiteur du CYP2C8, CYP2C9 et CYP2C19, un inducteur du CYP3A4 ainsi qu'un inhibiteur des transporteurs OAT1, OAT3, OCT2, MATE1, OATP1B1, OATP1B3, Pgp et BCRP. En raison de la faible dose et de la faible exposition systémique clinique par rapport à la puissance in vitro de l'inhibition et respectivement, de l'induction, le tramétinib n'est pas considéré comme un inhibiteur et respectivement, inducteur in vivo de ces enzymes et transporteurs.
Effet d'autres médicaments sur MEKINIST
Les données découlant de recherches in vitro et in vivo suggèrent que la pharmacocinétique du tramétinib n'est vraisemblablement pas influencée par d'autres médicaments. Le tramétinib n'est pas un substrat des enzymes du CYP, ni des transporteurs BCRP, OATP1B1, OATP1B3, OATP2B1, OCT1, MRP2 et MATE1. Le tramétinib est désacétylé par les carboxylestérases; des interactions médicamenteuses impliquant des estérases ne sont pas décrites dans la littérature. Le tramétinib est un substrat in vitro du transporteur d'efflux Pgp, une influence par inhibition de ces transporteurs est cependant peu vraisemblable étant donné la perméabilité passive élevée et la biodisponibilité élevée. Après administration simultanée du tramétinib avec le dabrafénib, un inducteur du CYP3A4, la Cmax et l'ASC du tramétinib après administration répétée correspondaient à l'exposition observée sous monothérapie; ceci montre qu'un inducteur du CYP3A4 n'a aucune influence sur l'exposition au tramétinib.
Grossesse, allaitementGrossesse
Mekinist peut nuire au fœtus lorsque le médicament est administré à une femme enceinte. Il n'existe aucune donnée sur Mekinist chez la femme enceinte. Des études réalisées sur des animaux ont montré une toxicité pour la reproduction (voir «Données précliniques»).
C'est pourquoi Mekinist en association avec le dabrafénib ne doit pas être administré pendant la grossesse, sauf en cas de nécessité absolue. Avant le début d'un traitement, un test de grossesse doit être effectué.
Aussi bien les hommes que les femmes doivent utiliser des méthodes de contraception efficaces:
Femmes: les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement et pendant les 4 premiers mois qui suivent la fin du traitement. Si Mekinist est utilisé avec le dabrafénib au cours d'une grossesse ou si une grossesse survient pendant le traitement, il faut informer la patiente du risque potentiel pour l'enfant à naître.
Les femmes en âge de procréer qui reçoivent Mekinist en association avec le dabrafénib doivent être informées que le partenaire de l'association, le dabrafénib, peut diminuer l'activité des moyens de contraception hormonaux et que des méthodes contraceptives alternatives doivent être utilisées (voir «Interactions»).
Hommes: les patients de sexe masculin (également ceux chez lesquels une vasectomie a été effectuée) ayant des partenaires sexuelles qui sont enceintes, qui le sont éventuellement ou qui peuvent tomber enceintes, doivent utiliser des préservatifs lors des rapports sexuels pendant la monothérapie avec Mekinist ou le traitement combiné par Mekinist et le dabrafénib, ainsi que pendant au moins 16 semaines après l'arrêt du traitement par Mekinist.
Allaitement
Il n'existe aucune donnée sur l'effet de Mekinist sur l'enfant allaité ou l'effet de Mekinist sur la production de lait. On ignore si le tramétinib est excrété dans le lait maternel. Un risque pour l'enfant allaité ne peut être exclu, car de nombreux médicaments sont excrétés dans le lait maternel. Mekinist ne doit pas être administré à la mère allaitante. En considérant l'importance de l'allaitement pour l'enfant et le bénéfice du traitement pour la mère, il conviendra de mettre fin soit à l'allaitement, soit au traitement par Mekinist.
Fertilité
Aucune donnée sur la fertilité chez l'être humain n'est disponible pour Mekinist. Des effets indésirables sur les organes reproducteurs mâles et femelles ont été observés chez l'animal (voir «Données précliniques»).
Il convient de signaler un risque éventuel de détérioration potentiellement irréversible de la spermatogenèse aux patients masculins.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude sur les effets de Mekinist sur l'aptitude à la conduite ou à l'utilisation de machines n'a été effectuée. Étant donné la pharmacologie du tramétinib, il ne devrait exister aucune influence défavorable sur ces activités. L'état clinique du patient et le profil des effets indésirables de Mekinist doivent être pris en compte lors de l'évaluation de la capacité du patient à effectuer des tâches qui font appel à la capacité de jugement ou à des aptitudes motrices ou cognitives.
Effets indésirablesLes effets indésirables médicamenteux (EIM) décrits ci-dessous prennent en compte différentes sources d'informations de sécurité, y compris celles des études cliniques, des rapports suivant l'introduction sur le marché et des rapports bibliographiques.
La fréquence des EIM décrits ci-dessous dans le tableau 4 est basée sur la population intégrée soumise à l'évaluation de l'innocuité comportant 1087 patients atteints d'un mélanome de stade III inopérable ou métastasique portant la mutation BRAF V600, un mélanome portant la mutation BRAF V600 après résection complète avec traitement adjuvant et un NSCLC avancé.
Tous les patients ont été traités par 2 mg de Mekinist une fois par jour et 150 mg de dabrafénib deux fois par jour. Parmi ces patients, 559 ont été traités dans deux études de phase III randomisées, MEK115306 (COMBId) et MEK116513 (COMBIv), avec la combinaison pour le mélanome portant la mutation BRAF V600. 435 ont été traités dans une étude de phase III randomisée BRF115532 (COMBI-AD) avec la combinaison pour le traitement adjuvant du mélanome de stade III portant la mutation BRAF V600 et 93 ont été traités avec la combinaison pour le NSCLC portant la mutation BRAF V600 dans une étude de cohortes multiples de phase II non randomisée BRF113928.
Les effets indésirables les plus fréquents (incidence > 20%) pour Mekinist en association avec le dabrafénib étaient: pyrexie, fatigue, nausées, frissons, céphalées, diarrhée, vomissements, arthralgie et éruption cutanée.
Les effets indésirables médicamenteux sont présentés selon le système de classe d'organe et classés par ordre décroissant de fréquence selon la convention suivante: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100); rares (≥1/10 000 à < 1/1000), très rares (< 1/10 000), y compris les cas isolés. Au sein de chaque groupe de fréquence, les effets indésirables sont classés par ordre décroissant de sévérité.
Tableau 4: effets indésirables ayant été rapportés parmi la population intégrée soumise à l'évaluation de l'innocuité de Mekinist en association avec le dabrafénib dans les études MEK115306, MEK116513a, BRF113928 et BRF115532 (n = 1076)
|
Infections et infestations
| |
Très fréquents
|
Rhinopharyngite (11%)
| |
Fréquents
|
Infection urinaire, cellulite, folliculite, paronychie, éruption cutanée pustuleuse
| |
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
| |
Fréquents
|
Carcinome épidermoïde cutané (squamous cell carcinoma, SCC)b, papillomec, kératose séborrhéique
| |
Occasionnels
|
Nouveau mélanome primitifd, acrochordon (fibrome)
| |
Affections hématologiques et du système lymphatique
| |
Fréquents
|
Neutropénie, anémie, thrombopénie, leucopénie
| |
Affections du système immunitaire
| |
Occasionnels
|
Hypersensibilitée, sarcoïdose
| |
Fréquence inconnue*
|
Lymphohistiocytose hémophagocytaire
| |
Troubles du métabolisme et de la nutrition
| |
Très fréquents
|
Perte d'appétit (14%)
| |
Fréquents
|
Déshydratation, hyponatrémie, hypophosphatémie, hyperglycémie
| |
Affections du système nerveux
| |
Très fréquents
|
Céphalées (33%), sensation vertigineuse (11%)
| |
Affections oculaires
| |
Fréquents
|
Vision floue, défauts visuels, uvéite
| |
Occasionnels
|
Choriorétinopathie, décollement de l'épithélium pigmentaire de la rétine/décollement de la rétine (RPED), œdème périorbital
| |
Affections cardiaques
| |
Fréquents
|
Fraction d'éjection diminuée
| |
Occasionnels
|
Bradycardie
| |
Rares
|
Myocardite*
| |
Affections vasculaires
| |
Très fréquents
|
Hypertension (19%), hémorragiesf (19%)
| |
Fréquents
|
Hypotension, lymphœdème
| |
Affections respiratoires, thoraciques et médiastinales
| |
Très fréquents
|
Toux (20%)
| |
Fréquents
|
Dyspnée
| |
Occasionnels
|
Pneumopathie inflammatoire
| |
Affections gastro-intestinales
| |
Très fréquents
|
Nausées (38%), diarrhée (32%), vomissements (29%), douleurs abdominales (17%)g, constipation (13%)
| |
Fréquents
|
Bouche sèche, stomatite
| |
Occasionnels
|
Pancréatite, colite
| |
Rares
|
Perforation gastro-intestinale
| |
Affections hépatobiliaires
| |
Très fréquents
|
Augmentation de l'alanine aminotransférase (14%), augmentation de l'aspartate aminotransférase (13%)
| |
Fréquents
|
Augmentation des phosphatases alcalines dans le sang, augmentation des gamma-glutamyltransférases
| |
Affections de la peau et du tissu sous-cutané
| |
Très fréquents
|
Éruption (24%), sécheresse cutanée (13%), prurit (10%), érythèmeh (10%)
| |
Fréquents
|
Dermatite, acnéiforme, kératose actinique, sueurs nocturnes, hyperkératose, alopécie, syndrome d'érythrodysesthésie palmoplantaire, lésion de la peau, hyperhidrose, fissures cutanées, panniculite, photosensibilitéi
| |
Affections musculosquelettiques et du tissu conjonctif
| |
Très fréquents
|
Arthralgie (26%), myalgie (15%), douleurs des extrémités (12%), crampes musculairesk (10%)
| |
Fréquents
|
Augmentation de la créatine phosphokinase dans le sang
| |
Affections du rein et des voies urinaires
| |
Fréquents
|
Insuffisance rénale
| |
Occasionnels
|
Néphrite
| |
Troubles généraux et anomalies au site d'administration
| |
Très fréquents
|
Pyrexie (58%), fatigue (38%), frissons (33%), œdème périphérique (16%), asthénie (15%), infection grippale (11%)
| |
Fréquents
|
Inflammation muqueuse, œdème du visage
|
a Le profil d'innocuité observé dans MEK116513 est dans l'ensemble similaire à celui observé dans MEK115306, avec les exceptions suivantes: 1) Les effets indésirables suivants ont une catégorie de fréquence plus élevée comparé à MEK115306: crampes musculaires (très fréquents); insuffisance rénale et lymphœdème (fréquents); insuffisance rénale aiguë (occasionnels); 2) Les effets indésirables suivants sont survenus dans MEK116513, mais pas dans MEK115306: insuffisance cardiaque, dysfonctionnement du ventricule gauche, pneumopathie interstitielle (occasionnel). 3) L'effet indésirable suivant est survenu dans MEK116513 et BRF115532, mais pas dans MEK115306 et BRF113928: rhabdomyolyse (occasionnels)
b Inclut SCC cutanés, SCC in situ (maladie de Bowen) et kératoacanthome
c Inclut papillomes, papillomes cutanés
d Inclut mélanome malin; mélanome métastasé malin, et mélanome à extension superficielle de stade III
e Inclut hypersensibilité aux médicaments
f Hémorragies à différents endroits, y compris hémorragies intracrâniennes et hémorragies mortelles
g Inclut douleurs abdominales, dans l'abdomen supérieur et inférieur
h Inclut érythème, érythème généralisé
i Des cas de photosensibilité ont également été observés pendant l'expérience acquise après la commercialisation. Tous les cas rapportés dans les études cliniques COMBId et COMBIv étaient de grade 1 et aucune adaptation posologique n'a été nécessaire.
k Inclut crampes musculaires et raideur musculo-squelettique
*Fréquence basée sur le rapport après l'introduction sur le marché
Populations particulières
Population pédiatrique
Mekinist en association avec le dabrafénib
La sécurité de Mekinist en association avec le dabrafénib a été étudiée auprès de 171 patients pédiatriques au cours de deux études (étude DRB436B2201; n = 123 et étude TMT212 X2101; n = 48), dont 159 avec gliome positif à la mutation BRAF V600E.
Le profil de sécurité général dans la population pédiatrique était similaire à celui observé pour l'adulte. Les effets indésirables médicamenteux rapportés le plus fréquemment (≥20%) étaient les suivants: fièvre (pyrexie) (65%), éruption cutanée (47%), céphalées (39%), vomissements (38%), sécheresse cutanée (34%), fatigue (33%), diarrhée (30%), saignements (29%), neutropénie (25%), nausées (25%), dermatite acnéiforme (25%), douleurs abdominales (23%) et toux (21%).
Dans l'ensemble pédiatrique de sécurité, un effet indésirable médicamenteux sous forme d'une prise de poids a été établi à la fréquence de 15,2% (très fréquents). Chez 51 patients sur 171 (29,8%), l'IMC a augmenté de ≥2 catégories de percentile de l'IMC par comparaison à la référence.
Dans l'étude DRB436G2201, une infection COVID-19 a été signalée chez 20,5% (15 sur 73) des patients pédiatriques traités par Mekinist en association avec le dabrafénib, dans un cas avec un degré de sévérité ≥3.
Dans l'ensemble pédiatrique de sécurité, une baisse de la fraction d'éjection du ventricule gauche (FEVG) de 10% ou plus par rapport à la valeur initiale et sous la limite inférieure de la normale (LIN) de l'institution a été observée chez 8,7% (14 sur 161) des patients pédiatriques sous l'association Mekinist avec dabrafénib.
D'autres effets indésirables médicamenteux, qui sont survenus plus fréquemment chez les patients pédiatriques par comparaison aux patients adultes, étaient la neutropénie, la dermatite acnéiforme, la paronychie, l'anémie, la leucopénie (très fréquents), la bradycardie, la dermatite exfoliative généralisée, l'hypersensibilité et la pancréatite (fréquents). En outre, dans la cohorte LGG de l'étude G2201, l'incidence relative des augmentations des lymphocytes, des augmentations du magnésium et d'hypotension systolique ont été plus élevées avec la thérapie ciblée que sous chimiothérapie.
Tableau 5: Effets indésirables médicamenteux les plus fréquents de grade 3/4 (≥2%) sous Mekinist en association avec le dabrafénib chez les patients pédiatriques
|
Effets indésirables médicamenteux
|
Mekinist en association avec le dabrafénib
N = 171
| |
Grade 3 et 4 (%)
| |
Neutropénie
|
25 (15)
| |
Fièvre (pyrexie)1
|
15 (9)
| |
Augmentation de l'alanine-aminotransférase²
|
10 (6)
| |
Augmentation de l'aspartate-aminotransférase3
|
6 (4)
| |
Prise de poids (augmentation du poids)
|
7 (4)
| |
Céphalées
|
5 (3)
| |
Vomissements
|
5 (3)
| |
Hypotension
|
4 (2)
| |
Éruption cutanée4
|
4 (2)
| |
Augmentation de la phosphatase alcaline dans le sang
|
4 (2)
| |
1
La neutropénie comprend la neutropénie, la réduction de la numération des neutrophiles et la neutropénie fébrile.
2 ALAT comprend l'augmentation de l'alanine-aminotransférase et l'augmentation des transaminases.
3 ASAT comprend l'augmentation de l'aspartate-aminotransférase et l'augmentation des transaminases.
4 L'éruption comprend l'éruption, l'éruption maculo-papuleuse, l'éruption papuleuse, l'éruption érythémateuse, l'éruption papuleuse et l'éruption maculeuse..
|
Chez les patients pédiatriques âgés de moins de 6 ans, plus d'événements graves ont été signalés sous traitement combiné de Mekinist et de dabrafénib que chez les patients âgés de 6 à 12 ans. De la fièvre a été observée plus fréquemment chez les enfants de moins de 6 ans que chez les patients pédiatriques plus âgés.
L'ensemble pédiatrique de sécurité ne comprend que quatre enfants âgés de 1 à 2 ans, de sorte que le profil de sécurité est insuffisamment caractérisé dans cette catégorie d'âge.
Allongement de l'intervalle QT
Pour des informations sur l'allongement de l'intervalle QT, voir «Propriétés/Effets».
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageDes cas de surdosage du tramétinib à plus de 4 mg par jour n'ont pas été signalés dans les études cliniques. Dans le cadre des études cliniques, des doses allant jusqu'à 4 mg par voie orale une fois par jour et des doses «choc» de 10 mg par voie orale une fois par jour sur deux jours successifs ont été examinées.
Traitement
Les mesures de traitement d'un surdosage dépendent des exigences cliniques et respectivement, des recommandations du centre d'information toxicologique, si disponibles. Dans le cas d'un surdosage du tramétinib, aucun traitement spécifique n'est disponible. Dans le cas d'un surdosage, le patient devra recevoir le traitement de soutien approprié pour ce cas précis et être surveillé en conséquence. Une hémodialyse ne devrait avoir aucun effet d'augmentation de l'élimination en raison de l'importante liaison aux protéines plasmatiques du tramétinib.
Propriétés/EffetsCode ATC
L01EE01
Mécanisme d'action
Le tramétinib est un inhibiteur allostérique, réversible et hautement sélectif de l'activation et de l'activité des kinases MEK 1 et MEK 2 (MEK = kinases régulées par des signaux extracellulaires, activées par un mitogène). Les protéines MEK sont des composants critiques de la voie des kinases régulées par des signaux extracellulaires (extracellular signal-regulated kinase, ERK). Dans les mélanomes et autres cancers, cette voie est fréquemment activée par des formes mutées de BRAF, ce qui active ensuite les MEK et stimule la croissance tumorale. Le tramétinib inhibe cette activation des MEK par BRAF, ainsi que l'activité de la kinase MEK. La prolifération de lignées cellulaires de mélanome exprimant la mutation BRAF V600 est inhibée par le tramétinib et dans les modèles animaux porteurs d'un mélanome avec une mutation BRAF V600, un effet antitumoral a pu être démontré.
Le dabrafénib est un inhibiteur efficace, sélectif, compétitif de l'ATP des kinases à mutation BRAF V600 et des kinases BRAF et CRAF de type sauvage. Les mutations oncogènes du gène BRAF conduisent à une activation constitutive de la voie RAS/RAF/MEK/ERK et à une stimulation de la croissance des cellules tumorales.
Le tramétinib et le dabrafénib inhibent les deux kinases MEK et BRAF au sein de cette chaine de transduction de signal; la combinaison des deux principes actifs conduit à une inhibition efficace, double de la chaine de transduction de signal. L'association du tramétinib et du dabrafénib s'est révélée synergique pour les lignées cellulaires de mélanome à mutation BRAF V600 in vitro et retarde l'apparition de résistance in vivo des xénogreffes de mélanome BRAF V600 mutées.
Pharmacodynamique
Le tramétinib a supprimé les concentrations d'ERK phosphorylée dans les lignées cellulaires de mélanome à mutation BRAF et dans les modèles de xénogreffe de mélanome.
Chez les personnes atteintes d'un mélanome porteur des mutations BRAF et NRAS, l'administration de tramétinib a conduit à des modifications dose-dépendantes des biomarqueurs tumoraux, en particulier une inhibition de l'ERK phosphorylée, une inhibition de Ki67 (un marqueur de prolifération cellulaire) ainsi qu'une augmentation de p27 (un marqueur de l'apoptose). Les concentrations moyennes de tramétinib observées à la suite d'une administration de doses répétées de 2 mg une fois par jour dépassent la concentration cible préclinique dans l'intervalle de dosage de 24 heures, permettant en conséquence une inhibition durable de la voie de signalisation MEK.
Électrophysiologie cardiaque
Étude MEK111054
L'action potentielle d'allongement du QT du tramétinib a été étudiée dans une étude indépendante de phase I, spécialement conçue à cet effet, comprenant 35 participants avec des tumeurs solides (dont 32 ont achevé l'étude).
Ceux-ci ont reçu 3 mg de placebo approprié au jour 1 de l'étude, avec suite à cela, une dose de 2 mg de tramétinib ainsi que 2 comprimés de 0,5 mg de placebo approprié une fois par jour aux jours 2 à 14 puis une dose unique de 3 mg de tramétinib le jour 15 de l'étude (dose suprathérapeutique).
Cette étude n'a pas permis de constater un allongement de l'intervalle QTcF dû au tramétinib après administration répétée de 2 mg de tramétinib, y compris l'administration unique de la dose suprathérapeutique de 3 mg au jour 15. L'analyse de la relation entre la modification de l'intervalle QTcF depuis le début de l'étude et les concentrations plasmatiques du tramétinib et la modification prédite du segment QTcF montre qu'il n'existait aucun rapport évident entre l'intervalle QTcF et les concentrations plasmatiques du tramétinib, avec une pente extrêmement faible de la droite de régression modélisée: tendance de 0,0577 msec/ng/ml (IC à 95% -0,124, 0,239).
La combinaison de tramétinib et de dabrafénib n'a pas fait l'objet d'études ciblées concernant l'influence sur l'intervalle QTc.
Mekinist ne prolonge pas l'intervalle QT de manière cliniquement significative lors de l'emploi de 1,5 fois la dose maximale recommandée.
Efficacité clinique
Mélanome non résécable ou métastatique
L'efficacité et la sécurité de la dose recommandée de Mekinist (2 mg une fois par jour) en association avec le dabrafénib (150 mg deux fois par jour) pour le traitement des patients adultes atteints d'un mélanome non résécable ou métastatique exprimant une mutation BRAF V600 ont été évaluées dans deux études pivots de phase III.
L'association du tramétinib 2 mg QD et du dabrafénib 150 mg BID a montré, dans une étude précoce, une activité clinique limitée chez un petit nombre de 26 patients dont la maladie avait progressé après un traitement avec un inhibiteur de BRAF. Le taux de réponse confirmé, évalué par l'investigateur, se situait à 15% (IC à 95%: 4,4, 34,9) et la survie sans progression (PFS) moyenne se situait à 3,6 mois (IC à 95%: 1,9, 5,2). Les résultats étaient similaires chez les 45 patients qui étaient passés du dabrafénib en monothérapie à l'association du tramétinib à 2 mg QD et du dabrafénib à 150 mg BID dans la partie C de l'étude. Chez ces patients, un taux de réponse confirmé de 13% (IC à 95%: 5,0, 27,0) a été observé avec une survie sans progression (PFS) moyenne de 3,6 mois (IC à 95%: 2, 4). Ce taux de réponse est nettement plus faible que chez les patients qui n'ont présenté aucune progression de la maladie sous traitement antérieur avec un inhibiteur de BRAF, de sorte qu'une utilisation du tramétinib en association avec le dabrafénib ne doit être envisagée chez ces patients qu'après évaluation des autres options thérapeutiques.
MEK115306 (COMBId)
MEK115306 (COMBId) était une étude randomisée, en double aveugle de phase III, destinée à comparer l'association dabrafénib et tramétinib avec le dabrafénib et placebo en première ligne de traitement chez des patients atteints d'un mélanome cutané non résécable (stade IIIC) ou métastatique (stade IV) positif pour la mutation BRAF V600E/K. Le critère d'évaluation principal de l'étude était la survie sans progression (PFS) évaluée par l'investigateur. Les critères d'évaluation secondaires étaient la survie globale (SG) médiane, le taux de réponse global (overall response rate, ORR) et la durée de la réponse (duration of response, DoR). Les participants de l'étude ont été stratifiés en fonction de l'activité de la lactate déshydrogénase (LDH) (> limite supérieure de la normale (LSN) versus ≤ LSN) et en fonction du type de mutation de BRAF (V600E par rapport à V600K).
Au total, 423 participants de l'étude ont été randomisés selon un rapport de 1:1 et inclus dans le bras de traitement combiné (tramétinib 2 mg une fois par jour et dabrafénib 150 mg deux fois par jour) (N = 211) ou le bras dabrafénib en monothérapie (150 mg deux fois par jour) (N = 212). Les caractéristiques à l'inclusion dans l'étude étaient similaires dans les deux groupes de traitement. Pour la plupart des patients, une mutation BRAF V600E était présente (85%); une mutation BRAF V600K était présente chez les 15% restants des patients.
Au moment de l'analyse primaire de la PFS, une PFS médiane de 9,3 mois a été observée sous l'association du tramétinib avec le dabrafénib et de 8,8 mois sous dabrafénib en monothérapie (HR = 0,75, IC à 95%: 0,57, 0,99, p = 0,035). L'ORR était de 67% contre 51% (p = 0,0014) et la DoR s'élevait à 9,2 contre 10,2 mois pour l'association versus dabrafénib en monothérapie. Dans une analyse ultérieure, qui a coïncidé avec l'analyse principale de la SG (voir ci-dessous), une nouvelle nette différence en faveur de l'association du tramétinib avec le dabrafénib a été observée, comme dans la première analyse principale de ce paramètre, avec 11,0 mois de PFS sous l'association (IC à 95%: 8,0, 13,9) et 8,8 mois (IC à 95%: 5,9, 9,3) sous dabrafénib en monothérapie (HR = 0,67, IC à 95%: 0,53, 0,84, p< 0,001). Dans cette analyse, l'ORR se situait à 69% versus 53% (p = 0,0014) et la DoR à 12,9 versus 10,6 mois pour le traitement combiné versus le traitement par dabrafénib en monothérapie.
Au moment de l'analyse principale de la SG, 222 décès (52,5%) ont été rapportés dans la population randomisée (ou ITT) [association 99 décès (47%) et dabrafénib 123 décès (58%)]. La durée médiane de suivi pour le traitement de l'étude se situait à 20 mois dans le bras de traitement combiné et à 16 mois dans le bras de dabrafénib en monothérapie. L'étude MEK115306 a montré une diminution statistiquement significative du risque de décès de 29% dans le bras de traitement combiné comparé au bras de dabrafénib en monothérapie (HR = 0,71, IC à 95%: 0,55, 0,92; p = 0,011). La SG médiane se situait à 25,1 mois dans le bras de traitement combiné et à 18,7 mois dans le bras de dabrafénib en monothérapie. Les valeurs de la SG déterminées à 12 mois (74%) et à 24 mois (51,4%) étaient plus élevées dans le bras de traitement combiné que dans le bras de dabrafénib en monothérapie (respectivement 67,6 et 42,1%).
Une analyse de la survie globale (SG) après 5 ans a révélé que la survie globale (SG) médiane sous traitement combiné avait augmenté de 7 mois environ par rapport à la survie globale (SG) médiane sous dabrafénib en monothérapie [25,8 mois (IC à 95%: 19,2, 38,2) versus 18,7 mois (IC à 95%: 15,2, 23,1)], avec un hazard ratio de 0,80 (IC à 95%: 0,63, 1,01). Le taux de survie globale à 5 ans était de 32% (IC à 95%: 25,1, 38,3) sous traitement combiné versus 27% (IC à 95%: 20,7, 33,0) sous dabrafénib en monothérapie. Au bout de 5 ans, la survie sans progression (PFS) médiane avec le traitement combiné était de 10,2 mois (IC à 95%: 8,1, 12,8) versus 8,8 mois (IC à 95%: 5,9, 9,3) avec le dabrafénib en monothérapie, avec un hazard ratio de 0,73 (IC à 95%: 0,59, 0,91).
MEK116513 (COMBI-v)
L'étude MEK116513 était une étude randomisée, ouverte, à 2 bras de phase III comparant l'association tramétinib et dabrafénib au vémurafénib en monothérapie en cas de mélanome métastatique positif à la mutation BRAF V600. Le critère d'évaluation principal de l'étude était la survie globale (SG). Les participants ont été stratifiés en fonction de l'activité de la lactate déshydrogénase (LDH) (> limite supérieure de la normale (LSN) versus ≤ LSN) et en fonction du type de mutation de BRAF (V600E versus V600K).
Au total, 704 participants ont été randomisés selon un rapport de 1:1 et inclus au bras de traitement combiné (tramétinib à 2 mg une fois par jour et dabrafénib à 150 mg deux fois par jour) ou au bras vémurafénib en monothérapie (960 mg deux fois par jour). La majorité des sujets de l'étude présentaient une mutation BRAF V600E (89%). 10% des patients avaient une mutation BRAF V600K et 1 patient (< 1%) présentait les deux mutations (BRAF V600E/K).
L'analyse de la SG a été réalisée lorsqu'au total, 222 décès ont été constatés (77% des résultats nécessaires pour l'analyse finale). Le Comité de contrôle des données indépendant (Independent Data Monitoring Committee, IDMC) a recommandé l'interruption de l'étude, car les résultats de SG avaient dépassé la limite d'efficacité statistique préalablement établie. Par la suite, l'analyse intermédiaire de la SG vaut comme analyse comparative finale de la SG.
L'analyse de la SG pour l'étude MEK116513 était basée sur 222 décès (32%) [association; 100 décès (28%) et vémurafénib 122 décès (35%)]. La durée médiane de suivi pour le traitement de l'étude a été de 11 mois dans le bras de traitement combiné et de 9 mois dans le bras vémurafénib. L'étude MEK116513 a montré une diminution statistiquement significative du risque de décès de 31% dans le bras de traitement combiné comparé au vémurafénib (HR = 0,69, IC 95%: 0,53, 0,89; p = 0,005). La SG médiane n'était pas encore atteinte pour le bras de traitement combiné et était de 17,2 mois pour le bras vémurafénib en monothérapie.
La PFS médiane observée était de 11,4 mois pour le traitement combiné du tramétinib et du dabrafénib, et de 7,3 mois sous vémurafénib en monothérapie (HR =0,56, IC à 95%: 0,46, 0,69, p < 0,001). L'ORR s'élevait à 64% versus 51% (p = 0,0005) et la DoR se situait à 13,8 versus 7,5 mois pour le traitement combiné par rapport au vémurafénib en monothérapie.
Une analyse de la survie globale (SG) après 5 ans a révélé que la survie globale (SG) médiane sous traitement combiné était plus longue de 8 mois environ par rapport à la survie globale (SG) médiane sous vémurafénib en monothérapie [26,0 mois (IC à 95%: 22,1, 33,8) versus 17,8 mois (IC à 95%: 15,6, 20,7)], avec un hazard ratio de 0,70 (IC à 95%: 0,58, 0,84). Le taux de survie globale à 5 ans était de 36% (IC à 95%: 30,5, 40,9) sous traitement combiné versus 23% (IC à 95%: 18,1, 27,4) sous vémurafénib en monothérapie.
BRF117277/DRB436B2204 (COMBI-MB) – Patients ayant des métastases cérébrales d'un mélanome métastasique
L'efficacité et la sécurité de Mekinist en association avec le dabrafénib chez les patients présentant un mélanome BRAFpositif avec des métastases cérébrales ont été étudiées lors d'une étude de phase II non randomisée, ouverte et multicentrique (étude COMBI-MB). Au total, 125 patients ont été inclus dans 4 cohortes:
Cohorte A: patients présentant un mélanome avec la mutation V600E sur le gène BRAF et des métastases cérébrales asymptomatiques sans traitement ciblé local préalable des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
Cohorte B: patients présentant un mélanome avec la mutation V600E sur le gène BRAF et des métastases cérébrales asymptomatiques avec traitement ciblé local antérieur des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
Cohorte C: patients présentant un mélanome avec la mutation V600D/K/R sur le gène BRAF et des métastases cérébrales asymptomatiques avec ou sans traitement ciblé local antérieur des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
Cohorte D: patients présentant un mélanome avec la mutation V600D/E/K/R sur le gène BRAF et des métastases cérébrales symptomatiques avec ou sans traitement ciblé local antérieur des métastases cérébrales avec un statut de performance ECOG de 0, 1 ou 2.
Au total, 104 des 125 patients présentaient une mutation V600E, 18 patients une mutation V600K et 3 patients seulement une mutation V600R. Aucun patient n'avait une mutation V600D.
Le critère d'évaluation principal de l'étude était la réponse intracrânienne dans la cohorte A, définie comme étant le pourcentage de patients avec une réponse intracrânienne confirmée évaluée par le médecin de l'étude au moyen des critères RECIST (Response Evaluation Criteria in Solid Tumors, critères d'évaluation de la réponse thérapeutique dans les tumeurs solides), version 1.1. Les résultats d'efficacité sont résumés dans le tableau 6. Les critères d'évaluation secondaires étaient la durée de la réponse intracrânienne, le taux de réponse global des manifestations tumorales intracrâniennes comme extracrâniennes, la PFS et la SG. Les résultats d'efficacité sont résumés dans le tableau 6.
Tableau 6: COMBI-MB – Résultats d'efficacité selon l'appréciation du médecin de l'étude
|
|
Tous les patients traités
| |
Critères/ Évaluation
|
Cohorte A
N = 76
|
Cohorte B
N = 16
|
Cohorte C
N = 16
|
Cohorte D
N = 17
| |
Taux de réponse intracrânienne, % (IC à 95%)
|
| |
|
59%
(47,3, 70,4)
|
56%
(29,9, 80,2)
|
44%
(19,8, 70,1)
|
59%
(32,9, 81,6)
| |
Durée de la réponse intracrânienne, valeur médiane, mois (IC à 95%)
| |
|
6,5
(4,9, 8,6)
|
7,3
(3,6, 12,6)
|
8,3
(1,3, 15,0)
|
4,5
(2,8, 5,9)
| |
Réponse globale (intra- et extracrânienne) ORR, % (IC à 95%)
| |
|
59%
(47,3, 70,4)
|
56%
(29,9, 80,2)
|
44%
(19,8, 70,1)
|
65%
(38,3, 85,8)
| |
Valeur médiane PFS, mois (IC à 95%)
| |
|
5,7
(5,3, 7,3)
|
7,2
(4,7, 14,6)
|
3,7
(1,7, 6,5)
|
5,5
(3,7, 11,6)
| |
Valeur médiane SG, mois (IC à 95%)
| |
Valeur médiane, mois
|
10,8
(8,7, 17,9)
|
24,3
(7,9, NR)
|
10,1
(4,6, 17,6)
|
11,5
(6,8, 22,4)
| |
IC = intervalle de confiance
NR = sans indication
|
Traitement adjuvant du mélanome
Étude BRF115532/DRB436F2301 (COMBI-AD)
L'efficacité et l'innocuité de Mekinist en association avec le dabrafénib ont été examinées après résection complète dans une étude de phase III multicentrique, randomisée, en double aveugle, contrôlée contre placebo, menée auprès de patients ayant un mélanome de stade III portant une mutation BRAF V600.
Les patients ont été randomisés selon un rapport de 1:1 et ont reçu soit un traitement combiné avec 2 mg de Mekinist une fois par jour et 150 mg de dabrafénib deux fois par jour, soit deux placebos sur une période de 12 mois. Seuls les patients ayant subi une résection totale du mélanome et une lymphadénectomie complète au cours des 12 semaines précédant la randomisation ont été acceptés dans l'étude. Un traitement systémique préalable du cancer, y compris une radiothérapie, n'était pas autorisé. Les patients ayant des antécédents de malignité antérieure ont été autorisés à participer s'ils avaient été sans maladie pendant au moins 5 ans. Les patients atteints de tumeurs malignes avec des mutations RAS activantes confirmées n'ont pas été autorisés à participer. Les patients ont été stratifiés en fonction du statut de la mutation BRAF (V600E ou V600K) et du stade de la maladie (en sous-stades du stade III, c'est-à-dire différentes atteintes des ganglions lymphatiques, taille primaire de la tumeur et ulcération) avant l'opération. Le critère d'évaluation primaire était la survie sans récidive évaluée par l'investigateur (relapse-free survival, RFS), définie comme étant la période de la randomisation à la réapparition de la tumeur ou au décès, quelle qu'en soit la cause. Une évaluation radiologique de la tumeur a été réalisée tous les 3 mois au cours des deux premières années, puis tous les 6 mois, jusqu'à ce que la première récidive soit observée. Les critères d'évaluation secondaires étaient la survie globale (SG; critère d'évaluation secondaire important) et la survie en l'absence de métastases distantes (distant metastasis-free survival, DMFS).
En tout, 870 patients ont été randomisés pour le traitement combiné (n = 438) et pour le placebo (n = 432). L'étude a inclus des patients ayant une maladie à tous les sous-stades du stade III avant la résection; 18% de ces patients présentaient des atteintes des ganglions lymphatiques seulement détectables au microscope et aucune ulcération tumorale primaire. La majorité des patients avait une mutation BRAF V600E (91%). Au moment de l'analyse principale, la durée médiane de l'observation qui a suivi (de la randomisation jusqu'au dernier contact ou à la mort) a été de 2,83 ans dans le groupe de traitement et 2,75 ans dans le groupe placebo.
Les résultats de l'analyse principale de la RFS sont représentés dans le tableau 7. L'étude a montré une différence statistiquement significative entre les bras de traitements pour le critère d'évaluation principal en ce qui concerne la RFS. Dans le groupe de traitement ayant reçu Mekinist et le dabrafénib comme traitement combiné, la réduction du risque était de 53% comparé au groupe placebo.
Tableau 7: Analyse principale de COMBI-AD - Résultats de la survie sans récidive
|
|
Mekinist + dabrafénib
|
Placebo
| |
Paramètres de RFS
|
N = 438
|
N = 432
| |
Nombre des évènements - n (%)
|
166 (38%)
|
248 (57%)
| |
Récidive
|
163 (37%)
|
247 (57%)
| |
Récidivé avec des métastases distantes
|
103 (24%)
|
133 (31%)
| |
Mort
|
3 (< 1%)
|
1 (< 1%)
| |
Valeur médiane (mois)
(IC à 95%)
|
NE
(44,5, non estimable [NE])
|
16,6
(12,7; 22,1)
| |
Hazard Ratio[1]
|
0,47
| |
(IC à 95%)
|
(0,39; 0,58)
| |
pValeur p[2]
|
1,53 × 10-14
| |
Taux sur 1 an (IC à 95%)
|
0,88 (0,85, 0,91)
|
0,56 (0,51, 0,61)
| |
Taux sur 2 ans (IC à 95%)
|
0,67 (0,63, 0,72)
|
0,44 (0,40, 0,49)
| |
Taux sur 3 ans (IC à 95%)
|
0,58 (0,54, 0,64)
|
0,39 (0,35, 0,44)
| |
[1]
Le Hazard Ratio résulte du modèle de Pike stratifié.
[2] La valeur p est calculée à partir du test du Logrank stratifié bilatéral (les facteurs de stratification étaient le stade de la maladie, IIIA vs IIIB vs IIIC, et le type de mutation BRAF V600 - V600E vs V600K).
NE = non estimable
|
Sur la base de 153 évènements [60 (14%) dans le bras de traitement combiné et 93 (22%) dans le bras placebo], ce qui correspond à une proportion d'information de 26% de l'objectif global de 597 évènements SG, le hazard ratio estimé pour la SG était de 0,57 (IC à 95%: 0,42, 0,79; p = 0,0006) au moment de l'analyse principale. Ces résultats n'ont pas correspondu à la limite préspécifiée pour obtenir une signification statistique dans cette première analyse intermédiaire de la SG (HR = 0,50; p = 0,000019). Les estimations de la survie 1 et 2 ans après la randomisation ont été de 97% et 91% dans le bras de traitement combiné et de 94% et 83% dans le bras placebo.
Sur la base des données actualisées issues de la période de suivi de 29 mois supplémentaires, par comparaison à l'analyse principale (suivi minimum de 59 mois), l'avantage de la survie sans récidive est maintenu avec un hazard ratio estimé de 0,51 (IC à 51%: 0,42, 0,61). Le taux de survie sans récidive à 5 ans a été de 52% (IC à 95%: 48, 58) dans le bras de traitement combiné, contre 36% (IC à 95%: 32, 41) dans le bras placebo.
NSCLC avancé
Étude BRF113928
L'efficacité et la sécurité de Mekinist en association avec le dabrafénib ont été examinées dans une étude multicentrique, non randomisée, en ouvert de phase II menée auprès de 57 patients atteints de NSCLC métastatique, positif pour la mutation BRAF V600E, précédemment traités par chimiothérapie. Le critère d'évaluation principal était le taux de réponse global évalué par le médecin (overall response rate, ORR) en utilisant les critères RECIST (Response Evaluation Criteria in Solid Tumors, critères d'évaluation de la réponse au traitement dans les tumeurs solides). Les critères d'évaluation secondaires comprenaient la durée de la réponse (duration of response, DoR), la survie sans progression (PFS) et la survie globale (SG). L'ORR, la DoR et la PFS ont été évaluées par un comité d'évaluation indépendant (Independant review Committee, IRC) sous forme d'une analyse de sensibilité.
Au moment de l'analyse principale, le taux de réponse global évalué par le médecin (overall response rate ORR), l'ORR dans la population précédemment traitée était de 66,7% (IC à 95%, 52,9%, 78,6%). La significativité statistique était ainsi atteinte avec rejet de l'hypothèse nulle, de sorte que l'ORR du dabrafénib en association avec Mekinist était de 30% au maximum pour cette population NSCLC. Les résultats du taux de réponse globale (overall response rate ORR) évaluée par un comité d'évaluation indépendant (Independant review Committee, IRC) correspondaient à l'évaluation par le médecin. L'analyse finale de l'efficacité, qui a été réalisée 5 ans après la première dose du dernier participant, est présentée dans le tableau 8.
La durée de la réponse se situait à une médiane de 9,8 (IC à 95%, 6,9, 16,0) mois, selon l'évaluation du médecin de l'étude.
Tableau 8: ORR par évaluation du médecin de l'étude et par examen radiographique indépendant
|
Groupe pour la radiographie
|
Testeur
|
Indépendant
| |
Catégorie
|
N = 57
|
N = 57
| |
Réponse globale – n (%)
| |
ORR (CR + PR)
|
39 (68,4%)
|
36 (63,2%)
| |
(IC à 95%)
|
(54,8, 80,1)
|
(49,3, 75,6)
| |
Durée de la réponse
| |
Nombre de répondeurs
|
39
|
36
| |
Nombre de patients avec progression de la maladie ou décès – n (%)
|
27 (71)
|
20 (56)
| |
DoR médiane, mois
(IC à 95%)
|
9,8
(6,9, 18,3)
|
12,6
(5,8, 26,2)
| |
Survie sans progression
| |
Progression de la maladie ou décès n (%)
|
41 (72)
|
38 (67)
| |
PFS médiane, mois
(IC à 95%)
|
10,2
(6,9, 16,7)
|
8,6
(5,2, 16,8)
| |
Survie globale
| |
Nombre de décès – n (%)
|
33 (58%)
| |
Médiane, mois
(IC à 95%)
|
18,2
(14,3, 28,6)
| |
L'intervalle de confiance (IC) est calculé par la méthode exacte (intervalle de Clopper-Pearson).
CR = rémission complète, PR = rémission partielle, SD = maladie stable, PD = progression de la maladie
|
Dans l'étude BRF113928, 36 patients atteints d'un NSCLC exprimant la mutation V600E de BRAF ayant reçu le dabrafénib en association avec le tramétinib comme traitement de première ligne contre une atteinte métastatique ont aussi été inclus. Lors de l'analyse finale après 5 ans pour tous les participants à l'étude ayant reçu le traitement de première ligne, le critère d'évaluation principal du taux de réponse global (ORR) évalué par l'investigateur a été de 63,9% (IC à 95%, 46,2%, 79,2%). Ainsi, la significativité statistique a été atteinte avec rejet de l'hypothèse nulle selon laquelle l'ORR du dabrafénib en association avec le tramétinib était de 30% au maximum pour cette population de patients avec NSCLC. La durée médiane de rémission (DoR) évaluée par l'investigateur a été de 10,2 mois (IC à 95%: 8,3, 15,2), la survie sans progression médiane évaluée par l'investigateur de 10,8 mois (IC à 95%: 7,0, 14,5), la survie globale médiane évaluée par l'investigateur de 17,3 mois (IC à 95%: 12,3, 40,2), avec 61% des cas de décès survenus au moment de l'analyse.
Gliome de bas grade (LGG)
Étude DRB436G2201
L'efficacité et la sécurité cliniques du traitement combiné Mekinist plus dabrafénib chez les patients pédiatriques âgés de 1 à < 18 ans avec gliome positif pour la mutation BRAF V600E ont été examinées dans une étude clinique de phase II, multicentrique, en ouvert CDRB436G2201. Des patients présentant un gliome de bas grade (grades 1 et 2 selon l'OMS), qui devaient recevoir un premier traitement systémique et n'avaient pas reçu de radiothérapie antérieurement, ont été randomisés selon un rapport 2:1 dans le groupe tramétinib plus dabrafénib (D+T) ou carboplatine plus vincristine (C+V). Environ 83% de tous les patients avaient subi une intervention chirurgicale; seuls deux patients (les deux du bras D+T) n'avaient aucune maladie résiduelle après l'intervention.
Les patients ayant un score de performance Karnofsky/Lansky < 50%, les patients n'ayant pas de fonctions médullaire, rénale, hépatique et cardiaque suffisantes ainsi que les patients avec maladies non contrôlées ou significatives, notamment des maladies cardiaques, le diabète, l'hypertension, des affections hépatiques ou des infections ont été exclus (voir «Mises en garde et précautions»).
Le statut de la mutation BRAF a été déterminé de manière prospective par un test local ou, si un test local n'est pas disponible, par un test PCR en temps réel par le laboratoire central. De plus, des tests rétrospectifs d'échantillons tumoraux disponibles ont été réalisés par le laboratoire central pour confirmer la mutation BRAF V600E.
Les doses de Tafinlar et de Mekinist dépendaient de l'âge et du poids, Tafinlar étant administré par voie orale à une dose de 2,625 mg/kg, deux fois par jour pour la tranche d'âges <12 ans et 2,5 mg/kg, deux fois par jour pour la tranche d'âges 12 ans et plus; Mekinist étant administré par voie orale à une dose de 0,032 mg/kg, une fois par jour pour la tranche d'âge < 6 ans et de 0,025 mg/kg, une fois par jour pour la tranche d'âges 6 ans et plus. La dose maximale de Tafinlar se situait à 150 mg deux fois par jour et celle de Mekinist, à 2 mg une fois par jour. Le carboplatine et la vincristine ont été administrés en fonction de l'âge et du poids corporel, à une dose de respectivement 175 mg/m² et 1,5 mg/m² en traitement d'induction de 10 semaines, suivi de huit cycles de 6 semaines en traitement d'entretien.
Le critère d'évaluation principal de l'efficacité dans les deux cohortes était le taux de réponse global (overall response rate [ORR], somme des patients avec rémission complète confirmée/RC et rémission partielle/RP) par évaluation indépendante sur la base des critères RANO (2017). L'analyse primaire a été réalisée lorsque tous les patients des deux cohortes ont eu terminé au moins 32 semaines de thérapie.
Dans la cohorte de gliome de bas grade (LGG) de l'étude G2201, 110 patients ont été choisis au hasard pour D+T (n = 73) ou C+V (n = 37). L'âge moyen était de 9,5 ans, dont 34 patients (30,9%) étaient âgés de 12 mois à 6 ans, 36 patients (32,7%) de 6 à < 12 ans et 40 patients (36,4%), de 12 à < 18 ans; 60% étaient des filles. Au moment de l'analyse primaire, la durée de traitement médiane dans le bras D+T était de 76 semaines, la durée de suivi médiane dans la cohorte LGG était de 18,9 mois. L'ORR dans le bras D+T (46,6%) montrait une amélioration statistiquement significative par rapport au bras C+V (10,8%), avec un risque relatif (IC à 95%) de 7,19 (2,3, 22,4) et une valeur p unilatérale < 0,001 (tableau 9). L'évaluation hiérarchisée ultérieure a également montré une amélioration de la survie sans progression (SSP) par rapport à la chimiothérapie, avec un risque relatif (IC à 95%) de 0,31 (0,17, 0,55) (valeur p unilatérale du test Log-Rang < 0,001). Une différence dans l'évaluation de l'efficacité a été observée entre l'évaluation indépendante et les centres d'étude, qui comportait également l'évaluation des rémissions complète et partielle, ainsi que la progression de la maladie. Le taux de surestimation s'élevait au total à 52% dans le bras D+T.
Comme paramètres fonctionnels, les modifications de fréquence de crise («seizure activity») et de la vue avant et après le traitement ont été examinées. Pour les deux paramètres, aucune amélioration sous traitement par D+T par comparaison au bras témoin n'a pu être décelée.
Tableau 9: Réponses et survie sans progression dans l'étude G2201 (cohorte LGG)
|
|
Dabrafénib + tramétinib
N = 73
|
Carboplatine plus vincristine
N = 37
| |
Meilleure réponse globale
| |
Rémission complète (RC), n (%)
|
2 (2,7)
|
1 (2,7)
| |
Rémission partielle (RP), n (%)
|
32 (43,8)
|
3 (8,1)
| |
Maladie stable (MS), n (%)
|
30 (41,1)
|
15 (40,5)
| |
Progression de la maladie (PM), n (%)
|
8 (11,0)
|
12 (32,4)
| |
Inconnu, n (%)
|
1 (1,4)
|
6 (16,2)
| |
Taux de réponse globale (ORR)
| |
ORR (RC+RP), IC à 95%, valeur p
|
46,6% (34,8 – 58,6%), p < 0,001
|
10,8% (3,0 - 25,4%)
| |
Risque relatif
|
7,19 (2,3 – 22,4)
| |
Survie sans progression
| |
Médiane (mois)
|
20,1 (12,8, NE)
|
7,4 (3,6, 11,8)
| |
Rapport des risques (IC à 95%), valeur p
|
0,31 (0,17-0,55), p < 0,001
|
PharmacocinétiqueAbsorption
Le tramétinib est absorbé oralement avec un temps médian pour atteindre la concentration plasmatique maximale de 1,5 heure après l'administration. La biodisponibilité absolue moyenne d'une dose unique sous forme d'un comprimé de 2 mg s'élève à 72% de celle après une microdose intraveineuse (IV). L'exposition (Cmax et ASC) a augmenté de manière proportionnelle à la dose après administration répétée. Après administration de 2 mg quotidiennement, les valeurs moyennes géométriques de Cmax, ASC(0-τ) et de la concentration avant administration se sont élevées à 22,2 ng/ml, 370 ng × h/ml et respectivement, 12,1 ng/ml, pour un faible rapport pic-vallée (1,8). La variabilité interindividuelle était faible (< 28%). Après administration en association avec le dabrafénib à 150 mg deux fois par jour, les moyennes géométriques de Cmax, d'ASC(0-τ) et de la concentration de tramétinib ont été avant administration de respectivement 22,6 ng/ml, 356 ng × h/ml et 10,9 ng/ml. L'administration d'une dose unique de tramétinib comprimés pelliculés avec un repas très calorique, riche en graisses, a conduit à une diminution de la Cmax et de l'ASC de respectivement 70% et 10% par rapport aux valeurs lors d'une prise à jeun (voir «Posologie/Mode d'emploi»). Les comprimés pelliculés et la poudre pour solution buvable de tramétinib sont des formes galéniques avec une libération immédiate, qui devrait avoir des effets similaires sur la pharmacocinétique.
Distribution
Le tramétinib se lie à 97,4% aux protéines plasmatiques humaines. Après administration d'une microdose intraveineuse de 5 µg, le tramétinib présente un volume de distribution de 1060 litres.
Métabolisme
Des études in vitro et in vivo ont montré que le tramétinib est métabolisé essentiellement par désacétylation seulement ou avec monooxygénation ou en combinaison avec des voies de biotransformation par glucuronidation. La désacétylation est médiée par des carboxylestérases (à savoir, les carboxylestérases 1b/c et 2) et peut être médiée également par d'autres enzymes hydrolytiques.
Élimination
En cas d'administration répétée d'une dose de 2 mg une fois par jour, le tramétinib s'accumule avec un rapport moyen d'accumulation de 6,0. La demi-vie terminale se situe en moyenne à 127 heures (5,3 jours) après une dose unique. L'équilibre est atteint au jour 15. La clairance plasmatique après administration intraveineuse est de 3,21 l/h.
Après l'administration par voie orale d'une dose unique de tramétinib radiomarqué en solution, la quantité totale de la dose récupérée est faible (< 50%) d'après les prélèvements effectués sur une période de 10 jours, du fait d'une longue demi-vie d'élimination.
Le médicament est essentiellement éliminé par voie fécale (≥81% de la radioactivité éliminée), seulement ≤19% dans les urines. Moins de 0,1% de la dose éliminée a été retrouvée sous forme de la substance mère dans les urines.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
Une analyse pharmacocinétique de population et les données d'une étude de pharmacologie clinique sur des patients présentant une fonction hépatique normale ou une augmentation légère, modérée ou sévère de la bilirubine et/ou de l'ASAT (selon la classification du National Cancer Institute [NCI]) montrent que la fonction hépatique influence de manière non significative la clairance orale du tramétinib.
Troubles de la fonction rénale
Une insuffisance rénale n'a vraisemblablement pas d'influence clinique importante sur la pharmacocinétique du tramétinib étant donné la faible élimination rénale de celui-ci. La pharmacocinétique du tramétinib a été examinée à l'aide d'une analyse pharmacocinétique de population sur 223 patients avec insuffisance rénale légère et 35 patients avec insuffisance rénale modérée inclus dans des études cliniques. Une insuffisance rénale légère à modérée n'a eu aucune influence sur l'exposition au tramétinib (< 6% dans les deux groupes). On ne possède pas de données concernant les patients avec insuffisance rénale sévère (voir «Posologie/Mode d'emploi»).
Patients âgés
Selon l'analyse pharmacocinétique de population, l'âge n'a eu aucune influence significative sur la pharmacocinétique du tramétinib.
Population pédiatrique
La pharmacocinétique du tramétinib en cas de gliomes et d'autres tumeurs solides a été étudiée chez 244 patients pédiatriques (1 à < 18 ans) après administration unique ou répétée, ajustée selon le poids. Les propriétés pharmacocinétiques (taux d'absorption, rapport des métabolites, clairance du médicament) du tramétinib chez les patients pédiatriques sont comparables à celles des patients adultes. Il a été établi que le poids influence la clairance orale du tramétinib. Les expositions pharmacocinétiques du tramétinib à la dose ajustée au poids corporel recommandée, chez les patients pédiatriques se situaient dans la plage des expositions observées chez l'adulte.
Données précliniquesTramétinib en association avec le dabrafénib
Chez des chiens auxquels ont été administrés le tramétinib et le dabrafénib en association pendant 4 semaines, des événements de toxicité ont été observés, similaires à ceux des études précliniques qui ont été réalisées avec les substances individuelles. Chez des chiens auxquels a été administrée l'association du tramétinib et du dabrafénib, les mêmes effets testiculaires, consistant en une dégénérescence et une oligospermie épididymaire secondaire, ont été observés que chez les chiens auxquels avait été administré le tramétinib ou le dabrafénib seuls. Voir également l'information professionnelle du dabrafénib.
Mutagénicité/Carcinogénicité
Le tramétinib s'est révélé non génotoxique sur des bactéries et des cultures de cellules de mammifère in vitro, ainsi que dans des tests sur micronoyaux de rongeurs in vivo. Aucune étude de carcinogénicité du tramétinib n'a été réalisée.
Toxicité sur la reproduction
Fertilité
Il n'existe aucune donnée pour le tramétinib sur l'homme. Aucune étude de fertilité du tramétinib n'a été réalisée. Dans des études avec administration répétée chez des rats juvéniles et adultes à ≥0,016 mg/kg/jour (environ 0,3 fois l'exposition clinique chez l'homme, sur la base de l'ASC), des modifications de la maturation folliculaire ont été observées, qui se sont manifestées par une augmentation des follicules vésicaux et la diminution des corps jaunes cystiques.
De plus, on a observé chez les rats juvéniles qui ont reçu le tramétinib une diminution du poids ovarien, un léger retard des caractéristiques de maturité sexuelle femelle (ouverture du vagin et taux d'incidence multiplié des bourgeons terminaux proéminents dans les glandes mammaires) et une légère hypertrophie de l'épithélium de surface de l'utérus. Tous ces effets ont été réversibles après une période sans traitement et attribuables à la pharmacologie. Aucun effet du traitement sur les tissus reproducteurs masculins n'a cependant été observé dans des études de toxicité avec des rats et des chiens d'une durée allant jusqu'à 13 semaines.
Gestation
Dans des études toxicologiques de reproduction sur des rats ont été observées, à des doses ≥0,031 mg/kg/jour (correspondant, sur la base de l'ASC, à environ 0,3 fois l'exposition clinique chez l'homme), une toxicité maternelle et une toxicité sur le développement du fœtus (poids fœtal réduit). Chez des lapines gravides ont été observées, à des doses ≥0,039 mg/kg/jour (correspondant, sur la base de l'ASC, à environ 0,1 fois l'exposition clinique chez l'homme), une toxicité maternelle et une toxicité sur le développement du fœtus (poids fœtal réduit et présence répétée de défauts d'ossification).
Autres données (toxicité locale, phototoxicité, immunotoxicité)
Dans des études avec administration répétée, des effets ont été observés essentiellement sur la peau, sur le tractus gastro-intestinal, sur le système hématologique, sur les os et sur le foie après exposition au tramétinib. La plupart des observations ont été réversibles après une période de récupération sans médicament. Dans des études avec administration répétée sur des rats, une nécrose de cellules hépatiques et une élévation des transaminases ont été observées après 8 semaines à une dose ≥0,062 mg/kg/jour (correspondant, sur la base de l'ASC, à environ 0,8 fois l'exposition clinique chez l'homme).
Chez des souris, une fréquence cardiaque diminuée, un poids du cœur diminué et une fonction ventriculaire gauche réduite sans signe d'histopathologie cardiaque ont été observés après 3 semaines à une dose ≥0,25 mg/kg/jour de tramétinib (correspondant, sur la base de l'ASC, à 3 fois l'exposition clinique chez l'homme) pendant une durée allant jusqu'à trois semaines. Chez des rats, une calcification et une nécrose du myocarde, se développant avec une élévation de la concentration en phosphate sérique, ont été observées à des doses ≥1 mg/kg/jour (correspondant, sur la base de l'ASC, à environ 12 fois l'exposition clinique chez l'homme). Chez des rats juvéniles, un poids cardiaque élevé sans observation histopathologique a été observé à une dose de 0,35 mg/kg/jour (sur base de l'ASC, environ 2 fois l'exposition clinique chez l'homme).
Chez les rats adultes, la minéralisation de plusieurs organes a été associée à des concentrations sériques élevées en phosphore et étroitement liée à des nécroses du cœur, du foie et des reins ainsi que des hémorragies pulmonaires, pour des expositions similaires à l'exposition humaine dans les études cliniques. Chez des rats, une hypertrophie du cartilage de croissance ainsi qu'un métabolisme accru des os ont été observés; l'hypertrophie du cartilage de croissance ayant cependant été estimée non cliniquement pertinente pour l'homme adulte.
Chez des rats et des chiens, auxquels des doses cliniques ou sous-cliniques de tramétinib ont été administrées, des nécroses de la moelle osseuse, une atrophie thymique lymphatique et du tissu lymphatique associé à l'intestin (GALT [Gut Associated Lymphoid Tissue]) ainsi que des nécroses lymphatiques dans les ganglions lymphatiques, la rate et le thymus ont été observées, lesquelles présentent un potentiel d'altération de la fonction immunitaire.
Chez des rats juvéniles, un poids cardiaque élevé sans observation histopathologique a été observé lors d'une exposition approximativement doublée par rapport à l'exposition clinique chez l'adulte, sur base de l'ASC.
Le tramétinib a été phototoxique dans un dosage de fixation du rouge neutre in vitro (Neutral Red Uptake, NRU) par des fibroblastes 3T3 de souris à des concentrations significativement plus élevées que les expositions cliniques (IC50 à 2,92 µg/ml, ≥130 fois l'exposition clinique, sur base de la Cmax), ce qui indique qu'il existe un faible risque de phototoxicité pour les patients qui utilisent le tramétinib.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Stabilité après première ouverture du flacon (comprimés pelliculés) ou après reconstitution de la poudre pour solution buvable:
Comprimés pelliculés
Après ouverture, le flacon peut être stocké au-dessous de 30 °C pendant 30 jours.
Poudre pour solution buvable
La solution reconstituée peut être conservée pendant 35 jours. Le flacon contenant la solution reconstituée doit être conservé dans le carton à 25 °C et ne doit pas être congelé.
La solution non utilisée 35 jours après la reconstitution doit être éliminée.
Remarques particulières concernant le stockage
Comprimés pelliculés
Ne pas conserver au-dessus de 25 °C.
Conserver dans l'emballage d'origine.
Conserver le récipient fermé.
Conserver bien fermé dans l'emballage d'origine pour le protéger de la lumière et de l'humidité.
Ne pas jeter ni ingérer l'agent desséchant.
Poudre pour solution buvable
Conserver au réfrigérateur (2–8 °C) jusqu'à reconstitution.
Conserver dans l'emballage d'origine pour protéger la poudre de la lumière et de l'humidité.
Conserver le récipient fermé.
Conserver hors de portée des enfants.
Numéro d’autorisation65883, 69134 (Swissmedic)
PrésentationMekinist 0,5 mg: emballages de 7 et de 30 comprimés pelliculés (A)
Mekinist 2 mg: emballages de 7 et de 30 comprimés pelliculés (A)
Mekinist 4,7 mg: 1 flacon contenant de la poudre pour solution buvable, 1 seringue doseuse pour administration orale et 1 adaptateur de flacon (A)
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz
Mise à jour de l’informationMars 2023
Autres indications - Instructions d'utilisation et de manipulation
|
MODE D'EMPLOI
MEKINIST 4,7 mg Poudre pour solution buvable
Tramétinib
Voie orale
| |
Ce mode d'emploi contient des informations pour la prise de Mekinist
| |
Informations importantes, dont vous devez prendre connaissance avant la prise de Mekinist
| |
·Veuillez lire attentivement ce mode d'emploi avant de commencer à utiliser Mekinist.
·Votre médecin vous expliquera comment prendre Mekinist de manière correcte. Prenez toujours Mekinist exactement selon les indications de votre médecin.
·Si vous avez des questions sur la préparation ou la prise de Mekinist, adressez-vous à votre médecin.
·Vous recevrez un flacon de Mekinist contenant de la poudre (vous pourrez alors préparer la solution vous-même) OU sous forme de solution prête à l'emploi. Vérifiez si vous avez reçu une poudre ou une solution. Suivez alors les indications appropriées ci-dessous.
·La solution préparée doit être utilisée dans les 35 jours.
·Ne jetez pas le médicament dans les eaux usées ou les ordures ménagères. Demandez à votre pharmacien comment éliminer le médicament si vous ne l'utilisez plus. Vous contribuerez ainsi à la protection de l'environnement.
| |
NETTOYAGE DES LIQUIDES RENVERSÉS
| |
Si Mekinist entre en contact avec la peau, lavez la zone avec de l'eau et du savon. Si Mekinist entre en contact avec les yeux, rincer vos yeux avec de l'eau.
Suivez les étapes suivantes si vous renversez Mekinist solution buvable:
·Enfilez des gants en plastique.
·Aspirez complètement la solution avec un matériau absorbant, par exemple, un mouchoir en papier.
·Introduisez le matériau absorbant dans un sachet en plastique.
·Essuyez avec un tampon imbibé d'alcool toutes les surfaces ayant été en contact avec la solution.
·Mettez le sachet, les gants et le tampon dans un deuxième sachet en plastique et fermez-le.
·Ne jetez pas le médicament dans les eaux usées ou les ordures ménagères. Demandez à votre pharmacien comment éliminer le médicament si vous ne l'utilisez plus. Vous contribuerez ainsi à la protection de l'environnement.
| |
Poudre pour solution
Si vous recevez Mekinist sous forme de Poudre, la boite doit contenir les éléments suivants:
1.1 flacon avec capuchon sécurisé contenant la poudre
2.1 seringue d'administration pour la préparation
3.1 adaptateur de flacon (fixé sur la seringue d'administration pour la préparation)
Mode d'emploi et notice (non représentés).
|
|

| |
Aperçu de la seringue d'administration pour la préparation, réutilisable:
|

N'insérez pas encore l'adaptateur de flacon.
Lisez les informations importantes sur Mekinist ci-dessus et poursuivez avec les indications de préparation de la section A.
| |
Poursuivez avec la section A
| |
OU
| |
Solution prête à l'emploi
Si vous recevez MEKINIST sous forme de solution prête à l'emploi, la boite doit contenir les éléments suivants:
1.1 flacon avec capuchon sécurisé contenant la solution prête à l'emploi
2.1 seringue d'administration pour la préparation
3.1 adaptateur de flacon (déjà en place)
|
|

| |
Aperçu de la seringue d'administration pour la préparation, réutilisable:
|
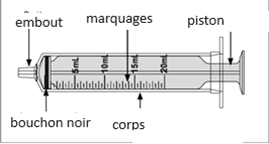
Ne retirez pas l'adaptateur de flacon.
Lisez les informations importantes sur Mekinist cidessus et poursuivez avec les indications d'administration de la section B.
| |
Poursuivez avec la section B
| |
SECTION A. PRÉPARATION
| |
Suivez les instructions de la section A pour la préparation lorsque vous recevez Mekinist sous forme de poudre.
En cas de renversement ou de contact de la solution de Mekinist avec la peau ou les yeux, suivez les instructions de la section «Nettoyage des liquides renversés».
Lavez et séchez vos mains avant de préparer Mekinist.
Pour préparer Mekinist, vous avez besoin de:
1.1 flacon avec capuchon sécurisé contenant la poudre
2.1 seringue d'administration pour la préparation
3.1 adaptateur de flacon (fixé sur la seringue d'administration pour la préparation)
Vous aurez besoin également d'eau plate en bouteille (non fournie).
Adressez-vous à votre pharmacien s'il manque un élément du matériel cité au point 1, 2 ou 3.
|
|

| |
1. Vérifiez si le flacon contient de la poudre ou une solution.
·Si le flacon contient de la poudre, passez à l'étape 2 ci-dessous.
·Si le flacon contient une solution, passez à l'étape 2 de la section B.
·Si le flacon contient une solution, n'y ajoutez pas d'eau supplémentaire.
|
|
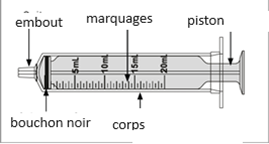
| |
2. N'agitez pas le flacon.
Tapotez fermement sur les parois du flacon pour désagglomérer la poudre.
|
|

| |
3. Retirez le capuchon doté d'une sécurité enfant en pressant et tournant dans le sens inverse des aiguilles d'une montre.
|
|
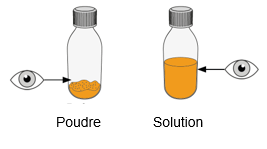
| |
4. Retirez l'adaptateur de la seringue.
|
|

| |
5. Remplissez un verre d'eau plate.*
·À l'aide de la seringue d'administration de la préparation, remplissez d'eau le flacon jusqu'à la flèche bleue imprimée sur l'étiquette du flacon. Cela correspond à l'ajout de 90 ml d'eau.
Remarque: si de la mousse se forme, remplissez jusqu'au bord supérieur de l'eau et pas jusqu'au bord supérieur de la mousse.
*Lors de la préparation en pharmacie, de l'eau stérile ou purifiée peut également être utilisée.
|
|

| |
6. Replacez le capuchon sécurisé sur le flacon et tournez dans le sens des aiguilles d'une montre pour fermer le flacon. Assurez-vous que le capuchon sécurisé est bien fixé sur le flacon avant de passer à l'étape suivante.
|
|
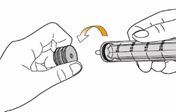
| |
7. Retournez plusieurs fois le flacon tête en bas puis en position normale jusqu'à ce que la poudre soit complètement dissoute. Vous pouvez également légèrement secouer le flacon.
Remarque: la dissolution complète de la poudre peut prendre 5 minutes ou plus.
|
|

| |
8. Laissez le flacon au repos pendant au moins 5 minutes sur une surface plane.
·Attendez qu'il n'y ait plus de mousse visible avant de passer à l'étape suivante.
|
|

| |
9. Une fois que la poudre est totalement dissoute, il est possible que le niveau d'eau dans le flacon se trouve sous la flèche bleue.*
·Si nécessaire, retirez le capuchon sécurisé et complétez d'eau claire jusqu'à la flèche bleue.
Remarque: au total, vous avez à présent ajouté 90 ml d'eau.
|
|

| |
10. Posez le flacon sur la table et maintenez-le fermement. Avec l'autre main, insérez l'adaptateur dans le flacon au moyen du pouce ou du plat de la main.
·L'adaptateur doit être complètement enfoncé dans le flacon, aucune rainure ne doit être visible. Appuyez fermement jusqu'à introduction complète.
Remarque: l'introduction de l'adaptateur peut demander beaucoup de force.
|
|

|
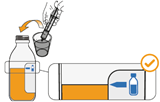
| |
11. Replacez le capuchon sécurisé sur le flacon et tournez dans le sens des aiguilles d'une montre pour fermer le flacon.
·Assurez-vous que le capuchon sécurisé est fermement fixé.
Remarque: si le capuchon sécurisé n'est pas fermement fixé, vérifiez que l'adaptateur est complètement inséré.
|
|
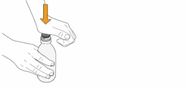
| |
12. Inscrivez la date de péremption sur la boite. La date de péremption se situe 35 jours après la date de préparation de la solution.
·S'il reste de la solution après 35 jours, demandez à votre pharmacien comment l'éliminer et prenez un nouveau flacon de Mekinist.
Ne jetez aucun médicament à l'égout ou dans les ordures ménagères.
Remarque: une fois l'étape de préparation terminée, n'ajoutez plus d'eau au flacon.
|
|

| |
13. Pour administrer la solution, suivez les étapes de la section B.
Pour administrer la solution par une sonde gastrique, suivez les étapes de la section C.
| |
SECTION B. UTILISATION
| |
Pour l'administration de Mekinist sous forme de solution prête à l'emploi, vous avez besoin de ce qui suit:
1.Solution dans le flacon
2.Seringue d'administration pour la préparation
3.Adaptateur (déjà inséré dans le col du flacon)
Adressez-vous à votre pharmacien si l'un ou plusieurs de ces éléments est/sont manquant(s).
N'administrez pas Mekinist si plus de 35 jours se sont écoulés depuis la date qui est indiquée sur la boite sous «Solution préparée le».
En cas de renversement ou de contact de la solution de Mekinist avec la peau ou les yeux, suivez les instructions de la section «Nettoyage de liquides renversés».
Lavez et séchez vos mains avant d'administrer Mekinist.
|
|

| |
1. Vérifiez que le flacon contient de la poudre ou une solution.
·Si le flacon contient de la poudre, passez à l'étape 2 ci-dessous.
·Si le flacon contient une solution, passez à l'étape 2 de la section B.
·Si le flacon contient une solution, n'y ajoutez pas d'eau supplémentaire.
|
|
| |
2. Vérifiez la date de la reconstitution (préparation) de la solution sur la boite. N'administrez pas Mekinist si la date de péremption est dépassée ou si aucune date n'est indiquée.
Remarque: la date de péremption imprimée sur le bord droit de l'étiquette du flacon ne vaut PAS pour la solution. Cette date de péremption imprimée s'applique à la poudre.
|
|
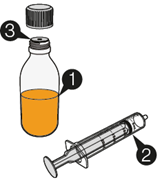
| |
3. Remuez légèrement le flacon pendant 30 secondes pour mélanger la solution.
·Si de la mousse se forme, laissez reposer le flacon jusqu'à disparition de la mousse.
|
|

| |
·Retirez le capuchon doté d'une sécurité enfants, en le pressant et tournant dans le sens inverse des aiguilles d'une montre.
|
|

| |
·Vérifiez qu'un adaptateur est inséré dans le col du flacon.
S'il n'est pas inséré, prenez l'adaptateur de la seringue et insérez-le. Pour plus d'informations, consultez les étapes 4 et 10 de la section A.
Adressez-vous à votre médecin en cas de doute ou si l'adaptateur est manquant.
|
|

| |
·Enfoncez totalement le piston dans la seringue d'administration de la préparation pour évacuer tout l'air.
|
|

| |
4. Posez le flacon sur une surface plane et maintenez-le verticalement.
·Insérez l'embout de la seringue d'administration dans l'orifice de l'adaptateur de flacon.
·Assurez-vous que la seringue d'administration est fermement fixée.
REMARQUE IMPORTANTE: en raison de la pression d'air, le piston pourrait bouger lorsque vous mesurez la dose à l'étape 7. Maintenez le piston pour qu'il ne bouge pas.
|
|

| |
5. Retournez avec précaution le flacon tête en bas et tirez sur le piston pour mesurer la dose. Avec la seringue vers le bas, la surface supérieure du bouchon noir doit coïncider avec la dose prescrite.
·S'il y a de grosses bulles d'air dans la seringue, renvoyez le médicament dans le flacon et prélevez à nouveau une dose. Répétez cela jusqu'à ce qu'il n'y ait plus de bulle d'air. Quelques petites bulles sont tolérées.
|
|
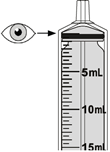
| |
6. Maintenez à nouveau fermement le piston, retournez le flacon tête en haut et posez-le sur une surface plane. En continuant à maintenir le piston, tirez avec précaution la seringue d'administration vers le haut pour la séparer du flacon.
|
|

| |
7. Assurez-vous que la surface supérieure du bouchon noir se trouve bien en face de la dose prescrite. Si ce n'est pas le cas, répétez les étapes 5 et 6.
·Si le médicament est pris directement dans la bouche (en avalant), poursuivez avec l'étape 8.
·Si le médicament est administré via une sonde gastrique, passez à la section C.
|
|

| |
8. REMARQUE IMPORTANTE: lors de l'administration à un enfant, veillez à ce qu'il soit assis.
·Placez l'extrémité de la seringue d'administration dans la bouche de manière à ce que la seringue touche la face interne de la joue.
·Poussez lentement sur le piston jusqu'à ce qu'il soit complètement enfoncé pour administrer la dose complète de Mekinist.
ATTENTION: si vous administrez Mekinist dans la gorge ou poussez trop vite sur le piston, l'enfant pourrait s'étouffer.
|
|
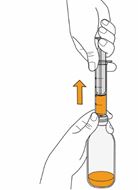
| |
9. Assurez-vous qu'il ne reste plus de solution dans la seringue.
·S'il reste de la solution dans la seringue, administrez-la.
Remarque: si votre dose est supérieure à la capacité de la seringue, répétez l'administration jusqu'à ce que le volume complet ait été donné.
|
|

| |
10. Ne retirez pas l'adaptateur.
·Replacez le capuchon sur le flacon et tournez dans le sens des aiguilles d'une montre.
·Assurez-vous de la bonne fermeture du flacon.
|
|

| |
11. Les instructions de lavage et de stockage sont données à la section D.
|
| |
SECTION C. ADMINISTRATION VIA UNE SONDE GASTRIQUE
| |
Veuillez suivre cette section uniquement si vous souhaitez administrer Mekinist via une sonde gastrique.
Pour l'administration via une sonde gastrique, veuillez lire les informations suivantes et poursuivez avec l'étape 1.
·La solution convient pour une administration par sonde gastrique.
·Utilisez une sonde nasogastrique (NG) ou gastrique (G) d'une taille minimale de 4 Ch.
·Utilisez toujours pour l'administration de Mekinist, la seringue d'administration de 20 ml fournie dans l'emballage.
·Si nécessaire, utilisez un adaptateur ENFIT (non fourni dans l'emballage), pour raccorder la seringue d'administration de 20 ml à la sonde gastrique.
| |
1. Rincez la sonde conformément aux instructions du fabricant, juste avant l'administration de Mekinist.
| |
2. Suivez les étapes 1 à 7 de la section B, puis passez à l'étape 3 de la présente section.
| |
3. Raccordez la seringue de 20 ml contenant Mekinist à la sonde. Si nécessaire, utilisez un adaptateur ENFIT pour raccorder la seringue à la sonde.
| |
4. Exercez une pression régulière pour injecter la solution dans la sonde gastrique.
| |
5. Assurez-vous qu'il ne reste pas de solution Mekinist dans la seringue. S'il reste de la solution dans la seringue, administrez-la.
| |
6. Rincez à nouveau la sonde conformément aux instructions du fabricant.
| |
7. Passez à la section D pour le nettoyage.
| |
SECTION D. NETTOYAGE
| |
En cas de renversement ou de contact de la solution de Mekinist avec la peau ou les yeux, suivez les instructions de la section «Nettoyage des liquides renversés».
Conservez la seringue d'administration de la préparation séparément des ustensiles de cuisine.
| |
1. Remplissez un verre d'eau savonneuse chaude.
|
|
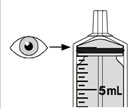
| |
2. Insérez la seringue d'administration dans le verre d'eau chaude. Aspirez et rejetez l'eau dans la seringue, quatre à cinq fois.
|
|

| |
3. Séparez le piston du corps de la seringue.
|
|
| |
4. Rincez le verre, le piston et le corps de la seringue à l'eau chaude.
|
|
| |
5. Laissez le piston et le corps de la seringue sécher à l'air sur une surface sèche jusqu'à l'utilisation suivante.
Conservez toujours la seringue hors de portée des enfants.
Remarque: utilisez une nouvelle seringue d'administration à chaque nouveau flacon de Mekinist.
|
|
| |
CONSERVATION
| |
Conservation de la poudre pour solution
Conservez Mekinist poudre hors de la portée et de la vue des enfants.
Jusqu'à la reconstitution, conserver au réfrigérateur (2 à 8 °C).
Conserver en position debout, dans l'emballage d'origine, pour protéger la poudre de la lumière et de l'humidité. Garder le flacon bien fermé.
Ne plus utiliser la poudre si la date de péremption imprimée sur le bord droit de l'étiquette est dépassée.
|
|

| |
Conservation de la solution prête à l'emploi
Conservez Mekinist solution hors de la portée et de la vue des enfants.
Conserver le flacon avec la solution dans la boite à une température inférieure à 25 °C.
Ne pas congeler le flacon.
Conserver la solution en position debout, dans la boite fournie, à l'abri de la lumière directe et avec le capuchon sécurisé fermement fixé.
Ne plus utiliser la solution si plus de 35 jours se sont écoulés depuis la date indiquée sur la boite sous «Solution préparée le».,
|
|

| |
Conservation de la seringue d'administration de la préparation
·Conservez la seringue d'administration hors de la portée et de la vue des enfants.
·Conserver la seringue d'administration dans la boite fournie, avec la poudre/solution.
|
|
| |
ÉLIMINATION
| |
·Demandez à votre pharmacien comment éliminer les médicaments périmés ou dont vous n'avez plus besoin.
·Pour chaque nouveau flacon de Mekinist, utilisez une nouvelle seringue. Demandez à votre pharmacien comment éliminer les seringues usagées.
·Ne jetez aucun médicament à l'égout ou dans les ordures ménagères.
·Vous contribuez ainsi à protéger l'environnement.
|
|