CompositionPrincipes actifs
Dabrafénib sous forme de mésylate de dabrafénib.
Excipients
Gélules:
Cellulose microcristalline, stéarate de magnésium, silice colloïdale anhydre, oxyde de fer rouge (E172), dioxyde de titane (E171), hypromellose (E464), gomme-laque, oxyde de fer noir (E172), propylène glycol, hydroxyde d'ammonium.
Comprimés dispersibles:
Mannitol (E421), cellulose microcristalline (E460), crospovidone (E1201), hypromellose (E464), acésulfame potassique (E950), stéarate de magnésium (E470b), arôme artificiel de fruits rouges (maltodextrine, propylène glycol 0,036 mg [E1520], arômes artificiels, citrate de triéthyle [E1505], alcool benzylique < 0,00078 mg [E1519]), silice colloïdale anhydre (E551).
Indications/Possibilités d’emploiIndication avec une autorisation à durée limitée
Gliome de bas grade (GBG)
Tafinlar en association avec le tramétinib est indiqué dans le traitement des patients pédiatriques âgés de 1 an et plus atteints d'un gliome de bas grade (GBG) présentant une mutation V600E du gène BRAF, qui nécessitent un traitement systémique.
Les données cliniques étant incomplètes au moment de l'expertise de la demande, l'autorisation de cette (ces) indication(s) est à durée limitée (art. 9a de la loi sur les produits pharmaceutiques). L'autorisation à durée limitée est obligatoirement liée au respect des conditions en temps voulu. Une fois ces conditions remplies, l'autorisation à durée limitée peut être convertie en autorisation ordinaire.
Indications avec une autorisation ordinaire
Mélanome non résécable ou métastatique
·Tafinlar en association avec le tramétinib est indiqué dans le traitement de patients adultes atteints de mélanome non résécable ou métastatique porteur de la mutation V600 du gène BRAF (V600E/K).
·Tafinlar est indiqué dans le traitement de patients adultes n'ayant jamais reçu de chimiothérapie, atteints de mélanome non résécable ou métastatique porteur de la mutation V600E du gène BRAF (voir «Propriétés/Effets»). Pour diagnostiquer la présence d'une mutation V600E, il est nécessaire de recourir à un test validé de la mutation BRAF.
Traitement adjuvant du mélanome
Tafinlar en association avec le tramétinib est indiqué dans le traitement adjuvant de patients atteints de mélanome de stade III porteur de la mutation V600 du gène BRAF après résection complète.
Cancer pulmonaire non à petites cellules avancé ou métastatique
Tafinlar en association avec le tramétinib peut être utilisé dans le traitement des patients adultes atteints d'un cancer pulmonaire non à petites cellules (NSCLC) métastatique porteur de la mutation V600E du gène BRAF.
Posologie/Mode d’emploiLe traitement par Tafinlar doit être instauré et surveillé par un médecin expérimenté dans l'utilisation de médicaments oncologiques.
Tafinlar est disponible sous deux formes pharmaceutiques: sous forme de gélules et sous forme de comprimés dispersibles.
Avant de prendre Tafinlar, la présence d'une mutation V600 du gène BRAF doit être confirmée au moyen d'un test validé, conformément à l'indication autorisée.
Posologie usuelle
Gélules
Chez les patients adultes, la dose recommandée de Tafinlar est de 150 mg (deux gélules de 75 mg) deux fois par jour (soit une dose quotidienne totale de 300 mg), que Tafinlar soit administré seul ou en association avec le tramétinib.
Chez les patients pédiatriques, la dose recommandée de comprimés dispersibles de Tafinlar dépend du poids corporel (tableau 1).
Tableau 1: Dosage recommandé des comprimés dispersibles de Tafinlar en fonction du poids
|
|
Dose recommandée
| |
Poids corporel
|
Dose journalière totale
|
Nombre de comprimés pour la solution orale
| |
8 à 9 kilos
|
20 mg deux fois par jour
|
deux comprimés dispersibles à 10 mg deux fois par jour
| |
10 à 13 kilos
|
30 mg deux fois par jour
|
trois comprimés dispersibles à 10 mg deux fois par jour
| |
14 à 17 kilos
|
40 mg deux fois par jour
|
quatre comprimés dispersibles à 10 mg deux fois par jour
| |
18 à 21 kilos
|
50 mg deux fois par jour
|
cinq comprimés dispersibles à 10 mg deux fois par jour
| |
22 à 25 kilos
|
60 mg deux fois par jour
|
six comprimés dispersibles à 10 mg deux fois par jour
| |
26 à 29 kilos
|
70 mg deux fois par jour
|
sept comprimés dispersibles à 10 mg deux fois par jour
| |
30 à 33 kilos
|
80 mg deux fois par jour
|
huit comprimés dispersibles à 10 mg deux fois par jour
| |
34 à 37 kilos
|
90 mg deux fois par jour
|
neuf comprimés dispersibles à 10 mg deux fois par jour
| |
38 à 41 kilos
|
100 mg deux fois par jour
|
dix comprimés dispersibles à 10 mg deux fois par jour
| |
42 à 45 kilos
|
110 mg deux fois par jour
|
onze comprimés dispersibles à 10 mg deux fois par jour
| |
46 à 50 kilos
|
130 mg deux fois par jour
|
treize comprimés dispersibles à 10 mg deux fois par jour
| |
≥51 kilos
|
150 mg deux fois par jour
|
quinze comprimés dispersibles à 10 mg deux fois par jour
|
Schéma d'administration
Tafinlar est pris au moins 1 heure avant, ou au moins 2 heures après un repas, en respectant un intervalle d'environ 12 heures entre les doses.
Tafinlar doit être pris chaque jour à la même heure environ.
Lorsque le tramétinib et Tafinlar sont pris en association, l'unique dose quotidienne de tramétinib doit être prise tous les jours à la même heure, soit avec la dose de Tafinlar du matin, soit avec celle du soir. La pharmacocinétique de la prise de tramétinib le soir n'a pas été étudiée, de sorte que la prise matinale doit être préférée.
Pour assurer la traçabilité des médicaments biotechnologiques, il convient de documenter pour chaque traitement le nom commercial et le numéro de lot.
Gélules
Les gélules de Tafinlar doivent être avalées sans être mâchées. Les gélules ne doivent pas être mâchées ni écrasées.
Comprimés dispersibles
Les comprimés dispersibles de Tafinlar doivent uniquement être pris sous forme de suspension et ne doivent pas être avalés entiers, mâchés ou écrasés.
La suspension orale est préparée dans un gobelet doseur fourni. Les suspensions de Tafinlar sous forme de comprimés dispersibles peuvent être prises de trois façons: en buvant la suspension dans le gobelet doseur, en avalant la solution de la seringue orale remplie de la suspension prélevée du gobelet doseur, ou en absorbant la suspension par une sonde gastrique.
Il faut veiller à prendre la totalité de la dose. Un délai de 3 minutes (ou plus long) peut s'écouler jusqu'à la dissolution complète des comprimés. Dès que ceux-ci sont dissous, la suspension doit avoir une apparence trouble-blanche.
La suspension ne doit pas être prise plus de 30 minutes après la dissolution des comprimés. Si plus de 30 minutes se sont écoulées, la suspension doit être éliminée conformément aux dispositions locales et il faut recommencer la préparation.
Vous trouverez les instructions d'utilisation illustrées complètes pour le comprimé dispersible dans la rubrique «Instructions d'utilisation».
Durée du traitement
Le traitement est maintenu jusqu'à constatation d'une progression de la tumeur ou jusqu'à l'apparition de symptômes de toxicité intolérables (voir tableaux 2, 3 et 4). L'arrêt du traitement est recommandé en cas de progression de la tumeur.
Dans le cas d'un traitement adjuvant du mélanome, les patients doivent être traités pendant 12 mois au maximum, sauf en cas de récurrence de la maladie ou d'une toxicité inacceptable.
La durée de traitement recommandée pour les patients pédiatriques présentant un GBG dure jusqu'à la progression de la maladie ou l'apparition d'une toxicité inacceptable. Il convient de noter que, dans les études cliniques menées auprès de patients pédiatriques présentant un GBG, des différences significatives dans l'évaluation de l'efficacité, qui incluaient également la constatation d'une progression de la maladie, ont été observées entre l'examen indépendant et les centres investigateurs (voir également «Efficacité clinique»). Par ailleurs, les données disponibles sur l'utilisation à long terme et la durée de traitement optimale du traitement par Tafinlar en association avec le tramétinib dans la population pédiatrique sont limitées (voir «Mises en garde et précautions»). La durée médiane de traitement dans les études cliniques pertinentes a été jusqu'à maintenant de 24 mois (voir «Efficacité clinique»). De plus, les données disponibles sur les patients présentant un GBG âgés de plus de 18 ans qui nécessitent un premier traitement systémique sont limitées. La poursuite du traitement jusqu'à l'âge adulte doit donc dépendre des avantages et des risques pour chacun des patients en fonction de l'évaluation du médecin.
Ajustement de la posologie du fait d'effets indésirables / d'interactions
En monothérapie et en association avec le tramétinib
En cas de réactions indésirables, une interruption du traitement, une réduction de la dose ou un arrêt du traitement peuvent s'avérer nécessaires (voir tableaux 2, 3 et 4).
Il n'est pas recommandé d'effectuer des ajustements posologiques ou d'interrompre le traitement en cas de réactions indésirables se traduisant par l'apparition d'un carcinome épidermoïde cutané (CEC) ou d'un nouveau mélanome primaire (voir «Mises en garde et précautions»).
Le traitement par dabrafénib doit être interrompu en cas de fièvre ≥38,5 °C. Les patients doivent faire l'objet d'une surveillance en vue de détecter tout signe ou symptôme évocateur d'une infection (voir «Mises en garde et précautions»).
Les recommandations sur les réductions par palier de la dose et les ajustements de la dose se trouvent respectivement dans les tableaux 2 et 3. Tafinlar sous forme de gélules doit être arrêté définitivement si la dose de 50 mg deux fois par jour n'est pas tolérée (tableau 2). On arrêtera le traitement chez les patients qui ne peuvent pas augmenter leur dose sous forme de gélules à > 50 mg deux fois par jour sur plusieurs semaines. Tafinlar sous forme de comprimés dispersibles doit être arrêté définitivement s'il n'est pas toléré à une dose de 10 mg deux fois par jour ou après trois réductions de la dose (tableau 3).
Tableau 2: Recommandations relatives à la réduction des doses de Tafinlar, gélules par palier chez les patients adultes
|
Réductions des doses
|
Dose/schéma d'administration
| |
Dose thérapeutique initiale complète
|
150 mg par voie orale deux fois par jour
| |
Première réduction de la dose
|
100 mg par voie orale deux fois par jour
| |
Deuxième réduction de la dose
|
75 mg par voie orale deux fois par jour
| |
Troisième réduction de la dose
|
50 mg par voie orale deux fois par jour
| |
Arrêter définitivement si Tafinlar 50 mg sous forme de gélule par voie orale deux fois par jour n'est pas toléré
|
Pour les instructions posologiques du tramétinib, consulter l'information professionnelle du tramétinib, «Posologie/Mode d'emploi».
Tableau 3: Recommandations relatives à la réduction des doses de Tafinlar, comprimés dispersibles, par palier en fonction du poids
|
Poids corporel (kg)
(dose initiale recommandée)
|
Première réduction de la dose
|
Deuxième réduction de la dose
|
Troisième réduction de la dose
| |
Comprimés pour suspension orale deux fois par jour
| |
8 à 9 kg
(20 mg deux fois par jour)
|
20 mg deux fois par jour
|
N/A
|
N/A
| |
10 à 13 kg
(30 mg deux fois par jour)
|
20 mg deux fois par jour
|
10 mg deux fois par jour
|
N/A
| |
14 à 17 kg
(40 mg deux fois par jour)
|
30 mg deux fois par jour
|
20 mg deux fois par jour
|
10 mg deux fois par jour
| |
18 à 21 kg
(50 mg deux fois par jour)
|
30 mg deux fois par jour
|
20 mg deux fois par jour
|
10 mg deux fois par jour
| |
22 à 25 kg
(60 mg deux fois par jour)
|
40 mg deux fois par jour
|
30 mg deux fois par jour
|
20 mg deux fois par jour
| |
26 à 29 kg
(70 mg deux fois par jour)
|
50 mg deux fois par jour
|
40 mg deux fois par jour
|
20 mg deux fois par jour
| |
30 à 33 kg
(80 mg deux fois par jour)
|
50 mg deux fois par jour
|
40 mg deux fois par jour
|
30 mg deux fois par jour
| |
34 à 37 kg
(90 mg deux fois par jour)
|
60 mg deux fois par jour
|
50 mg deux fois par jour
|
30 mg deux fois par jour
| |
38 à 41 kg
(100 mg deux fois par jour)
|
70 mg deux fois par jour
|
50 mg deux fois par jour
|
30 mg deux fois par jour
| |
42 à 45 kg
(110 mg deux fois par jour)
|
70 mg deux fois par jour
|
60 mg deux fois par jour
|
40 mg deux fois par jour
| |
46 à 50 kg
(130 mg deux fois par jour)
|
90 mg deux fois par jour
|
70 mg deux fois par jour
|
40 mg deux fois par jour
| |
≥51 kg
(150 mg deux fois par jour)
|
100 mg deux fois par jour
|
80 mg deux fois par jour
|
50 mg deux fois par jour
| |
Arrêter définitivement Tafinlar s'il n'est pas toléré à la dose de 10 mg deux fois par jour sous forme de comprimés dispersibles ou après trois réductions de la dose maximum.
|
Tableau 4: Recommandations de modifications du dosage de Tafinlar en cas d'effets indésirables
|
Sévérité de l'effet indésirable [voir Mises en garde et précautions] a
|
Modification du dosage de Tafinlarb
| |
Nouvelles tumeurs malignes primitives
| |
Tumeurs malignes non cutanées RAS mutées
|
Arrêter définitivement Tafinlar
| |
Myocardiopathie
| |
·Insuffisance cardiaque congestive symptomatique
·Diminution absolue de la fraction d'éjection du ventricule gauche (FEVG) de plus de 20% par rapport à la valeur initiale qui est inférieure à la limite inférieure de la normale (LIN).
|
Suspendre la prise de Tafinlar jusqu'à ce que la FEVG se soit améliorée à au moins la LIN institutionnelle et que la réduction de la FEVG absolue soit rétablie à 10% ou moins par rapport à la valeur initiale, puis poursuivre le traitement avec la même dose.
| |
Uvéite
| |
·Uvéite, y compris iritis et iridocyclite
|
En cas d'uvéite légère ou modérée qui ne répond pas à un traitement, ou en cas d'uvéite sévère, suspendre la prise de Tafinlar jusqu'à 6 semaines.
·En cas d'amélioration au grade 0–1, reprendre Tafinlar à la même dose ou à une dose inférieure.
·En l'absence d'amélioration, arrêter définitivement Tafinlar.
| |
Réactions fébriles
| |
·Fièvre de 38 °C à 40 °C (ou premiers symptômes en cas de récidive)
|
Interrompre le traitement par Tafinlar jusqu'à ce que la fièvre soit tombée, puis poursuivre le traitement à la même dose ou à une dose inférieure.
| |
·Fièvre supérieure à 40 °C
·Fièvre, compliquée par des frissons, une hypotension, une déshydratation ou une insuffisance rénale
|
·Interrompre le traitement par Tafinlar jusqu'à ce que les réactions fébriles aient disparu pendant au moins 24 heures, puis poursuivre le traitement à une dose inférieure.
Ou
·Arrêter définitivement Tafinlar.
| |
Toxicités cutanées
| |
·Sévérité de grade 2, si non tolérable
·Sévérité de grade 3 ou 4
|
Interrompre Tafinlar pour une durée allant jusqu'à 3 semaines.
·En cas d'amélioration, reprendre Tafinlar à une dose inférieure.
·En l'absence d'amélioration, arrêter définitivement Tafinlar.
| |
·Réactions cutanées indésirables graves (SCAR)
|
Arrêter définitivement Tafinlar.
| |
Autres effets indésirables, y compris hémorragies
| |
·Sévérité de grade 2, si non tolérable
·Sévérité de grade 3
|
Suspendre la prise de Tafinlar.
·En cas d'amélioration au grade 0–1, reprendre Tafinlar à une dose inférieure.
·En l'absence d'amélioration, arrêter définitivement Tafinlar.
| |
·Première apparition d'une sévérité de grade 4
|
·Interrompre le traitement par Tafinlar jusqu'à l'amélioration du traitement au grade 0–1, puis poursuivre le traitement à une dose inférieure.
Ou
·Arrêter définitivement Tafinlar.
| |
·Récidive du grade 4
|
Arrêter définitivement Tafinlar.
| |
a
National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 4.0.
b Voir tableaux 2 et 3 pour les recommandations de réductions des doses de Tafinlar.
c Pour les effets indésirables suivants, aucun ajustement posologique de Tafinlar n'est recommandé lorsqu'il est administré avec le tramétinib: occlusion veineuse rétinienne (OVR), décollement de l'épithélium pigmentaire de la rétine (DEP), pneumopathie interstitielle/pneumopathie inflammatoire et thromboembolie veineuse non compliquée. Un ajustement posologique de Tafinlar n'est pas nécessaire en cas de nouvelles tumeurs malignes primitives.
|
Veuillez respecter l'information professionnelle du tramétinib pour les ajustements posologiques en cas d'effets indésirables associés au tramétinib.
Lorsque les effets indésirables d'un patient sont efficacement contrôlés, une nouvelle augmentation progressive de la dose peut être envisagée en respectant les paliers utilisés au moment de la réduction de la dose. La dose de Tafinlar ne doit pas dépasser 150 mg deux fois par jour chez les patients adultes, voir le tableau 2 pour les paliers de réduction de la dose recommandés pour les gélules de Tafinlar. Chez les patients pédiatriques, la réduction de la dose de Tafinlar, comprimés dispersibles dépend du poids corporel (tableau 3).
Lorsque des réactions de toxicité liées au traitement surviennent au cours du traitement concomitant par Tafinlar et le tramétinib, il convient de réduire simultanément les doses des deux médicaments, ou d'interrompre ou d'arrêter complètement les deux traitements, à l'exception des cas suivants.
Exceptions pour lesquelles seul un ajustement de la posologie de Tafinlar est nécessaire:
·Uvéite
Traitement de la pyrexie
Le traitement doit être interrompu (Tafinlar lorsque celui-ci est utilisé en monothérapie ou Tafinlar et le tramétinib lorsque ceux-ci sont utilisés en association) si la température corporelle d'un patient est supérieure ou égale à 38 °C (≥100,4 °F). En cas de récidive, le traitement peut également être interrompu dès l'apparition du premier symptôme d'une pyrexie. Un traitement antipyrétique, p.ex. par l'ibuprofène ou le paracétamol, sera initié. Les patients doivent être surveillés à la recherche de signes et de symptômes d'une infection (voir «Mises en garde et précautions»). Le traitement par Tafinlar ou le traitement par Tafinlar et le tramétinib, lorsque ceux-ci sont utilisés en association, doit être repris si le patient ne présente pas de symptômes depuis au moins 24 heures, à savoir:
·au même palier de dose
·ou à une dose réduite d'un palier si la pyrexie réapparaît et/ou s'accompagne de symptômes sévères tels qu'une déshydratation, une hypotension ou une insuffisance rénale.
L'utilisation de corticostéroïdes doit être envisagée dans les cas où les antipyrétiques sont insuffisants.
Traitement de l'uvéite
Il convient d'essayer de maîtriser l'uvéite par un traitement local dans l'œil sans modifier la posologie de Tafinlar. Si l'uvéite ne répond pas à un traitement oculaire local, interrompre le traitement par dabrafénib jusqu'à guérison de l'inflammation oculaire, puis reprendre avec une dose de Tafinlar réduite d'un palier. Aucun ajustement de la posologie du tramétinib n'est nécessaire lorsque Tafinlar est pris en association avec le tramétinib.
Pour les ajustements recommandés de la posologie du tramétinib, voir l'information professionnelle de Mekinist.
Traitement associé
En cas d'administration de Tafinlar en association avec le tramétinib, consulter l'information professionnelle de Mekinist pour les instructions posologiques de Mekinist (voir «Posologie/Mode d'emploi» de l'information professionnelle de Mekinist).
Instructions posologiques particulières
Patients présentant des troubles de la fonction hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance hépatique légère. Une analyse pharmacocinétique de population indique qu'une insuffisance hépatique légère n'a pas d'effet significatif sur la clairance orale du dabrafénib ni sur la concentration de ses métabolites (voir «Pharmacocinétique»). Aucune donnée clinique n'est disponible chez des sujets présentant une insuffisance hépatique modérée à sévère. Il n'est donc pas possible de donner des indications sur la nécessité d'un ajustement de la dose. Le métabolisme hépatique et la sécrétion biliaire sont les principales voies d'élimination du dabrafénib et de ses métabolites. Par conséquent, il est possible que les patients atteints d'insuffisance hépatique modérée à sévère soient plus fortement exposés et présentent un risque plus élevé d'effets indésirables en raison d'une plus forte exposition au dabrafénib. La prudence est de mise lors de l'administration de Tafinlar à des patients atteints d'insuffisance hépatique modérée à sévère.
Patients présentant des troubles de la fonction rénale
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance rénale légère à modérée. Une analyse pharmacocinétique de population indique qu'une insuffisance rénale légère à modérée n'a pas d'effet significatif sur la clairance orale du dabrafénib ni sur la concentration de ses métabolites (voir «Pharmacocinétique»). Aucune donnée clinique n'est disponible chez des sujets présentant une insuffisance rénale sévère. Il n'est donc pas possible de donner des indications sur la nécessité d'un ajustement de la dose. La prudence est de mise lors de l'administration de Tafinlar à des patients atteints d'insuffisance rénale sévère.
Patients âgés
Aucun ajustement de la dose n'est nécessaire chez les patients âgés de plus de 65 ans (voir «Pharmacocinétique»).
Population pédiatrique
L'administration de Tafinlar aux patients pédiatriques âgés de moins de 1 an n'est pas autorisée. En raison de la toxicité rénale de Tafinlar dans les études de toxicité juvénile réalisées chez le rat, ces patients ont été exclus des études cliniques avec Tafinlar (voir «Toxicité générale»). Pour les enfants âgés de 1 à 2 ans, seules des données limitées sur la sécurité et l'efficacité sont disponibles (voir «Mises en garde et précautions»).
Génotype / polymorphismes génétiques
L'efficacité et la sécurité de Tafinlar n'ont pas été établies chez les patients atteints de mélanome à gène BRAF de type sauvage ou de NSCLC à gène BRAF de type sauvage; par conséquent, Tafinlar ne doit pas être utilisé chez les patients atteints de mélanome ou de NSCLC à gène BRAF de type sauvage (voir «Propriétés/Effets»).
Prise retardée
Une dose oubliée de Tafinlar ne doit pas être rattrapée s'il reste moins de 6 heures avant la prise de la dose suivante prévue.
Contre-indicationsHypersensibilité au principe actif ou à l'un des autres excipients.
Mises en garde et précautionsÉvénements fébriles graves non infectieux
Des cas de fièvre ont été rapportés dans les essais cliniques réalisés avec Tafinlar seul ou en association avec le tramétinib (voir «Effets indésirables»).
La fréquence et la sévérité de la pyrexie étaient accrues avec le traitement combiné. La majorité des événements de pyrexie est survenue au cours du premier mois de traitement. Trois événements ou plus sont survenus chez environ un tiers des patients traités par l'association qui ont présenté une pyrexie.
La pyrexie peut s'accompagner de frissons, de déshydratation et d'hypotension sévères, ce qui peut entraîner une insuffisance rénale aiguë dans certains cas. Pendant et après un épisode de fièvre sévère, il faut contrôler les taux sériques de créatinine et vérifier si le patient présente d'autres signes d'une insuffisance rénale.
Des événements fébriles, sévères, non infectieux ont été observés. Dans les études cliniques, ces événements ont bien répondu à une interruption du traitement et/ou à une réduction posologique et à des mesures de soutien (incluant l'administration d'antipyrétiques non stéroïdiens et stéroïdiens).
Une comparaison effectuée au moyen de plusieurs études chez 1810 patients ayant été traités par l'association a montré une diminution de l'incidence d'une pyrexie sévère et d'autres événements indésirables en rapport avec une pyrexie lorsque le traitement par Tafinlar et par le tramétinib a été interrompu, en comparaison avec une interruption de Tafinlar uniquement. Par conséquent, il est recommandé d'interrompre Tafinlar et le tramétinib si la température corporelle du patient est supérieure ou égale à 38 °C (≥100,4 °F). En cas de récidive, le traitement peut également être interrompu dès l'apparition du premier symptôme d'une pyrexie (voir «Posologie/Mode d'emploi» et «Efficacité clinique»).
Pour le traitement de la pyrexie, voir les directives relatives à l'ajustement posologique (voir «Posologie/Mode d'emploi»).
Déficit en glucose-6-phosphate déshydrogénase
Tafinlar, qui contient des sulfonamides, présente un risque potentiel d'anémie hémolytique chez les patients ayant un déficit en glucose-6phosphate déshydrogénase (G6PD). Pendant le traitement par Tafinlar, veuillez surveiller les patients qui ont un déficit en G6PD pour détecter les signes d'une anémie hémolytique.
Hyperglycémie
Des taux de glycémie élevés ont été mesurés dans des études cliniques chez quelques patients traités par Tafinlar. Chez les patients diabétiques ou atteints d'hyperglycémie, la glycémie doit être étroitement surveillée pendant le traitement par Tafinlar.
Carcinome épidermoïde cutané (CEC)
Des cas de CEC (incluant ceux classés comme appartenant au kératoacanthome ou au sous-type, le kératoacanthome mixte) ont été rapportés chez des patients traités aussi bien par Tafinlar seul que par l'association de Tafinlar et de tramétinib (voir «Effets indésirables»).
Dans l'étude de phase III MEK115306, un CEC est survenu chez 10% (22/211) des patients ayant reçu Tafinlar en monothérapie, le délai médian jusqu'au diagnostic de la première apparition ayant été d'environ 8 semaines. Un CEC est survenu chez 3% (6/209) des patients ayant reçu Tafinlar en association avec le tramétinib; les événements sont apparus plus tard ici, le délai médian jusqu'au diagnostic de la première apparition ayant été de 20 à 32 semaines. Plus de 90% des patients traités par Tafinlar et ayant présenté un CEC ont poursuivi le traitement par Tafinlar sans ajustement posologique. Dans l'étude de phase III du traitement adjuvant du mélanome, un carcinome épidermoïde cutané (CEC) est survenu chez 1% (6/435) des patients sous traitement avec Tafinlar en association avec le tramétinib contre 1% (5/432) des patients ayant reçu un placebo. Le délai médian jusqu'à la première apparition d'un CEC était d'environ 18 semaines dans le bras traité par l'association.
Un examen dermatologique est recommandé avant l'instauration d'un traitement par Tafinlar, puis tous les 2 mois au cours du traitement. Jusqu'à six mois après l'arrêt du traitement par Tafinlar ou jusqu'à l'instauration d'un autre traitement anti-néoplasique, les contrôles dermatologiques doivent être poursuivis régulièrement tous les 2–3 mois.
Les CEC devront être retirés par exérèse chirurgicale et le traitement par Tafinlar sera poursuivi sans ajustement de la posologie. Les patients doivent être informés de la nécessité de signaler immédiatement à leur médecin l'apparition de toute nouvelle lésion.
Nouveau mélanome primaire
L'apparition de nouveaux mélanomes primaires a été rapportée au cours du traitement par Tafinlar dans les études cliniques. Ces mélanomes ont été observés au cours des 5 premiers mois de traitement et ont été traités par exérèse sans nécessiter aucune modification du traitement. Dans l'étude clinique de phase III du traitement adjuvant du mélanome, des nouveaux mélanomes primaires sont survenus chez moins de 1% (1/435) des patients sous traitement combiné de Tafinlar et du tramétinib contre 1% (6/432) des patients ayant reçu un placebo.
Comme pour le carcinome épidermoïde cutané, les patients doivent être contrôlés afin d'identifier toute apparition de lésions cutanées.
Pour toute information supplémentaire concernant les mises en garde et précautions en rapport avec le traitement par le tramétinib, consulter l'information professionnelle de Mekinist.
Tumeurs malignes non cutanées secondaires/récidivantes
Des expérimentations in vitro ont montré une activation paradoxale de la voie de signalisation de la protéine kinase MAPK lors de l'exposition des cellules à gène BRAF de type sauvage présentant des mutations NRAS à des inhibiteurs de BRAF. Chez les patients sous Tafinlar, cela peut entraîner un risque accru de tumeurs malignes non cutanées. Des cas de tumeurs malignes associées à RAS ont été observés sous inhibiteurs de BRAF.
Lors de la comparaison de l'association de Tafinlar et du tramétinib avec le placebo, des tumeurs malignes secondaires non cutanées ou des tumeurs malignes récidivantes ont été observées dans l'étude de phase III du traitement adjuvant du mélanome chez 1% (5/435) des patients sous traitement actif contre 1% (3/432) des patients ayant reçu un placebo.
Les patients doivent être surveillés en fonction de l'état clinique. Chez les patients atteints d'une malignité non cutanée caractérisée par une mutation RAS, évaluer le rapport risque/bénéfice avant de poursuivre le traitement par Tafinlar. Aucun ajustement de la posologie du tramétinib n'est nécessaire au cours du traitement combiné par Tafinlar et le tramétinib.
Avant le début du traitement, les patients doivent être soumis à un examen de la région de la tête et du cou avec inspection visuelle de la muqueuse buccale et palpation des ganglions lymphatiques (comme contrôle minimal) ainsi qu'à un examen tomodensitométrique du thorax/de l'abdomen. Au cours du traitement, les patients doivent être surveillés comme cliniquement indiqué, y compris examens de la tête et du cou tous les 3 mois et TDM du thorax/abdomen tous les 6 mois. Il est recommandé de procéder à un examen par toucher rectal – et chez la femme à un examen du bassin – avant le début du traitement et à la fin du traitement ou chaque fois que ces examens sont cliniquement indiqués. Un hémogramme complet doit être fait lorsqu'il est cliniquement indiqué.
Jusqu'à 6 mois après la fin du traitement par Tafinlar ou jusqu'à l'instauration d'un autre traitement anti-néoplasique, le patient doit encore être contrôlé régulièrement quant à l'apparition de tumeurs malignes non cutanées secondaires/récidivantes.
Uvéite
Le traitement par Tafinlar a été associé au développement d'uvéites (y compris d'iritis). Les patients doivent être surveillés régulièrement pendant le traitement afin de détecter des signes et symptômes visuels (tels que des troubles de la vision, une photophobie et des douleurs oculaires) (voir «Posologie/Mode d'emploi»).
Pancréatite
Des cas de pancréatite ont été rapportés chez moins de 1% des participants à l'étude atteints d'un mélanome métastatique et traités par Tafinlar. Des cas de pancréatite aiguë ont été rapportés chez 1% des participants à l'étude atteints d'un NSCLC et traités par Tafinlar. Un de ces événements s'est produit le premier jour du traitement, puis à nouveau après la reprise du traitement à dose réduite. Dans l'étude du traitement adjuvant du mélanome, des cas de pancréatite ont été rapportés chez 1% des patients sous traitement avec Tafinlar en association avec le tramétinib ainsi que chez moins de 1% des patients ayant reçu un placebo.
Toute douleur abdominale inexpliquée doit rapidement faire l'objet d'examens, incluant un dosage de l'amylase et de la lipase sériques. Les patients doivent être étroitement surveillés lors de la reprise de Tafinlar après un épisode de pancréatite.
Hémorragies
Des événements hémorragiques, pour certains sévères, dans des zones corporelles et organes critiques, sont survenus chez des patients traités par Tafinlar en association avec le tramétinib (voir «Effets indésirables»). Dans le cadre d'études cliniques évaluant cette association, des hémorragies ont été signalées chez 17% des patients; 3% des patients ont eu des hémorragies gastro-intestinales. Sur 559 patients présentant un mélanome non résécable ou métastatique qui ont reçu Tafinlar en association avec le tramétinib, six (1%) ont eu des événements hémorragiques intracrâniens mortels. Trois cas ont été observés dans le cadre d'une étude MEK115306 (COMBI-d) et trois dans le cadre d'une étude MEK116513 (COMBI-v). Dans le cas du traitement adjuvant du mélanome, aucun événement hémorragique d'issue fatale n'est survenu dans le cadre d'une étude de phase III.
Deux des 93 participants (2%) qui ont reçu Tafinlar en association avec le tramétinib dans le cadre d'une étude de phase II portant sur le cancer pulmonaire non à petites cellules (NSCLC) ont présenté des événements hémorragiques d'issue fatale (un cas d'hémorragie sous-arachnoïdienne et un cas d'hémorragie rétropéritonéale).
Les patients développant des symptômes d'une hémorragie intracrânienne requièrent une prise en charge médicale immédiate.
L'administration concomitante d'un traitement antiplaquettaire ou anticoagulant peut augmenter le risque d'événements hémorragiques. En cas d'hémorragies, les patients doivent être traités en fonction de l'indication clinique.
En cas d'hémorragies de grade 4, Tafinlar doit être définitivement arrêté; il en est de même pour les hémorragies de grade 3 qui ne s'améliorent pas.
Thromboembolies veineuses (TEV)
Lors de l'administration de Tafinlar en association avec le tramétinib, des thromboembolies veineuses (TEV), y compris une thrombose veineuse profonde (TVP) et une embolie pulmonaire, peuvent survenir. Les patients doivent recevoir l'instruction de se faire soigner immédiatement en cas de survenue de symptômes d'une TEV. En cas dְ'embolie pulmonaire potentiellement mortelle, le traitement par le tramétinib et le dabrafénib doit être définitivement arrêté.
Réactions cutanées indésirables graves (SCAR)
Des rapports faisant état de réactions cutanées indésirables graves, y compris syndrome de Stevens-Johnson et réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), qui peuvent mettre le pronostic vital en jeu ou être fatales, ont été émis pendant le traitement par le dabrafénib en association avec le tramétinib. Les patients doivent être informés des signes et symptômes avant le début du traitement.
Pendant le traitement, les patients doivent être examinés de près pour déceler d'éventuelles réactions cutanées. En cas de soupçon de réaction cutanée indésirable grave, le dabrafénib et le tramétinib doivent être arrêtés.
InteractionsLe dabrafénib est un substrat des cytochromes CYP2C8 et CYP3A4. La co-administration de puissants inducteurs de ces enzymes doit donc être si possible évitée, l'efficacité de Tafinlar risquant d'être altérée.
Le dabrafénib est un inducteur d'enzymes métabolisantes et peut être un inducteur des transporteurs.
La co-administration de Tafinlar avec des médicaments qui sont des substrats sensibles des cytochromes CYP3A4, CYP2C8, CYP2C9, CYP2C19 ou CYP2B6 (p.ex. contraceptifs oraux) ou des substrats de protéines de transport, peut entraîner une perte d'efficacité de ces médicaments et doit généralement être évitée s'il n'est pas possible d'adapter la posologie (voir «Interactions»).
L'administration concomitante du dabrafénib et de la warfarine entraîne une diminution de l'exposition à la warfarine. Lors de l'instauration ou de l'arrêt d'un traitement par le dabrafénib en cas d'administration concomitante de warfarine, d'acénocoumarol et de phenprocoumone, la prudence et éventuellement une surveillance renforcée de l'INR (International Normalized Ratio) sont de rigueur.
Excipients d'un intérêt particulier (comprimés dispersibles)
Chaque comprimé dispersible contient < 0,00078 mg d'alcool benzylique. L'alcool benzylique peut provoquer des réactions allergiques graves.
N'utilisez pas ce médicament plus longtemps qu'une semaine chez les enfants en bas âge (âgés de moins de 3 ans), sauf sur avis de votre médecin ou de votre pharmacien. Chez les enfants en bas âge, il existe un risque accru dû à l'accumulation.
Indiquez aux patientes qui sont enceintes ou qui pourraient tomber enceintes le risque potentiel que représente l'excipient alcool benzylique pour le fœtus, qui peut s'accumuler au cours du temps et provoquer une acidose métabolique. La prudence est de mise lors de l'utilisation chez les patients présentant une insuffisance hépatique ou rénale, car l'alcool benzylique s'accumule au cours du temps et peut provoquer une acidose métabolique.
La prudence est de mise en cas de changement de traitement vers une autre forme pharmaceutique et/ou un autre médicament contenant le même principe actif. Le patient doit alors faire l'objet d'une surveillance adaptée.
Lymphohistiocytose hémophagocytaire (LHH)
Une LHH a été observée lors du suivi post-marketing de Tafinlar en association avec Mekinist (tramétrinib). La LHH est un syndrome potentiellement mortel s'accompagnant d'une activation pathologique des défenses immunitaires. En l'absence de diagnostic et de traitement précoces, la LHH a fréquemment une évolution létale. Cette maladie se caractérise par des signes et symptômes cliniques d'une inflammation systémique sévère, tels que fièvre, éruption cutanée, hépatosplénomégalie, cytopénies, lymphadénopathie, symptômes neurologiques, taux élevé de ferritine sérique, hypertriglycéridémie ainsi que troubles de la fonction hépatique et de la coagulation. Les symptômes surviennent généralement dans les deux mois suivant le début du traitement, mais peuvent également se manifester plus tard. En cas de suspicion de LHH, le traitement doit être interrompu. Après confirmation du diagnostic de LHH, le traitement doit être arrêté et un traitement adapté de la LHH instauré.
Population pédiatrique
Les patients pédiatriques en cours de développement sont soumis à un traitement potentiel à long terme par Tafinlar en association avec le tramétinib. Ces deux médicaments agissent ensemble sur une chaîne de transduction du signal qui joue un rôle important dans la régulation du développement des cellules et des tissus (voir «Mécanisme d'action»). Dans ce contexte, les données disponibles dans la population pédiatrique présentent des limitations (voir également «Posologie/Mode d'emploi»).
Ainsi, il n'existe pas suffisamment de données sur la sécurité et l'efficacité de l'utilisation à long terme et la durée optimale de traitement par Tafinlar en association avec le tramétinib dans la population pédiatrique. La durée médiane de traitement et de suivi dans les études cliniques pertinentes était jusqu'à maintenant d'environ 24 et 26 mois, respectivement (voir «Efficacité clinique»). Les conséquences à long terme des gains de poids indésirables observés très fréquemment sous traitement combiné ne sont pas claires à l'heure actuelle. Même si jusqu'à présent aucune augmentation des fréquences de néoplasies malignes secondaires n'a été observée dans la population pédiatrique, une évaluation finale n'est pas possible à l'heure actuelle (voir «Effets indésirables»).
Pour les enfants âgés de 1 à 2 ans, seules des données limitées sur la sécurité et l'efficacité sont disponibles.
Étant donné qu'il n'existe pas de comparaison directe entre l'association et les monothérapies dans une population suffisamment grande de patients, la contribution de Tafinlar et du tramétinib à l'efficacité de l'association chez les patients pédiatriques présentant un GBG n'est pas claire.
Patients exclus des études cliniques
Les critères d'exclusion communs à toutes les études sur Tafinlar étaient: BRAF de type sauvage ou une mutation non V600; antécédents ou indications concernant un risque cardiovasculaire; statut de performance plus faible; déficit connu en glucose-6-phosphate déshydrogénase (G6PD). Vous trouverez les critères d'exclusion spécifiques à l'étude dans la section «Efficacité clinique».
Interactions
Tafinlar en monothérapie
Effet de TAFINLAR sur d'autres médicaments
Dans des hépatocytes humains, le dabrafénib a entraîné une augmentation dose-dépendante de la concentration en ARNm des cytochromes CYP2B6 et du CYP3A4 allant jusqu'à 32 fois les valeurs des contrôles et s'est avéré in vivo un inducteur du CYP3A4 et du CYP2C9. Une étude clinique menée auprès de 12 participants qui recevaient une dose unique de midazolam, un substrat du cytochrome CYP3A4, et qui prenaient régulièrement du dabrafénib, a mis en évidence une diminution de l'ASC et la Cmax du midazolam de respectivement 74% et de 61% par rapport à leurs valeurs pour une dose unique de midazolam sans dabrafénib. Dans une autre étude réalisée auprès de 14 participants, l'ASC de la warfarine-S (substrat du CYP2C9) administrée en dose unique et l'ASC de la warfarine-R (substrat des CYP3A4/CYP1A2) administrée en dose unique ont été réduites de 37% et de 33% respectivement lors d'une administration répétée de dabrafénib, tandis que la Cmax était légèrement augmentée (de 18% et de 19% respectivement). Le dabrafénib peut également avoir des effets inducteurs sur d'autres enzymes, dont les CYP2B6, CYP2C8 et CYP2C19, l'UDP glucuronosyltransférase (UGT) et des transporteurs (p.ex. glycoprotéine, P-gp).
L'administration concomitante de Tafinlar et de médicaments qui sont influencés par l'induction de ces enzymes sous Tafinlar, p.ex. des contraceptifs hormonaux (voir «Grossesse, Allaitement»), des anticoagulants (p.ex. acénocoumarol, warfarine, rivaroxaban), des corticostéroïdes (p.ex. dexaméthasone), des antirétroviraux (p.ex. amprénavir, atazanavir, darunavir), des statines métabolisées par le cytochrome CYP3A4 (p.ex. atorvastatine, simvastatine) ou des immunosuppresseurs (p.ex. ciclosporine, tacrolimus, sirolimus) peut entraîner une diminution des concentrations et une perte d'efficacité. On envisagera, dans la mesure du possible, une substitution de ces médicaments. L'effet du dabrafénib sur l'exposition à la phenprocoumone et à l'acénocoumarol n'a pas été étudié à ce jour. Si une co-administration ne peut être évitée, les patients concernés doivent être surveillés dans l'éventualité d'une certaine perte d'efficacité.
In vitro, le dabrafénib est un inhibiteur du polypeptide de transport d'anions organiques (OATP) 1B1 (OATP1B1) et de l'OATP1B3 et un impact clinique de cette propriété ne peut pas être exclu. La prudence est par conséquent recommandée en cas de co-administration du dabrafénib avec des substrats de l'OATB1B1 ou de l'OATP1B3, p.ex. des statines.
Bien que le dabrafénib et ses métabolites, l'hydroxy-dabrafénib, le carboxy-dabrafénib et le déméthyle-dabrafénib soient des inhibiteurs in vitro du polypeptide humain de transport d'anions organiques (OATP) 1B1 (OATP1B1), de l'OATP1B3, du polypeptide de transport d'anions organiques (OAT) 1 et de l'OAT3, le risque d'une interaction médicamenteuse lors d'une exposition clinique est minime. Le dabrafénib et le déméthyl-dabrafénib se sont en outre révélés être des inhibiteurs modérés de la protéine de résistance du cancer du sein humain (BCRP); lors d'une exposition clinique, le risque d'interaction médicamenteuse est toutefois minime.
Ni le dabrafénib ni ses 3 principaux métabolites ne se sont révélés être des inhibiteurs de la Pgp in vitro. Des interactions avec de nombreux médicaments éliminés par métabolisation ou par un transporteur actif sont prévisibles. Si leur effet thérapeutique revêt une grande importance pour le patient et que des ajustements posologiques ne peuvent pas se faire simplement sur la base de la surveillance de l'efficacité ou des concentrations plasmatiques, ces médicaments ne doivent être utilisés qu'avec prudence. Il semblerait que le risque de lésions hépatiques après administration de paracétamol soit plus important chez les patients recevant un traitement concomitant par inducteurs enzymatiques.
Un grand nombre de médicaments concernés par des interactions potentielles est attendu, bien que l'importance des interactions puisse être variable. Les catégories de médicaments pouvant être concernées incluent (liste non exhaustive):
·analgésiques (p.ex. fentanyl, méthadone)
·antibiotiques (p.ex. clarithromycine, doxycycline)
·agents anti-cancéreux (p.ex. cabazitaxel)
·anticoagulants (p.ex. acénocoumarol, phenprocoumone, warfarine)
·antiépileptiques (p.ex. carbamazépine, phénytoïne, primidone, acide valproïque)
·antipsychotiques (p.ex. halopéridol)
·inhibiteurs des canaux calciques (p.ex. diltiazem, félodipine, nicardipine, nifédipine, vérapamil)
·glycosides cardiaques (p.ex. digoxine)
·corticoïdes (p.ex. dexaméthasone, méthylprednisolone)
·médicaments antiviraux contre le VIH (p.ex. amprénavir, atazanavir, darunavir, delavirdine, éfavirenz, fosamprénavir, indinavir, lopinavir, nelfinavir, saquinavir, tipranavir)
·contraceptifs hormonaux
·hypnotiques (p.ex. diazépam, midazolam, zolpidem)
·immunosuppresseurs (p.ex. ciclosporine, tacrolimus, sirolimus)
·statines métabolisées par le cytochrome CYP3A4 (p.ex. atorvastatine, simvastatine).
L'induction est susceptible de survenir après 3 jours d'administration répétée du dabrafénib. À l'arrêt du dabrafénib, l'induction disparaît graduellement, les concentrations des cytochromes sensibles CYP3A4, CYP2B6, CYP2C8, CYP2C9 et CYP2C19, de l'UDP-glucuronosyltransférase (UGT) et des substrats de transport peuvent augmenter; l'apparition de toute toxicité chez les patients doit aussi être surveillée et la dose de ces médicaments doit être ajustée en cas de nécessité. L'association avec des médicaments augmentant le pH gastrique tels que les inhibiteurs de la pompe à protons, des antagonistes des récepteurs H2 et des antiacides n'a pas été étudiée. L'association avec de tels médicaments risquant de compromettre l'absorption du dabrafénib, en raison de la dépendance du pH de la solubilité du dabrafénib, il convient donc de l'éviter.
Effet d'autres médicaments sur TAFINLAR
Des essais précliniques in vitro ont démontré que le dabrafénib est principalement métabolisé par les cytochromes CYP2C8 et CYP3A4, alors que l'hydroxy-dabrafénib et le déméthyl-dabrafénib sont des substrats du CYP3A4. Des données pharmacocinétiques ont montré une augmentation de 33% de la Cmax et de 71% de l'ASC (aire sous la courbe/AUC) du dabrafénib en doses répétées, co-administré avec le kétoconazole (inhibiteur du CYP3A4), ainsi qu'une augmentation de l'ASC des métabolites hydroxy-dabrafénib et déméthyl-dabrafénib (augmentations respectives de 82 et 68%). Une diminution de 16% de l'ASC a été observée pour le carboxy-dabrafénib. Lors d'une co-administration de gemfibrozil (inhibiteur du CYP2C8) dans cette étude, l'ASC du dabrafénib a augmenté de 47%, mais la Cmax n'a pas augmenté. Le gemfibrozil n'a pas eu d'effet cliniquement significatif sur l'exposition systémique aux métabolites du dabrafénib (≤13%).
Les puissants inhibiteurs ou inducteurs des cytochromes CYP2C8 ou CYP3A4 sont par conséquent susceptibles de respectivement augmenter ou diminuer les concentrations du dabrafénib. Dans la mesure du possible, des agents alternatifs doivent être envisagés pendant le traitement par le dabrafénib en cas de co-administration. Une attention particulière est requise lorsque des inhibiteurs puissants (par exemple: le kétoconazole, la néfazodone, la clarithromycine, le ritonavir, le saquinavir, la télithromycine, l'itraconazole, le voriconazole, le posaconazole, l'atazanavir) ou des inducteurs puissants (tel que la rifampicine, la phénytoïne, la carbamazépine, le phénobarbital ou le millepertuis (Hypericum perforatum)) des cytochromes CYP2C8 ou CYP3A4 sont co-administrés avec le dabrafénib.
Le dabrafénib est un substrat de la glycoprotéine P humaine (Pgp) et de la protéine BCRP 1 murine in vitro. Toutefois, ces protéines de transport ont un impact minime sur la biodisponibilité orale et l'élimination du dabrafénib; c'est pourquoi le risque d'interactions médicamenteuses cliniquement significatives avec les inhibiteurs de la Pgp ou de la BCRP est faible.
Effets de médicaments modifiant le pH intragastrique
Les médicaments qui influencent le pH intragastrique (p.ex. inhibiteurs de la pompe à protons, antihistaminiques H2, antiacides) peuvent modifier la solubilité du dabrafénib et réduire ainsi sa biodisponibilité. Cependant, aucune étude clinique spécifique correspondante n'a été effectuée. L'influence sur l'efficacité de Tafinlar est inconnue.
Tafinlar en association avec le tramétinib
L'administration concomitante de doses répétées de dabrafénib 150 mg deux fois par jour et de tramétinib 2 mg une fois par jour n'a pas provoqué de modifications cliniquement significatives des Cmax et des ASC du dabrafénib ou du tramétinib, les élévations de la Cmax et de l'ASC du dabrafénib ayant été respectivement de 16% et 23%. À l'aide d'une analyse pharmacocinétique de population, une diminution minime de la biodisponibilité du tramétinib, correspondant à une réduction de 12% de l'ASC, a été estimée pour l'administration concomitante du dabrafénib et du tramétinib.
Grossesse, allaitementGrossesse
Les patients, hommes et femmes, doivent utiliser des méthodes de contraception efficaces:
Femmes: les femmes en âge de procréer doivent utiliser des méthodes contraceptives efficaces (méthodes conduisant à moins de 1% de grossesses) pendant le traitement et au cours des deux premières semaines qui suivent la fin du traitement par Tafinlar et pendant au moins 16 semaines après la dernière prise du tramétinib.
Si Tafinlar seul ou en association avec le tramétinib est administré pendant la grossesse ou dans le cas où une grossesse surviendrait pendant le traitement par le dabrafénib, la patiente doit être informée du risque potentiel pour le fœtus.
Les femmes en âge de procréer qui reçoivent Tafinlar en association avec le tramétinib doivent être informées que Tafinlar peut diminuer l'efficacité des contraceptifs hormonaux et qu'une autre méthode de contraception doit être utilisée (voir «Interactions»).
Hommes: les patients de sexe masculin (également ceux chez lesquels une vasectomie a été effectuée) ayant des partenaires sexuelles qui sont enceintes, qui le sont éventuellement ou qui pourraient tomber enceintes, doivent utiliser des préservatifs lors des rapports sexuels pendant la monothérapie avec Tafinlar, ainsi que pendant au moins 2 semaines après l'arrêt du traitement par Tafinlar. Lorsque les patients de sexe masculin reçoivent un traitement combiné par Tafinlar et le tramétinib, ils doivent utiliser des préservatifs lors des rapports sexuels pendant la durée du traitement, ainsi que pendant au moins 16 semaines après l'arrêt du traitement par Tafinlar.
Allaitement
Il n'existe aucune donnée concernant l'effet de Tafinlar sur l'enfant allaité ou sur la production de lait. Le passage du dabrafénib dans le lait maternel n'est pas connu. De nombreux médicaments étant excrétés dans le lait maternel, un risque pour l'enfant allaité ne peut être exclu. Tafinlar seul ou en association avec le tramétinib ne doit pas être utilisé chez la femme qui allaite. La décision d'interrompre soit l'allaitement soit le traitement par Tafinlar seul ou en association avec le tramétinib doit prendre en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Aucune donnée concernant la fertilité n'est disponible chez l'être humain pour le dabrafénib en monothérapie ou en association avec le tramétinib. Chez l'animal, des effets indésirables sur les organes reproducteurs mâles et femelles ont été observés (voir «Données précliniques»). Les patients de sexe masculin doivent être informés du risque potentiel d'une atteinte de la spermatogénèse potentiellement irréversible.
Effet sur l’aptitude à la conduite et l’utilisation de machinesAucune étude n'a été effectuée sur les effets de Tafinlar sur l'aptitude à la conduite ou l'utilisation de machines. Au vu de la pharmacologie de Tafinlar, il ne devrait pas y avoir d'effet défavorable sur ce type d'activités. L'état clinique du patient et le profil d'effets secondaires de Tafinlar doivent être pris en compte lors de l'évaluation de la capacité du patient à effectuer des tâches qui font appel au discernement ou à des aptitudes motrices ou cognitives.
Effets indésirablesMélanome non résécable ou métastatique
Tafinlar en monothérapie
Les effets indésirables médicamenteux (EIM) décrits ci-dessous prennent en compte différentes sources d'informations de sécurité, y compris celles des études cliniques, des rapports suivant l'introduction sur le marché et des rapports bibliographiques.
La fréquence des EIM décrits ci-dessous a été établie à partir des données provenant de cinq études cliniques menées en monothérapie chez en tout 578 patients atteints de mélanome. Environ 30% des patients ont reçu un traitement par Tafinlar de plus de 6 mois. Les effets indésirables médicamenteux (EIM) les plus fréquemment observés et rapportés sous traitement avec Tafinlar (≥15%) étaient: hyperkératose, céphalées, pyrexie, myalgie, arthralgie, abattement, nausées, diarrhée, papillome, alopécie, éruptions cutanées et vomissements.
Les EI rapportés chez des patients atteints de mélanome sont présentés ci-dessous, selon la terminologie MedDRA, par classes de systèmes d'organes et par fréquence. Fréquence: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100), rares (≥1/10 000 à < 1/1000); très rares (< 1/10 000), y compris cas isolés.
Infections et infestations
Fréquents: Rhinopharyngite.
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Très fréquents: Papillomea (24%), carcinome épidermoïde cutanéb CEC (11%).
Fréquents: Kératose séborrhéique, acrochordons (verrues pédiculées).
Occasionnels: Nouveau mélanome primaire.
Affection du système immunitaire
Occasionnels: Hypersensibilité.
Fréquence inconnue: Lymphohistiocytose hémophagocytaire.
Troubles du métabolisme et de la nutrition
Très fréquents: Diminution de l'appétit (14%).
Fréquents: Hypophosphatémie, hyperglycémie.
Affection du système nerveux
Très fréquents: Céphalées (30%).
Affections oculaires
Occasionnels: Uvéite.
Affections respiratoires, thoraciques et médiastinales
Très fréquents: Toux (13%).
Affections gastro-intestinales
Très fréquents: Nausées (25%), vomissements (18%), diarrhée (16%), constipation (10%).
Occasionnels: Pancréatite.
Affections de la peau et du tissu sous-cutané
Très fréquents: Hyperkératose (32%), alopécie (23%), éruption cutanée (20%), syndromed'érythrodysesthésie palmo-plantaire (14%).
Fréquents: Sécheresse cutanée, kératose actinique, lésions cutanées, érythème, prurit, réactions de photosensibilité.
Occasionnels: Panniculite.
Affections musculo-squelettiques et du tissu conjonctif
Très fréquents: Arthralgie (29%), myalgie (15%), douleurs des extrémités (16%).
Affections du rein et des voies urinaires
Fréquents: Insuffisance rénale, insuffisance rénale aiguë.
Occasionnels: Néphrite tubulo-interstitielle.
Troubles généraux et anomalies au site d'administration
Très fréquents: Pyrexie (30%), abattement (26%), frisson (13%), asthénie (11%).
Fréquents: Troubles pseudo-grippaux.
Investigations
Fréquents: Baisse de la FEVG, allongement de l'intervalle QT.
Désignations génériques
a Papillome, papillome cutané
b Carcinome épidermoïde cutané: CEC, CEC de la peau, CEC in situ (maladie de Bowen) et kératoacanthome
c Des cas de réactions de photosensibilité ont également été observés après la mise sur le marché. Tous les cas qui ont été rapportés dans les études cliniques ont été de grade 1 et n'ont pas nécessité d'ajustement posologique.
Tafinlar en association avec le tramétinib
Les effets indésirables médicamenteux (EIM) décrits ci-dessous prennent en compte différentes sources d'informations de sécurité, y compris celles des études cliniques, des rapports suivant l'introduction sur le marché et des rapports bibliographiques.
La fréquence des EIM décrits ci-dessous a été établie sur la base de la population intégrée soumise à l'évaluation de l'innocuité comportant 1087 patients avec un mélanome de stade III inopérable ou métastasique portant la mutation V600 du gène BRAF, un mélanome portant la mutation V600 du gène BRAF après résection complète avec traitement adjuvant et un NSCLC avancé.
Tous les patients ont été traités par 2 mg de tramétinib une fois par jour et 150 mg de Tafinlar deux fois par jour. Parmi ces patients, 559 ont été traités dans deux études de phase III randomisées, MEK115306 (COMBI-d) et MEK116513 (COMBIv)a, avec l'association pour le mélanome portant la mutation V600 du gène BRAF. 435 ont été traités dans une étude de phase III randomisée BRF115532 (COMBI-AD) avec l'association dans le traitement adjuvant du mélanome de stade III portant la mutation V600 du gène BRAF après résection complète et 93 ont été traités avec l'association pour le NSCLC portant la mutation V600 du gène BRAF dans une étude de cohortes multiples de phase II non randomisée BRF113928.
Les effets indésirables les plus fréquents (incidence > 20%) pour Tafinlar en association avec le tramétinib étaient: pyrexie, fatigue, nausées, frissons, céphalées, diarrhée, vomissements, arthralgie et éruption cutanée.
Fréquence: très fréquents (≥1/10); fréquents (≥1/100 à < 1/10); occasionnels (≥1/1000 à < 1/100); rares (≥1/10 000 à < 1/1000); très rares (< 1/10 000), y compris cas isolés.
Infections et infestations
Très fréquents: Rhinopharyngite (11%).
Fréquents: Infection urinaire, cellulite, folliculite, paronychie, éruption cutanée pustuleuse.
Tumeurs bénignes, malignes et non précisées (incl. kystes et polypes)
Fréquents: Carcinome épidermoïde cutané (squamous cell carcinoma, SCC)b, papillomec, kératose séborrhéique.
Occasionnels: Nouveau mélanome primaired, acrochordons (fibromes).
Troubles hématologiques et du système lymphatique
Fréquents: Neutropénie, anémie, thrombocytopénie, leucopénie.
Troubles du système immunitaire
Occasionnels: Hypersensibilitée, sarcoïdose
Troubles du métabolisme et de la nutrition
Très fréquents: Perte d'appétit (14%).
Fréquents: Déshydratation, hyponatrémie, hypophosphatémie, hyperglycémie.
Troubles du système nerveux
Très fréquents: Céphalées (33%), sensation vertigineuse (11%).
Maladies oculaires
Fréquents: Vue floue, défauts visuels, uvéite.
Occasionnels: Choriorétinopathie, décollement de l'épithélium pigmentaire de la rétine/décollement de la rétine (RPED), œdème périorbitaire.
Affections cardiaques
Fréquents: Diminution de la fraction d'éjection.
Occasionnels: Bradycardie.
Rares: Myocardite*.
Troubles vasculaires fonctionnels
Très fréquents: Hypertension (19%), hémorragie (19%)f.
Fréquents: Hypotension, lymphœdème.
Affections respiratoires, thoraciques et médiastinales
Très fréquents: Toux (20%).
Fréquents: Dyspnée.
Occasionnels: Pneumonie.
Affections gastro-intestinales
Très fréquents: Nausées (38%), diarrhée (32%), vomissements (29%),douleurs abdominales (17%)g, constipation (13%).
Fréquents: Sécheresse buccale, stomatite.
Occasionnels: Pancréatite, colite.
Rares: Perforation gastro-intestinale.
Troubles hépatobiliaires fonctionnels
Très fréquents: Élévation de l'alanine aminotransférase (14%), élévation de l'aspartate aminotransférase (13%).
Fréquents: Élévation de la phosphatase alcaline dans le sang, élévation des concentrations de gamma-glutamyltransférase.
Troubles fonctionnels de la peau et du tissu sous-cutané
Très fréquents: Éruption (24%), sécheresse cutanée (13%), prurit (10%), érythèmeh (10%).
Fréquents: Dermatite acnéiforme, kératose actinique, sueurs nocturnes, hyperkératose, alopécie, syndrome d'érythrodysesthésie palmo-plantaire, lésion cutanée, hyperhidrose, crevasses cutanées, panniculite, photosensibilité.
Très rares: Syndrome de Steven-Johnson, réaction médicamenteuse avec éosinophilie et symptômes systémiques (DRESS), dermatite exfoliative généralisée..
Troubles fonctionnels de l'appareil locomoteur, du tissu conjonctif et des os
Très fréquents: Arthralgie (26%), myalgie (15%), douleurs des extrémités (12%), crampes musculairesi (10%).
Fréquents: Élévation de la créatine phosphokinase dans le sang.
Affections du rein
Fréquents: Insuffisance rénale.
Occasionnels: Néphrite.
Troubles généraux
Très fréquents: Pyrexie (58%), fatigue (38%), frissons (33%), œdème périphérique (16%), asthénie (15%), infection grippale (11%).
Fréquents: Inflammation des muqueuses, œdème facial.
a Le profil de sécurité de MEK116513 est dans l'ensemble similaire à celui de MEK115306, avec les exceptions suivantes: 1) Les effets indésirables suivants ont une catégorie de fréquence plus élevée comparé à MEK115306: crampes musculaires (très fréquent); défaillance rénale et lymphœdème (fréquent); défaillance rénale aiguë (occasionnel); 2) Les effets indésirables suivants sont survenus dans MEK116513, mais pas dans MEK115306: insuffisance cardiaque, dysfonctionnement du ventricule gauche, pneumopathie interstitielle (occasionnel). 3) L'effet indésirable suivant est survenu dans MEK116513 et BRF115532, mais pas dans MEK115306 et BRF113928: rhabdomyolyse (occasionnel)
b Inclut SCC cutanés, SCC in situ (maladie de Bowen) et kératoacanthome
c Inclut papillomes, papillomes cutanés
d Inclut mélanome malin; mélanome métastasé, malin, et mélanome à extension superficielle de stade III
e Inclut hypersensibilité aux médicaments
f Hémorragies à différents endroits, y compris hémorragies intracrâniennes et hémorragies mortelles
g Inclut douleurs abdominales, dans l'abdomen supérieur et inférieur
h Inclut érythème, érythème généralisé
i Inclut crampes musculaires et raideur musculo-squelettique
*Fréquence basée sur le rapport après l'introduction sur le marché
Description d'effets indésirables spécifiques et informations complémentaires
Pyrexie
Des cas de fièvre ont été rapportés dans des études cliniques. Chez 1% des patients inclus dans les études cliniques, des événements fébriles sévères non infectieux ont été identifiés comme de la fièvre accompagnée d'une rigidité sévère, d'une déshydratation, d'une hypotension et/ou d'une insuffisance rénale aiguë d'origine pré-rénale chez des sujets dont la fonction rénale à l'inclusion était normale. Ces événements fébriles graves non infectieux sont généralement survenus au cours du premier mois de traitement. Les patients ayant présenté des épisodes fébriles graves non infectieux ont bien répondu à une interruption du traitement et/ou une réduction de la dose avec prise en charge symptomatique.
Carcinome épidermoïde cutané
Des cas de CEC (incluant ceux classés comme appartenant au kératoacanthome ou au sous-type, le kératoacanthome mixte) sont survenus chez 9% des patients sous traitement par Tafinlar. Environ 70% de ces événements sont apparus au cours des 12 premières semaines de traitement, avec un temps d'apparition médian de 8 semaines. 96% des patients ayant développé des CEC ont poursuivi le traitement sans modification de la dose.
Nouveaux mélanomes primaires
Des cas de nouveaux mélanomes primaires ont été rapportés dans des études cliniques avec Tafinlar. Ces cas ont été traités par exérèse, un ajustement de la dose de dabrafénib n'a pas été nécessaire.
Tumeur maligne non cutanée
L'activation de la voie de signalisation des MAP-kinases dans des cellules à gène BRAF de type sauvage exposées à des inhibiteurs de BRAF peut entraîner une augmentation du risque de tumeurs malignes non cutanées, dont certaines avec mutation RAS. Des cas de tumeurs malignes induites par une mutation RAS ont été rapportés dans des études cliniques sous Tafinlar lorsque celui-ci était administré en association avec le tramétinib (inhibiteur de MEK). Les patients doivent bénéficier d'une surveillance clinique appropriée.
Diminution de la FEVG
Une diminution de la FEVG, asymptomatique et réversible dans la plupart des cas, a été rapportée chez 1% des patients. Les patients dont la FEVG était inférieure à la limite basse de la normale de l'institution respective n'ont pas été inclus dans les essais cliniques réalisés avec le dabrafénib.
Arthralgies
Des cas d'arthralgies principalement de sévérité de grade 1 ou 2 ont été rapportés très fréquemment (25%) dans les études cliniques réalisées avec Tafinlar, les arthralgies de sévérité de grade 3 survenant peu fréquemment (< 1%); aucun cas de grade 4 n'a été rapporté.
Hypophosphatémie
Dans les études cliniques avec Tafinlar, des cas d'hypophosphatémie (7%) ont été fréquemment rapportés. Pour environ la moitié de ces cas (4%), la sévérité était de grade 3.
Pancréatite
Des cas de pancréatite ont été rapportés chez des patients traités par Tafinlar. Les douleurs abdominales inexpliquées doivent faire l'objet d'investigations immédiates, en incluant le dosage de l'amylase et de la lipase sériques. Lors de la reprise du traitement par Tafinlar, ces patients doivent être étroitement surveillés.
Insuffisance rénale
Les insuffisances rénales consécutives à une azotémie prérénale associée à une pyrexie ou à une néphrite granulomateuse étaient rares, mais Tafinlar n'a pas été étudié chez les patients atteints d'insuffisance rénale (définie par une créatinine > 1,5× la limite supérieure de la normale [LSN]). La prudence est de rigueur chez cette population de patients.
Populations particulières de patients
Patients pédiatriques
Tafinlar en association avec le tramétinib
La sécurité de Tafinlar en association avec le tramétinib a été évaluée dans deux études (étude DRB436G2201; n = 123 et étude TMT212 X2101; n = 48) auprès de 171 patients pédiatriques dont 159 présentant un gliome porteur de la mutation V600E du gène BRAF.
Le profil de sécurité général dans la population pédiatrique était comparable à celui observé chez les adultes. Les effets indésirables médicamenteux signalés le plus fréquemment (≥20%) ont été de la fièvre (pyrexie) (65%), une éruption cutanée (47%), des céphalées (39%), des vomissements (38%), une sécheresse cutanée (34%), une fatigue (33%), des diarrhées (30%), des hémorragies (29%), une neutropénie (25%), des nausées (25%), une dermatite acnéiforme (25%), des douleurs abdominales (23%) et une toux (21%).
Dans le groupe pédiatrique faisant l'objet d'une évaluation de la sécurité, un effet indésirable médicamenteux sous la forme d'un gain de poids a été constaté avec une fréquence de 15,2% (très fréquent). Sur 171 patients, 51 (29,8%) ont présenté une augmentation de l'IMC de ≥2 catégories de percentile d'IMC par rapport à la référence.
Dans l'étude DRB436G2201, une infection par le virus de la COVID-19, dont un cas de sévérité de grade ≥3, a été rapportée chez 20,5% (15 sur 73) des patients pédiatriques ayant été traités par Tafinlar en association avec le tramétinib.
Dans le groupe pédiatrique faisant l'objet d'une évaluation de la sécurité, une diminution de la fraction d'éjection du ventricule gauche (FEVG) de 10% ou plus par rapport à la valeur initiale et en-dessous de la limite inférieure institutionnelle de la normale (LIN) a été observée chez 8,7% (14 sur 161) des patients pédiatriques sous traitement par l'association de Tafinlar et du tramétinib.
D'autres effets indésirables médicamenteux qui sont survenus plus fréquemment chez les patients pédiatriques que chez les patients adultes ont été une neutropénie, une dermatite acnéiforme, une paronychie, une anémie, une leucopénie (très fréquents); une bradycardie, une dermatite exfoliative généralisée, une hypersensibilité et une pancréatite (fréquents). Par ailleurs, dans la cohorte GBG de l'étude G2201, l'incidence relative des augmentations de lymphocytes, des augmentations de magnésium et d'une tension artérielle systolique basse était plus élevée dans le cas d'un traitement ciblé que dans le cas d'une chimiothérapie.
Tableau 5: Effets indésirables médicamenteux de grade 3/4 les plus fréquents (≥2%) dans le cas de l'association de Tafinlar et du tramétinib chez les patients pédiatriques
|
Effets indésirables médicamenteux
|
Tafinlar en association avec le tramétinib
N = 171
| |
Grade 3/4
n (%)
| |
Neutropénie1
|
25 (15)
| |
Fièvre (pyrexie)
|
15 (9)
| |
Alanine aminotransférase augmentée2
|
10 (6)
| |
Aspartate aminotransférase augmentée3
|
6 (4)
| |
Gain de poids (poids augmenté)
|
7 (4)
| |
Céphalées
|
5 (3)
| |
Vomissements
|
5 (3)
| |
Hypotension
|
4 (2)
| |
Éruption cutanée4
|
4 (2)
| |
Phosphatase alcaline sanguine augmentée
|
4 (2)
| |
1. Neutropénie inclut neutropénie, nombre de neutrophiles diminué et neutropénie fébrile.
2. ALAT inclut alanine aminotransférase augmentée et transaminases augmentées.
3. ASAT inclut aspartate aminotransférase augmentée et transaminases augmentées.
4. Éruption inclut éruption cutanée, éruption maculo-papuleuse, éruption pustuleuse, éruption érythémateuse, éruption papuleuse et éruption maculaire.
|
Sous traitement par l'association de Tafinlar et du tramétinib, un plus grand nombre d'événements graves ont été signalés chez les patients pédiatriques âgés de moins de 6 ans que chez les patients âgés de 6 à 12 ans. L'apparition de fièvre a été plus fréquente chez les enfants âgés de moins de 6 ans que chez les patients pédiatriques plus âgés.
Le groupe pédiatrique faisant l'objet d'une évaluation de la sécurité ne comportant que quatre enfants âgés de 1 à 2 ans, le profil de sécurité dans cette catégorie d'âge est caractérisé de manière incomplète.
L'annonce d'effets secondaires présumés après l'autorisation est d'une grande importance. Elle permet un suivi continu du rapport bénéfice-risque du médicament. Les professionnels de santé sont tenus de déclarer toute suspicion d'effet secondaire nouveau ou grave via le portail d'annonce en ligne ElViS (Electronic Vigilance System). Vous trouverez des informations à ce sujet sur www.swissmedic.ch.
SurdosageLes expériences relatives à un surdosage de Tafinlar sont actuellement très limitées. La dose maximale de dabrafénib administrée pendant les études cliniques était de 600 mg (300 mg deux fois par jour).
Traitement
Aucun traitement spécifique en cas de surdosage de Tafinlar n'est disponible. En cas de surdosage, le patient doit recevoir un traitement symptomatique approprié et une surveillance adéquate. La suite de la prise en charge du patient se fera en fonction de l'indication clinique ou le cas échéant avec l'aide du centre national d'information toxicologique.
Propriétés/EffetsCode ATC
L01EC02
Mécanisme d'action
Tafinlar en monothérapie
Le dabrafénib est un puissant inhibiteur sélectif, compétitif de l'ATP, des kinases RAF, avec des valeurs de CI50 de 0,65, 0,5 et 1,84 nM pour les enzymes BRAF V600E, BRAF V600K et BRAF V600D. Des mutations oncogènes de BRAF entraînent une activation constitutive de la cascade de transduction du signal RAS/RAF/MEK/ERK, stimulant la croissance de cellules tumorales. Des mutations BRAF sont observées très fréquemment dans certaines sortes de cancers, notamment dans environ 50% de tous les mélanomes. La mutation BRAF V600E, la plus fréquemment observée, représente environ 90% des mutations BRAF observées chez des patients atteints de mélanome. On trouve en outre une série de substitutions plus rares, p.ex. les mutations V600K, V600D, V600G, V600M et V600R.
De surcroît, le dabrafénib inhibe les enzymes BRAF et CRAF de type sauvage avec des valeurs de CI50 de 3,2 et 5,0 nM. Le dabrafénib inhibe in vitro et in vivo la croissance cellulaire de mélanome positif à la mutation BRAF V600.
Dans des lignées cellulaires de mélanome exprimant la mutation BRAF V600, la suppression par le dabrafénib d'un biomarqueur pharmacodynamique en aval (ERK phosphorylée) a été démontrée in vitro et dans des modèles animaux.
Chez des participants atteints d'un mélanome positif à la mutation BRAF V600, l'administration de dabrafénib a entraîné l'inhibition de la phosphorylation d'ERK dans les cellules tumorales par rapport à la valeur mesurée avant le traitement.
Tafinlar en association avec le tramétinib - mélanome métastatique et NSCLC
Le tramétinib est un inhibiteur allostérique, réversible et hautement sélectif de l'activation et de l'activité des kinases MEK 1 et MEK 2 (MEK = kinases régulées par des signaux extracellulaires, activées par un mitogène). Les protéines MEK sont des composants critiques de la voie des kinases régulées par des signaux extracellulaires (extracellular signal-regulated kinase, ERK).
Le tramétinib et le dabrafénib inhibent les deux kinases MEK et BRAF au sein de cette chaîne de transduction de signal; la combinaison des deux principes actifs conduit à une inhibition efficace, double de la chaîne de transduction de signal. L'association de tramétinib et de dabrafénib s'est révélée synergique pour les lignées cellulaires de mélanome et de NSCLC porteurs de la mutation BRAF V600 in vitro et retarde l'apparition de résistance in vivo des xénogreffes de mélanome porteur de la mutation BRAF V600.
Étude MEK111054
Allongement de l'intervalle QT
Un allongement de l'intervalle QT > 60 ms dans les cas les plus défavorables a été observé chez 3% des patients traités par Tafinlar (dont un cas > 500 ms dans la population totale pour l'analyse des données de sécurité).
Le potentiel du dabrafénib d'allongement de l'intervalle QT a été évalué dans le cadre d'une étude portant spécifiquement sur l'intervalle QT sous administration répétée. Une dose suprathérapeutique de 300 mg de dabrafénib a été administrée deux fois par jour aux 32 participants atteints de tumeurs porteuses de la mutation V600 du gène BRAF. Aucun effet cliniquement significatif du dabrafénib ou des métabolites du dabrafénib sur l'intervalle QTc n'a été observé.
Dans les cas défavorables, des allongements de l'intervalle QTc de plus de 60 millisecondes (ms) ont été observés chez 3% des patients traités par le dabrafénib (dans un cas > 500 ms dans la population intégrée pour l'analyse des données de sécurité). Dans l'étude de phase III MEK115306, le cas défavorable d'un allongement de l'intervalle QTcB > 500 ms ne s'est présenté chez aucun des patients traités par le tramétinib en association avec le dabrafénib; l'intervalle QTcB a été allongé de plus de 60 ms par rapport à la valeur initiale chez 1% (3/209) des patients. Dans l'étude de phase III MEK116513, quatre patients (1%) traités par le tramétinib en association avec le dabrafénib ont présenté un allongement du QTcB de grade 3 (> 500 ms). Deux de ces patients ont présenté un allongement du QTcB de grade 3 (> 500 ms) qui représentait simultanément une augmentation > 60 ms par rapport à la valeur initiale.
Pharmacodynamique
Efficacité clinique
Tafinlar en monothérapie
L'efficacité de Tafinlar dans le traitement de patients adultes atteints de mélanome non résécable ou métastatique exprimant la mutation BRAF V600 a été examinée dans 3 études (BRF113683 [BREAK-3], BRF113929 [BREAK-MB] et BRF113710 [BREAK-2]) chez des patients porteurs de la mutation BRAF-V600E et/ou BRAF V600K. L'étude pivot BREAK-3 a été exclusivement réalisée chez des patients porteurs de la mutation V600E. Dans ces études, 402 patients au total porteurs de la mutation BRAF V600E et 49 patients porteurs de la mutation BRAF V600K ont été inclus. Selon les résultats des études de phase II, en présence d'une mutation V600K, l'efficacité est plus faible que sur les tumeurs positives à la mutation V600E.
L'efficacité de Tafinlar chez des patients prétraités avec un inhibiteur de la protéine kinase n'a pas été étudiée.
Patients non traités préalablement (résultats de l'étude de phase III BREAK-3)
L'efficacité et la sécurité de Tafinlar ont été évaluées dans une étude de phase III randomisée, ouverte (BREAK 3) comparant Tafinlar à la dacarbazine (DTIC) chez des patients atteints de mélanome avancé (non résécable, stade III) ou métastatique (stade IV) positif à la mutation BRAF V600E et non traités préalablement.
L'objectif principal de l'étude consistait à évaluer l'efficacité de Tafinlar par rapport à celle de la DTIC en termes de survie sans progression (progression-free survival, PFS). Après la première confirmation radiographique indépendante d'une progression, les patients du bras DTIC ont pu recevoir Tafinlar. Les caractéristiques à l'inclusion ont été réparties de manière homogène entre les groupes de traitement. 60% des patients étaient de sexe masculin, 99,6% de type caucasien; l'âge médian était de 52 ans, 21% étant âgés de ≥65 ans; 98,4% avaient un indice ECOG de 0 ou 1 et 97% avaient un cancer métastatique.
L'analyse prédéfinie incluant les données recueillies jusqu'au 19 décembre 2011 montrait une amélioration significative du critère principal de PFS (HR = 0,30; IC à 95% 0,18, 0,51; p < 0,0001). Les résultats d'efficacité d'une analyse post hoc avec 6 mois supplémentaires de suivi sont résumés dans le tableau 5. La survie globale (OS) d'une autre analyse post-hoc reposant sur les données recueillies le 18 décembre 2012 est présentée dans le tableau 6 et dans la figure 1.
Tableau 6: Efficacité chez des patients non traités préalablement selon les médecins-investigateurs (étude BREAK-3, 25 juin 2012)
|
|
Population Intention-to-treat
| |
|
Tafinlar
N = 187
|
DTIC
N = 63
| |
Survie sans progression (selon les médecins-investigateurs)
| |
Médiane (mois) (IC à 95%)
|
6,9 (5,2; 9,0)
|
2,7 (1,5; 3,2)
| |
HR (IC à 95%)
|
0,37 (0,24; 0,58)
P < 0,0001
| |
Taux de réponse globala
| |
% (IC à 95%)b
|
59 (51,4; 66,0)
|
24 (21,4; 36,2)
| |
P < 0,0001
| |
Durée de la réponse
| |
|
N = 110
|
N = 15
| |
Médiane (mois) (IC à 95%)
|
8,0 (6,6; 11,5)
|
7,6 (5,0; 9,7)
| |
Abréviations: IC: intervalle de confiance; DTIC: dacarbazine; HR: Hazard Ratio
a. Défini comme réponse complète + partielle.
b. Réponse confirmée.
|
Au 25 juin 2012, date de collecte des données, 35 participants sur 63 (55,6%), randomisés pour recevoir de la DTIC, sont passés dans le groupe sous Tafinlar. La PFS médiane après le changement de groupe de traitement a été de 4,4 mois.
Tableau 7: Données de survie de l'analyse post hoc (18 décembre 2012).
|
Traitement
|
Nombre de cas de décès (%)
|
Taux de survie globale à 12 mois
|
Hazard Ratio
(IC à 95%)
| |
DTIC
|
28 (44%)
|
63%
|
0,76 (0,48, 1,21) (a)
| |
Tafinlar
|
78 (42%)
|
70%
| |
(a) Les patients n'étaient pas censurés au moment du changement de groupe de traitement
|
Figure 1: Courbe de Kaplan-Meier de la survie globale (BREAK-3) (18 décembre 2012)

Patients avec métastases cérébrales (résultats de l'étude de phase II BREAK-MB)
BREAK-MB était une étude multicentrique, ouverte de phase II, composée de deux cohortes, conçue pour évaluer la réponse intracrânienne à Tafinlar chez les patients de l'étude atteints de mélanome confirmé histologiquement (stade IV) avec métastases cérébrales et une mutation de BRAF (V600E ou V600K). Les participants à l'étude ont été inclus dans la cohorte A (sujets n'ayant pas reçu de traitement local préalable pour les métastases cérébrales) ou dans la cohorte B (sujets ayant reçu un traitement local préalable pour les métastases cérébrales).
Le critère primaire de cette étude était le taux de réponse intracrânienne global (Overall Intracranial Response Rate, OIRR), évalué par les médecins-investigateurs, dans la population présentant la mutation V600E. L'OIRR confirmé ainsi que d'autres résultats relatifs à l'efficacité sont présentés dans le tableau 8.
Tableau 8: Données sur l'efficacité chez des patients présentant des métastases cérébrales (étude BREAK-MB)
|
Population totale incluant tous les participants à l'étude traités
| |
|
BRAF V600E (principal)
|
BRAF V600K
| |
|
Cohorte A
N = 74
|
Cohorte B
N = 65
|
Cohorte A
N = 15
|
Cohorte B
N = 18
| |
Taux de réponse intracrânienne global, % (IC à 95%)a
| |
|
39%
(28,0, 51,2)
P < 0,001b
|
31%
(19,9, 43,4)
P < 0,001b
|
7%
(0,2, 31,9)
|
22%
(6,4, 47,6)
| |
Durée de la réponse intracrânienne, médiane, mois (IC à 95%)
| |
|
N = 29
4,6 (2,8, NA)
|
N = 20
6,5 (4,6, 6,5)
|
N = 1
2,9 (NA, NA)
|
N = 4
3,8 (NA, NA)
| |
Taux de réponse global, % (IC à 95%)a
| |
|
38%
(26,8, 49,9)
|
31%
(19,9, 43,4)
|
0
(0, 21,8)
|
28%
(9,7, 53,5)
| |
Durée de la réponse, médiane, mois (IC à 95%)
| |
|
N = 28
5,1 (3,7, NA)
|
N = 20
4,6 (4,6, 6,5)
|
S/O
|
N = 5
3,1 (2,8, NA)
| |
Survie sans progression, médiane, mois (IC à 95%)
| |
|
3,7 (3,6, 5,0)
|
3,8 (3,6, 5,5)
|
1,9 (0,7, 3,7)
|
3,6 (1,8, 5,2)
| |
Survie globale, médiane, mois (IC à 95%)
| |
Médiane, mois
|
7,6 (5,9, NA)
|
7,2 (5,9, NA)
|
3,7 (1,6, 5,2)
|
5,0 (3,5, NA)
| |
Abréviations: IC: Intervalle de confiance; NA: non atteint; s/o: sans objet
a – Réponse confirmée.
b – Cette étude a été conçue pour confirmer ou rejeter l'hypothèse nulle d'un OIRR ≤10% (sur la base de résultats antérieurs) en faveur de l'hypothèse alternative d'un OIRR ≥30% chez des sujets porteurs de la mutation BRAF-V600E
|
Patients non traités préalablement ou ayant connu un échec avec au moins un traitement systémique antérieur (résultats de la phase II [BREAK-2])
L'étude BRF113710 (BREAK-2) était une étude multicentrique à un seul bras, portant sur 92 sujets atteints d'un mélanome métastatique (stade IV) avec une mutation BRAF-V600E ou V600K confirmée.
Le taux de réponse confirmé évalué par les médecins-investigateurs chez des patients présentant un mélanome métastatique et porteurs de la mutation BRAF V600E (n = 76) était de 59% (IC à 95%: 48,2; 70,3) et la durée médiane de la réponse était de 5,2 mois (IC à 95%: 3,9; non évaluable), sur la base d'un temps de suivi d'observation médian de 6,5 mois depuis le début du traitement. Chez les patients présentant un mélanome métastatique et porteurs de la mutation BRAF V600K (n = 16), le taux de réponse était de 13% (2 patients sur 16) (IC à 95%: 0,0; 28,7), avec une durée médiane de réponse de 5,3 mois (IC à 95%: 3,7; 6,8).
Tafinlar en association avec le tramétinib
L'efficacité et la sécurité de la dose de Tafinlar recommandée (150 mg deux fois par jour) en association avec le tramétinib (2 mg une fois par jour) pour le traitement des patients adultes atteints d'un mélanome non résécable ou métastatique porteur de la mutation BRAF-V600 ont été évaluées dans deux études pivots de phase III.
Dans une étude précoce, l'association de Tafinlar 150 mg deux fois par jour et de tramétinib 2 mg une fois par jour a montré une activité clinique limitée chez un petit nombre (26) de patients qui avaient présenté une progression de la maladie après un traitement par un inhibiteur de BRAF. Le taux de réponse confirmé, évalué par l'investigateur, était de 15% (IC à 95%: 4,4, 34,9) et la PFS moyenne de 3,6 mois (IC à 95%: 1,9, 5,2). Les résultats étaient similaires chez les 45 patients qui étaient passés de Tafinlar en monothérapie à l'association de tramétinib 2 mg une fois par jour et de Tafinlar 150 mg deux fois par jour dans la partie C de l'étude. Chez ces patients, un taux de réponse confirmé de 13% (IC à 95%: 5,0, 27,0) a été observé, avec une PFS moyenne de 3,6 mois (IC à 95%: 2,4). Ce taux de réponse est nettement plus faible que chez les patients qui n'avaient présenté aucune progression de la maladie sous traitement préalable par un inhibiteur de BRAF, de sorte que l'administration du tramétinib en association avec Tafinlar ne doit être envisagée chez ces patients qu'après avoir considéré d'autres options thérapeutiques.
MEK115306 (COMBI-d)
MEK115306 (COMBI-d) était une étude randomisée, en double aveugle de phase III, destinée à comparer l'association de Tafinlar et de tramétinib avec Tafinlar et un placebo en première ligne de traitement chez des patients atteints d'un mélanome cutané non résécable (stade IIIC) ou métastatique (stade IV) porteur de la mutation BRAF-V600E/K. Le critère d'évaluation principal de l'étude était la survie sans progression (PFS) évaluée par l'investigateur. Les critères d'évaluation secondaires étaient la survie globale (overall survival, OS) médiane, le taux de réponse global (overall response rate, ORR) et la durée de la réponse (duration of response, DOR). Les participants à l'étude ont été stratifiés en fonction de l'activité de la lactate déshydrogénase (LDH) (> limite supérieure de la normale (LSN) versus ≤ LSN) et de la mutation BRAF (V600E versus V600K).
Au total, 423 participants à l'étude ont été randomisés selon un ratio 1:1 et répartis soit dans le bras traité par l'association (Tafinlar 150 mg deux fois par jour et tramétinib 2 mg une fois par jour) (N = 211) soit dans le bras traité par Tafinlar seul (150 mg deux fois par jour) (N = 212). Les caractéristiques à l'inclusion dans l'étude étaient équilibrées entre les deux groupes de traitement. Chez la plupart des patients, une mutation BRAF-V600E était présente (85%); une mutation BRAF-V600K était présente chez les 15% restants des patients.
Au moment de l'analyse primaire de la PFS, la PFS médiane était de 9,3 mois avec l'association de Tafinlar et de tramétinib et de 8,8 mois avec Tafinlar seul (HR = 0,75, IC à 95%: 0,57, 0,99, p = 0,035). L'ORR était de 67% vs 51% (p = 0,0014) et la DOR s'élevait à 9,2 vs 10,2 mois avec l'association par rapport à Tafinlar seul. Dans une analyse ultérieure qui a coïncidé avec l'analyse principale de l'OS (voir ci-dessous), la différence de PFS en faveur de l'association de tramétinib et de Tafinlar a été plus nette que lors de la première analyse principale, avec une PFS de 11,0 mois avec l'association (IC à 95%: 8,0, 13,9) et encore de 8,8 mois (IC à 95%: 5,9, 9,3) avec Tafinlar seul (HR = 0,67, IC à 95%: 0,53, 0,84, p < 0,001). Dans cette analyse, l'ORR était de 69% vs 53% (p = 0,0014) et la DOR de 12,9 vs 10,6 mois avec l'association par rapport au traitement par Tafinlar seul.
Au moment de l'analyse principale de l'OS, 222 décès (52,5%) ont été rapportés dans la population randomisée (ou ITT) [association: 99 décès (47%) et Tafinlar seul: 123 décès (58%)]. La durée médiane de suivi pour le traitement de l'étude était de 20 mois dans le bras traité par l'association et de 16 mois dans le bras traité par Tafinlar seul. L'étude MEK115306 a montré une diminution statistiquement significative du risque de décès de 29% dans le bras traité par l'association par rapport au bras traité par Tafinlar seul (HR = 0,71, IC à 95%: 0,55, 0,92; p = 0,011). L'OS médiane était de 25,1 mois dans le bras traité par l'association et de 18,7 mois dans le bras traité par Tafinlar seul. Les valeurs de l'OS déterminées à 12 mois (74%) et à 24 mois (51,4%) étaient en outre plus élevées dans le bras traité par l'association que dans le bras traité par Tafinlar seul (respectivement 67,6 et 42,1%).
Une analyse de la survie globale (OS) après 5 ans a révélé que la survie globale (OS) médiane avec le traitement combiné était environ 7 mois plus longue que la survie globale (OS) médiane avec le traitement par Tafinlar seul (25,8 mois (IC à 95%: 19,2; 38,2) contre 18,7 mois (IC à 95%: 15,2; 23,1)) avec un hazard ratio de 0,80 (IC à 95%: 0,63; 1,01). Le taux de survie globale à 5 ans était de 32% (IC à 95%: 25,1; 38,3) avec le traitement combiné contre 27% (IC à 95%: 20,7; 33,0) avec le traitement par Tafinlar seul. Après 5 ans, la survie sans progression (PFS) médiane pour le traitement combiné était de 10,2 mois (IC à 95%: 8,1; 12,8) contre 8,8 mois (IC à 95%: 5,9; 9,3) pour le traitement par Tafinlar seul avec un hazard ratio de 0,73 (IC à 95%: 0,59; 0,91).
MEK116513 (COMBI-v)
L'étude MEK116513 était une étude randomisée, ouverte, à 2 bras, de phase III destinée à comparer l'association de Tafinlar et de tramétinib avec le vémurafénib en monothérapie dans le mélanome métastatique porteur de la mutation BRAF-V600. Le critère d'évaluation principal de l'étude était la survie globale (OS). Les participants à l'étude ont été stratifiés en fonction de l'activité de la lactate déshydrogénase (LDH) (> limite supérieure de la normale (LSN) versus ≤ LSN) et de la mutation BRAF (V600E versus V600K).
Au total, 704 participants à l'étude ont été randomisés selon un ratio 1:1 et répartis soit dans le bras traité par l'association (Tafinlar 150 mg deux fois par jour et tramétinib 2 mg une fois par jour) soit dans le bras traité par le vémurafénib en monothérapie (960 mg deux fois par jour). La majorité des participants à l'étude présentaient une mutation BRAF-V600E (89%). 10% des patients avaient une mutation BRAF-V600K et 1 patient (< 1%) présentait les deux mutations (BRAF-V600E/K).
L'analyse de l'OS a été réalisée lorsque 222 décès au total ont été constatés (77% des résultats nécessaires pour l'analyse finale). Le comité d'examen indépendant (Independent Data Monitoring Committee, IDMC) a recommandé d'interrompre l'étude, car les résultats de l'OS avaient dépassé la limite d'efficacité statistique préalablement établie. Par la suite, l'analyse intermédiaire de l'OS a été considérée comme l'analyse comparative finale de l'OS.
L'analyse de l'OS pour l'étude MEK116513 était basée sur 222 décès (32%) [association: 100 décès (28%) et vémurafénib: 122 décès (35%)]. La durée médiane de suivi pour le traitement de l'étude était de 11 mois dans le bras traité par l'association et de 9 mois dans le bras traité par le vémurafénib. L'étude MEK116513 a montré une diminution statistiquement significative du risque de décès de 31% dans le bras traité par l'association par rapport au bras traité par le vémurafénib (HR = 0,69, IC à 95%: 0,53, 0,89; p = 0,005). L'OS médiane n'était pas encore atteinte pour le bras traité par l'association et était de 17,2 mois pour le bras traité par le vémurafénib seul.
La PFS médiane observée était de 11,4 mois pour l'association de Tafinlar et de tramétinib et de 7,3 mois pour le vémurafénib en monothérapie (HR = 0,56, IC à 95%: 0,46, 0,69, p < 0,001). L'ORR était à 64% vs 51% (p = 0,0005) et la DOR de 13,8 vs 7,5 mois pour l'association par rapport au vémurafénib seul.
Une analyse de la survie globale (OS) après 5 ans a révélé que la survie globale (OS) médiane avec le traitement combiné était environ 8 mois plus longue que la survie globale (OS) médiane avec le traitement par le vémurafénib seul (26,0 mois (IC à 95%: 22,1; 33,8) contre 17,8 mois (IC à 95%: 15,6; 20,7) avec un hazard ratio de 0,70 (IC à 95%: 0,58; 0,84). Le taux de survie globale à 5 ans était de 36% (IC à 95%: 30,5; 40,9) avec le traitement combiné contre 23% (IC à 95%: 18,1; 27,4) avec le traitement par le vémurafénib seul.
BRF117277/DRB436B2204 (COMBI-MB) – Patients ayant des métastases cérébrales d'un mélanome métastatique
L'efficacité et la sécurité de Tafinlar en association avec le tramétinib chez les patients présentant un mélanome BRAF-positif avec des métastases cérébrales ont été étudiées lors d'une étude de phase II non randomisée, ouverte et multicentrique (étude COMBI-MB). Au total, 125 patients ont été admis dans 4 cohortes:
·Cohorte A: patients présentant un mélanome avec la mutation V600E sur le gène BRAF et des métastases cérébrales asymptomatiques sans traitement ciblé local préalable des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
·Cohorte B: patients présentant un mélanome avec la mutation V600E sur le gène BRAF et des métastases cérébrales asymptomatiques avec traitement ciblé local préalable des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
·Cohorte C: patients présentant un mélanome avec la mutation V600D/K/R sur le gène BRAF et des métastases cérébrales asymptomatiques avec ou sans traitement ciblé local préalable des métastases cérébrales avec un statut de performance ECOG de 0 ou 1.
·Cohorte D: patients présentant un mélanome avec la mutation V600D/E/K/R sur le gène BRAF et des métastases cérébrales symptomatiques avec ou sans traitement ciblé local préalable des métastases cérébrales avec un statut de performance ECOG de 0, 1 ou 2.
Au total, 104 des 125 patients avaient une mutation V600E, 18 patients une mutation V600K et 3 patients seulement une mutation V600R. Aucun patient n'avait une mutation V600D.
Le critère d'évaluation principal de l'étude était la réponse intracrânienne dans la cohorte A, définie comme étant le pourcentage de patients avec une réponse intracrânienne confirmée évaluée par le médecin de l'étude au moyen des critères RECIST (Response Evaluation Criteria in Solid Tumors, critères d'évaluation de la réponse thérapeutique dans les tumeurs solides), version 1.1. Les résultats d'efficacité sont résumés dans le tableau 6. Les critères d'évaluation secondaires étaient la durée de la réponse intracrânienne, le taux de réponse global des manifestations tumorales intracrâniennes comme extracrâniennes, la PFS et l'OS. Les résultats d'efficacité sont regroupés dans le tableau 9.
Tableau 9: COMBI-MB – Résultats d'efficacité selon l'estimation du médecin-investigateur
|
|
Tous les patients traités
| |
Critères/Évaluation
|
Cohorte A
N = 76
|
Cohorte B
N = 16
|
Cohorte C
N = 16
|
Cohorte D
N = 17
| |
Taux de réponse intracrânienne, % (IC à 95%)
|
| |
|
59%
(47,3, 70,4)
|
56%
(29,9, 80,2)
|
44%
(19,8, 70,1)
|
59%
(32,9, 81,6)
| |
Durée de la réponse intracrânienne, valeur médiane, mois (IC à 95%)
| |
|
6,5
(4,9, 8,6)
|
7,3
(3,6, 12,6)
|
8,3
(1,3, 15,0)
|
4,5
(2,8, 5,9)
| |
Taux de réponse globale (intra et extracrânienne) ORR, % (IC à 95%)
| |
|
59%
(47,3, 70,4)
|
56%
(29,9, 80,2)
|
44%
(19,8, 70,1)
|
65%
(38,3, 85,8)
| |
Valeur médiane de la PFS, mois (IC à 95%)
| |
|
5,7
(5,3, 7,3)
|
7,2
(4,7, 14,6)
|
3,7
(1,7, 6,5)
|
5,5
(3,7, 11,6)
| |
Valeur médiane de l'OS, MOIS (IC à 95%)
| |
Valeur médiane, mois
|
10,8
(8,7, 17,9)
|
24,3
(7,9, NR)
|
10,1
(4,6, 17,6)
|
11,5
(6,8, 22,4)
| |
IC = intervalle de confiance
NR = sans indication
|
Traitement adjuvant du mélanome
Étude BRF115532/CDRB436F2301 (COMBI-AD)
L'efficacité et la sécurité de Tafinlar en association avec le tramétinib ont été étudiées dans une étude de phase III multicentrique, randomisée, en double aveugle, contrôlée contre placebo, menée auprès de patients ayant un mélanome de stade III portant une mutation V600 du gène BRAF après résection complète.
Les patients ont été randomisés dans un rapport 1:1 et ont reçu soit un traitement combiné avec 150 mg de Tafinlar deux fois par jour et 2 mg de tramétinib une fois par jour, soit deux placebos sur une période de 12 mois. Seuls des patients ayant subi une résection complète du mélanome et une lymphadénectomie complète au cours des 12 semaines précédant la randomisation ont été acceptés dans l'étude. Un traitement systémique préalable du cancer, y compris une radiothérapie, n'était pas autorisé. Les patients ayant des antécédents de malignité antérieure ont été autorisés à participer s'ils n'étaient pas tombés malades pendant au moins 5 ans. Les patients atteints de tumeurs malignes avec des mutations RAS activantes confirmées n'ont pas été autorisés à participer. Les patients ont été stratifiés en fonction du statut de la mutation BRAF (V600E ou V600K) et du stade de la maladie avant l'opération (en sous-stades du stade III, c'est-à-dire différentes atteintes des ganglions lymphatiques, taille primaire de la tumeur et ulcération). Le critère d'évaluation principal était la survie sans récidive évaluée par l'investigateur (relapse-free survival, RFS), définie comme étant la période de la randomisation à la réapparition de la tumeur ou à la mort, quelle qu'en soit la cause. Une évaluation radiologique de la tumeur a été réalisée tous les 3 mois au cours des deux premières années, puis tous les 6 mois, jusqu'à ce que la première récidive ait été observée. Les critères d'évaluation secondaires étaient la survie globale (OS; critère secondaire important) et la survie en l'absence de métastases distantes (distant metastasis-free survival, DMFS).
En tout, 870 patients ont été randomisés pour le traitement combiné (n = 438) et pour le placebo (n = 432). L'étude a inclus des patients ayant une maladie à tous les sous-stades du stade III avant la résection; 18% de ces patients ont présenté des atteintes des ganglions lymphatiques seulement détectables au microscope et aucune ulcération tumorale primaire. La majorité des patients avait une mutation V600E du gène BRAF (91%). La durée médiane du suivi du traitement (temps écoulé de la randomisation jusqu'au dernier contact ou à la mort) était de 2,83 ans dans le groupe de traitement et 2,75 ans dans le groupe placebo.
Les résultats de l'analyse primaire sont représentés dans le tableau 10. L'étude a montré une différence statistiquement significative entre les bras de traitements pour le critère d'évaluation principal concernant la RFS. Dans le groupe de traitement ayant reçu Tafinlar et le tramétinib comme traitement combiné, la réduction du risque a été de 53% comparée au groupe placebo.
Tableau 10: COMBI-AD - Résultats de la survie sans récidive
|
|
Tafinlar + tramétinib
|
Placebo
| |
Paramètres de RFS
|
N = 438
|
N = 432
| |
Nombre d'évènements-n (%)
|
166 (38%)
|
248 (57%)
| |
Récidive
|
163 (37%)
|
247 (57%)
| |
Récidive avec des métastases distantes
|
103 (24%)
|
133 (31%)
| |
Mort
|
3 (< 1%)
|
1 (< 1%)
| |
Valeur médiane (mois)
|
NE
|
16,6
| |
(CI à 95%)
|
(44,5, non estimable (NE))
|
(12,7; 22,1)
| |
Hazard ratio[1]
|
0,47
| |
(IC à 95%)
|
(0,39; 0,58)
| |
Valeur de P[2]
|
1,53×10-14
| |
Taux sur 1 an (IC à 95%)
|
0,88 (0,85, 0,91)
|
0,56 (0,51, 0,61)
| |
Taux sur 2 ans (IC à 95%)
|
0,67 (0,63, 0,72)
|
0,44 (0,40, 0,49)
| |
Taux sur 3 ans (IC à 95%)
|
0,58 (0,54, 0,64)
|
0,39 (0,35, 0,44)
| |
[1]
Le hazard ratio résulte du modèle de Pike stratifié.
[2] La valeur de P est calculée à partir du test du Logrank stratifié bilatéral (les facteurs de stratification étaient le stade de la maladie, IIIA vs. IIIB vs. IIIC, et le type de mutation V600 sur le gène BRAF - V600E vs. V600K).
NE = non estimable
|
Sur la base de 153 évènements (60 (14%) dans le bras de traitement combiné et 93 (22%) dans le bras placebo), ce qui correspond à une proportion d'information de 26% de l'objectif global de 597 évènements OS, le hazard ratio estimé pour l'OS était de 0,57 (IC à 95%: 0,42, 0,79; p = 0,0006). Ces résultats n'ont pas correspondu à la limite pré-spécifiée pour obtenir une significativité statistique dans cette première analyse intermédiaire de l'OS (HR = 0,50; p = 0,000019). Les estimations de la survie 1 et 2 ans après la randomisation étaient de 97% et 91% dans le bras de traitement combiné et de 94% et 83% dans le bras placebo.
NSCLC avancé
Étude BRF113928
L'efficacité et la sécurité de Tafinlar en association avec le tramétinib ont été évaluées dans une étude de phase II multicentrique, ouverte et non randomisée menée auprès de 57 patients atteints d'un NSCLC métastatique porteur de la mutation BRAF V600E et préalablement traités par chimiothérapie. Le critère d'évaluation principal était le taux de réponse globale (overall response rate, ORR) évalué par le médecin investigateur à l'aide des critères d'évaluation de la réponse au traitement en cas de tumeurs solides (Response Evaluation Criteria in Solid Tumors, RECIST). Les critères d'évaluation secondaires incluaient la durée de la réponse (duration of response, DoR), la survie sans progression (progression-free survival, PFS) et la survie globale (overall survival, OS). L'ORR, la DoR et la PFS ont été évalués par un comité d'évaluation indépendant (Independent Review Committee, IRC) sous la forme d'une analyse de sensibilité.
Pour le critère d'évaluation principal, à savoir le taux de réponse globale (overall response rate, ORR) évalué par le médecin investigateur, l'ORR dans la population préalablement traitée a atteint 66,7% (IC à 95%: 52,9%–78,6%). Ainsi, la significativité statistique a été atteinte, permettant de rejeter l'hypothèse nulle correspondant à un ORR inférieur ou égal à 30% pour le traitement par Tafinlar en association avec le tramétinib pour cette population de patients atteints d'un NSCLC. Les résultats de l'évaluation du taux de réponse globale (overall response rate, ORR) par un comité d'évaluation indépendant (Independent Review Committee, IRC) concordaient avec l'évaluation faite par l'investigateur (tableau 11).
La durée de la réponse a atteint une médiane de 9,8 mois (IC à 95%: 6,9–16,0), selon l'évaluation du médecin investigateur.
Tableau 11: ORR selon l'évaluation du médecin investigateur et l'évaluation radiologique indépendante
|
Groupe pour lequel l'évaluation radiologique a été réalisée
|
Investigateur:
|
Évaluation indépendante
| |
Catégorie
|
N = 57*
|
N = 57*
| |
Réponse globale – n (%)
| |
ORR (CR + PR)
|
38 (66,7),
|
36 (63.2)
| |
(IC à 95%)
|
(52,9; 78,6),
|
(49.3; 75.6)
| |
Durée de la réponse
| |
Nombre de répondeurs
|
38
|
36
| |
Nombre de patients présentant une progression de la maladie ou décédés - n (%)
|
27 (71)
|
20 (56)
| |
DOR médiane, en mois
(IC à 95%)
|
9,8
(6,9; 16,0)
|
12,6
(5.8; NR)
| |
Survie sans progression
| |
Progression de la maladie ou décès - n (%)
|
41 (72)
|
38 (67)
| |
PFS médiane, en mois
(IC à 95%)
|
10,2
(6,9; 16,7)
|
8,6
(5.2; 16.8)
| |
Survie globale
| |
Nombre de décès - n (%)
|
33 (58)
| |
Médiane, en mois
(IC à 95%)
|
18,2
(14,3; not reached (NR))
| |
Intervalle de confiance (IC) calculé avec la méthode exacte (appelé intervalle de Clopper-Pearson).
CR (complete remission) = rémission complète; PR (partial remission) = rémission partielle; SD (stable disease) = maladie stable; PD (progressive disease) = progression de la maladie; NR (not reached) = non atteint
|
Dans l'étude BRF113928, 36 patients atteints d'un NSCLC exprimant la mutation V600E de BRAF ayant reçu le dabrafénib en association avec le tramétinib comme traitement de première ligne contre une atteinte métastatique ont aussi été inclus. Pour une période d'observation d'au moins 3 ans pour tous les participants à l'étude traités avec le traitement de première ligne (date butoir des données: 22 juin 2019), le critère d'évaluation principal du taux de réponse global évalué par l'investigateur (ORR) a été de 63,9% (IC à 95%, 46,2%, 79,2%). Ainsi, la significativité statistique a été atteinte avec rejet de l'hypothèse nulle selon laquelle l'ORR du dabrafénib en association avec le tramétinib était de 30% au maximum pour cette population de patients avec NSCLC. La durée médiane de rémission (DoR) évaluée par l'investigateur a été de 10,2 mois (IC à 95%: 8,3, 15,2), la survie sans progression médiane évaluée par l'investigateur de 10,8 mois (IC à 95%: 7,0, 14,5), la survie globale médiane évaluée par l'investigateur de 17,3 mois (IC à 95%: 12,3, 40,2), avec 61% des cas de décès survenus au moment de l'analyse.
Gliome de bas grade (GBG)
Étude DRB436G2201
L'efficacité clinique et la sécurité du traitement par l'association de Tafinlar et du tramétinib chez les patients pédiatriques âgés de 1 à < 18 ans présentant un gliome porteur de la mutation V600E du gène BRAF ont été évaluées dans l'étude clinique de phase II multicentrique et ouverte CDRB436G2201. Les patients présentant un gliome de bas grade (de grades 1 et 2 selon la classification de l'OMS) qui ont nécessité un premier traitement systémique et qui n'avaient pas reçu de radiothérapie auparavant ont été randomisés selon un rapport de 2:1 pour recevoir dabrafénib plus tramétinib (D + T) ou carboplatine plus vincristine (C + V). Environ 83% de tous les patients avaient subi une intervention chirurgicale préalable; Deux patients seulement (tous deux dans le bras D + T) n'avaient pas de maladie résiduelle après l'intervention. Les patients présentant un score de performance de Karnofsky/Lansky inférieur à 50%, les patients dont les fonctions médullaire, rénale, hépatique et cardiaque étaient insuffisantes ainsi que les patients souffrant d'affections incontrôlées ou significatives, y compris les affections cardiaques, le diabète sucré, l'hypertension, les maladies hépatiques ou les infections, étaient exclus de l'étude (voir «Mises en garde et précautions»).
Le statut de la mutation du gène BRAF a été déterminé de manière prospective par un test local ou, lorsqu'un test local n'était pas disponible, par un test de PCR en temps réel du laboratoire central. Par ailleurs, des tests rétrospectifs sur des échantillons de tumeurs disponibles ont été effectués par le laboratoire central afin de confirmer la mutation V600E du gène BRAF.
La posologie de Tafinlar et du tramétinib dépendait de l'âge et du poids, Tafinlar ayant été administré par voie orale à la posologie de 2,625 mg/kg deux fois par jour pour le groupe d'âge < 12 ans et de 2,5 mg/kg deux fois par jour pour le groupe d'âge 12 ans et plus; le tramétinib a été administré par voie orale à la posologie de 0,032 mg/kg une fois par jour pour le groupe d'âge < 6 ans et de 0,025 mg/kg une fois par jour pour le groupe d'âge 6 ans et plus. La dose maximale de Tafinlar a été limitée à 150 mg deux fois par jour et celle du tramétinib à 2 mg une fois par jour. Le carboplatine et la vincristine ont été administrés en fonction de l'âge et de la surface corporelle à une posologie de 175 mg/m2 et 1,5 mg/m2, respectivement, en tant que traitement d'induction de 10 semaines, suivi d'un cycle de 6 semaines d'un traitement d'entretien.
Le critère d'évaluation de l'efficacité principal dans les deux cohortes était le taux de réponse globale (ORR, somme des patients présentant une rémission complète/CR et une rémission partielle/PR confirmées) par un examen indépendant sur la base des critères RANO (2017). L'analyse primaire a été effectuée lorsque tous les patients des deux cohortes avaient terminé au moins 32 semaines de traitement.
Dans la cohorte des gliomes de bas grade (GBG) de l'étude G2201, 110 patients ont été choisis de manière aléatoire pour D + T (n = 73) ou C + V (n = 37). L'âge moyen était de 9,5 ans, 34 patients (30,9%) ayant entre 12 mois et moins de 6 ans, 36 patients (32,7%) entre 6 ans et moins de 12 ans et 40 patients (36,4%) entre 12 ans et moins de 18 ans; 60% étaient de sexe féminin. Au moment de l'analyse primaire, la durée médiane de traitement était de 76 semaines dans le bras D + T, la durée médiane de suivi dans la cohorte GBG de 18,9 mois. L'ORR dans le bras D + T (46,6%) a montré une amélioration statistiquement significative par rapport au bras C + V (10,8%), avec un odds ratio (IC à 95%) de 7,19 (2,3; 22,4) et une valeur de p unilatérale < 0,001 (tableau 12). L'examen hiérarchique ultérieur a également révélé une amélioration de la survie sans progression (PFS) par rapport à la chimiothérapie, avec un hazard ratio (IC à 95%) de 0,31 (0,17; 0,55) (valeur de p log-rank unilatérale du test du log-rank < 0,001). Des différences dans l'évaluation de l'efficacité, qui comprenaient également la constatation de rémissions complètes et partielles ainsi que des progressions de la maladie, ont été observées entre l'examen indépendant et les centres investigateurs. Le taux de concordance dans le bras D + T était de 52% en tout.
Les modifications de la fréquence des convulsions («seizure activity») et de l'acuité visuelle («visual acuity») ont été étudiées en tant que paramètres fonctionnels avant et après le traitement. Pour les deux paramètres, aucune amélioration n'a été démontrée sous traitement par D + T par rapport au bras témoin.
Tableau 12: Réponse et survie sans progression dans l'étude G2201 (cohorte GBG)
|
|
Dabrafénib + tramétinib
N = 73
|
Carboplatine plus vincristine
N = 37
| |
Meilleure réponse globale
| |
Rémission complète (CR), n (%)
|
2 (2,7)
|
1 (2,7)
| |
Rémission partielle (PR), n (%)
|
32 (43,8)
|
3 (8,1)
| |
Maladie stable (SD), n (%)
|
30 (41,1)
|
15 (40,5)
| |
Progression de la maladie (PD), n (%)
|
8 (11,0)
|
12 (32,4)
| |
Inconnu, n (%)
|
1 (1,4)
|
6 (16,2)
| |
Taux de réponse globale (ORR)
| |
ORR (CR + PR), IC à 95%, valeur de p
|
46,6% (34,8–58,6%), p < 0,001
|
10,8% (3,0–25,4%)
| |
Odds ratio
|
7,19 (2,3–22,4)
| |
Survie sans progression
| |
Valeur médiane (mois)
|
20,1 (12,8; NA)
|
7,4 (3,6; 11,8)
| |
Hazard ratio (IC à 95%), valeur de p
|
0,31 (0,17–0,55), p < 0,001
|
PharmacocinétiqueLa pharmacocinétique de Tafinlar a été déterminée chez des patients atteints de mélanome métastatique à mutation BRAF-V600 après l'administration d'une dose unique et après administrations répétées de 150 mg deux fois par jour à 12 heures d'intervalle environ.
Absorption
Le dabrafénib est absorbé par voie orale, avec un temps médian pour atteindre la concentration plasmatique maximale de 2 heures après l'administration. La biodisponibilité absolue moyenne du dabrafénib administré par voie orale est de 95% (IC à 90%: 81; 110%). L'exposition au dabrafénib (Cmax et ASC) a augmenté proportionnellement à la dose après administration d'une dose unique entre 12 et 300 mg; après une administration deux fois par jour, répétée, l'augmentation était en revanche moins proportionnelle à la dose. Lors d'une administration répétée, une diminution de l'exposition a été observée, probablement en raison de l'induction de son propre métabolisme par le dabrafénib. Le rapport moyen d'accumulation pour l'ASC jour 18/jour 1 était de 0,73. Après l'administration de 150 mg deux fois par jour, la moyenne géométrique de la Cmax, de l'ASC (0-τ) et de la concentration avant l'administration (Cτ) était respectivement de 1478 ng/ml, 4341 ng*h/ml et 26 ng/ml.
Après administration du tramétinib en association avec le dabrafénib 150 mg deux fois par jour, les moyennes géométriques de la Cmax et de l'ASC(0-τ) du tramétinib et de la concentration en tramétinib avant administration s'élevaient à respectivement 22,6 ng/ml, 356 ng*h/ml et 10,9 ng/ml.
L'administration d'une gélule de dabrafénib accompagnée d'une prise d'aliments a réduit la biodisponibilité (Cmax et ASC ont diminué respectivement de 51% et 31%) et retardé l'absorption des gélules de dabrafénib en comparaison avec une administration à jeun. Les gélules et comprimés dispersibles de dabrafénib sont des formes pharmaceutiques avec libération immédiate qui devraient avoir des effets similaires sur la pharmacocinétique.
Distribution
Le dabrafénib se lie à 99,7% aux protéines plasmatiques humaines. Le volume de distribution à l'état d'équilibre après administration intraveineuse est de 46 l.
Métabolisme
Le dabrafénib est principalement métabolisé par les cytochromes CYP2C8 et CYP3A4 en hydroxy-dabrafénib, qui est ensuite oxydé par le CYP3A4 pour former le carboxy-dabrafénib. Le carboxy-dabrafénib peut être décarboxylé par un processus non-enzymatique pour former le déméthyl-dabrafénib. Le carboxy-dabrafénib est excrété dans la bile et l'urine. Le déméthyl-dabrafénib peut aussi se former dans l'intestin et être réabsorbé. Le déméthyl-dabrafénib est métabolisé par le CYP3A4 en métabolites oxydants. La demi-vie terminale de l'hydroxy-dabrafénib est de 10 heures et correspond à celle de la substance mère, tandis que les métabolites carboxy et déméthyle ont des demi-vies plus longues (21–22 heures). Le rapport moyen des ASC entre les métabolites et la substance mère après administration répétée était de 0,9 pour l'hydroxy-dabrafénib, de 11 pour le carboxy-dabrafénib et de 0,7 pour le déméthyl-dabrafénib. Sur la base de l'exposition, de la puissance relative et des propriétés pharmacocinétiques, il est probable que l'hydroxy- et le déméthyl- contribuent à l'activité clinique du dabrafénib, alors qu'il est peu probable que l'activité du carboxy-dabrafénib soit significative.
Élimination
La demi-vie terminale après administration d'une microdose intraveineuse s'élève à 2,6 heures. La demi-vie terminale du dabrafénib après une administration orale est de 8 heures, en raison d'une phase terminale ralentie. La clairance plasmatique après administration intraveineuse est de 12 l/h.
L'excrétion dans les selles est la voie principale d'élimination après administration orale; 71% d'une dose radioactive ont été retrouvés dans les selles, contre seulement 23% dans l'urine.
Cinétique pour certains groupes de patients
Troubles de la fonction hépatique
La pharmacocinétique de Tafinlar a été déterminée à l'aide d'une analyse de population chez 65 patients inclus dans les études cliniques et atteints d'une insuffisance hépatique légère (selon la classification du National Cancer Institute [classification NCI]). Chez ces patients, la clairance orale du dabrafénib ne présentait pas de différence significative par rapport à celle des participants avec des valeurs hépatiques normales (foie sain) (différence de 4%). En outre, une insuffisance hépatique légère n'a pas influencé de façon significative les concentrations plasmatiques des métabolites du dabrafénib. On ne dispose pas de données concernant les patients atteints d'insuffisance hépatique modérée à sévère (voir «Posologie/Mode d'emploi»).
Troubles de la fonction rénale
Dans les études cliniques, la pharmacocinétique de Tafinlar a été déterminée à l'aide d'une analyse de population chez 233 patients atteints d'insuffisance rénale légère (DFG 60–89 ml/min/1,73 m2) et chez 30 patients atteints d'insuffisance rénale modérée (DFG 30–59 ml/min/1,73 m2). Une insuffisance rénale légère à modérée n'a que peu d'effet sur la clairance orale du dabrafénib (< 6% dans les deux catégories) et n'était pas cliniquement significative. En outre, une insuffisance rénale légère à modérée n'a pas eu d'influence significative sur les concentrations plasmatiques des métabolites hydroxy, carboxy et déméthyle du dabrafénib. Aucune donnée n'est disponible chez des patients atteints d'insuffisance rénale sévère (voir «Posologie/Mode d'emploi»).
Population pédiatrique
La pharmacocinétique du dabrafénib dans les gliomes et autres tumeurs solides a été étudiée chez 243 patients pédiatriques (âgés de 1 à moins de 18 ans) après un dosage unique ou répété adapté au poids. Les propriétés pharmacocinétiques (taux d'absorption, rapport des métabolites, clairance du médicament) du dabrafénib chez les patients pédiatriques sont comparables à celles des adultes. Il a été constaté que le poids a un impact sur la clairance orale du dabrafénib. L'exposition pharmacocinétique au dabrafénib à la posologie recommandée adaptée au poids chez les patients pédiatriques se situait dans la plage de l'exposition observée chez les adultes.
Patients âgés
Sur la base de l'analyse pharmacocinétique de population, l'âge n'a pas eu d'influence significative sur la pharmacocinétique du dabrafénib. Un âge supérieur à 75 ans était un facteur prédictif significatif de concentrations plasmatiques de carboxy- et déméthyl-dabrafénib, les participants âgés d'au moins 75 ans ayant une exposition de 40% supérieure à celle des participants de moins de 75 ans.
Poids corporel et sexe
Dans le cadre de l'analyse pharmacocinétique de population réalisée chez des adultes, on a constaté que le sexe et le poids corporel avaient une influence sur la clairance orale du dabrafénib; le poids avait en outre un impact sur le volume de distribution après administration orale et sur la clairance de distribution. Ces différences pharmacocinétiques n'ont pas été considérées comme cliniquement pertinentes.
Origine ethnique
L'analyse de pharmacocinétique de population du dabrafénib n'a pas montré de différences significatives entre les patients asiatiques et les patients caucasiens. Aucun ajustement posologique n'est nécessaire chez les patients d'origine asiatique.
On ne dispose pas de données suffisantes pour évaluer un éventuel effet de l'ethnicité sur la pharmacocinétique de Tafinlar.
Données précliniquesDabrafénib en association avec le tramétinib
Chez des chiens ayant reçu le dabrafénib et le tramétinib en association pendant 4 semaines, les réactions de toxicité ont été similaires à celles observées dans les études précliniques réalisées avec les substances individuelles. Chez les chiens ayant reçu l'association de dabrafénib et de tramétinib, les effets testiculaires observés, consistant en une dégénérescence et une oligospermie épididymaire secondaire, étaient les mêmes que chez les chiens ayant reçu le dabrafénib ou le tramétinib seuls. Voir également l'information professionnelle du tramétinib à ce propos.
Pharmacologie de sécurité
Chez le rat et le chien, une élévation de la fréquence cardiaque dose-dépendante a été observée dans des études avec dose unique; en revanche, aucune anomalie sur le tracé de l'ECG, ni arythmies ni aucun effet sur la tension artérielle ou la température corporelle n'a pu être constaté. Le dabrafénib a entraîné une légère inhibition de la repolarisation par les canaux hERG in vitro (CI25 de 11,7 μM; 6,1 μg/ml), ses 3 métabolites principaux n'induisant toutefois pas d'inhibition du canal hERG (CI50> 30 μM; 15,6 μg/ml). Lors d'une étude de perfusion myocardique en coupes du ventricule gauche de lapin (Wedge-Assay), un raccourcissement de l'intervalle QT (30% à 30 μM) sans changements de l'intervalle QRS et sans potentiel d'induction de torsades de pointes a été observé. Dans des études avec des doses uniques et répétées chez le chien, aucun effet sur l'intervalle QTc n'a été observé (durée jusqu'à 13 semaines; ≥9 fois l'exposition clinique chez l'homme, sur la base de l'ASC).
Mutagénicité/carcinogénicité
Le dabrafénib s'est révélé non mutagène ou clastogène in vitro dans des bactéries et des cultures de cellules de mammifères et in vivo dans le test du micronoyau chez des rongeurs. Aucune étude de carcinogénicité n'a pas été réalisée avec le dabrafénib.
Toxicité sur la reproduction
Dans des études combinées sur la fertilité féminine et le développement précoce embryonnaire et embryo-fœtal, menées chez des rats, le nombre de corps jaunes dans les ovaires de femelles gravides a été réduit à 300 mg/kg/jour (ce qui correspond à environ trois fois l'exposition clinique chez l'être humain, sur la base de l'ASC), sans effets décelables sur le cycle, le comportement d'accouplement ou la fertilité. Des symptômes de toxicité sur le développement, p.ex. une mortalité embryonnaire et des malformations du septum inter-ventriculaire, ont été observés à des doses de 300 mg/kg/jour; un ralentissement du développement du squelette et une réduction du poids des fœtus sont apparus à partir de doses de 20 mg/kg/jour (ce qui correspond à ≥0,5 fois l'exposition clinique chez l'être humain sur la base de l'ASC).
Aucune étude sur la fertilité masculine n'a pas été réalisée avec le dabrafénib. Dans des études avec administration répétée, une dégénérescence/atrophie des testicules a toutefois été observée chez le rat et le chien (à des doses correspondant à ≥0,2 fois l'exposition clinique chez l'être humain sur la base de l'ASC). Les lésions testiculaires chez le rat et le chien étaient toujours présentes au bout d'une période de rétablissement de 4 semaines.
Toxicité générale
Des effets cardiovasculaires, notamment des altérations dégénératives, nécrotiques et/ou hémorragiques des artères coronaires, une hypertrophie/hémorragie des valvules auriculo-ventriculaires et une prolifération fibrovasculaire auriculaire ont été observées chez le chien, à des doses correspondant à ≥2 fois l'exposition clinique chez l'être humain sur la base de l'ASC. Chez la souris, des foyers d'événements inflammatoires artériels/périvasculaires ont été observés dans divers types de tissus et chez le rat, une dégénérescence de l'artère hépatique ainsi qu'une dégénérescence des cardiomyocytes spontanée avec altérations inflammatoires (myocardiopathie spontanée) a été observée (à des doses correspondant chez le rat à ≥0,5 fois l'exposition clinique chez l'être humain, rapport qui était de 0,6 chez la souris).
Chez la souris, des effets sur le foie ont été observés, notamment une nécrose et une inflammation hépatocellulaires (exposition clinique ≥0,6 fois).
Chez plusieurs chiens, une inflammation broncho-alvéolaire des poumons associée à une respiration superficielle et/ou une dyspnée est apparue à des doses ≥20 mg/kg/jour (ce qui correspond, sur la base de l'ASC, à ≥9 fois l'exposition clinique chez l'être humain).
Des lésions épithéliales caractérisées essentiellement par une hyperplasie, une dégénérescence et/ou une hyperkératose épithéliales, ont été observées sur la peau des rats et des chiens ainsi que dans le proventricule non glandulaire de rats (≥0,7 fois l'exposition clinique humaine chez le rat; 1,5 fois l'exposition clinique humaine chez le chien, toujours sur la base de l'ASC).
Des effets hématologiques réversibles ont été observés chez le chien et le rat sous dabrafénib. Dans des études d'une durée maximale de 13 semaines, une baisse de la numération réticulocytaire et/ou du volume des érythrocytes circulants a été constatée chez le chien et le rat (à des doses correspondant respectivement ≥10 fois et à 1,4 fois l'exposition clinique chez l'être humain).
Dans des études de toxicité juvénile chez le rat, des effets sur la croissance (diminution de la longueur des os longs), une toxicité rénale (dépôts dans les tubules rénaux, incidence accrue de kystes rénaux corticaux et de basophilie tubulaire, ainsi qu'une augmentation réversible de l'urée et/ou des concentrations de créatinine) et une toxicité testiculaire (dégénérescence et dilatation des tubes) ont été observés (≥0,2 fois l'exposition clinique de la dose d'un adulte sur la base de l'ASC).
In vitro, dans un test NRU (Neutral Red Uptake) 3T3 avec une lignée de fibroblastes de souris, ainsi qu'in vivo dans une étude de phototoxicité sur des souris imberbes à des dosages administrés par voie orale ≥100 mg/kg (> 44 fois l'exposition clinique sur la base de la Cmax), le dabrafénib s'est avéré phototoxique. Bien que le dabrafénib se soit avéré phototoxique dans des études non cliniques, les données cliniques relatives à la sécurité pour les patients laissent conclure à l'existence d'un faible risque de phototoxicité pour les patients prenant du dabrafénib.
Remarques particulièresStabilité
Le médicament ne doit pas être utilisé au-delà de la date figurant après la mention «EXP» sur le récipient.
Remarques particulières concernant le stockage
Gélules
Ne pas conserver au-dessus de 30 °C.
Comprimés dispersibles
Ne pas conserver au-dessus de 30 °C. Conserver dans le flacon d'origine pour protéger le contenu de l'humidité. Ne pas retirer le dessicant.
Conserver hors de portée des enfants.
Numéro d’autorisation62781 (Swissmedic)
69135 (Swissmedic)
PrésentationGélules 50 mg: 28 (A)
Gélules 50 mg: 120 (A)
Gélules 75 mg: 28 (A)
Gélules 75 mg: 120 (A)
Comprimés dispersibles 10 mg: 210 comprimés et 2 gobelets doseurs (A)
Titulaire de l’autorisationNovartis Pharma Schweiz AG, Risch; Domicile: 6343 Rotkreuz
Mise à jour de l’informationMars 2023
Remarques particulières
Remarques concernant l'utilisation et la manipulation
|
Tafinlar®
(dabrafénib)
Comprimés dispersibles
| |
Ces «instructions» contiennent les informations concernant la prise ou l'administration de TAFINLAR, comprimés dispersibles sous forme de suspension buvable.
| |
Informations importantes qui doivent être prises en compte avant la prise ou l'administration des comprimés dispersibles de TAFINLAR (comprimés de TAFINLAR).
| |
·Veuillez lire attentivement les présentes instructions avant d'utiliser Tafinlar pour la première fois et à chaque fois que vous recevez une nouvelle boîte. Celles-ci peuvent contenir de nouvelles informations.
·Ces instructions d'utilisation ne remplacent pas un entretien avec votre médecin ou votre pharmacien sur votre maladie ou celle de votre enfant et son traitement.
·Votre médecin ou votre pharmacien doit vous montrer comment prendre ou administrer correctement une dose de Tafinlar. Vous devez toujours prendre ou administrer Tafinlar exactement tel que votre médecin vous dit de le faire.
·Si vous avez des questions concernant la préparation, la prise ou l'administration de Tafinlar, veuillez vous adresser à votre médecin ou à votre pharmacien.
·Utilisez toujours le gobelet doseur inclus dans l'emballage de Tafinlar. Si votre emballage ne contient pas de gobelet doseur, veuillez contacter votre médecin ou votre pharmacien.
·Si la suspension buvable de Tafinlar entre en contact avec votre peau ou celle de votre enfant à un moment donné, lavez soigneusement la zone concernée à l'eau et au savon.
·Si la suspension buvable de Tafinlar entre en contact avec vos yeux ou ceux de votre enfant à un moment donné, rincez abondamment les yeux à l'eau froide.
·Si vous renversez la suspension buvable de Tafinlar, suivez les instructions figurant à la fin des présentes instructions dans la rubrique «Comment nettoyer la suspension de Tafinlar renversée».
·Vous recevez votre prescription de Tafinlar ou celle de votre enfant dans un flacon scellé qui contient des comprimés solubles. Vous devez dissoudre les comprimés dans de l'eau avant de prendre ou d'administrer Tafinlar. Veuillez suivre les instructions ci-dessous pour dissoudre les comprimés dans l'eau.
·Votre emballage de Tafinlar doit contenir:
|
1.1 flacon contenant des comprimés de Tafinlar
2.2 gobelets doseurs réutilisables
Notice et information destinée aux patients (le présent document)De plus, vous avez besoin d'eau potable.Pour l'utilisation par voie orale, aller à la section A. Pour l'utilisation par sonde alimentaire ou seringue orale, aller à la section B.
|
|
SECTION A. Préparation et administration d'une dose par voie orale directement dans le gobelet doseur
Si la solution de Tafinlar a été renversée ou est entrée en contact avec la peau ou les yeux, suivez les instructions de la section E, Élimination de la suspension buvable de Tafinlar renversée.
Pour la préparation et l'administration de Tafinlar, voici ce dont vous avez besoin:
·nombre de comprimés prescrits;
·1 gobelet doseur;
·1 cuillère à café;
·eau potable plate.
Pour l'utilisation par voie orale (c.-à-d. pour l'ingestion de la solution), vous ou votre enfant pouvez boire celle-ci directement dans le gobelet doseur.
| |
Étape 1. Lavez-vous et séchez-vous les mains avant de procéder à la préparation de Tafinlar.Ajoutez de l'eau potable fraîche jusqu'au repère du gobelet tel que cela est indiqué dans le tableau ci-dessous.
Remarque: la quantité d'eau ne doit pas être mesurée exactement.
·Si la dose prescrite pour vous ou votre enfant comporte 1 à 4 comprimés, vous avez besoin d'environ 5 ml d'eau.
·Si la dose prescrite pour vous ou votre enfant comporte 5 à 15 comprimés, vous avez besoin d'environ 10 ml d'eau.
|
|
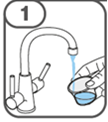
|
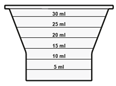
| |
Étape 2. Retirez le capuchon en appuyant dessus et en le tournant dans le sens inverse des aiguilles d'une montre (voir illustration).
Ne pas jeter le capuchon.
Si vous ouvrez le flacon pour la première fois, retirez le scellage du flacon.
|
|
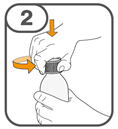
| |
Étape 3. Comptez le nombre de comprimés prescrits dans votre main.
|
|
| |
Mettez le nombre de comprimés prescrits dans l'eau qui se trouve dans le gobelet doseur.
|
|
| |
Le flacon contient 2 récipients en plastique qui permettent de garder les comprimés secs. Si l'un des deux récipients tombe lorsque vous retirez vos comprimés du flacon, remettez celui-ci dans le flacon.
|
|
| |
Étape 4. Remettez le capuchon sur le flacon et tournez-le dans le sens des aiguilles d'une montre dans le sens indiqué par la flèche comme montré pour fermer le flacon.
|
|
| |
Étape 5. Avec une main, inclinez le gobelet doseur.
·Avec l'autre main, remuez délicatement l'eau et les comprimés à l'aide du manche d'une cuillère à café jusqu'à ce que les comprimés soient complètement dissous.
·La dissolution complète des comprimés peut durer 3 minutes (ou plus longtemps). Dès qu'ils sont dissous, la solution doit avoir un aspect trouble-blanc, mais elle peut contenir des petits morceaux.
Ne pas administrer la solution plus de 30 minutes après la dissolution des comprimés.
Si plus de 30 minutes se sont écoulées, éliminez la solution conformément aux dispositions locales et reprenez les instructions à partir du début de la section A. Si vous n'êtes pas sûr(e) de savoir comment éliminer la suspension buvable de Tafinlar, demandez à votre médecin ou à votre pharmacien.
|
|
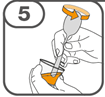
|
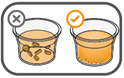
| |
Étape 6. Buvez la solution dans le gobelet doseur.
IMPORTANT: après avoir bu la solution, des résidus du médicament restent dans le gobelet. Il se peut que ces résidus soient difficiles à reconnaître. Procédez aux étapes 7 à 9 pour ingérer tous les résidus et recevoir une dose complète.
|
|

| |
Étape 7. Mettez environ 5 ml d'eau dans le gobelet doseur vide.
|
|
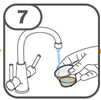
| |
Étape 8. Remuez à l'aide du manche d'une cuillère à café pour dissoudre les résidus restants.
|
|
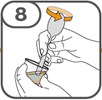
| |
Étape 9. Buvez la solution.
|
|
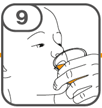
| |
·Pour 1 à 3 comprimés: exécutez les étapes 7 à 9 une fois
·Pour 4 à 15 comprimés: exécutez les étapes 7 à 9 deux fois
Il est important d'ingérer ou d'administrer tous les résidus du médicament afin que vous ou votre enfant receviez la dose complète de TAFINLAR
| |
Étape 10. Poursuivez avec les étapes de nettoyage figurant dans la section C.
| |
SECTION B. Préparation et administration de Tafinlar par une sonde d'alimentation ou une seringue orale
| |
CONSEILS D'UTILISATION IMPORTANTS
Assurez-vous que tous les comprimés sont dissous complètement avant d'administrer la solution.
Taille minimale de la sonde d'alimentation que vous pouvez utiliser pour administrer la suspension buvable de TAFINLAR:
·Si la dose qui vous a été prescrite est de 1 à 3 comprimés, la taille minimale de la sonde d'alimentation à utiliser est de 10 French.
·Si la dose qui vous a été prescrite est de 4 à 15 comprimés, la taille minimale de la sonde d'alimentation à utiliser est de 12 French.
·Si la suspension buvable de Tafinlar entre en contact avec votre peau ou vos yeux pendant que vous exécutez les étapes suivantes, suivez les instructions contenues dans la section «Informations importantes à prendre en considération avant de prendre ou d'administrer Tafinlar sous forme de suspension buvable.»
·Si la suspension buvable de Tafinlar est renversée, suivez les instructions contenues dans la section «Comment nettoyer la suspension de Tafinlar renversée».
·Lavez-vous et séchez-vous les mains avant d'administrer une dose de la suspension buvable de Tafinlar.
| |
Étape 1. Exécutez les étapes 1 à 5 de la section A pour dissoudre les comprimés. Si vous utilisez une sonde d'alimentation, rincez la sonde avec de l'eau potable plate, puis passez à l'étape 2 de la section B.
| |
Étape 2. Aspirez la totalité de la suspension buvable de TAFINLAR du gobelet doseur dans une seringue en tirant sur le piston. Veillez à utiliser une seringue pouvant être utilisée avec la sonde d'alimentation ou permettant d'administrer par voie orale la suspension buvable de Tafinlar. Si vous avez des doutes quant à la seringue à utiliser, demandez à votre médecin.
|
|
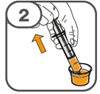
| |
Étape 3. En cas d'administration par une sonde d'alimentation, mettez la solution dans la sonde d'alimentation selon les indications du fabricant de la sonde.
En cas d'administration orale, placez l'extrémité de la seringue dans la bouche de telle façon que la pointe touche l'intérieur d'une joue. Lors de l'administration à un enfant, veillez à ce que celui-ci soit assis bien droit.
Poussez lentement sur le piston jusqu'au bout pour administrer la dose complète.
Attention: l'administration directe de Tafinlar dans la gorge ou le fait de pousser trop vite sur le piston peut entraîner un étouffement.
|
|
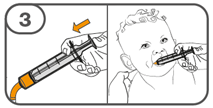
| |
Étape 4. Mettre environ 5 ml d'eau dans le gobelet doseur vide.
|
|
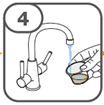
| |
Étape 5. Remuer pour dissoudre le reste.
|
|
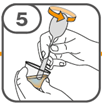
| |
Étape 6. Aspirer la solution.
|
|
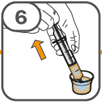
| |
Étape 7. Mettre la solution dans la sonde d'alimentation ou à l'intérieur de la joue.
|
|
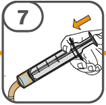
| |
Répéter 3 fois
La totalité des résidus de médicament doit être utilisée. Répétez les étapes 4 à 7 trois fois en tout afin d'administrer une dose complète.
| |
Étape 8. Une fois que vous avez répété les étapes 4 à 7 trois fois en tout, rincez la sonde d'alimentation avec de l'eau potable plate. Poursuivez ensuite avec les étapes de nettoyage de la section C.
| |
SECTION C. Nettoyage du gobelet doseur et de la seringue (s'ils ont été utilisés)
| |
UTILISER UNIQUEMENT DE L'EAU PROPRE POUR NETTOYER LE GOBELET DOSEUR. NE PAS UTILISER DE SAVON OU DE PRODUIT VAISSELLE POUR NETTOYER LE GOBELET DOSEUR.
| |
Étape 1. Rincez le gobelet doseur immédiatement après le dosage sous l'eau propre et fraîche.Secouez le gobelet pour enlever l'excédent d'eau et séchez-le avec une serviette en papier propre.
Remarque: toujours tenir le gobelet doseur à l'écart des ustensiles de cuisine.
|
|
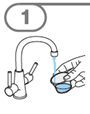
| |
Étape 2. Rincez la cuillère à café dans de l'eau propre et froide, puis lavez-la dans de l'eau chaude savonneuse et séchez-la avec une serviette en papier propre.Vous pouvez également laver la cuillère à café dans le lave-vaisselle.
|
|
| |
Étape 3. Nettoyez la seringue orale ou la seringue entérale (si elles ont été utilisées) selon les indications du fabricant.
|
|
| |
SECTION D. Comment éliminer les comprimés de Tafinlar périmés ou dont vous n'avez plus besoin, ou les vieux gobelets doseurs
| |
Éliminez tous les comprimés de Tafinlar dont la date de péremption est dépassée ou dont vous n'avez plus besoin de manière sûre.En cas de doute, demandez à votre médecin ou à votre pharmacien comment éliminer les comprimés de Tafinlar de manière sûre.
| |
SECTION E. Que dois-je faire si la solution buvable de Tafinlar entre en contact avec ma peau ou celle de mon enfant ou si elle pénètre dans les yeux? NETTOYAGE SUITE À UN RENVERSEMENT
| |
Si la solution de Tafinlar entre en contact avec votre peau ou celle de votre enfant, lavez soigneusement la zone concernée avec de l'eau et du savon.
Si la solution de Tafinlar pénètre dans vos yeux ou ceux de votre enfant, rincez soigneusement les yeux avec de l'eau froide.
Si vous avez renversé la solution buvable de Tafinlar, exécutez les étapes suivantes:
1.Mettez des gants en plastique.
2.Absorbez complètement la solution avec un matériel absorbant, par exemple avec des serviettes en papier imbibées soit d'un mélange d'eau et de désinfectant, soit d'éthanol à 70% (ou plus).
3.Répétez le nettoyage avec un matériel absorbant et fraîchement imbibé au moins trois fois jusqu'à ce que la zone soit propre.
4.Séchez la zone avec des serviettes en papier.
5.Mettez tous les matériels jetables ayant été utilisés pour nettoyer le liquide renversé dans un sachet en plastique refermable.
6.Éliminez le sachet conformément aux dispositions locales.
7.Lavez-vous soigneusement les mains à l'eau et au savon.
| |
Comment dois-je conserver Tafinlar
| |
·Conservez le flacon de Tafinlar de telle manière que les deux récipients en plastique se trouvent à l'intérieur et que le flacon soit bien refermé avec le capuchon. Les récipients maintiennent le médicament au sec et le protègent de l'humidité.
·Conservez le flacon et les gobelets doseurs dans l'emballage original.
·Conservez entre pas au-dessus de 30 °C.
·Conservez ce médicament hors de portée des enfants.
|
|